
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ring-locking enables selective anhydrosugar synthesis from carbohydrate pyrolysis†
Li
Chen
a,
Jinmo
Zhao
b,
Sivaram
Pradhan
a,
Bruce E.
Brinson
b,
Gustavo E.
Scuseria
b,
Z. Conrad
Zhang
*c and
Michael S.
Wong
 *abde
*abde
aDepartment of Chemical and Biomolecular Engineering, Rice University, Houston, TX 77005, USA. E-mail: mswong@rice.edu
bDepartment of Chemistry, Rice University, Houston, TX 77005, USA
cDalian National Laboratory of Clean Energy, Dalian Institute of Chemical Physics, Dalian, Liaoning 116023, China. E-mail: zczhang@dicp.ac.cn
dDepartment of Civil and Environmental Engineering, Rice University, Houston, TX 77005, USA
eDepartment of Materials Science and NanoEngineering, Rice University, Houston, TX 77005, USA
First published on 21st July 2016
Abstract
The selective production of platform chemicals from thermal conversion of biomass-derived carbohydrates is challenging. As precursors to natural products and drug molecules, anhydrosugars are difficult to synthesize from simple carbohydrates in large quantities without side products, due to various competing pathways during pyrolysis. Here we demonstrate that the nonselective chemistry of carbohydrate pyrolysis is substantially improved by alkoxy or phenoxy substitution at the anomeric carbon of glucose prior to thermal treatment. Through this ring-locking step, we found that the selectivity to 1,6-anhydro-β-D-glucopyranose (levoglucosan, LGA) increased from 2% to greater than 90% after fast pyrolysis of the resulting sugar at 600 °C. DFT analysis indicated that LGA formation becomes the dominant reaction pathway when the substituent group inhibits the pyranose ring from opening and fragmenting into non-anhydrosugar products. LGA forms selectively when the activation barrier for ring-opening is significantly increased over that for 1,6-elimination, with both barriers affected by the substituent type and anomeric position. These findings introduce the ring-locking concept to sugar pyrolysis chemistry and suggest a chemical-thermal treatment approach for upgrading simple and complex carbohydrates.
1. Introduction
Biomass-derived carbohydrates are an abundant but underutilized carbon source for the production of fuels, chemicals and materials.1–5 Pyrolysis is a widely studied thermal process for carbohydrate conversion,1 but its usefulness in synthesis chemistry is limited by the low structural stability and high reactivity of carbohydrates.6 Anhydrosugars are primarily prepared from carbohydrate pyrolysis. Their fixed conformation, protected stereoselectivity, and retention of most of their chemical functionality make these sugar derivatives highly desirable precursors. As a model anhydrosugar, LGA is a chiral building block to antibiotics, antiparasitic agents, and other biologically active compounds.7–12 It also has strong potential as a sugar-based biorefinery feedstock chemical, with the US Department of Energy identifying it as a top-15 candidate.13 Found in smoke particles from forest fires,14 LGA is currently prepared in small quantities by extensive purification from cellulose-derived pyrolysis oil.1,11,15 Large-scale production of LGA remains elusive.To address this, we considered the counter-intuitive approach of pyrolyzing glucose. As the constituent sugar monomer of cellulose, glucose is precursor to a growing number of valuable chemicals, such as 5-hydroxymethylfurfural,16,17 lactic acid,18 butanol,19 and γ-valerolactone (GVL).5 However, glucose pyrolysis generates mostly cracked products and char, with negligible amounts of LGA.20 In principle, synthesizing pure LGA from glucose pyrolysis can be accomplished by (i) carrying out the dehydration step that leads to intramolecular C1–O6 bond formation, (ii) de-selecting the other dehydration pathways, and (iii) avoiding ring-opening of the monosaccharide.
The length of cellulose plays a telling role in LGA formation in pyrolysis: longer chains generated more LGA than shorter ones.20,21 Mettler et al. carefully showed that cellulose with ∼130 sugar units yielded 27% LGA after pyrolysis, cellodextrins with 2–6 units yielded ∼8–18%, and D-glucose yielded 3%.20 Longer chains contain a larger proportion of sugar units bound to another unit via the β-1,4-glycosidic linkage, from which we conjectured that a substituent group at the C1 position improves LGA selectivity. In this work, we report that the pyrolysis of β-D-glucose (“β-G”) generates LGA with excellent selectivity after its chemical modification at the C1 position (Scheme 1). Using a combination of flash pyrolysis experiments and density functional theory (DFT) calculations, we show how LGA forms preferentially when ring-opening of the sugar unit is hindered by the presence of the C1 substituent group and describe how the chemical environment around the C1 atom impacts 1,6-anhydro bridge formation.
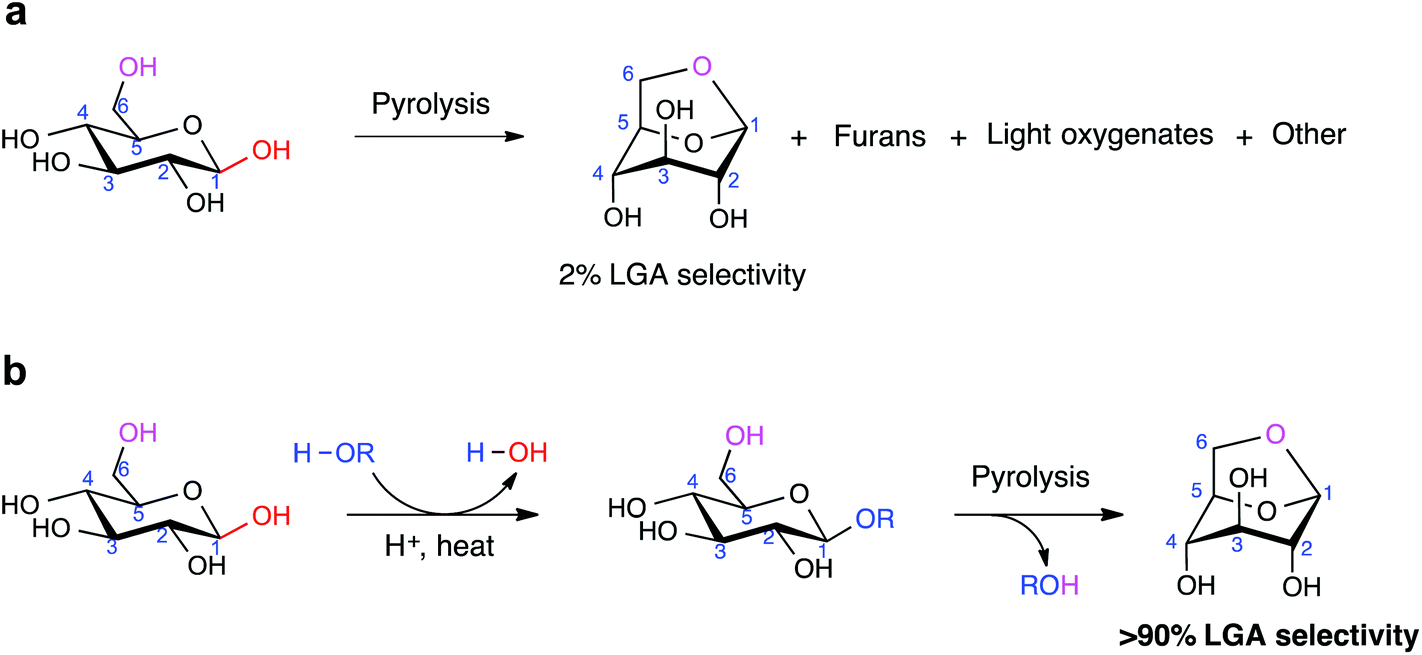 | ||
| Scheme 1 General schemes of sugar pyrolysis to form levoglucosan (“LGA”). a, Pyrolysis of β-D-glucose (“β-G”) leads to LGA after elimination of one water molecule and formation of the 1,6-anhydro bridge, among many other products formed. b, Pyrolysis of functionalized β-G leads to LGA selectively after elimination of ROH. | ||
2. Experimental
2.1 Materials
β-D-Glucose (>80.0%, containing α-D-glucose), phenyl-α-D-glucoside (>97%) and methyl-α-D-galactoside (>97% GC) were purchased from TCI America. Methyl-α-D-glucoside (>99%), methyl-β-D-glucoside (>99%), levoglucosan (99%, 1,6-anhydro-β-D-glucose), phenyl-β-D-glucoside (97%), D-mannose (≥99%), D-galactose (>99%) and methyl-α-D-mannoside (≥99% GC) were purchased from Sigma Aldrich. Microcrystalline cellulose was purchased from Alfa Aesar. Other compounds were purchased for column calibrations: DL-glyceraldehyde (>90% GC), glycolaldehyde dimer, furfural (99%), 5-(hydroxymethyl)-furfural (>99%), acetone (>99.9%), and methanol (>99.9%).2.2 Thin-film for pyrolysis experiments
β-D-Glucose, methyl-β-D-glucoside, and methyl-α-D-glucoside were dissolved into deionized water (18.2 MΩ, Barnstead Nano-pure Diamond System), while phenyl-β-D-glucoside and phenyl-α-D-glucoside were dissolved into 1-propanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:3 by volume) solvent mixture. 4 μL of the solution (8 mg mL−1) was transferred into an open-end cylinder quartz tube (1.9 × 25 mm, diameter × length) by microliter syringe (Hamilton 700 series, 10 μL). Solvent was removed in a vacuum oven at 0.7 atm at 37 °C for 3 h, leading to the formation of a thin-film (containing 32 μg of sugar). The thin-film thickness is determined from Scanning Electron Microscopy (SEM) which is equipped with Energy Dispersive X-ray Spectrometer (EDS) (see ESI for details†). SEM imaging with EDS indicated that the film thickness for all of the samples was less than 10 μm (Fig. S1†).
:H2O (1:3 by volume) solvent mixture. 4 μL of the solution (8 mg mL−1) was transferred into an open-end cylinder quartz tube (1.9 × 25 mm, diameter × length) by microliter syringe (Hamilton 700 series, 10 μL). Solvent was removed in a vacuum oven at 0.7 atm at 37 °C for 3 h, leading to the formation of a thin-film (containing 32 μg of sugar). The thin-film thickness is determined from Scanning Electron Microscopy (SEM) which is equipped with Energy Dispersive X-ray Spectrometer (EDS) (see ESI for details†). SEM imaging with EDS indicated that the film thickness for all of the samples was less than 10 μm (Fig. S1†).
To confirm appropriate reaction conditions, we followed the analysis of Mettler et al.22 using the Pyrolysis number (Py) and Biot number (Bi) to evaluate our thin-film pyrolysis. Our calculations showed that the thin-film experimentation satisfied the criteria for kinetically limited, isothermal pyrolysis reactions (see the ESI†).
2.3 Pyroprobe coupled with GC/FID system
Thin-films pyrolysis experiments were conducted using a Model 5150 pyroprobe analytical pyrolyzer (CDS Analytical Inc.). The pyrolyzer was coupled to an Agilent 6890N GC/FID through a transfer line for online sampling (Fig. S2, see the ESI† for details of reaction and analysis system). The peaks eluted in the chromatogram was identified by mass spectroscopy, compared with retention time of standard compounds and verified with the previous literatures.21–23 The GC column was calibrated by injecting standard solution of methanol and phenol into the GC/FID system. LGA was calibrated by evaporating the LGA thin film sample directly through the pyrolyzer upon heating at pyrolysis temperature (600 °C), which appears as a single peak in the GC chromatogram.212.4 Pyrolysis data evaluation
In order to monitor product distribution of glucose pyrolysis, the yields in this paper are reported in terms of molar carbon yield where the moles of carbon in the product are divided by the moles of carbon in the glucose moiety.The following equations were used to calculate carbon yield of LGA (YLGA), conversion of different substrates (Xsugar) and selectivity to LGA (SLGA).
LGA carbon yield (YLGA) for substituted glucose:
 | (1) |
LGA carbon yield (YLGA) from glucose was calculated by the same equation.
We used methanol and phenol as in situ generated internal standards to quantify glucoside conversion. Conversion (XMG) for methyl-β-glucoside and methyl-α-glucoside were defined as follows:
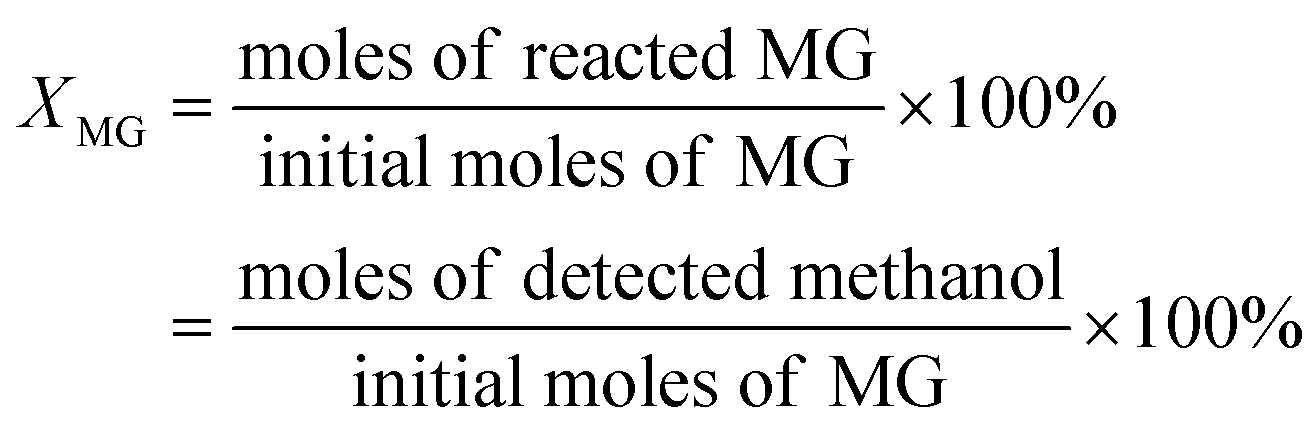 | (2) |
Conversion (XPG) for phenyl-β-glucoside and phenyl-α-glucoside:
 | (3) |
For eqn (2) and (3), we confirmed that (i) methanol/phenol was thermally stable under pyrolysis condition (no side reaction); (ii) methanol/phenol did not react with other pyrolysis products to form ether; and (iii) pyrolysis products did not decompose to form methanol/phenol.
Conversion (XG) for glucose was quantified using HPLC (details in next section):
 | (4) |
The LGA yield, conversion of starting precursor, and selectivity to LGA were related in the following manner:
| SLGA = YLGA ÷ Xi | (5) |
If i = MG, selectivity to LGA (SLGA) is:
 | (6) |
If i = PG, selectivity to LGA (SLGA) is:
 | (7) |
If i = G, selectivity to LGA (SLGA) is:
 | (8) |
2.5 Methyl-D-glucoside synthesis using glucose
The synthesis was carried out in a microwave reactor (Anton Paar Monowave 300), adapted from published procedures.24 90 mg of glucose, 21.5 mg of Amberlyst 15 (dry) and 2 mL of methanol were transferred into a 30 mL reaction tube, which was then heated to 130 °C, and held for 30 min under stirring at 1200 rpm. After cooling, the solution was diluted with DI water to a total volume 10 mL, and then the solid resin acid catalyst was removed using a 0.2 mm microfiber syringe filter (25 mm, VWR). The filtrate containing crude methyl-glucoside was analyzed for methyl-α-D-glucoside (“α-MG”) and methyl-β-D-glucoside (“β-MG”) content by ion-exclusion high-performance liquid chromatography (HPLC), using a Shimadzu Prominence SIL 20 system (Shimadzu Scientific Instruments, Inc., Columbia, MD, USA) equipped with an HPX-87H organic acid column (Bio-Rad, Hercules, CA, USA) and a refractive index detector (RID). The HPLC operation conditions were as follow: the column was operated at 42 °C, and 30 mM H2SO4 mobile phase was flowing through the column at 0.3 mL min−1. Pure methyl-α-glucoside, methyl-β-glucoside, and D-glucose were used to calibrate the column.Four μL of filtrate was transferred into a quartz tube and dried under vacuum to form the crude methyl-glucoside thin-film for the pyrolysis studies.
Glucose conversion in the synthesis step (XG_synthesis):
 | (9) |
Methyl-glucosides yield (Yα-MG, Yβ-MG):
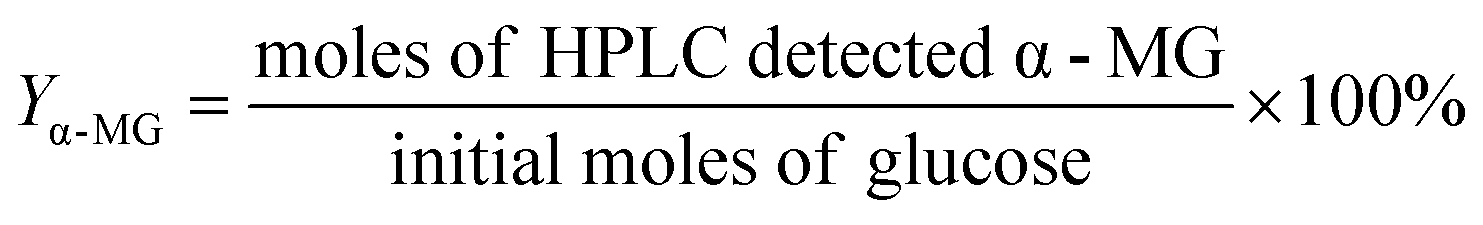 | (10) |
Y β-MG was calculated by following similar equations.
Methyl-D-glucoside has also been synthesized from cellulose, filter paper and cotton (see ESI† for details).
2.6 Computation method
Quantum mechanical and statistical mechanical computations were performed using the Gaussian 09 program package (rev C.01).25 The classical B3LYP functional was used with the electronic wave function expanded by the 6-31G(d,p) basis set. The Gibbs free energy was evaluated at 600 °C based on frequency analysis at the same level of theory. This level of theory is at a relatively lower level compared to some previous studies. To compare with published results, the Gibbs activation energy (the free energy difference between the highest-energy transition state and the lowest-energy reactant26) was computed to be 47.0 kcal mol−1 for LGA formation reaction, which was reasonably close to the value of 48.2 kcal mol−1 calculated from M06-2X/6-311+G (2df,p) level of theory27 and the value of 52.5 kcal mol−1 from MP4(SDTD) single-point energy calculation based on geometries from B3LYP functional.283. Results and discussion
As an initial test, we synthesized a methyl substituted glucose (MG) by replacing the C1 hydroxyl group of glucose (β-G and α-G mixture) with a methoxy group through Fischer glycosylation chemistry.29 The resulting product contained 84 wt% methyl-D-glucoside, of which 44% was the β anomeric form. We heated the unpurified MG to 600 °C rapidly (flash pyrolysis ramp rate of ∼20000 °C s−1), held it at 600 °C for 20 s, and analyzed the gas-phase effluent for pyrolysis products (Fig. S2†). LGA selectivity from crude MG pyrolysis was ∼64%, significantly higher than the experimentally determined ∼2% selectivity from glucose pyrolysis.
We next carried out the pyrolysis of high-purity methyl-β-D-glucoside (“β-MG”) and methyl-α-D-glucoside (“α-MG”) under the same experimental conditions. We chose 600 °C for our studies, which is the higher end of the temperature range used in fast pyrolysis processes.30 At lower pyrolysis temperatures, the selectivity for anhydrosugar increases and the selectivity for light oxygenates and permanent gases (CO2 and CO) decreases (Table S1†). We adopted the thin-film pyrolysis technique of Dauenhauer and co-workers22 to ensure the various sugar compounds did not experience concentration and temperature gradients during heating and to ensure consistent substrate conversion and LGA yield measurements. Fig. 1 shows representative chromatograms of the detected products from the pyrolysis of glucose, β-MG, and α-MG, as well as the β and α forms of phenyl-D-glucoside (PG). Pyrolysis of glucose led to dehydrated (LGA, 1,6-anhydro-β-D-glucofuranose, and HMF) and cracked (furfural, glyceraldehyde, glycolaldehyde, and acetone) products.21–23 In comparison, pyrolysis of the substituted sugars led to mostly LGA (with methanol or phenol eliminated as a co-product) with decreased amounts of the other pyrolysis products.
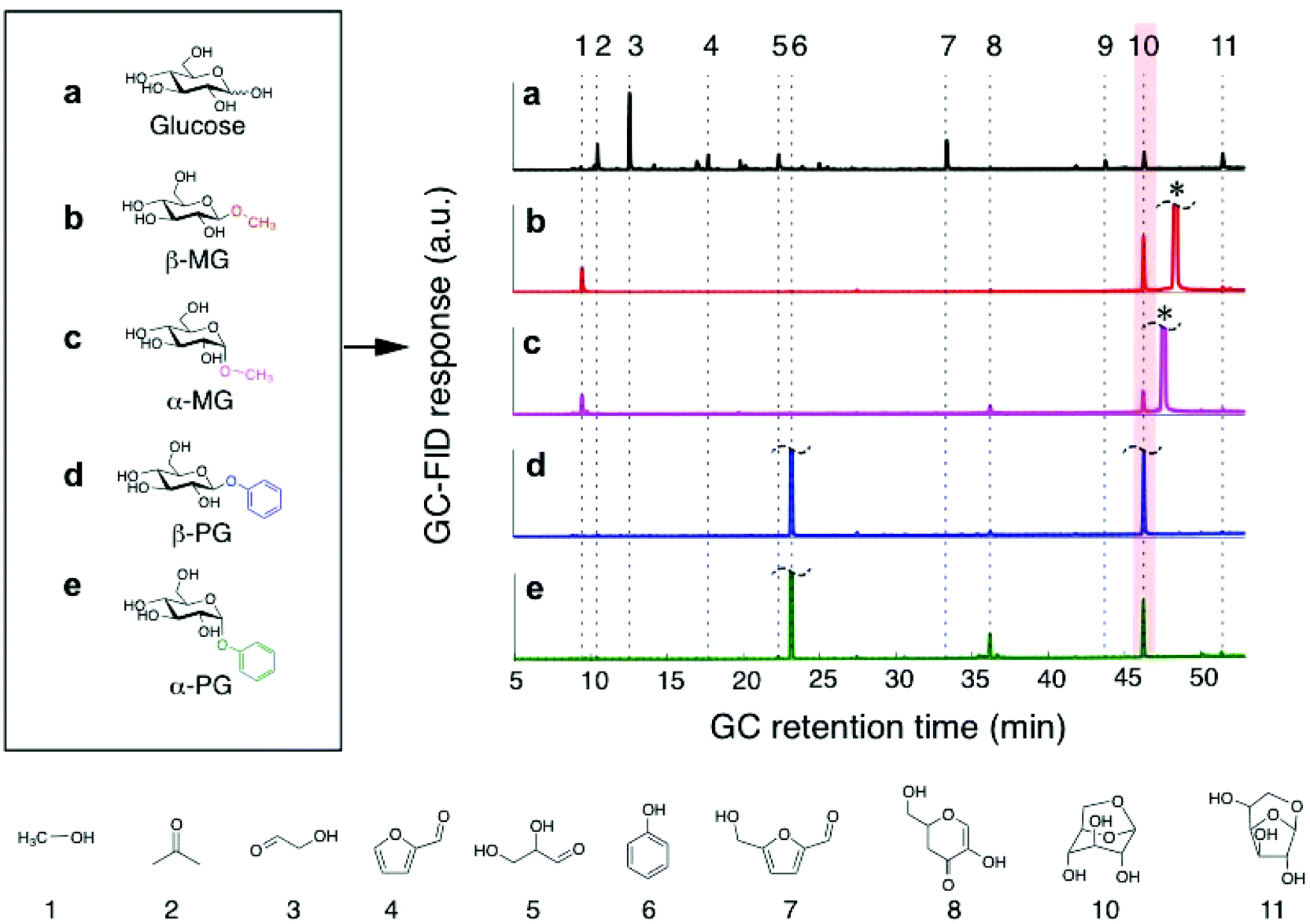 | ||
| Fig. 1 Typical product distributions from glucose and glucoside pyrolysis at 600 °C. a–e, The multiplicity of products from (a) glucose pyrolysis is not found in the pyrolysis of (b) β-MG, (c) α-MG, (d) β-PG, or (e) α-PG. LGA product (compound #10) is highlighted. Details about the detected species are provided in Table S2.† The unreacted β-MG or α-MG precursor is marked with *. Flash pyrolysis reaction conditions: 600 °C, thin-film sample size = 32 μg, film thickness <10 μm (Fig. S1†), heating time of 20 s, carrier gas split ratio = 100:1. | ||
A direct comparison of LGA yields provided an incomplete analysis, because the sugar compounds volatilized to different extents during pyrolysis treatment and did not undergo complete conversion (<60%). Instead, we quantified and compared LGA selectivity values, which accounts for incomplete conversion of the parent sugar (Table 1). Fig. 2 shows LGA selectivities from MG and PG pyrolysis, which readily exceeded 20%. In comparison, glucose pyrolysis had very low LGA selectivity (∼1–4%) at all conversions observed (∼30–100%). LGA selectivity from β-MG pyrolysis was in the 80–85% range at <20% conversion and it decreased to ∼48% near 50% conversion. Furan derivatives, light oxygenates, and char residue formed at higher conversions. α-MG pyrolysis showed a similar trend but selectivities were roughly half as much. Pyrolysis of the phenoxy-functionalized glucoses gave similar trends, with LGA selectivities measured as high as 93 ± 4% for β-PG. The highest LGA yield (39%) came from β-PG pyrolysis at 54% conversion (Table 1). At a sugar/glucoside conversion of 35%, LGA selectivity decreased in the following order: β-PG > β-MG > α-PG > α-MG ≫ glucose, showing the wide-ranging effects of substituent type and anomeric position.
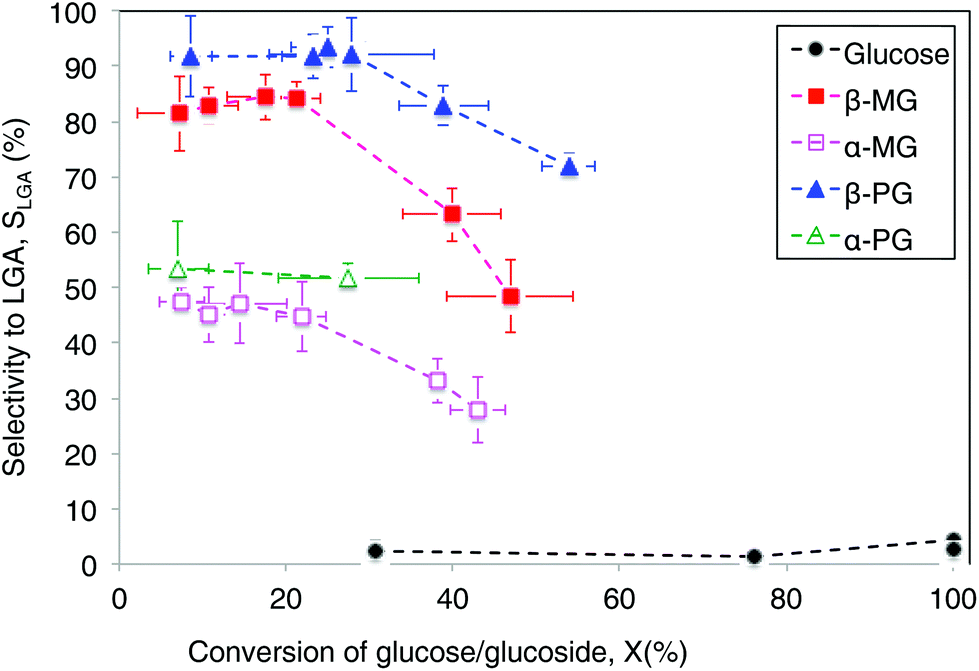 | ||
| Fig. 2 LGA carbon selectivity-conversion analysis for the different sugar compounds. Each data point represents a minimum of 3 experiments, and the error bar is ±1 standard deviation. LGA selectivity values (SLGA = YLGA ÷ Xsugar) were determined from yields YLGA measured at different sugar conversions (Xsugar). Conversion varied with carrier gas contact time at 600 °C. | ||
| Compound | Split ratio | Probe heating time (second) | Interface temperature (°C) | Conversion (%) | Carbon yield (%) | Carbon selectivity (%) |
|---|---|---|---|---|---|---|
| β-G (decomposition temperature >152 °C)31 | 100 | 0.3 | 150 | 31 | 0.7 ± 0.6 | 2 ± 2 |
| 0.4 | 150 | 76 | 0.9 ± 0.3 | 1 ± 0.4 | ||
| 20 | 150 | 100 | 4 ± 0.2 | 4 ± 0.2 | ||
| 20 | 250 | 100 | 3 ± 0.4 | 2 ± 0.4 | ||
| β-MG (predicted boiling point = 389.1 ± 42.0 °C)32 | 100 | 0.5 | 250 | 7 ± 5 | 6 ± 5 | 81 ± 7 |
| 1 | 250 | 11 ± 4 | 9 ± 2 | 83 ± 3 | ||
| 20 | 250 | 17 ± 4 | 15 ± 4 | 84 ± 4 | ||
| 50 | 20 | 250 | 21 ± 3 | 18 ± 3 | 84 ± 3 | |
| 20 | 20 | 250 | 40 ± 6 | 25 ± 5 | 63 ± 5 | |
| 10 | 20 | 250 | 47 ± 8 | 21 ± 6 | 48 ± 6 | |
| α-MG (predicted boiling point = 389.1 ± 42.0 °C)32 | 100 | 0.5 | 250 | 7 ± 3 | 4 ± 1 | 47 ± 3 |
| 1 | 250 | 11 ± 4 | 5 ± 2 | 45 ± 5 | ||
| 20 | 250 | 15 ± 5 | 7 ± 3 | 47 ± 7 | ||
| 50 | 20 | 250 | 22 ± 5 | 10 ± 2 | 44 ± 6 | |
| 20 | 20 | 250 | 38 ± 2 | 13 ± 2 | 33 ± 4 | |
| 10 | 20 | 250 | 43 ± 5 | 12 ± 2 | 28 ± 4 | |
| β-PG (predicted boiling point = 482.1 ± 45.0 °C)32 | 100 | 0.1 | 250 | 9 ± 2 | 8 ± 3 | 92 ± 7 |
| 0.5 | 250 | 18 ± 4 | 22 ± 4 | 92 ± 8 | ||
| 20 | 250 | 23 ± 3 | 24 ± 4 | 93 ± 4 | ||
| 50 | 20 | 250 | 28 ± 7 | 26 ± 11 | 92 ± 6 | |
| 20 | 20 | 250 | 39 ± 5 | 32 ± 5 | 83 ± 4 | |
| 10 | 20 | 250 | 54 ± 3 | 39 ± 3 | 72 ± 2 | |
| α-PG (predicted boiling point = 482.1 ± 45.0 °C)32 | 100 | 0.1 | 250 | 7 ± 4 | 12 ± 6 | 53 ± 8 |
| 20 | 250 | 27 ± 8 | 4 ± 3 | 52 ± 3 | ||
We next studied the energy profiles of several key reaction pathways in β-G, β-MG, and β-PG pyrolysis, through DFT computation using the B3LYP functional and 6-31G(d,p) basis set. β-G can dehydrate into LGA and other anhydrosugars.27,33 It can also mutarotate to the open-chain aldehyde form, which then goes through isomerization, retro-aldol cleavage, and dehydration reactions. Consistent with observations of numerous cracked products and low LGA selectivity, the ring-opening pathway for β-G pyrolysis has the lowest activation energy (P-1; 37.3 kcal mol−1), followed by 1,6-dehydration to form LGA (P-2; 47.0 kcal mol−1) and then by dehydration via β-eliminations (P-3 to P-5; ∼59–71 kcal mol−1) (Fig. 3).
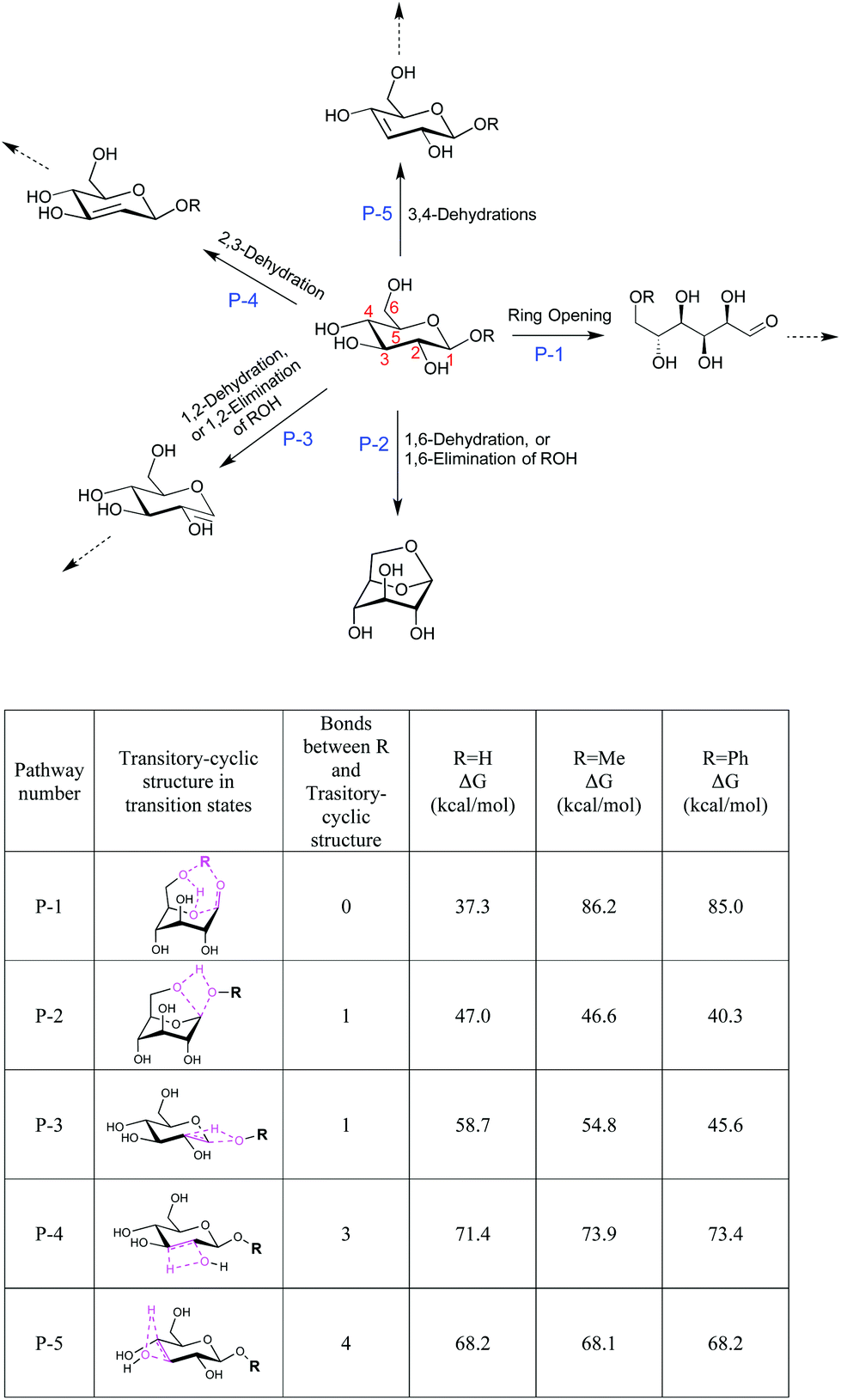 | ||
| Fig. 3 Summary of key reactions and energy barriers in β-G, β-MG, and β-PG pyrolysis. | ||
Of particular interest are the lowest-barrier P-1 and P-2 pathways, which proceed through a common series of conformational changes of the pyranose ring. β-G flips from its stable chair conformation I into a boat structure III and then into an inverted chair V; the energy barriers are sufficiently small (∼8 kcal mol−1) such that these structures can be considered in pre-equilibrium with one another (Fig. 4a, Table S3†). Through a concerted set of bond cleavage and formation steps, structure V then converts into either the open-chain form via a six-atom-containing transition state VI34 or into LGA via a four-atom-containing transition state vi.35
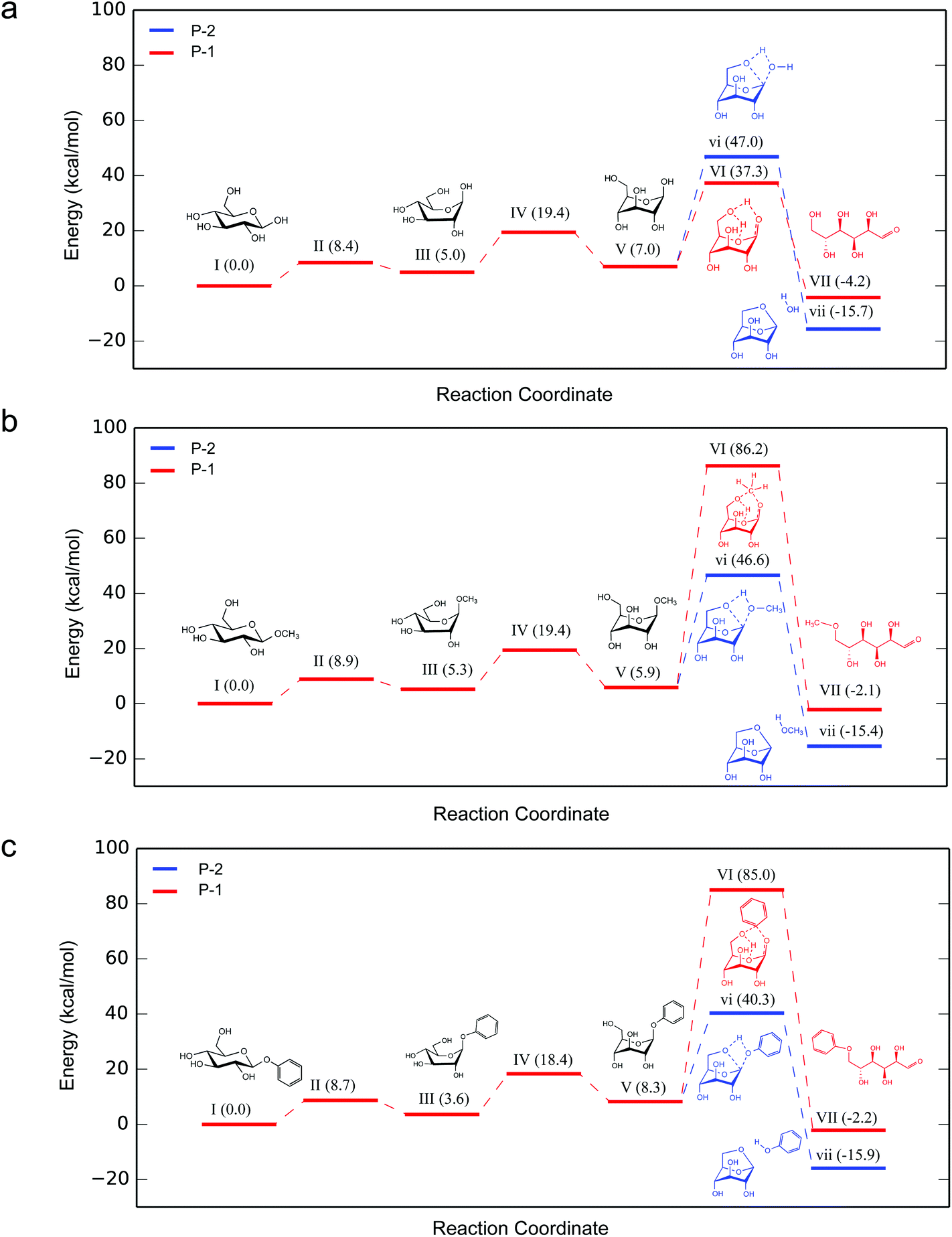 | ||
| Fig. 4 Relative Gibbs free energy profiles for ring-opening (P-1) and LGA formation (P-2) pathways for (a) β-G, (b) β-MG and (c) β-PG pyrolysis at 600 °C. Structures vi and VI are the rate-determining transition states containing transient four-atom and six-atom cyclic structures, respectively. 3-D structures for (a) is shown in Fig. 5, for (b) and (c) are shown in Fig. S3.† | ||
 | ||
| Fig. 5 Representative 3-D structures used in β-G ring opening and LGA formation pathway calculation, optimized at the B3LYP/6-31G(d,p) level of theory. | ||
With the C1 methoxy substituent, β-MG goes through P-1 and P-2 pathways via geometries very similar to those of β-G (Fig. 4b, Table S3†). The relative energies of the various states are essentially the same as in the β-G case, except for transition state VI. The activation barrier height increases substantially (by ∼50 kcal mol−1 to 86.2 kcal mol−1), leading to a ring-locking effect. Ring-opening becomes very difficult because the C1 methoxy impedes the formation of the transitory six-atom cyclic structure. Whereas the 1s orbital of the H atom on O1 of β-G has ample overlap with the O6 sp3 orbital (Fig. 6a), the methyl carbon sp3 orbital on O1 of β-MG does not (Fig. 6b). The methoxy group lowers the P-2 and P-3 barriers somewhat, and has no effect on the much higher P-4 and P-5 barriers. The observed high LGA selectivities directly result when 1,6-elimination becomes the energetically favored pathway after ring-opening is greatly suppressed.
 | ||
| Fig. 6 Transition states showing the six-atom ring structure of (a) β-G, (b) β-MG, and (c) β-PG undergoing ring-opening, and the four-atom ring structures of (d) β-G, (e) β-MG, and (f) β-PG undergoing 1,6-dehydration/1,6-elimination. | ||
The C1 phenoxy substituent also impedes the pyranose ring from opening, with the P-1 barrier increasing to 85.0 kcal mol−1 (Fig. 4c and 6c; Table S3†). The P-2 barrier decreases by ∼7 kcal mol−1, reflecting an inductive effect of the phenyl group on the adjacent four-atom cyclic structure of transition state vi (Fig. 3 and 6d–f). The P-3 barrier decreases also (by ∼13 kcal mol−1) due to the inductive effect, generating 1,2-anhydro-β-D-glucopyranose that then isomerizes to LGA.36 P-2 and P-3 pathways are kinetically favored over P-1, P-4, and P-5, contributing to the >90% LGA selectivities seen for β-PG pyrolysis.
We found that high LGA selectivities come primarily from the ring-locking effect by the C1 group, even with the α anomers. Ring-opening of α-G, α-MG and α-PG proceeds through a different pathway involving a four-atom cyclic structure (Fig. S4 and S5†).34 The activation energy increases from 44.9 kcal mol−1 to 76.2 and 70.1 kcal mol−1 when α-G is functionalized at the C1 with methoxy and phenoxy groups, respectively.
LGA formation is more difficult when the C1 OH/OR groups and C6 hydroxymethyl groups are on opposite sides of the pyranose ring, with the α anomers going through a two-step carbene mechanism instead of the four-atom cyclic structure of the P-2 pathway.27 The C1 substituent abstracts the hydrogen at C1 to form a three-center transition state (structure b), resulting in a carbene intermediate (structure c); the latter then forms LGA via another three-center transition state (structure d). The LGA formation activation energy is relatively large, changing little after methoxy functionalization (from 66.2 to 69.0 kcal mol−1) and decreasing after phenoxy functionalization (to 51.4 kcal mol−1) (Fig. S4 and S5†). The lower LGA selectivities for α-MG and α-PG compared to β-MG and β-PG are due to the smaller activation barrier differences between ring-opening and 1,6-dehydration/elimination.
We observed the ring-locking effect in other C1-functionalized monosaccharides (Fig. S6†). D-Mannose and D-galactose are common stereoisomers of D-glucose, in which their respective C2 and C4 OH groups are in the axial position. Pyrolysis of D-mannose (a mixture of α and β forms) led to 5% yield of the corresponding 1,6-anhydrosugar mannosan; in comparison, pyrolysis of methyl-α-D-mannoside led to 23% yield. Mannosan selectivities were 5% and 50%, respectively, after accounting for the difference in conversion. Similarly, pyrolysis of D-galactose and methyl-α-D-galactoside led to 5% and 11% galactosan yields and to selectivities of 5% and 23%.
Ring-locking can explain the higher LGA yields from longer cellulose chains than shorter ones. Cellulose pyrolysis generates LGA through depolymerization, in which the glycosidic bond between the C1 of one sugar unit and the O4 of an adjacent unit cleaves, and C1 forms the intramolecular bond with O6. The adjacent unit at the C1 position helps hinder ring-opening during depolymerization. It can also help explain how lignocellulose pyrolysis leads to higher LGA selectivities normalized to cellulose content (∼48% for poplar wood v. ∼27% for Avicel cellulose37). The cellulose fraction contains longer chains and has additional glycosidic linkages to hemicellulose and lignin, both of which contribute to the ring-locking effect.
We studied if cellulose solids can be pre-treated in such a way to improve LGA selectivity prior to pyrolysis. As a proof-of-concept, three different cellulose sources (microcrystalline cellulose, filter paper and cotton) were methoxylated into mixtures of α-MG and β-MG (Table S4†). Flash pyrolysis of the cellulose-derived methyl-D-glucoside crude gave a LGA yield of 15%. Pyrolysis of untreated cellulose gave a similar yield, though needing complete cellulose conversion. Accounting for the differences in conversion, LGA selectivities were 59% and 14% for treated and untreated cellulose, respectively. LGA selectivities (∼50–60%) from the various methyl-D-glucoside crudes mapped between the selectivity-conversion curves for α-MG and for β-MG (Fig. S7†). Chemical modification of the carbohydrate source reduces the atom-inefficiency of cellulose pyrolysis.
4. Conclusions
Ring-locking is established as an efficient means to achieve substantially improved LGA selectivities from the pyrolysis of sugars. The presence of a substituent group at the anomeric carbon impedes pyranose ring-opening by raising the activation barrier of this pathway. The barrier for 1,6-elimination to form LGA decreases when the substituent group has a strong inductive effect. The multitude of reaction pathways in pyrolysis may be selectively redirected for substantially improved selectivities to anhydrosugars by simply ring-locking the anomeric carbon of the carbohydrates. Similar methods may also be applicable to monosaccharides with furanose ring structures.Author contributions
L. C., M. S. W. and Z. C. Z. conceived the idea and designed the experiments. L.C. performed the pyrolysis experiments. L. C. and S. P. synthesized methyl-glucoside. J. M. Z. performed computations. B. E. B. collected SEM data. The manuscript was drafted by L. C. and M. S. W., with contributions and input from all authors.Acknowledgements
This work was supported by the National Science Foundation (CBET-1153232, CBET-1247347 and CHE-1462434) and the CAS/SAFEA International Partnership Program for Creative Research Teams. L. Chen acknowledges support from the Kobayashi Fellowship Fund. We thank Prof. R. Gonzalez (Rice) for use of the high-performance liquid chromatography, and Dr J. M. Clomburg (Rice) for assistance. We also thank Dr P. Yan (DICP) and Mr Y. Gao (UT Austin) for experimental assistance, and Dr H. Qian (Rice), Dr H. Yu (DICP), Mr Y. Yin (Rice), and Mr Y. Zhou (Rice) for helpful discussions. We also thank additional support from KiOR, Inc.Notes and references
- R. C. Brown and T. R. Brown, Biorenewable resources: engineering new products from agriculture, John Wiley & Sons, 2013 Search PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- P. Anbarasan, Z. C. Baer, S. Sreekumar, E. Gross, J. B. Binder, H. W. Blanch, D. S. Clark and F. D. Toste, Nature, 2012, 491, 235–239 CrossRef CAS PubMed.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2011, 13, 520 RSC.
- C.-H. Zhou, X. Xia, C.-X. Lin, D.-S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- S. Czernik and A. V. Bridgwater, Energy Fuels, 2004, 18, 590–598 CrossRef CAS.
- D. Radlein, in Fast pyrolysis of biomass: a handbook, ed. A. Bridgwater, 2002, pp. 205–241 Search PubMed.
- M. Bols, Carbohydrate building blocks, John Wiley & Sons, 1996 Search PubMed.
- C. J. Longley and D. P. C. Fung, in Advances in Thermochemical Biomass Conversion, 1994, vol. 2, pp. 1484–1494 Search PubMed.
- C. M. Lakshmanan, B. Gal-or and H. E. Hoelscher, Ind. Eng. Chem. Prod. Res. Dev., 1969, 8, 261–267 CAS.
- J. P. Henschke, C. W. Lin, P. Y. Wu, W. S. Tsao, J. H. Liao and P. C. Chiang, J. Org. Chem., 2015, 80, 5189–5195 CrossRef CAS PubMed.
- US Department of Energy and E. E. and R. E. Energy, 2004.
- B. R. T. Simoneit, J. J. Schauer, C. G. Nolte, D. R. Oros, V. O. Elias, M. P. Fraser, W. F. Rogge and G. R. Cass, Atmos. Environ., 1999, 33, 173–182 CrossRef CAS.
- F. Shafizadeh, R. H. Furneaux, T. G. Cochran, J. P. Scholl and Y. Sakai, J. Appl. Polym. Sci., 1979, 23, 3525–3539 CrossRef CAS.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS PubMed.
- H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597–1600 CrossRef CAS PubMed.
- M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602–605 CrossRef CAS PubMed.
- C. Dellomonaco, J. M. Clomburg, E. N. Miller and R. Gonzalez, Nature, 2011, 476, 355–359 CrossRef CAS PubMed.
- M. S. Mettler, A. D. Paulsen, D. G. Vlachos and P. J. Dauenhauer, Green Chem., 2012, 14, 1284 RSC.
- P. R. Patwardhan, J. A. Satrio, R. C. Brown and B. H. Shanks, J. Anal. Appl. Pyrolysis, 2009, 86, 323–330 CrossRef CAS.
- M. S. Mettler, S. H. Mushrif, A. D. Paulsen, A. D. Javadekar, D. G. Vlachos and P. J. Dauenhauer, Energy Environ. Sci., 2012, 5, 5414 CAS.
- Y.-C. Lin, J. Cho, G. A. Tompsett, P. R. Westmoreland and G. W. Huber, J. Phys. Chem. C, 2009, 113, 20097–20107 CAS.
- Y. Yang, M. M. Abu-Omar and C. Hu, Appl. Energy, 2012, 99, 80–84 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Norman, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi and N. Rega, 2009.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
- H. B. Mayes, M. W. Nolte, G. T. Beckham, B. H. Shanks and L. J. Broadbelt, ACS Sustainable Chem. Eng., 2014, 2, 1461–1473 CrossRef CAS.
- T. Hosoya, H. Kawamoto and S. Saka, J. Anal. Appl. Pyrolysis, 2009, 84, 79–83 CrossRef CAS.
- F. W. Lichtenthaler, in Ullmann's Encyclopedia of Industrial Chemistry, 2010, pp. 617–642 Search PubMed.
- P. Basu, Biomass gasification and pyrolysis: practical design and theory, Academic press, 2010 Search PubMed.
- M. Hurtta, I. Pitkänen and J. Knuutinen, Carbohydr. Res., 2004, 339, 2267–2273 CrossRef CAS PubMed.
- ACD/Percepta, Adv. Chem. Dev. Inc., Toronto, Canada, 2014. http//www.acdlabs.com.
- R. Vinu and L. J. Broadbelt, Energy Environ. Sci., 2012, 5, 9808 CAS.
- V. Seshadri and P. R. Westmoreland, J. Phys. Chem. A, 2012, 116, 11997–12013 CrossRef CAS PubMed.
- T. Hosoya, Y. Nakao, H. Sato, H. Kawamoto and S. Sakaki, J. Org. Chem., 2009, 74, 6891–6894 CrossRef CAS PubMed.
- R. S. Assary and L. A. Curtiss, ChemCatChem, 2012, 4, 200–205 CrossRef CAS.
- J. Piskorz, D. S. A. G. Radlein, D. S. Scott and S. Czernik, J. Anal. Appl. Pyrolysis, 1989, 16, 127–142 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc01600f |
| This journal is © The Royal Society of Chemistry 2016 |