
A computational functional genomics based self-limiting self-concentration mechanism of cell specialization as a biological role of jumping genes†
Jörn
Lötsch
*ab and
Alfred
Ultsch
c
aInstitute of Clinical Pharmacology, Goethe-University, Theodor-Stern-Kai 7, 60590 Frankfurt am Main, Germany. E-mail: j.loetsch@em.uni-frankfurt.de; Fax: +49-69-6301-4354; Tel: +49-69-6301-4589
bFraunhofer Institute of Molecular Biology and Applied Ecology-Project Group Translational Medicine and Pharmacology (IME-TMP), Theodor-Stern-Kai 7, 60590 Frankfurt am Main, Germany
cDataBionics Research Group, University of Marburg, Hans-Meerwein-Straβe, D-35032 Marburg, Germany
First published on 10th December 2015
Abstract
Specialization is ubiquitous in biological systems and its manifold mechanisms are active research topics. Although clearly adaptive, the way in which specialization of cells is realized remains incompletely understood as it requires the reshaping of a cell's genome to favor particular biological processes in the competition on a cell's functional capacity. Here, a self-specialization mechanism is identified as a possible biological role of jumping genes, in particular LINE-1 retrotransposition. The mechanism is self-limiting and consistent with its evolutionary preservation despite its likely gene-breaking effects. The scenario we studied was the need for a cell to process a longer exposition to an extraordinary situation, for example continuous exposure to the nociceptive input or the intake of addictive drugs. Both situations may evolve toward chronification. The mechanism involves competition within a gene set in which a subset of genes cooperating in particular biological processes. The subset carries a piece of information, consisting of the LINE-1 sequence, about the destruction of their functional competitor genes which are not involved in that process. During gene transcription, an active copy of LINE-1 is co-transcribed. At a certain low probability, a subsequently transcribed and thus actually exposed gene can be rendered nonfunctional by LINE-1 retrotransposition in a relevant gene part. As retrotransposition needs time it is unlikely that LINE-1 retrotranspose into its own carrier gene. This reshapes the cell genome toward self-specializing of those biological processes that are carried out with a high number of LINE-1 containing genes. Self-termination of the mechanism is achieved by allowing LINE-1 to also occasionally jump into the coding region of itself, thus destroying the information about competitor destruction by successively decreasing the number of LINE-1 until the mechanism ceases. Employing a computational functional genomics approach, we demonstrate the biological plausibility in functional genomic datasets of potentially chronifying situations and interpret our results in relation to a biological mechanism of self-specialization of complex systems in response to a persistent challenge as met in chronifying traits.
Insight, innovation, integrationBiological systems such as cells adapt to persistent challenges by reshaping their genome toward more specialized biological processes needed to cope with the situation, for example during the development of chronic pain or addiction following persistent exposure to nociceptive inputs or drugs. The way in which specialization of cells is realized remains incompletely understood. This simulation proposes a role of jumping genes. It is demonstrated that sets of genes containing a substantial subset of genes carrying active LINE-1 retrotransposable elements implement a self-limiting self-concentration process, which over time decreases the number of active genes, however, more likely of those not carrying LINE-1, which finally favors biological processes that are carried out with a high number of LINE-1 containing genes. |
Introduction
Transposable elements (“jumping genes”) in the DNA1 form 45% of the human genome2 and have proliferated during the past 80 million years.3 They use a copy and paste mechanism4 and belong to the two major classes of DNA transposons and retrotransposons.5,6 Retrotransposons move via an RNA intermediate and are classified into LTR-containing retroviral like and non-LTR retrotransposons.7 Among the latter, long interspersed nuclear elements (LINE-1) are the only autonomously moving retrotransposons. They account for approximately 17% of the human genome2,4,8 of which the majority are nonfunctional although active LINE-1 is carried by a seventh of all human genes.9 The insertion of LINE-1 into active genes (Textbox 1) has often deleterious consequences, i.e., gene disruption or splicing defects10 that inhibit the transcription or valid translation of the affected gene. However, despite its potentially gene-breaking effect, LINE-1 has been evolutionary preserved. This suggests a substantial biological role that confers a survival advantage to its carrier. In the present work, a potential role of LINE-1 and its mechanism (Fig. 1), providing a possible motive for its evolutionary preservation, was investigated using a computer simulation (Fig. 2). The results suggested that LINE-1 is involved in a self-terminating gene competition mechanism that enables cell self-specialization toward biologically plausible consequences, as shown in datasets comprising functional genomics of chronifying traits.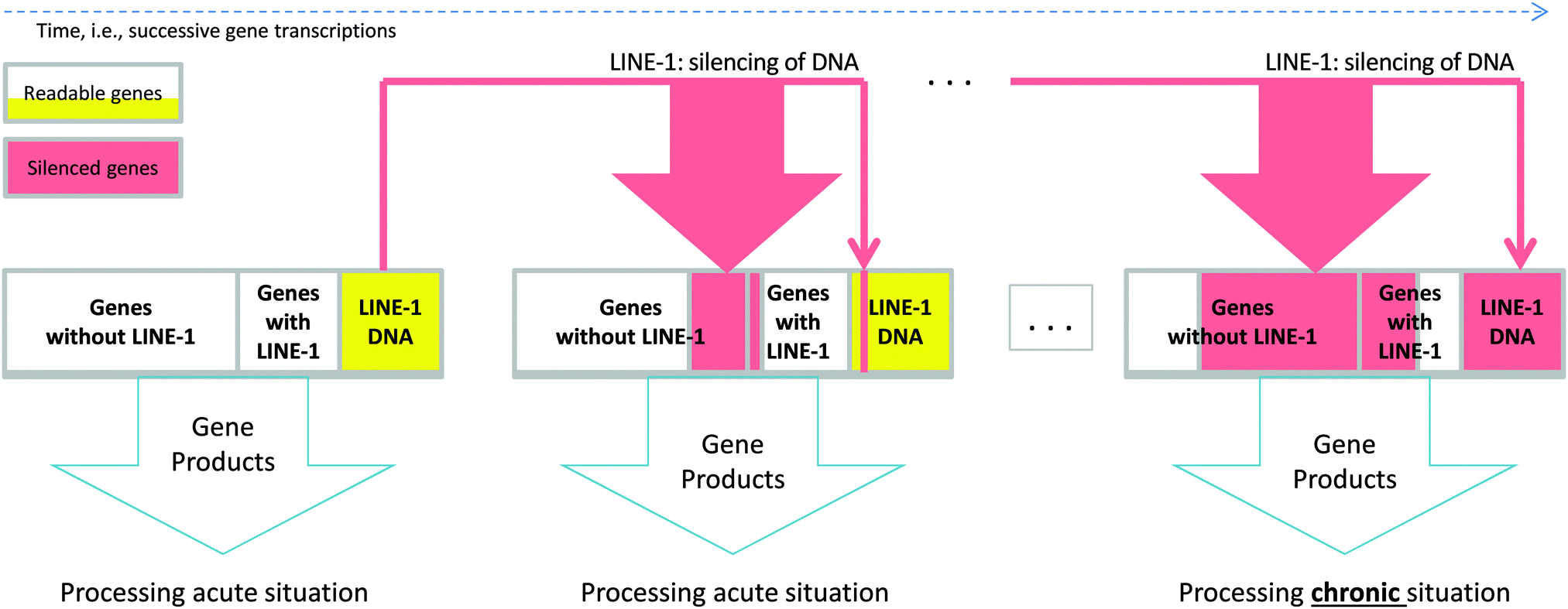 | ||
| Fig. 1 Schematic representation of the proposed mechanism showing how LINE-1 retrotransposition changes the processing of a biological situation. During gene transcription of a gene set, which is the cell's genetic response to a longer exposition to an extraordinary situation, an active copy of LINE-1 is co-transcribed (see the red arrows in Fig. 1). Subsequently transcribed and thus actually exposed genes can be rendered nonfunctional by LINE-1 retrotransposition (red areas in the next genome). Probabilities of LINE-1 of silencing genes are different for genes without LINE-1 since there are about 10 times more such genes in a given set than LINE-1 genes, and this is shown by the thickness of the red down arrows. As retrotransposition needs time it is unlikely that LINE-1 retrotranspose into its own carrier gene. Self-termination of the mechanism is achieved by allowing LINE-1 to also occasionally jump into the coding region of itself (represented as yellow areas). This ultimately terminates the mechanism, as can be seen in the rightmost genome in Fig. 1. Over time, shown from left to right, this leads to a self-specialization of the processes associated with the gene set from processing the acute situation towards dealing with the chronified situation. | ||
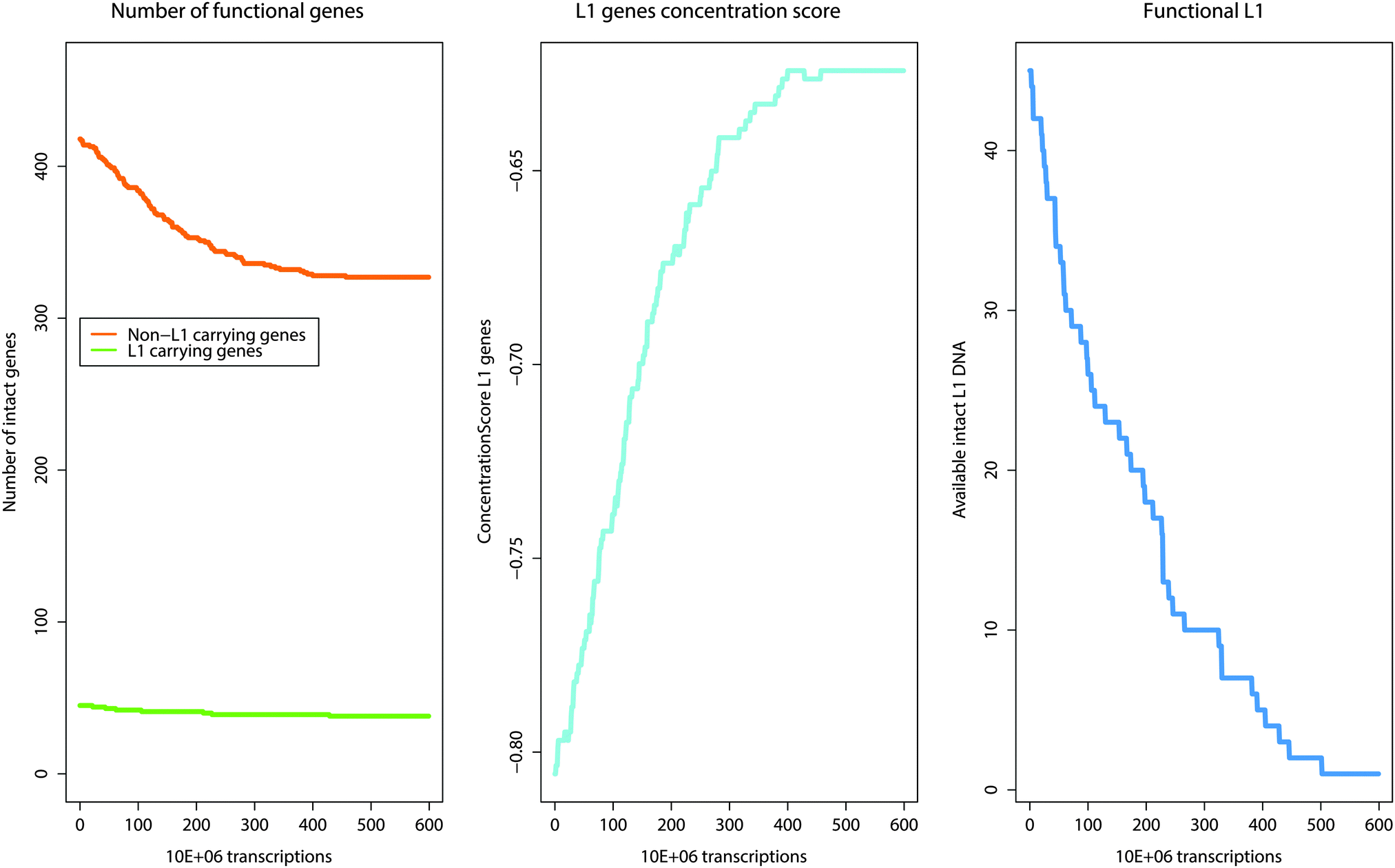 | ||
Fig. 2 Changes and shifts in the number of active genes that result over time from the activity of the self-limiting self-specialization mechanism of a cell's genome as a biological role of jumping genes. The example shown contains pain genes (n = 463)15,16 of which 45 carry putatively active LINE-1. The mechanism involves competition of a gene set in which a subset of genes cooperating in particular biological processes carries a piece of information, consisting of the LINE-1 sequence, about the destruction of their functional competitor genes not involved in that process. During gene transcription, an active copy of LINE-1 is co-transcribed. At a certain low probability, a subsequently transcribed and thus actually exposed gene can be rendered nonfunctional by LINE-1 retrotransposition in a relevant gene part. As retrotransposition needs time it is unlikely that LINE-1 retrotranspose into its own carrier gene. I.e., the original number of genes not carrying LINE-1 (left panel, green line) decreases much more than the number of LINE-1 carrying genes (left, green line). This reshapes the cells genome toward self-specializing toward those biological processes that are carried out with a high number of LINE-1 containing genes. This is described as the concentration score (middle panel, light blue line) of genes toward those that contain LINE-1 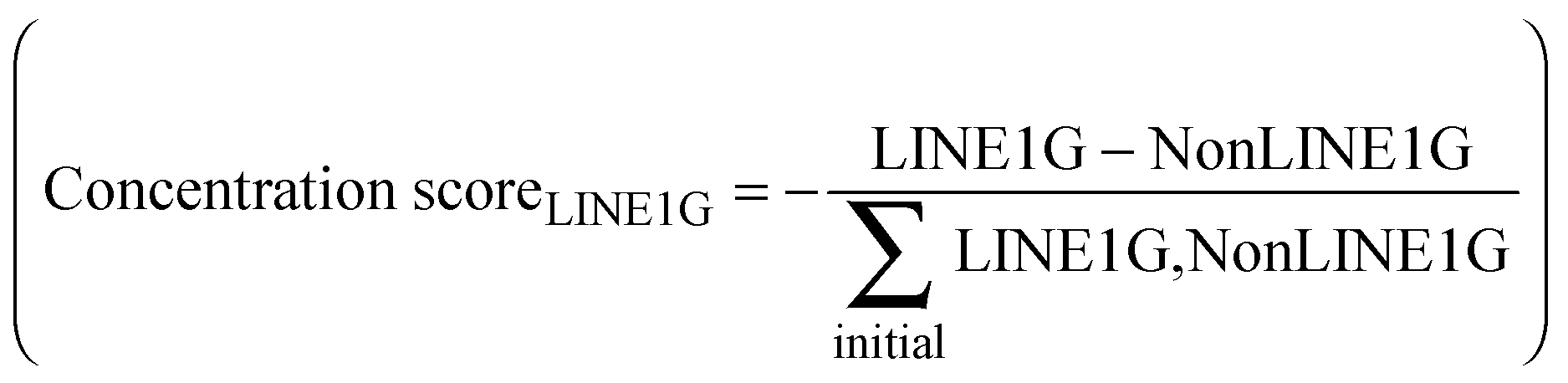 . Self-termination of the mechanism is achieved by allowing LINE-1 to also occasionally jump into the coding region of itself, thus destroying the information about competitor destruction by successively decreasing the number of LINE-1 until the mechanism ceases (right panel, blue line). . Self-termination of the mechanism is achieved by allowing LINE-1 to also occasionally jump into the coding region of itself, thus destroying the information about competitor destruction by successively decreasing the number of LINE-1 until the mechanism ceases (right panel, blue line). | ||
|
Textbox 1: Molecular mechanisms of LINE-1 retrotransposition.
LINE-1 has a length of approximately 6 kb and consists of a 5′ untranslated region (UTR), two open reading frames (ORF 1 and 2) and a 3′-UTR. The 5′ UTR contains an internal RNA polymerase II promoter that initiates the transcription of the LINE-1 element. In addition, it contains sense and, in addition, antisense promoters with binding motifs for transcription factors (e.g., the SRY-family or RUNX346). Following LINE-1 RNA transcription, mRNA is transported to the cytoplasm where the two LINE-1-encoded proteins ORF1 and ORF2 are translated, which are both required for complete LINE-1 retrotransposition. In detail, ORF1 seems to be important for the binding to nucleic acids47 and for LINE-1 integration,48 while ORF2 contains endonuclease13 and reverse transcriptase activities. The LINE-1 proteins form with the RNA ribonucleoprotein (RNP) complexes that are partly transported back to the nucleus. There, the ORF2 endonuclease domain nicks genomic DNA causing a free 3′-OH that serves as a primer for the reverse transcription of the LINE-1 RNA, a mechanism called “target-site primed reverse transcription” (TPRT). However, the processes required for second-strand cleavage, cDNA synthesis and the completion of LINE-1 integration still need more clarification.7 The remaining cytoplasmic RNPs are supposed to be hijacked by non-autonomous transposable elements (e.g. Alu) to mediate their mobilization.49 Depending on where they hit the DNA, the inserted LINE-1 elements may influence a gene in various ways, e.g., may disrupt the exonic sequence, induce missplicing or exon skipping and other often deleterious effects.4 |
Methods
Computer simulation of the mechanism by which jumping genes facilitate gene competition
A computer simulation of the proposed mechanism of LINE-1 mediated facilitation of gene competition was implemented using the LabVIEW® programming environment (version 2014 for Linux, National Instruments, Houston, TX, USA;11 for another simulation example, see ref. 12). LabVIEW® is a tool for data acquisition and processing available for multiple platforms including Windows, Mac OS X, and Linux. A LabVIEW® program consists of a front panel that contains graphical elements of the user interface, including controls and data visualizing elements (Fig. 3), and of a block diagram that contains the programming code (Fig. 4). The latter is created using the programming language “G” that employs a wiring analogy resembling an electrical circuit diagram, i.e., objects are connected graphically by drawing lines between specific connectors (Fig. 4). The abstract program code is not needed, which is a qualitative difference to classical programming languages such as C or FORTRAN. Available elements provide basic to advanced mathematical functions for data processing, of which for the present simulation sufficed basic arithmetic operations, random number generators [0…1] and numerical array structures. Program execution is controlled, for example, by while-loops, conditional if-then-else structures or sequences [1,2…n].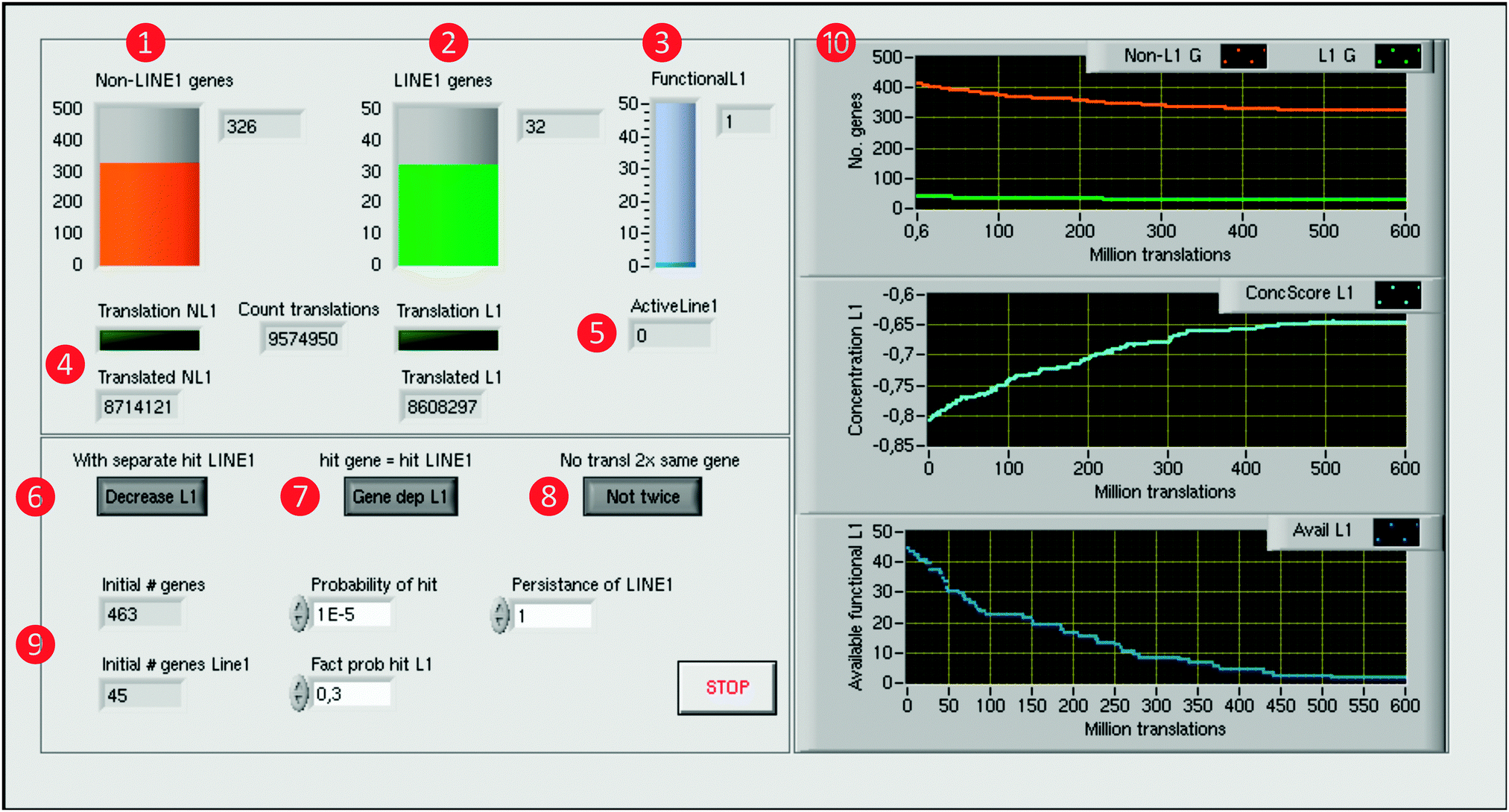 | ||
Fig. 3 Computer simulation of the self-limiting self-specialization mechanism of a cell's genome as a biological role of jumping genes. The example shown contains pain genes (n = 463)15,16 of which 45 carry putatively active LINE-1. The front panel of the LabVIEW-implemented simulation displays on the left side the numbers of genes, arbitrarily set probabilities and program control elements, and on the right side the results consisting of the time courses of gene numbers and the derived score. Specifically, on the left side, the numbers of functional pain genes ( orange), either containing LINE-1 or not ( orange), either containing LINE-1 or not ( green), and the number of still functional LINE-1 elements ( green), and the number of still functional LINE-1 elements ( blue) are displayed as containers with the numerical counts at the right (upper part, orange, green and blue, respectively). Below are indicators ( blue) are displayed as containers with the numerical counts at the right (upper part, orange, green and blue, respectively). Below are indicators ( ) of the translation of genes along with counters of the total number of gene translations and of currently active LINE-1 ( ) of the translation of genes along with counters of the total number of gene translations and of currently active LINE-1 ( ). At the lower part, control elements are available to modify details (switch on/off) of the simulation process, i.e., whether LINE-1 DNA can be separately hit without affecting the coding part of its carrier gene ( ). At the lower part, control elements are available to modify details (switch on/off) of the simulation process, i.e., whether LINE-1 DNA can be separately hit without affecting the coding part of its carrier gene ( ), whether or not when the coding gene part is hit by LINE-1 then the LINE-1 content of the gene, if present, will also be excluded from future transcription ( ), whether or not when the coding gene part is hit by LINE-1 then the LINE-1 content of the gene, if present, will also be excluded from future transcription ( ), and whether a just translated gene can immediately be translated again or first, another gene is translated before the gene can be retranslated ( ), and whether a just translated gene can immediately be translated again or first, another gene is translated before the gene can be retranslated ( ). Initial gene numbers and the chosen set of probabilities of hits by LINE-1 are displayed at the lower left part ( ). Initial gene numbers and the chosen set of probabilities of hits by LINE-1 are displayed at the lower left part ( ). The right side displays the results of the computer simulation as xy-graphs with the abscissa scaled for million translations ( ). The right side displays the results of the computer simulation as xy-graphs with the abscissa scaled for million translations ( ). From top to bottom is shown: (i) the time courses of the number of genes carrying active LINE-1 (green) or not carrying LINE-1 (orange), (ii) the concentration score (light blue) of genes toward those that contain LINE-1 and (iii) the number of available functional LINE-1 that when the carrying gene is translated can still retrotranspose (blue). ). From top to bottom is shown: (i) the time courses of the number of genes carrying active LINE-1 (green) or not carrying LINE-1 (orange), (ii) the concentration score (light blue) of genes toward those that contain LINE-1 and (iii) the number of available functional LINE-1 that when the carrying gene is translated can still retrotranspose (blue). | ||
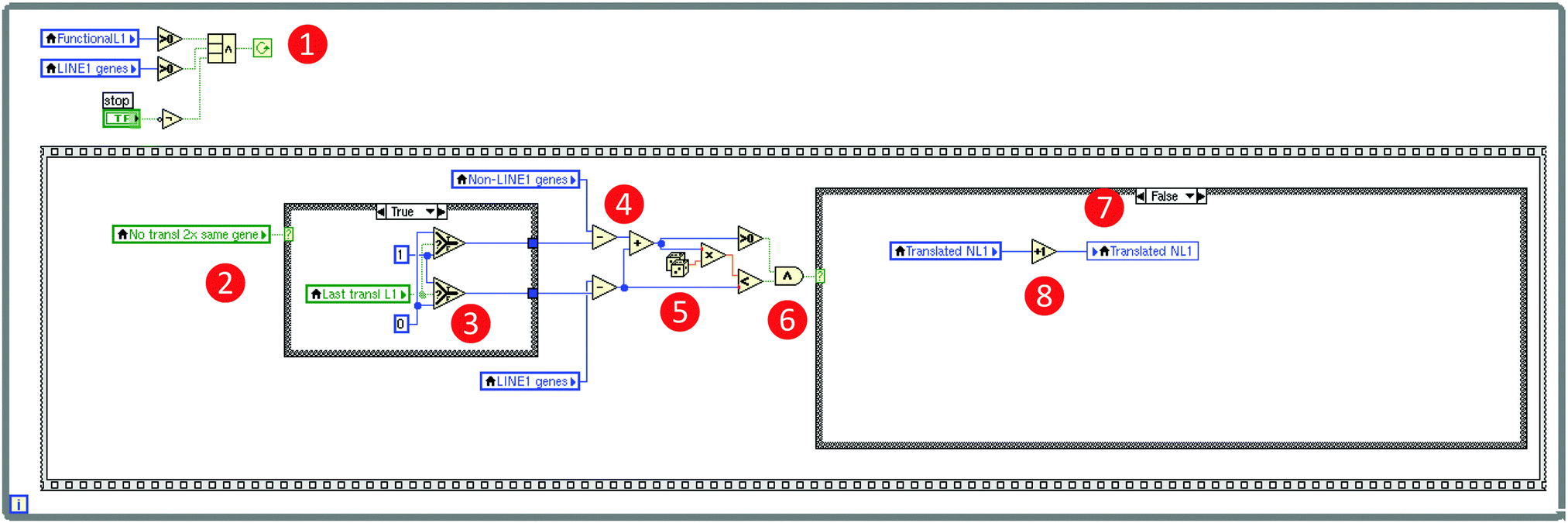 | ||
Fig. 4 Technical details of the computer simulation of the self-limiting self-specialization mechanism of a cell's genome as a biological role of jumping genes (Fig. 3). The figure shows a snapshot of a relevant part of the so-called block diagram of the LabVIEW based computer simulation, displaying the global mechanism to let the program run in loops (the frame structure surrounding the whole code) symbolizing gene translations and the implementation of the selection of either a LINE-1 carrying or a non-LINE-1 carrying gene for “transcription”. The programming is done by “wiring” pre-built elements provided by LabVIEW® resembling an electric circuit diagram as exemplified using the program code of the implementation of the selection of either a non-LINE-1 or a LINE-1 carrying gene for “transcription”. Specifically, the mechanism's loop runs as long as ( ) (i) LINE-1 carrying genes including functional retrotransposons are available and (logical operator) a “Stop” button has not been pressed on the program's front panel (Fig. 3). The inner frame is one of the successions and shows the mechanism if the condition is true that a just translated gene cannot be immediately translated again ( ) (i) LINE-1 carrying genes including functional retrotransposons are available and (logical operator) a “Stop” button has not been pressed on the program's front panel (Fig. 3). The inner frame is one of the successions and shows the mechanism if the condition is true that a just translated gene cannot be immediately translated again ( ), which reduces either the number of LINE-1 carrying genes or the number of not LINE-1 carrying genes (decision depending on the last translated gene ), which reduces either the number of LINE-1 carrying genes or the number of not LINE-1 carrying genes (decision depending on the last translated gene  ) by one (arithmetic operator ) by one (arithmetic operator  ). Which subgroup gene is translated is random (random number generator ). Which subgroup gene is translated is random (random number generator  ) and the probability is equal, i.e., depends on the ratio of the number of genes per subgroup. The example code shows a translation of a non-LINE-1 carrying gene because the random number was larger than the probability of translation of LINE-1 genes ( ) and the probability is equal, i.e., depends on the ratio of the number of genes per subgroup. The example code shows a translation of a non-LINE-1 carrying gene because the random number was larger than the probability of translation of LINE-1 genes ( ), i.e., the conditional structure ( ), i.e., the conditional structure ( ) was directed to perform the “false” condition and increased the number of translated non-LINE-1 genes by one ( ) was directed to perform the “false” condition and increased the number of translated non-LINE-1 genes by one ( ). The program is available as a download in the ESI† of this paper. ). The program is available as a download in the ESI† of this paper. | ||
The continuous cellular process of gene transcription was implemented by letting the program run in loops that recalculate the number of genes, separately for those carrying LINE-1 or not, the number of currently functional copies of LINE-1, and the number of still retrotransposable and LINE-1 that are still available for the mechanism, regardless of their current state of translation. Each loop has the meaning of the transcription of one gene. If a gene carrying active LINE-1 is transcribed, the latter is considered to also be transcribed at this occasion, which increases the pool of currently active LINE-1 retrotransposable elements by one. Each active LINE-1 is assigned to a “lifetime” consisting of the number of subsequent gene transcriptions during which the translated copy of LINE-1 can jump into a currently transcribed gene; in the present simulation this lifetime has been arbitrarily set at a value of 1. With also an arbitrarily set probability (p = 10−5), LINE-1 can jump into a relevant part of the gene actually transcribed during the lifetime of this LINE-1 element, which leads to the functional destruction to the gene. This was implemented by decreasing the number of LINE-1 containing or not LINE-1 containing (pain) genes by 1, depending on the group to which the currently transcribed gene belongs, which was randomly chosen at probabilities depending on the actual ratio of LINE-1 carrying to non-LINE-1 carrying genes.
However, the simulation was programmed so that LINE-1 cannot jump into the gene from which it has been transcribed as LINE-1 needs mRNA transport out of the nucleus, translation of its components, for example, ORF2 that contains the code for the endonuclease13 and confers reverse transcriptase activities, re-entering of the nucleus, which are time-consuming processes unlikely to occur during transcription of the gene from which the actual copy of LINE-1 originated. According the proposed mechanism, LINE-1 is able to jump only when the transcription and thus the exposure of its original gene has been ended. As this process would nevertheless result in the destruction of all genes over time, which is biologically not plausible as an evolutionary preserved mechanism, a self-limitation of the mechanism had to be implemented by creating the possibility that LINE-1 can also jump into a DNA sequence part that carries the genetic information of active LINE-1 in a currently transcribed gene. This was implemented by decreasing the number of LINE-1 by one, finally leading to the cessation of the LINE-1 mediated gene competition.
As LINE-1 can be anywhere in a gene and its relevant sequence is smaller than that of the average pain gene, the probability of this event can be a fraction of the probability of hitting a relevant functional part of a protein coding gene, which was implemented as a factor (> 0 and ≤ 1; arbitrarily set at 0.3) by which the aforementioned probability was multiplied. A further modulation of this process was implemented by alternatively allowing that the destruction of a LINE-1 containing gene could either stop the transcription of that gene including that of LINE-1, or the possibility of transcription of the included LINE-1 could continue, which merely changed the speed of the whole process without further qualitative consequences. Finally, the simulation stops when no transposable LINE-1 is left due to the self-limitation mechanism. The result of the gene-competition enhancing mechanism was measured as the so-called “concentration scorer of LINE-1 carrying genes” defined as: 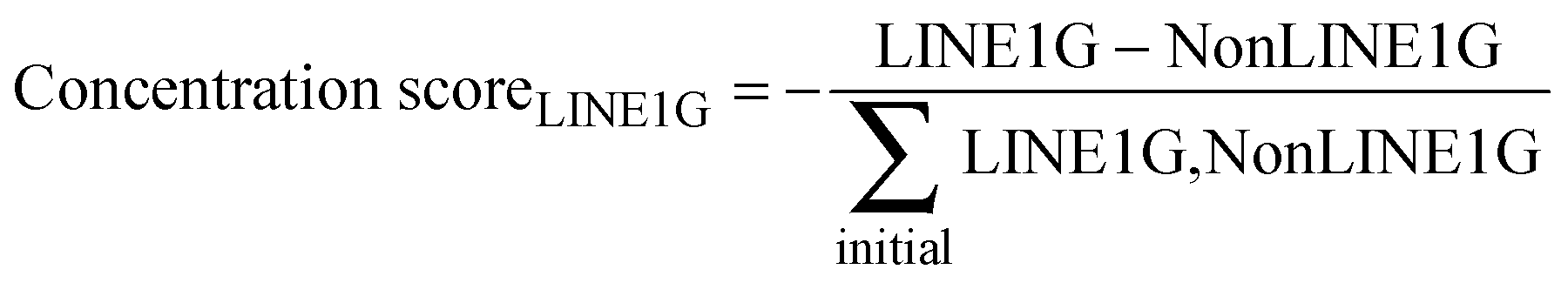 , where LINE1G and NonLINE1G denote the numbers of LINE-1 carrying or not LINE-1 carrying genes, respectively, (actual in the counter and initial in the denominator) that are functional.
, where LINE1G and NonLINE1G denote the numbers of LINE-1 carrying or not LINE-1 carrying genes, respectively, (actual in the counter and initial in the denominator) that are functional.
Data mining for functional genomics of traits
To assess the consequences of the proposed LINE-1 mechanism, topical sets of genes were analyzed for the occurrence of LINE-1. The information on LINE-1 carrying genes was available from a list of n = 1454 genes9 with intragenic insertions of full-length (>6000 bp) and putatively active LINE-1, i.e., containing regulatory elements. This was available from a previous publication where these genes had been identified using the L1Base database14 at http://line1.bioapps.biozentrum.uni-wuerzburg.de/L1base.php (supplementary table S1, ESI† of ref. 9).The consequences of the mechanism were studied for pain, which required a set of “pain genes”. This set is an updated version from,15 mainly based on the PainGenes Database.16 The pain genes can be considered as coding for important players in pain (referred to as “pain genes”). From the intersection of pain genes with the LINE-1 containing genes, a subset of n = 45 pain genes (Table 1) was identified to contain putatively active intragenomic LINE-1 on the basis of the L1Base database.14
| Pain genes | ||||
| ABCB1 | CHRNA7 | GCH1 | LYST | PLCL1 |
| ABCC4 | CXCL13 | GLRA2 | MAO A | PRKCA |
| AR | DAB1 | GLRA3 | MME | PRKG1 |
| ASIC2 | DISC1 | GRIA1 | NCAM1 | RELN |
| BDNF | DLG2 | GRM5 | NMU | SLC12A6 |
| CAMK2D | DTNBP1 | HCN1 | OPRM1 | SLC15A2 |
| CD38 | EDNRB | KCNJ3 | PCSK2 | SPTLC1 |
| CHRM2 | ESR1 | KCNK2 | PLCB1 | SV2B |
| CHRNA3 | GABBR2 | LEPR | PLCB4 | SYN2 |
| Addiction genes | ||||
| APP | GABRB1 | OPRM1 | RB1 | TMOD2 |
| BDNF | GRM5 | PCSK2 | RTN1 | |
| CHRNA7 | MAPK10 | PLCB1 | SCHIP1 | |
| CTNND2 | MEF2C | PRKCA | SYN2 | |
A second set of genes was identified containing n = 386 genes reported to underlie substance addiction.17 This trait was chosen as addiction has been reported to involve in LINE-1.18,19 Moreover, it shares some properties with chronic pain, such as the presence of a persistent stimulus triggering chronification that involves neuronal plasticity. An intersection with the LINE-1 containing gene set identified n = 21 LINE-1 containing genes within this set.
The biological roles of trait-associated gene sets
The biological roles of a gene set were queried from the Gene Ontology knowledgebase (GO; http://www.geneontology.org/).20 In the GO database the knowledge on the biological processes, the molecular function and the cellular components of genes is formulated using a controlled and clear defined vocabulary of GO terms that are annotated21 to the genes.22 Here, the GO category of biological processes was used. GO terms are related to each other by “is-a”, “part-of”, “regulates” and “subclass of” relationships forming a polyhierarchy that is organized in a directed acyclic graph (DAG,23 knowledge representation graph). In this graph, the polyhierarchy of GO terms starts at the root usually displayed at the top and containing terms with the broadest definition, and specializes toward the leaves. Leaves represent GO terms of the narrowest definition.Particular biological roles exerted the set of pain genes, among all human genes, were found by means of an over-representation analysis (ORA24) using the web-based tool GeneTrail25 (Table 2). This compared the occurrence (annotation) of a set of genes annotated at GO terms with the expected occurrence of all human genes at these terms. The significance of a GO term associated with a gene set was determined by means of a Fisher's exact test that calculated p-values for the GO terms. Subsequently, α correction for multiple testing was applied and only terms with a p-value lower than a preset threshold tp were considered as significant. The result was a representation of the complete knowledge on the biological processes of the gene set (complete DAG). Usually this DAG contains several hundred GO terms, called the ORA set. To transform this information into a more intelligible form, functional abstraction26 was applied. This identifies a special subset of GO terms, i.e., “functional areas”. Functional areas cover the entire knowledge in the complete DAG. Each functional area describes one particular aspect of the knowledge contained in the complete DAG at a maximum of coverage, certainty, information value and conciseness.26
| Site name | URL | Ref. |
|---|---|---|
| AmiGO (search utility for GO) | http://amigo.geneontology.org/ | 50 |
| Gene ontology (GO) | http://www.geneontology.org/ | 20 |
| Gene trail | http://genetrail.bioinf.uni-sb.de/ | 25 |
| HUGO gene nomenclature committee | http://www.genenames.org/ | 51 |
| L1Base database | http://line1.bioapps.biozentrum.uni-wuerzburg.de/L1base.php | 14 |
| Pain genes database | http://www.jbldesign.com/jmogil/enter.html | 16 |
| PubMed | http://www.ncbi.nlm.nih.gov/pubmed | |
| R software (version 3.0.2) | http://CRAN.R-project.org/ |
Excess or deficit of LINE-1 containing genes in the ORA term set
LINE-1 containing genes are a subset of the gene sets of pain and drug addiction. The percentage of LINE-1 containing genes with respect to the number of genes in a gene set annotated to a particular term was calculated. As expected,27,28 the distribution of this percentage followed a hypergeometric distribution. For a given percentage pT of LINE-1 containing genes within the set of genes annotated to a term T this allowed the calculation of p-values for pT as the probability prob(p > pT) for probabilities larger than the mean expected percentage and prob(p < pT) for probabilities smaller than the mean expected percentage. This allowed the identification of terms in the ORA set which contained either an excess or a deficit of LINE-1 carrying genes with respect to the statistical expectation. The result could be assigned to the above-mentioned functional areas.26 This provided precise information about those functional areas and subsumed GO terms that will be functionally favored and respectively neglected following the concentration toward LINE-1 containing genes.Results
A mechanism of LINE-1 facilitated genome self-specialization
The mechanism involves an advanced form gene competition in which a subset of genes cooperating in particular biological processes carries a piece of information, consisting of the LINE-1 sequence, about the destruction of their functional competitor genes not involved in that process. The mechanism proposes that if a set of genes is read over a longer period of time and contains a substantial subset of genes that carry active LINE-1 retrotransposable elements, then over time a preferential destruction of non-LINE-1 carrying genes takes place. In particular, when the subset of LINE-1 carrying genes is initially comparatively smaller by probabilistic reasoning more non-LINE-1 carrying genes than carrying genes will be destroyed. Thus favoring LINE-1 containing genes, the action of LINE-1 will finally lead to reshaping of the genome of individual cells toward a self-specialization29,30 in those biological processes that are carried out with a high number of LINE-1 containing genes.The biological mechanism is proposed as follows (Fig. 1). If a gene that contains an active copy of LINE-1 is transcribed, the retrotransposon can also be transcribed and thus retrotranspose. As insertion of LINE-1 into active genes is more likely to have deleterious functional consequences.10 At a certain low probability, a subsequently transcribed and thus actually exposed gene can be rendered nonfunctional by LINE-1 retrotransposition in a relevant gene part, as its DNA is exposed for transcription but also for retrotransposition. Since the latter needs time for translation of LINE-1, formation of the ORF2 coded the endonuclease13 and re-entering the nucleus, it is less likely that LINE-1 retrotranspose into its own carrier gene. Usually the number of non-LINE carrying genes is substantially bigger than the number of LINE-1 carrying genes in an active gene set. These two effects lead to a preference for LINE-1 mediated destruction of non-LINE carrying genes. As a consequence, this leads to a cell specialization toward LINE-1 containing genes and the biological processes exerted by this subset of genes. Self-termination of the mechanism is achieved by allowing LINE-1 to also occasionally jump into the coding region of itself, thus destroying the information about competitor destruction by successively decreasing the number of LINE-1 until the mechanism ceases. As the process is time consuming, i.e., many gene translations are needed to satisfy the low probability of retrotransposition into a relevant gene part, and the biologically plausible setting of the mechanism is the chronification of traits.
Simulation of the gene-competition mechanism
The proposed mechanism of LINE-1 mediated gene competition was functionally assessed in a computer simulation (Fig. 3). The number of genes is continuously recalculated separately either for those carrying LINE-1 or not carrying it. Transcription of a gene carrying active LINE-1 includes transcription of the latter increasing the pool of currently active LINE-1 retrotransposable elements by one. After a short while LINE-1 will be destroyed and this mechanistic feature also agrees with molecular evidence.31 However, during its active life span it can retrotranspose into a gene transcribed after translation of LINE-1 and, at an arbitrarily set probability of p = 10−5, hit a relevant part implemented by decreasing the number of active genes by 1 in the group of the currently transcribed gene. The simulation indeed resulted in more deactivated non-LINE-1 carrying genes than deactivated LINE-1 carrying genes. However, the simulation ended in a biologically implausible complete destruction of the cell's genome. This emphasized the need for implementing a self-limitation. This was simply achieved by allowing LINE-1 to also jump into the coding region of LINE-1, thus destroying itself and successively decreasing the number of LINE-1 until the mechanism ceases.The satisfactory model consisted of a self-limiting LINE-1-mediated gene competition mechanism. This leads to an asymptotically increasing ConcentrationScoreLINE1G that finally favored the genes containing functional LINE-1 (Fig. 3, right part). As a consequence, this will favor biological processes in which these genes are mainly involved. Hence, the cells undergo genomic re-programming toward a self-limiting self-specialization on a particular function(s). This mechanism of gene self-specialization on biological processes, predominately exerted by LINE-1 containing genes, represents an important biological mechanism provided that it can be shown that the role of the active genes changes toward a meaningful function for the cell.
Application of the mechanism to functional genomics of traits
The simulation showed that the proposed mechanism can efficiently enhance gene competition in settings where the cell is confronted with the need to process a persistent situation, for example continuous exposure to nociceptive inputs or to chemical noxes. According to the proposed mechanism, a self-specialization on biological processes associated with genes which carry particularly large proportions of LINE-1 will take place. If this implements an important biological mechanism these genes should be particularly needed for a longer lasting cellular challenge. To explore the biological role of the proposed LINE-1 mediated cell specialization, the nature of these processes was analyzed in the functional genomics of pain and also the intake of addictive drugs. These biological conditions share the presence of a persistent stimulus (nociceptive inputs, respectively chemical exposure).Over-representation analysis (ORA24) using a Bonferroni α-corrected p-value threshold, tp, of 0.05 resulted in 377 significant GO terms. Subsequent functional abstraction26 provided 12 terms (“functional areas”) as a comprehensive coverage of the biological functions of the pain genes (Table 3). Based on the content of LINE-1 carrying genes expected from the hypergeometric distribution and the number of pain genes annotated to a particular term, n = 23 terms with significantly more than expected LINE-1 carrying genes were found at p < 0.05 (Table 4). Regarding these terms within the functional areas showed that the largest subset belonged to the GO polyhierarchy of the functional area “nervous system development” (Fig. 5). Specifically, LINE-1 carrying pain genes were found more frequently than expected in GO terms “neuron development”, “regulation of neurogenesis”, “generation of neurons”, and “neurogenesis” (Table 4). This pointed to processes related to neuronal restructuring or neuroplasticity which seems to accommodate an interpretation such as pain chronification.32–34 By contrast, underrepresentation of LINE-1 carrying pain genes was found in the GO polyhierarchy of “transport” and comprises ion transport processes, that are for example more needed in acute signal transduction that according to the proposed model would be disfavored in a cell specialized toward chronification.
| GO term ID | Functional area (GO term of category biological process) | Expected no. of genes | Observed no. of genes | −Log 10 p-value |
|---|---|---|---|---|
| GO:0007399 | Nervous system development | 21 | 52 | 5.4 |
| GO:0007268 | Synaptic transmission | 8 | 53 | 26.2 |
| GO:0007610 | Behavior | 8 | 52 | 24 |
| GO:0040011 | Locomotion | 13 | 37 | 4.9 |
| GO:0009987 | Cellular process | 263 | 317 | 4.3 |
| GO:0006810 | Transport | 57 | 132 | 17.6 |
| GO:0007165 | Signal transduction | 58 | 152 | 28 |
| GO:0035295 | Tube development | 4 | 14 | 1.3 |
| GO:0002376 | Immune system process | 26 | 59 | 5.1 |
| GO:0046879 | Hormone secretion | 2 | 13 | 3.7 |
| GO term ID | GO term of category biological process | Expected no. of genes | Observed no. of genes in term | No. of INE-1 genes | Percent LINE-1 genes in term | p-Value |
|---|---|---|---|---|---|---|
| GO:0035094 | Response to nicotine | 0 | 5 | 2 | 40 | 0 |
| GO:0043279 | Response to alkaloid | 1 | 7 | 2 | 28.57 | 0 |
| GO:0014070 | Response to organic cyclic substance | 1 | 7 | 2 | 28.57 | 0 |
| GO:0006939 | Smooth muscle contraction | 1 | 11 | 3 | 27.27 | 0 |
| GO:0035095 | Behavioral response to nicotine | 0 | 4 | 1 | 25 | 0 |
| GO:0007200 | Activation of phospholipase C activity by G-protein coupled receptor protein signaling pathway coupled to IP3 second messenger | 1 | 14 | 3 | 21.43 | 0 |
| GO:0030595 | Leukocyte chemotaxis | 2 | 10 | 2 | 20 | 0 |
| GO:0048015 | Phosphoinositide-mediated signaling | 2 | 16 | 3 | 18.75 | 0 |
| GO:0060326 | Cell chemotaxis | 2 | 11 | 2 | 18.18 | 0 |
| GO:0050900 | Leukocyte migration | 2 | 11 | 2 | 18.18 | 0 |
| GO:0090257 | Regulation of muscle system process | 2 | 12 | 2 | 16.67 | 6.1 × 10−5 |
| GO:0042417 | Dopamine metabolic process | 0 | 6 | 1 | 16.67 | 6.1 × 10−5 |
| GO:0006584 | Catecholamine metabolic process | 0 | 6 | 1 | 16.67 | 6.1 × 10−5 |
| GO:0009712 | Catechol metabolic process | 0 | 6 | 1 | 16.67 | 6.1 × 10−5 |
| GO:0034311 | Diol metabolic process | 0 | 6 | 1 | 16.67 | 6.1 × 10−5 |
| GO:0048699 | Generation of neurons | 9 | 24 | 4 | 16.67 | 6.1 × 10 −5 |
| GO:0018958 | Phenol metabolic process | 1 | 6 | 1 | 16.67 | 6.1 × 10−5 |
| GO:0051899 | Membrane depolarization | 1 | 6 | 1 | 16.67 | 6.1 × 10−5 |
| GO:0022008 | Neurogenesis | 9 | 24 | 4 | 16.67 | 6.1 × 10 −5 |
| GO:0050767 | Regulation of neurogenesis | 3 | 12 | 2 | 16.67 | 6.1 × 10 −5 |
| GO:0048666 | Neuron development | 6 | 19 | 3 | 15.79 | 0.000582 |
| GO:0007399 | Nervous system development | 21 | 52 | 8 | 15.38 | 0.000582 |
| GO:0006813 | Potassium ion transport | 2 | 13 | 2 | 15.38 | 0.000582 |
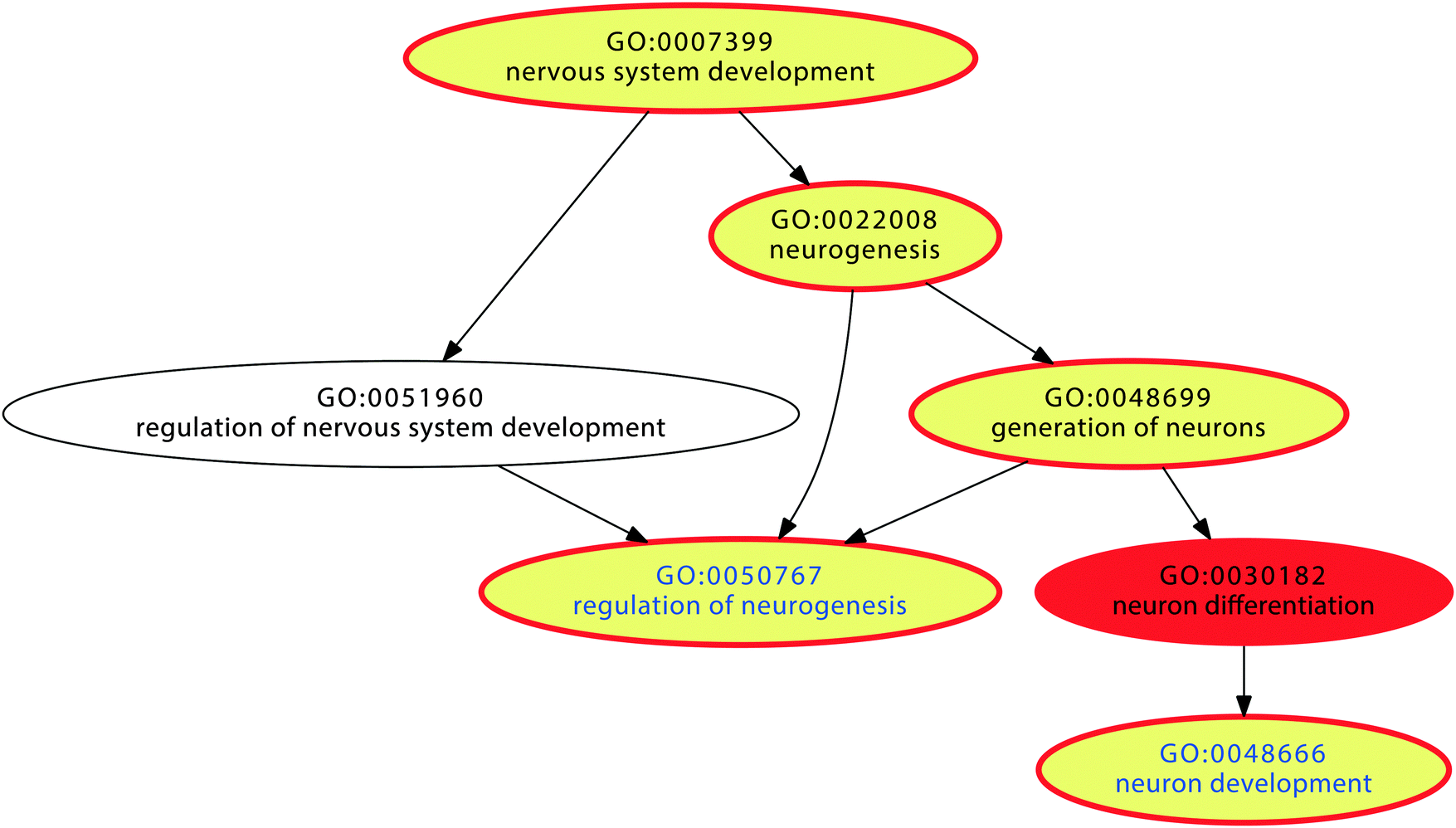 | ||
| Fig. 5 Graphical representation of the significant Gene Ontology (GO) terms of the GO category “biological process” (red). Non-significant GO terms are shown in uncolored circles. The polyhierarchy of GO terms assigned to those pain genes that are annotated with the functional area26 of “nervous system development” is shown. Terms containing significantly (at p < 0.05) more than expected LINE-1 carrying genes are shown as yellow colored circles. The vertical succession reflects the detailization of the terms in the GO polyhierarchy. | ||
| GO term ID | GO term of category biological process | Expected no. of genes | Observed no. of genes in term | No. of LINE-1 genes | Percent LINE-1 genes in term | Log p-value |
|---|---|---|---|---|---|---|
| GO:0051641 | Cellular localization | 26 | 74 | 3 | 4.054054 | 12.6 |
| GO:0006810 | Transport | 61 | 125 | 4 | 3.2 | 12 |
| GO:0000902 | Cell morphogenesis | 9 | 36 | 2 | 5.555556 | 8.1 |
| GO:0008283 | Cell proliferation | 25 | 73 | 4 | 5.479452 | 12.8 |
| GO:0008219 | Cell death | 27 | 86 | 2 | 2.325581 | 18.6 |
| GO:0007399 | Nervous system development | 25 | 85 | 2 | 2.352941 | 19.8 |
| GO:0023052 | Signaling | 76 | 188 | 8 | 4.255319 | 34.2 |
| GO:0007610 | Behavior | 11 | 67 | 5 | 7.462687 | 30.6 |
| GO:0050896 | Response to stimulus | 80 | 176 | 5 | 2.840909 | 24.3 |
| GO:0050789 | Regulation of biological process | 140 | 244 | 7 | 2.868852 | 23.4 |
Discussion
In this paper we present a self-limiting self-concentration process that provides a plausible explanation of the biological role of the so far incompletely understood jumping genes, i.e., of the function of LINE-1 retrotransposition accommodating its evolutionary preservation in the genome. The mechanism bears the potential of reshaping a cell's genome toward biological processes required for the response to persistently present environmental conditions. In this mechanism, self-concentration is achieved by implementing a competitive element, here in a gene set, that favors a subset of elements or genes which cooperate in the achievement of a particular (biological) process. The mechanism runs, including its self-limitation, without the need for any external influence. However, it can be modulated as implemented in the simulation by, for example, modulators of the probability of hits or of the life-span of the competitive elements.The starting point of the simulation was the need for a cell to process a given extraordinary situation, for example, the necessity to process exposure to nociceptive input or to addictive drugs. Furthermore, the situation should be persisting. The genetic response of a cell to such a situation is the transcription of a specific set of genes which allows the cell to cope with the particular situation. The computer simulation was restricted to the mechanism of hitting genes, either those not carrying LINE-1, or those carrying LINE-1, or the LINE-1 genetic code itself, without any further implementation of a possible outcome. The mechanism involves an advanced form gene competition in which a subset of genes cooperating in particular biological processes carries a piece of information, consisting of the LINE-1 sequence, about the destruction of their functional competitor genes not involved in that process. A main result was that after a certain period of time the cell's genome specializes. This means that the number of genes not containing LINE-1 was substantially reduced favoring the fraction of LINE-1 containing genes, which was only slightly affected. As a consequence, the response of the cell to a longer lasting condition will emphasize the biological processes corresponding to the LINE-1 carrying genes and suppress the biological processes corresponding to the genes that do not carry LINE-1.
The self-specialization mechanism needs time to outplay its effects and is therefore likely to play its role in trait chronification, i.e., the putative role of jumping genes is self-specialization of the genetic response to a longer lasting situation by providing useful functions for the organism. In pain and drug exposure, following an immediate response a persistent condition has to be handled if the exposure, i.e., nociceptive input or drug intake, persists. It turned out that in both traits the processes to which the cells specialize, i.e., those containing more LINE-1 carrying genes than expected by chance, are mainly concerned with restructuring of neural networks in the brain. Estimation of the real time in which this process finishes as indicated by the curves in Fig. 2 has to be regarded with caution as in the present approach several assumptions of probabilities, such as those of how likely a LINE-1 element retrotranspose in a functionally relevant exomic part of an active gene were made arbitrarily. The present simulation came close to a stable state of the process after approximately 200 million gene expressions. When assuming a transcription rate of up to 70 nucleotides per second in humans (see Table 1 in;35http://book.bionumbers.org/what-is-faster-transcription-or-translation/), 183![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 960000 genes would be transcribed in a year. This is not an implausible duration for the chronification of pain, and adjusting the probabilities slightly would easily provide a shorter period of perhaps not less than half a year. The estimated timelines agree with the clinical definition of chronic pain for which intervals from 3–12 months from onset are recognized time estimates (e.g., http://https://en.wikipedia.org/wiki/Chronic_pain).
960000 genes would be transcribed in a year. This is not an implausible duration for the chronification of pain, and adjusting the probabilities slightly would easily provide a shorter period of perhaps not less than half a year. The estimated timelines agree with the clinical definition of chronic pain for which intervals from 3–12 months from onset are recognized time estimates (e.g., http://https://en.wikipedia.org/wiki/Chronic_pain).
Once the stable state of the process, i.e., chronification, has been reached, the cell specialization toward chronification can be considered as established and the mechanism plays no further active role. This agrees with the clinical experience. This convergence to a stable state required the implementation of a self-limiting component in the proposed mechanism, which prevented its continuation toward the extinction of all active genes. Indeed, simulations with deactivated self-limiting components mostly ended in a number of active genes close to or equaling zero, which contrasts with the pathophysiological state of disease chronification that requires active cells. Thus, the inclusion of active LINE-1 segments as possible targets of the retrotransposition as a self-limiting mechanism leads to biologically more plausible results. The absence of this self-limitation would be possible, however, under the scenario of a far slower gene transcription rate to avoid extinction of the cell's active genome, which, while remaining a possibility would require the additional assumption of a protection of active LINE-1 segments from retrotransposition and would contrasts to the rather realistic temporal scenario as elaborated in the previous paragraph.
A mechanistic involvement of LINE-1 in gene regulation and in chronification of pain would be supported by several lines of molecular evidence. DNA demethylation including that of transposons has been proposed to be involved in the epigenetic regulation controlling the dosage of active genes36 and hypomethylated intragenic LINE-1s are nuclear cis-regulatory elements shown to repress genes.9 Moreover, roles of LINE-1 in pain or addiction are supported by molecular evidence including its role in neuronal reshaping,37 its presumed transcription in neural progenitor cells,38 the reported genomic change of individual neurons29 leading to somatic mosaicism in the nervous system30 following its release from epigenetic suppression,39 the reported association of retrotransposition with DNA hypermethylation,9 which is met in neuropathic pain40 and the reduced LINE-1 histone methylation in the N. accumbens following cocaine exposure,19 the established role of neural plasticity in chronic pain41 to which epigenetic mechanisms are recognized as important contributors,42 and the critical importance of the brain-derived neurotrophic factor (BDNF) for adult neurogenesis,43,44 which is coded by a LINE-1 containing pain and addiction gene (Table 1).
The reproducible involvement in trait chronification suggests possible LINE-1 related drug targets of a novel class of analgesics against chronification of pain. Pending toxicity assessments, LINE-1 or related enzymes including the ORF2 (LINE-1's open reading frame 2, see Textbox 1) coded endonuclease or APOBEC3A (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3A) involved in anti-LINE-1 defense31 potentially as such targets. The derived drugs may serve as preventive treatments of the development of chronic pain when the causative process persists, for which currently only a few substances such as tricyclic antidepressants of calcium channel modulators are available. Moreover, the mechanism seems not to be restricted to the chosen example traits. An exploratory analysis of a chronic lymphatic leukemia gene set identified apoptosis as among processes with underrepresented LINE-1 (details not shown), which is consistent with both, modified apoptosis of cancer cells and a reported role of LINE-1 in the genetic regulation of cancer cells.9
Taking these generalization considerations a step further, an ORA of all 1454 genes9 with intragenic insertions of full-length (>6000 bp) and putatively active LINE-1 gains all human genes known to the GO database, applying a p-value threshold of 0.02 and false discovery rate (FDR) correction followed by functional abstraction to identify the main relevant biological processes exerted by a given set of genes resulting in “learning and memory” (GO:0007611) and “neuron development” (GO:0048666) as the main processes in which functional LINE-1 carrying genes were overrepresented, i.e., more frequently found that expected from a random set of 1454 genes. Importantly, using a random set of n = 1454 human genes for an ORA with the same statistical parameters, no significant GO term emerged. This verifies the specificity of the present findings for the roles of LINE-1 in pain and addiction and at the same time strongly hints at a general biological role of LINE-1 retrotransposition in cell specialisation during biological processes that can be interpreted as indicating chronification. Indeed, leaning and neuronal plasticity are recognised features of chronifying pain.32
The present mechanism of a LINE-1 mediated cell specialization is proposed as a possible motive of the evolutionary preservation of active functional LINE-1 elements in 1454 genes across the human genome. It implies a few assumptions such as (i) the localization of LINE-1 within a subset of genes that are not particularly needed under general lenient conditions, hence having less effect on the biological fitness of individuals. This requires that (ii) LINE-1 mainly affects genes that are predominately needed under certain non-lenient conditions, i.e., the genes that host LINE-1 have been already specialized in certain functions. While these points have been shown, by means of ORA, to possibly apply to the traits of chronification of pain or addiction, they also emphasize that the proposed mechanism addresses the reshaping of an actual living cell toward more specialized functions, which can only function after a precedent evolutionary process has led to the selection of preserved LINE-1 segments in genes that meet the above criteria. In this respect, the proposed mechanism provides an evolutionary reason for this development, but should not be understood as the evolutionary mechanism toward the selection of genes carrying LINE-1. A further assumption (iii) was the biological advantage conferred by the LINE-1 preservation in certain genes. An alternative model could imply a population of organisms in which some may have LINE-1 in important genes, such individuals being deleterious in nature, and other individuals having this transposon in non-important genes, with the latter individuals not being affected by the presence of LINE-1 genes. This would exceed the present focus on the development of a cell specialization mechanism. This limitation, however, does not hamper the functionality of the proposed mechanism. Establishing a possible evolutionary advantage in humans, it requires future research to analyze the distribution of LINE-1 containing genes, including comparative analyses in other organisms with different population sizes, as, for example, Drosophila melanogaster, to further explore the generic role of such a genetic mechanism.
LINE-1 retrotransposons have been proposed as modulators of quantity and quality of mammalian gene expression 7. We present databionics, i.e., data processing methods learned from biology, a derived biologically plausible mechanism of this role that regulates both, the gene quantity as shown in the computer simulation as well as the gene quality as shown in the systems biology analyses. This resulted in the clear evidence that particular functional areas representing aspects of biological processes are favored or disfavored, providing a possible motive for the evolutionary preservation of LINE-1 across the (human) genome. Overall, we present a self-limiting mechanism of self-specialization that is important both to explain a fundamental role of retrotransposition as an evolutionary preserved property of the genome, as well as to advance the field of databionics based artificial intelligence.45 The latter is a generalization which implies that a subset of elements contains information that initiates a mechanism that preferentially destroys their competitors. It is self-limiting by also slowly destroying the necessary information itself, resulting in a reshaping of the system toward more specialized tasks.
Funding
This work has been funded by the Landesoffensive zur Entwicklung wissenschaftlich-ökonomischer Exzellenz (LOEWE), Schwerpunkt: Anwendungsorientierte Arzneimittelforschung (JL) and in addition, by the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 602919 (JL, GLORIA). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.Conflict of interest statement
The authors have declared that no further conflicts of interest exist.References
- B. McClintock, Proc. Natl. Acad. Sci. U. S. A., 1950, 36, 344–355 CrossRef CAS.
- E. S. Lander, L. M. Linton, B. Birren, C. Nusbaum, M. C. Zody, J. Baldwin, K. Devon, K. Dewar, M. Doyle, W. FitzHugh, R. Funke, D. Gage, K. Harris, A. Heaford, J. Howland, L. Kann, J. Lehoczky, R. LeVine, P. McEwan, K. McKernan, J. Meldrim, J. P. Mesirov, C. Miranda, W. Morris, J. Naylor, C. Raymond, M. Rosetti, R. Santos, A. Sheridan, C. Sougnez, N. Stange-Thomann, N. Stojanovic, A. Subramanian, D. Wyman, J. Rogers, J. Sulston, R. Ainscough, S. Beck, D. Bentley, J. Burton, C. Clee, N. Carter, A. Coulson, R. Deadman, P. Deloukas, A. Dunham, I. Dunham, R. Durbin, L. French, D. Grafham, S. Gregory, T. Hubbard, S. Humphray, A. Hunt, M. Jones, C. Lloyd, A. McMurray, L. Matthews, S. Mercer, S. Milne, J. C. Mullikin, A. Mungall, R. Plumb, M. Ross, R. Shownkeen, S. Sims, R. H. Waterston, R. K. Wilson, L. W. Hillier, J. D. McPherson, M. A. Marra, E. R. Mardis, L. A. Fulton, A. T. Chinwalla, K. H. Pepin, W. R. Gish, S. L. Chissoe, M. C. Wendl, K. D. Delehaunty, T. L. Miner, A. Delehaunty, J. B. Kramer, L. L. Cook, R. S. Fulton, D. L. Johnson, P. J. Minx, S. W. Clifton, T. Hawkins, E. Branscomb, P. Predki, P. Richardson, S. Wenning, T. Slezak, N. Doggett, J. F. Cheng, A. Olsen, S. Lucas, C. Elkin, E. Uberbacher, M. Frazier, R. A. Gibbs, D. M. Muzny, S. E. Scherer, J. B. Bouck, E. J. Sodergren, K. C. Worley, C. M. Rives, J. H. Gorrell, M. L. Metzker, S. L. Naylor, R. S. Kucherlapati, D. L. Nelson, G. M. Weinstock, Y. Sakaki, A. Fujiyama, M. Hattori, T. Yada, A. Toyoda, T. Itoh, C. Kawagoe, H. Watanabe, Y. Totoki, T. Taylor, J. Weissenbach, R. Heilig, W. Saurin, F. Artiguenave, P. Brottier, T. Bruls, E. Pelletier, C. Robert, P. Wincker, D. R. Smith, L. Doucette-Stamm, M. Rubenfield, K. Weinstock, H. M. Lee, J. Dubois, A. Rosenthal, M. Platzer, G. Nyakatura, S. Taudien, A. Rump, H. Yang, J. Yu, J. Wang, G. Huang, J. Gu, L. Hood, L. Rowen, A. Madan, S. Qin, R. W. Davis, N. A. Federspiel, A. P. Abola, M. J. Proctor, R. M. Myers, J. Schmutz, M. Dickson, J. Grimwood, D. R. Cox, M. V. Olson, R. Kaul, N. Shimizu, K. Kawasaki, S. Minoshima, G. A. Evans, M. Athanasiou, R. Schultz, B. A. Roe, F. Chen, H. Pan, J. Ramser, H. Lehrach, R. Reinhardt, W. R. McCombie, M. de la Bastide, N. Dedhia, H. Blocker, K. Hornischer, G. Nordsiek, R. Agarwala, L. Aravind, J. A. Bailey, A. Bateman, S. Batzoglou, E. Birney, P. Bork, D. G. Brown, C. B. Burge, L. Cerutti, H. C. Chen, D. Church, M. Clamp, R. R. Copley, T. Doerks, S. R. Eddy, E. E. Eichler, T. S. Furey, J. Galagan, J. G. Gilbert, C. Harmon, Y. Hayashizaki, D. Haussler, H. Hermjakob, K. Hokamp, W. Jang, L. S. Johnson, T. A. Jones, S. Kasif, A. Kaspryzk, S. Kennedy, W. J. Kent, P. Kitts, E. V. Koonin, I. Korf, D. Kulp, D. Lancet, T. M. Lowe, A. McLysaght, T. Mikkelsen, J. V. Moran, N. Mulder, V. J. Pollara, C. P. Ponting, G. Schuler, J. Schultz, G. Slater, A. F. Smit, E. Stupka, J. Szustakowski, D. Thierry-Mieg, J. Thierry-Mieg, L. Wagner, J. Wallis, R. Wheeler, A. Williams, Y. I. Wolf, K. H. Wolfe, S. P. Yang, R. F. Yeh, F. Collins, M. S. Guyer, J. Peterson, A. Felsenfeld, K. A. Wetterstrand, A. Patrinos, M. J. Morgan, J. Szustakowki, P. de Jong, J. J. Catanese, K. Osoegawa, H. Shizuya, S. Choi and Y. J. Chen, Nature, 2001, 409, 860–921 CrossRef CAS PubMed.
- A. F. Smit, G. Toth, A. D. Riggs and J. Jurka, J. Mol. Biol., 1995, 246, 401–417 CrossRef CAS PubMed.
- C. R. Beck, J. L. Garcia-Perez, R. M. Badge and J. V. Moran, Annu. Rev. Genomics Hum. Genet., 2011, 12, 187–215 CrossRef CAS PubMed.
- R. Cordaux and M. A. Batzer, Nat. Rev. Genet., 2009, 10, 691–703 CrossRef CAS PubMed.
- D. C. Hancks and H. H. Kazazian, Jr., Curr. Opin. Genet. Dev., 2012, 22, 191–203 CrossRef CAS PubMed.
- J. S. Han and J. D. Boeke, BioEssays, 2005, 27, 775–784 CrossRef CAS PubMed.
- K. Kaer and M. Speek, Gene, 2013, 518, 231–241 CrossRef CAS PubMed.
- C. Aporntewan, C. Phokaew, J. Piriyapongsa, C. Ngamphiw, C. Ittiwut, S. Tongsima and A. Mutirangura, PLoS One, 2011, 6, e17934 CAS.
- T. Singer, M. J. McConnell, M. C. Marchetto, N. G. Coufal and F. H. Gage, Trends Neurosci., 2010, 33, 345–354 CrossRef CAS PubMed.
- P. A. Blume, The LabVIEW Style Book, Prentice Hall, Upper Saddle River, NJ, USA, 2007 Search PubMed.
- J. Lötsch, G. Kobal and G. Geisslinger, Int. J. Clin. Pharmacol. Ther., 2004, 42, 15–22 CrossRef.
- Q. Feng, J. V. Moran, H. H. Kazazian, Jr. and J. D. Boeke, Cell, 1996, 87, 905–916 CrossRef CAS PubMed.
- T. Penzkofer, T. Dandekar and T. Zemojtel, Nucleic Acids Res., 2005, 33, D498–D500 CrossRef CAS PubMed.
- J. Lötsch, A. Doehring, J. S. Mogil, T. Arndt, G. Geisslinger and A. Ultsch, Pharmacol. Ther., 2013, 139, 60–70 CrossRef PubMed.
- M. L. Lacroix-Fralish, J. B. Ledoux and J. S. Mogil, Pain, 2007, 131(1–2), 3.e1–e4 CrossRef PubMed.
- C.-Y. Li, X. Mao and L. Wei, PLoS Comput. Biol., 2008, 4, e2 Search PubMed.
- A. Doehring, B. G. Oertel, R. Sittl and J. Lötsch, J. Pain, 2013, 154, 15–23 CrossRef CAS PubMed.
- I. Maze, J. Feng, M. B. Wilkinson, H. Sun, L. Shen and E. J. Nestler, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3035–3040 CrossRef CAS PubMed.
- M. Ashburner, C. A. Ball, J. A. Blake, D. Botstein, H. Butler, J. M. Cherry, A. P. Davis, K. Dolinski, S. S. Dwight, J. T. Eppig, M. A. Harris, D. P. Hill, L. Issel-Tarver, A. Kasarskis, S. Lewis, J. C. Matese, J. E. Richardson, M. Ringwald, G. M. Rubin and G. Sherlock, Nat. Genet., 2000, 25, 25–29 CrossRef CAS PubMed.
- E. Camon, M. Magrane, D. Barrell, D. Binns, W. Fleischmann, P. Kersey, N. Mulder, T. Oinn, J. Maslen, A. Cox and R. Apweiler, Genome Res., 2003, 13, 662–672 CrossRef CAS PubMed.
- E. Camon, M. Magrane, D. Barrell, V. Lee, E. Dimmer, J. Maslen, D. Binns, N. Harte, R. Lopez and R. Apweiler, Nucleic Acids Res., 2004, 32, D262–D266 CrossRef CAS PubMed.
- K. Thulasiraman and M. N. S. Swamy, Graphs: theory and algorithms, Wiley, New York, 1992 Search PubMed.
- C. Backes, A. Keller, J. Kuentzer, B. Kneissl, N. Comtesse, Y. A. Elnakady, R. Muller, E. Meese and H. P. Lenhof, Nucleic Acids Res., 2007, 35, W186–W192 CrossRef PubMed.
- A. Keller, C. Backes, M. Al-Awadhi, A. Gerasch, J. Kuntzer, O. Kohlbacher, M. Kaufmann and H. P. Lenhof, BMC Bioinf., 2008, 9, 552 CrossRef PubMed.
- A. Ultsch and J. Lötsch, PLoS One, 2014, 9, e90191 Search PubMed.
- P. Khatri and S. Draghici, Bioinformatics, 2005, 21, 3587–3595 CrossRef CAS PubMed.
- C. Ju, Phd thesis, BA, Scripps, 2011.
- A. R. Muotri, V. T. Chu, M. C. Marchetto, W. Deng, J. V. Moran and F. H. Gage, Nature, 2005, 435, 903–910 CrossRef CAS PubMed.
- J. K. Baillie, M. W. Barnett, K. R. Upton, D. J. Gerhardt, T. A. Richmond, F. De Sapio, P. M. Brennan, P. Rizzu, S. Smith, M. Fell, R. T. Talbot, S. Gustincich, T. C. Freeman, J. S. Mattick, D. A. Hume, P. Heutink, P. Carninci, J. A. Jeddeloh and G. J. Faulkner, Nature, 2011, 479, 534–537 CrossRef CAS PubMed.
- S. R. Richardson, I. Narvaiza, R. A. Planegger, M. D. Weitzman and J. V. Moran, eLife, 2014, 3, e02008, DOI:10.7554/eLife.02008.
- A. R. Mansour, M. A. Farmer, M. N. Baliki and A. V. Apkarian, Restor. Neurol. Neurosci., 2014, 32, 129–139 CAS.
- T. J. Price and K. E. Inyang, Prog. Mol. Biol. Transl. Sci., 2015, 131, 409–434 Search PubMed.
- A. May, J. Pain, 2008, 137, 7–15 CrossRef PubMed.
- R. Milo and R. Philips, Cell Biology by the Numbers, 2015, http://book.bionumbers.org Search PubMed.
- S. Preuss and C. S. Pikaard, Biochim. Biophys. Acta, 2007, 1769, 383–392 CrossRef CAS PubMed.
- N. G. Coufal, J. L. Garcia-Perez, G. E. Peng, G. W. Yeo, Y. Mu, M. T. Lovci, M. Morell, K. S. O'Shea, J. V. Moran and F. H. Gage, Nature, 2009, 460, 1127–1131 CrossRef CAS PubMed.
- C. A. Thomas, A. C. Paquola and A. R. Muotri, Annu. Rev. Cell Dev. Biol., 2012, 28, 555–573 CrossRef CAS PubMed.
- V. P. Belancio, A. M. Roy-Engel, R. R. Pochampally and P. Deininger, Nucleic Acids Res., 2010, 38, 3909–3922 CrossRef CAS PubMed.
- M. Tajerian, S. Alvarado, M. Millecamps, P. Vachon, C. Crosby, M. C. Bushnell, M. Szyf and L. S. Stone, PLoS One, 2013, 8, e55259 CAS.
- M. Zhuo, G. Wu and L. J. Wu, Mol. Brain, 2011, 4, 31 CrossRef CAS PubMed.
- F. Denk and S. B. McMahon, Neuron, 2012, 73, 435–444 CrossRef CAS PubMed.
- J. K. Zhu, Annu. Rev. Genet., 2009, 43, 143–166 CrossRef CAS PubMed.
- D. K. Ma, M. H. Jang, J. U. Guo, Y. Kitabatake, M. L. Chang, N. Pow-Anpongkul, R. A. Flavell, B. Lu, G. L. Ming and H. Song, Science, 2009, 323, 1074–1077 CrossRef CAS PubMed.
- E. Ferrante, A. E. Turgut, E. Duéñez-Guzmán, M. Dorigo and T. Wenseleers, PLoS Comput. Biol., 2015, e1004273, DOI:10.1371/journal.pcbi.1004273.
- J. Lee, S. Mun, T. J. Meyer and K. Han, Comp. Funct. Genomics, 2012, 2012, 129416 Search PubMed.
- K. Januszyk, P. W. Li, V. Villareal, D. Branciforte, H. Wu, Y. Xie, J. Feigon, J. A. Loo, S. L. Martin and R. T. Clubb, J. Biol. Chem., 2007, 282, 24893–24904 CrossRef CAS PubMed.
- S. L. Martin, M. Cruceanu, D. Branciforte, P. Wai-Lun Li, S. C. Kwok, R. S. Hodges and M. C. Williams, J. Mol. Biol., 2005, 348, 549–561 CrossRef CAS PubMed.
- M. Dewannieux, C. Esnault and T. Heidmann, Nat. Genet., 2003, 35, 41–48 CrossRef CAS PubMed.
- S. Carbon, A. Ireland, C. J. Mungall, S. Shu, B. Marshall, S. Lewis, G. O. H. Ami and G. Web Presence Working, Bioinformatics, 2009, 25, 288–289 CrossRef CAS PubMed.
- R. L. Seal, S. M. Gordon, M. J. Lush, M. W. Wright and E. A. Bruford, Nucleic Acids Res., 2011, 39, D514–D519 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Two lists of (i) n = 463 “pain genes” (file name “S1_Table.txt”) and (ii) n = 386 “addiction genes” (file name “S2_Table.txt”) used for the present functional genomics analyses, and the simulation program as LabVIEW implementation given as the source code and complied for the Linux operating system (File name “S1_Software.zip”, see included file “Readme.rtf” for instructions). See DOI: 10.1039/c5ib00203f |
| This journal is © The Royal Society of Chemistry 2016 |