
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethyl vinyl glycolate as a diverse platform molecule†
Amanda
Sølvhøj
ab,
Esben
Taarning
b and
Robert
Madsen
*a
aDepartment of Chemistry, Technical University of Denmark, 2800 Kgs. Lyngby, Denmark. E-mail: rm@kemi.dtu.dk
bHaldor Topsøe A/S, Haldor Topsøes Allé 1, 2800 Kgs. Lyngby, Denmark
First published on 2nd August 2016
Abstract
Methyl vinyl glycolate (methyl 2-hydroxybut-3-enoate, MVG) is available by zeolite catalyzed degradation of mono- and disaccharides and has the potential to become a renewable platform molecule for commercially relevant catalytic transformations. This is further illustrated here by the development of four reactions to afford industrially promising structures. Catalytic homo metathesis of MVG using Grubbs-type catalysts affords the crystalline dimer dimethyl (E)-2,5-dihydroxyhex-3-enedioate in excellent yield and with meso stereochemical configuration. Cross metathesis reactions between MVG and various long-chain terminal olefins give unsaturated α-hydroxy fatty acid methyl esters in good yields. [3,3]-Sigmatropic rearrangements of MVG also proceed in good yields to give unsaturated adipic acid derivatives. Finally, rearrangement of the allylic acetate of MVG proceeds in acceptable yield to afford methyl 4-acetoxycrotonate.
Introduction
Platform molecules, or base chemicals, are molecules which play a central role in the infrastructure of the chemical industry, i.e. intermediates which are used for the production of several other chemical products.1 Conventional fossil-derived platform molecules include ethylene, propylene and benzene. In recent years, much attention has been directed towards developing renewable platform molecules.2 Some of the most important characteristics of a future renewable platform molecule are that it can be produced at low cost from biomass, and that it can be used for the production of several other useful chemical products. Examples of potential platform molecules which have been investigated previously include ethanol, furfural and 5-hydroxymethylfurfural. Ethanol has been envisaged as a platform molecule for the production of ethylene, ethyl acetate and acetic acid.3 Furfural has been investigated as a platform molecule for the production of furan and methyl tetrahydrofuran.4 Finally, 5-hydroxymethylfurfural has been investigated for the production of dimethyl furan, levulinic acid and terephthalic acid.5Another interesting candidate as a future platform molecule is methyl 2-hydroxybut-3-enoate (methyl vinyl glycolate, MVG). It is a small molecule with a simple structure, and yet it possesses several functional groups, providing it with ample handles for many different chemical transformations. It was first observed as a byproduct in the stannosilicate catalyzed formation of methyl lactate from various mono- and disaccharides in methanol solution.6 MVG is believed to be formed in a dehydration–esterification reaction catalyzed by the stannosilicate catalyst from tetroses which are formed in low amounts from glucose in a retro-aldol reaction. A mechanistic study of the conversion of tetroses to MVG and other α-hydroxy acid derivatives confirms this assumption.7 The theory is further substantiated by the fact that the yield of MVG ranges from 3–11% when employing pentoses or hexoses as substrates,6 but when employing a tetrose as the substrate the yield increases to 50–56%.6,8 A subsequent study has found that the yield of MVG from glucose can be further improved to around 18% by the presence of alkali metal salts.9 Under these reaction conditions the co-product methyl lactate (ML) is obtained in close to 50% yield.9 The various sugars that can be used as feed for the production of ML and MVG may be derived from 2nd generation biomass like corn cobs. From one ton of corn cobs it is possible to obtain about 265 kg of ML and 62 kg of MVG.6,9–11
Although MVG fulfills the requirements for a good bio-based platform molecule, its applications are relatively unexplored. MVG dimerizes under alkaline conditions via a cascade reaction commencing with the isomerization to methyl 2-oxobutanoate (Scheme 1). This isomer enolizes and undergoes an aldol condensation and ultimately forms a cyclic dimer, which upon heating decarboxylates to 5-ethyl-2-hydroxy-3-methyl-2-furanone (also known as maple furanone – an important food-flavoring compound).12
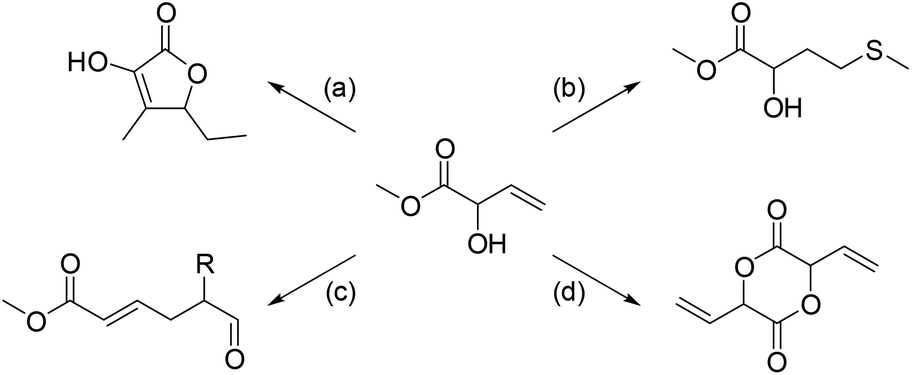 | ||
| Scheme 1 Previously reported transformations of MVG. (a) Alkaline dimerization to form “maple furanone”, 87%,12 (b) radical addition of methanethiol to form the hydroxy analog of methionine, 85%,13 (c) claisen rearrangement to yield 6-oxohex-2-enoates, 46–64%14 and (d) zeolite catalyzed formation of the glycolide dimer, 16–24%.15 | ||
In the presence of a free radical initiator, like AIBN or NBS, methanethiol adds to the double bond of MVG giving rise to methyl 2-hydroxy-4-(methylthio)butanoate,13 which is the methyl ester of the hydroxy analog of the α-amino acid methionine. Both the amino acid and its hydroxy analog are produced synthetically on a large scale and used e.g. as a dietary supplement in poultry feed.
With acid catalysis the hydroxy group of MVG adds to various aldehydes forming hemiacetals, which upon dehydration give rise to allyl vinyl ethers. The latter rearrange in a Claisen [3,3]-sigmatropic shift to the corresponding 6-oxohex-2-enoates.14 The 1,6-dioxo compounds are interesting synthetic motifs particularly as precursors to large scale polyester and polyamide monomers such as adipic acid, caprolactone and caprolactam.
Recently the formation of a vinyl glycolide dimer from 2-hydroxybut-3-enoic acid has been achieved in up to 24% yield by employing a shape selective zeolite catalyst.15 MVG has also been copolymerized with lactic acid (LA) to tune the properties of PLA-based polymers. This can be done either by varying the ratio between MVG and LA or through functionalization of the reactive vinyl side chain of the MVG units.16
Apart from these very product-specific applications of MVG, other examples include various functional group manipulations like the Larock quinolone synthesis,17 the Heck reaction,18 the Tsuji–Trost reaction,19 olefin cross metathesis reactions,20 1,3-dipolar additions,21 reduction of the ester group22 and alkylations of the alcohol.23
Although the examples are few in number, the reaction types are versatile emphasizing that MVG is a molecule for which many chemical transformations are possible. In the present work the focal point was reactions that would transform MVG into industrially important compounds either directly or through few and simple manipulations. Four general strategies were devised, as outlined in Scheme 2.
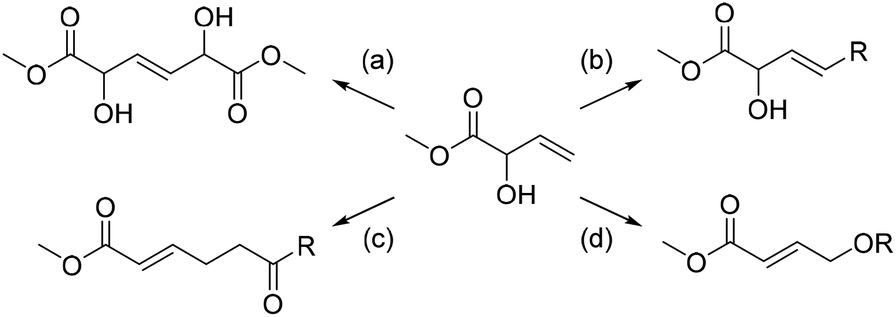 | ||
| Scheme 2 Four general strategies for transformation of MVG into valuable compounds. (a) Homo metathesis of MVG to form a 1,6 diester. (b) Cross metathesis of MVG to form unsaturated α-hydroxy FAME's. (c) Claisen-type rearrangements to form 6-oxo-substituted hex-2-enoic acid derivatives. (d) Oxygen-centered rearrangement to form 4-oxybut-2-enoic acid compounds. | ||
A homo metathesis reaction24 would yield a dimer of MVG, which could be an interesting monomer for polyester production, due to its 1,6-diester skeleton which is reminiscent of adipic acid, and because of the inherent possibility for further functionalization by manipulation of the α-hydroxy groups. A cross metathesis reaction with long straight-chain terminal olefins would result in the formation of unsaturated α-hydroxy fatty acid methyl esters (FAME's). These may be easily reduced to their saturated counterparts, which are well known surfactants with a variety of applications.25
The allylic alcohol moiety of MVG makes it a precursor for various allyl vinyl ethers and derivatives hereof, which may be rearranged in a [3,3]-sigmatropic shift yielding 1,6-diesters or other 1,6-dioxo structures as products. These compounds may serve as direct precursors for valuable polyester and polyamide monomers like adipic acid, caprolactone or caprolactam. Finally, the secondary allylic alcohol may be transformed into its primary allylic alcohol isomer, i.e. a 1,4-dioxygenated motif, which could serve as a precursor for 1,4-butanediol (BDO) or γ-butyrolactone (GBL). This transformation may occur by acetylation of the allylic alcohol followed by a rearrangement of the allylic acetate.
Herein, we emphasize the potential of MVG by describing several new transformations of MVG into a range of industrially promising structures by the use of homo and cross metathesis reactions as well as Claisen and allylic alcohol rearrangements.
Results and discussion
Homo metathesis of MVG
Preliminary experiments had shown that a solid precipitated when heating MVG with a metathesis catalyst in the absence of a solvent. The solid was identified as the homo metathesis product dimethyl 2,5-dihydroxyhex-3-enedioate. Thus, a series of experiments were performed in order to optimize the yield of the reaction (Table 1). Comparison of Grubbs 1st and 2nd generation catalysts (Fig. 1) showed that the 2nd generation catalyst was far superior to the 1st generation complex both in terms of isolated yield and the amount of catalyst. The 2nd generation catalyst gave an isolated yield of 75% with a 0.4% loading of catalyst on a 10 mmol scale (entry 6), whereas the 1st generation catalyst gave only 8% isolated yield with a 5% loading on the same scale (entry 1).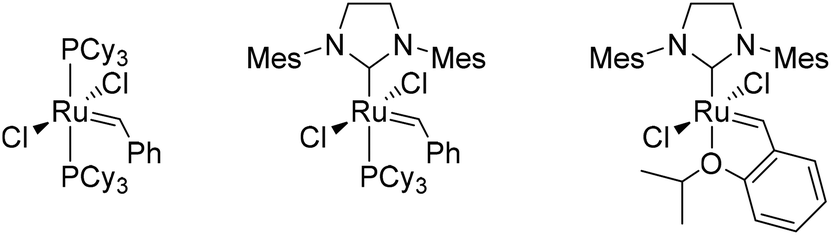 | ||
| Fig. 1 From left to right: Grubbs 1st generation catalyst, Grubbs 2nd generation catalyst and Hoveyda–Grubbs 2nd generation catalyst. | ||
|
|
|||||
|---|---|---|---|---|---|
| Catalyst | Loading (mol%) | Scaleb (mmol) | Solvent & vol.c | Yieldd (%) | |
| a The experiments were performed in a Schlenk flask under inert atmosphere at 80 °C. b Amount of MVG. c Ratio = volume solvent: MVG. d Isolated yield. e Reaction temperature 40 °C. f Reaction temperature 70 °C. g GC yield. | |||||
| 1e | Grubbs 1st gen. | 5 | 10 | — | 8 |
| 2 | Grubbs 2nd gen. | 0.3 | 5 | — | 74 |
| 3 | Grubbs 2nd gen. | 0.3 | 2.5 | Toluene 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
55 |
| 4 | Grubbs 2nd gen. | 0.3 | 5 | Toluene 2:1 |
63 |
| 5f | Grubbs 2nd gen. | 2.2 | 2.5 | EtOAc 4:1 |
46g |
| 6 | Grubbs 2nd gen. | 0.4 | 10 | — | 75 |
| 7 | Grubbs 2nd gen. | 0.4 | 19 | — | 85 |
| 8 | Grubbs 2nd gen. | 0.4 | 39 | — | 88 |
| 9 | Hoveyda–Grubbs 2nd gen. | 0.4 | 2.5 | — | 73 |
| 10 | Hoveyda–Grubbs 2 nd gen. | 0.4 | 19 | — | 93 |
| 11 | Hoveyda–Grubbs 2nd gen. | 0.2 | 19 | — | 90 |
| 12 | Hoveyda–Grubbs 2nd gen. | 0.05 | 20 | — | 77 |
| 13 | Hoveyda–Grubbs 2nd gen. | 0.045 | 39 | — | 80 |
The product is crystalline and the reaction mixture quickly solidifies completely, as it is run without the presence of a solvent. This hampers efficient stirring and may hinder full conversion. Attempts to run the reaction in a solvent (toluene or ethyl acetate) were less successful and resulted in a lowering of the yield (entries 2–5).
Increasing the scale of the reaction from 10 to 19 mmol (entry 7) resulted in an increase of the yield from 75% to 85%. Further increasing the scale to 39 mmol gave a slight improvement of the yield to 88% (entry 8). The catalyst was then successfully exchanged for the less expensive and more stable Hoveyda–Grubbs 2nd generation catalyst (Fig. 1) with an increase in yield as an additional bonus (entry 10). With only 0.4% catalyst loading on a 19 mmol scale the isolated yield reached 93%. The catalyst loading was subsequently reduced to 0.2% without any significant decrease in the yield (entry 11). A further reduction of the catalyst loading to 0.05% resulted in a noticeable lowering of the yield to 77% (entry 12), although this seems in part to be overcome by a further upscaling of the reaction (entry 13).
It should be noted that while all the reactions were run at 80 °C the reaction mixture solidified already at 30 °C when employing the Hoveyda–Grubbs 2nd generation catalyst. With the Grubbs 2nd generation catalyst immediate precipitation was first observed at temperatures above 60 °C. All the reactions were run overnight (16 h), although complete solidification of the mixture seems to occur within the first hour of the reaction.
The crude product may be purified by direct crystallization from ethyl acetate, which gave 71% yield of the pure product in the first crystallization, when employing the reaction conditions in entry 10, Table 1. More of the product is available in the mother liquor, but must be purified by chromatography or collected in larger amounts in order to be isolated by a further crystallization.
As can be seen from the reaction scheme in Table 1 the homo metathesis reaction of MVG forms equimolar amounts of ethylene gas as a co-product, which may easily be separated from the main product. This increases the amount of utilized carbon atoms in the reaction from 80 to 100%, since ethylene is valuable as a bio-based monomer on its own.
The product has 2 stereocenters, corresponding to three different stereoisomers, of which one is a meso form. Taking into account the possibility of forming both the (E)- and the (Z)-isomer, the total number of different isomers amounts to six. Surprisingly, the reaction yields only one isomeric form of the product, which was determined by X-ray diffraction to be the meso form of the (E)-isomer (Fig. 2). NMR spectra of the crude reaction mixture show no signals from a (Z)-double bond, and a GC of the crude reaction mixture shows no presence of other diastereomeric forms. Most likely, the high crystallinity of the meso (E)-isomer is the main driving force for the exclusive formation of this compound since the metathesis reaction is a reversible transformation.
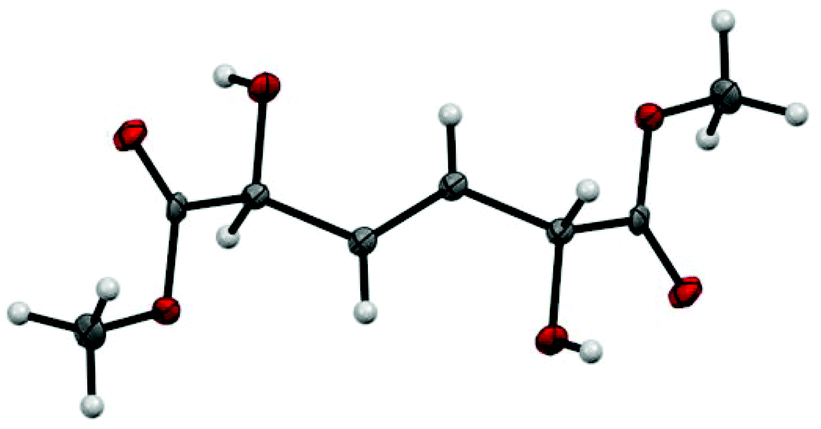 | ||
| Fig. 2 Crystal structure of dimethyl 2,5-dihydroxyhex-3-enedioate. Only the meso-form of the (E)-isomer is formed. | ||
The dimer is a structurally interesting molecule for which many applications can be envisioned. The 1,6-diester structure is reminiscent of adipic acid, making it reasonable to assume that the MVG dimer can be utilized in similar applications. Unlike adipic acid though, the metathesis dimer possesses two hydroxy substituents which introduces the possibility of a functionalized polyester monomer. In fact, very recently Sels and coworkers performed a study where 2,5-dihydroxyhex-3-enedioic acid (DHHDA) was synthesized and copolymerized with lactic acid to yield a crosslinked polyester, which showed improved thermal stability as compared to pure PLA.26 Furthermore, DHHDA was also reacted with hexamethylenediamine to give a partly bio-based polyamide, which in both structure and properties resembles the adipic acid-based polyamide nylon-6,6.26 The meso form of the MVG dimer is achiral but possesses two stereocenters, making both positions pro-chiral. This makes the molecule a potential precursor for asymmetric synthesis.
Cross metathesis of MVG
MVG can also be brought to react with long chain terminal olefins in a cross metathesis reaction, yielding unsaturated α-hydroxy FAME's. These can be hydrogenated to give their fully saturated counterparts, which can be employed as surfactants either directly or after transformation into their alkali metal salts, sulfonates or other derivatives. Bio-based long chain terminal olefins can be obtained from unsaturated fatty acids, like oleic or linoleic acid from palm oil, by reacting these with ethylene in a metathesis reaction.27 For optimization of the reaction conditions MVG was reacted with 1.5 equivalent of dodec-1-ene in refluxing dichloromethane under an inert atmosphere in the presence of a suitable metathesis catalyst to afford methyl (E)-2-hydroxytetradec-3-enoate. The results are shown in Table 2.|
|
|||||
|---|---|---|---|---|---|
| Catalyst | Loading (mol%) | Olefin | Yielda (%) | Yielda Dimerb (%) | |
| a Isolated yield. b Dimer = dimethyl (E)-2,5-dihydroxyhex-3-enedioate. c Not isolated. d With ethyl acetate as the solvent. | |||||
| 1 | Grubbs 1st gen. | 5 | Dodec-1-ene | 25 | —c |
| 2 | Grubbs 2nd gen. | 5 | Dodec-1-ene | 62 | —c |
| 3 | Hoveyda–Grubbs 2nd gen. | 5 | Dodec-1-ene | 44 | 24 |
| 4 | Grubbs 2nd gen. | 1 | Dodec-1-ene | 68 | 19 |
| 5 | Grubbs 2nd gen. | 0.5 | Dodec-1-ene | 65 | 14 |
| 6d | Grubbs 2nd gen. | 1 | Dodec-1-ene | 40 | —c |
| 7 | Grubbs 2nd gen. | 1 | Dec-1-ene | 63 | 21 |
| 8 | Grubbs 2nd gen. | 1 | Tetradec-1-ene | 63 | 20 |
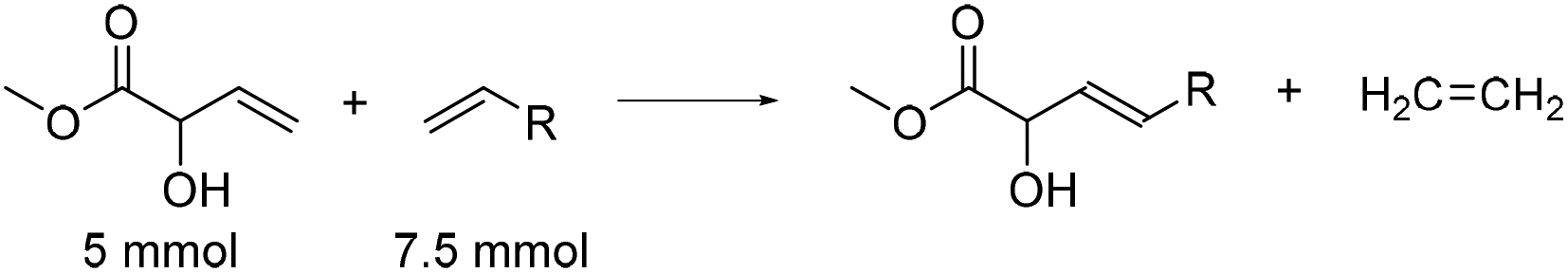
In analogy to the homo metathesis reaction, the Grubbs 2nd generation catalyst proved to be far superior to the Grubbs 1st generation complex. The former gave 62% isolated yield of methyl (E)-2-hydroxytetradec-3-enoate (entry 2), whereas the latter gave only 25% yield (entry 1). As for the homo metathesis reaction the Hoveyda–Grubbs 2nd generation catalyst seems to be more active, and an intense development of ethylene gas was observed already upon mixing the reagents with the catalyst at room temperature. Regardless, the desired cross metathesis product was only isolated in 44% yield (entry 3) and the byproduct from the homo metathesis of MVG was isolated in 24% yield. Lowering the loading of Grubbs 2nd generation catalyst to 1 mol% while increasing the reaction time to two days resulted in a slight increase of the yield to 68% (entry 4), while the yield of the byproduct was reduced to 19%. A further lowering of the catalyst loading to 0.5 mol% gave a slight decrease in the yield of the cross metathesis product (65%, entry 5), but also resulted in a further reduction in the formation of the byproduct. The solvent may be exchanged for ethyl acetate, but at the expense of a significant reduction in the yield (40%, entry 6).
The reaction conditions are applicable to other olefinic substrates. MVG was reacted with dec-1-ene to afford methyl (E)-2-hydroxydodec-3-enoate in 63% isolated yield (entry 7) and the same yield was obtained upon reaction with tetradec-1-ene to afford methyl (E)-2-hydroxyhexadec-3-enoate (entry 8). In all cases in Table 2, traces of the corresponding (Z)-isomers of the cross metathesis products were also detected, but the amounts were not further quantified.
In all the experiments, full conversion of the long chain terminal olefin was achieved and formation of the corresponding internal alkene dimer was observed which accounts for the fate of the excess olefin. This dimerization is faster than the cross metathesis reaction, as expected for this type of substrate,28 but the dimer is easily consumed in an ensuing cross metathesis reaction with MVG and constitutes no hindrance for completion of the desired reaction. The long chain alkene dimer is removed during the work-up and could in principle be reused as a substitute for the long chain terminal olefin.
[3,3]-Sigmatropic rearrangements
Attaching a vinyl group to the allylic alcohol gives rise to a substituted allyl vinyl ether which can rearrange in a [3,3]-sigmatropic shift as outlined in Scheme 3. One example is known from the literature, namely the reaction between MVG and diethoxyethane as shown in reaction (a).14 In this case the resulting trans-acetal eliminates ethanol to yield the unsubstituted allyl vinyl ether, which rearranges to afford methyl 6-oxohex-2-enoate in 46% yield.14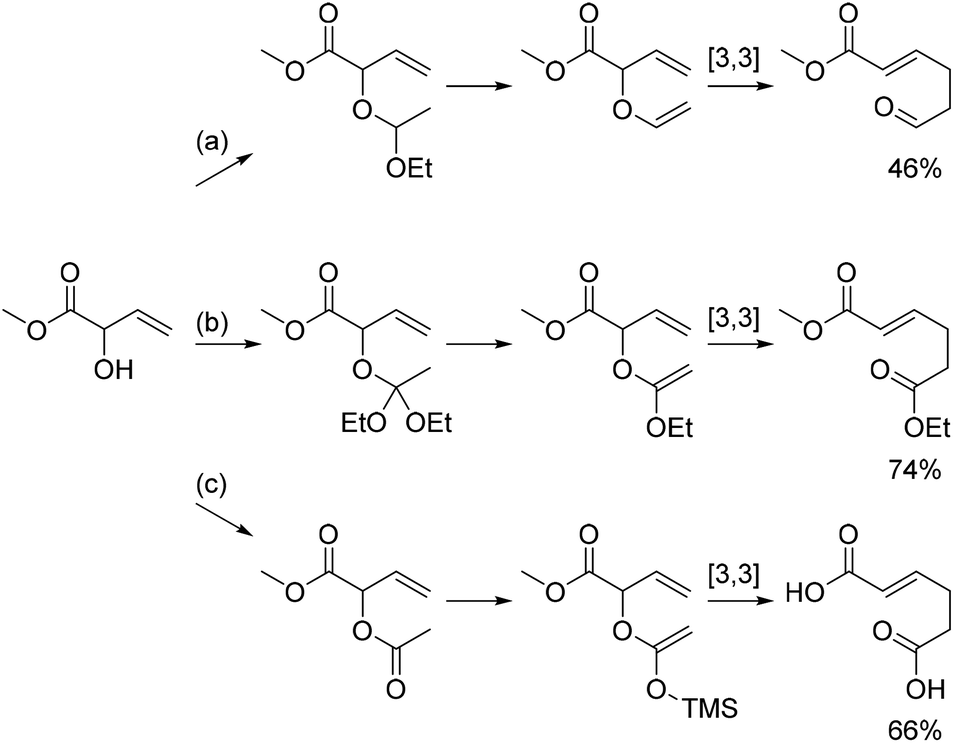 | ||
| Scheme 3 Variations of the [3,3]-sigmatropic rearrangement of vinylic derivatives of MVG. (a) Vinylation with CH3CH(OEt)2 and Claisen rearrangement affords methyl 6-oxohex-2-enoate as shown by Motherwell et al.14 (b) Johnson–Claisen rearrangement with triethoxyacetate affords 6-ethyl 1-methyl hex-2-enedioate. (c) Acetylation of MVG followed by an Ireland–Claisen rearrangement affords hex-2-enedioic acid. | ||
A similar reaction between MVG and an orthoacetate would give a new orthoacetate, which upon elimination of an alcohol would yield an alkoxy substituted allyl vinyl ether. Rearrangement of the latter, known as a Johnson–Claisen rearrangement,29 would then lead to the formation of a diester of hex-2-enoic acid. Yet another possible variation of this reaction, the Ireland–Claisen rearrangement,30 is based on the acylation of the allylic alcohol, followed by formation of the corresponding silyl enol ether and finally a [3,3]-sigmatropic rearrangement leading to the free acid.
The Johnson–Claisen and the Ireland–Claisen reaction were both investigated with MVG. The most successful transformation was the Johnson–Claisen rearrangement. The reaction procedure is simple and consists of heating MVG and the orthoacetate in the presence of catalytic amounts of acetic acid. The temperature is kept just below the boiling point of the orthoacetate until all MVG has been converted, at which point the temperature is raised to 155 °C to facilitate the rearrangement. A distillation column is attached throughout the reaction, and the alcohol is continuously distilled off, driving the reaction towards completion. In the reaction between MVG and triethyl orthoacetate, the rearranged product 6-ethyl 1-methyl (E)-hex-2-enedioate was obtained in 74% isolated yield. For the reaction between MVG and trimethyl orthoacetate the yield was slightly lower and the product dimethyl (E)-hex-2-enedioate was obtained in 72% yield.
For the Ireland–Claisen reaction the first step is the formation of the acetate of MVG, which was formed by reacting MVG with acetyl chloride. The acetylation gave a near quantitative yield in dichloromethane while the product was obtained in 76% yield in the more benign solvent ethyl acetate. The silyl enol ether of the acetate of MVG was formed by treatment with LDA and TMSCl at low temperatures. Subsequent heating to 90 °C in a sealed vessel facilitated the rearrangement, and the silyl as well as the methyl ester were cleaved off during workup, resulting in 66% isolated yield of the free diacid.
The Johnson–Claisen reaction is performed in the absence of a solvent with only a minuscule amount of acidic acid catalyst and requires no distillation of the final product. The atom economy is excellent since the alcohol from the orthoacetate is recovered before the actual rearrangement. However, the temperature of the transformation is rather high and the reaction time long for forming the orthoacetate with MVG and achieving the rearrangement. The Ireland–Claisen reaction illustrates that the rearrangement can be performed at a lower temperature and in a shorter time, but at the expense of the atom economy.
Allylic alcohol transposition
It is well known that palladium(II) complexes catalyze the equilibration between the regioisomers of allylic acetates.31 This also turned out to be the case for the acetate of MVG, which can be rearranged into the isomer methyl 4-acetoxybut-2-enoate with catalytic amounts of palladium(II) species (Scheme 4). The best results were obtained by treating the MVG acetate with catalytic amounts of Pd(MeCN)2Cl2 in refluxing dry THF under nitrogen. The reaction was monitored by GC/MS and showed full conversion of the starting material to give the rearranged product in 59% isolated yield.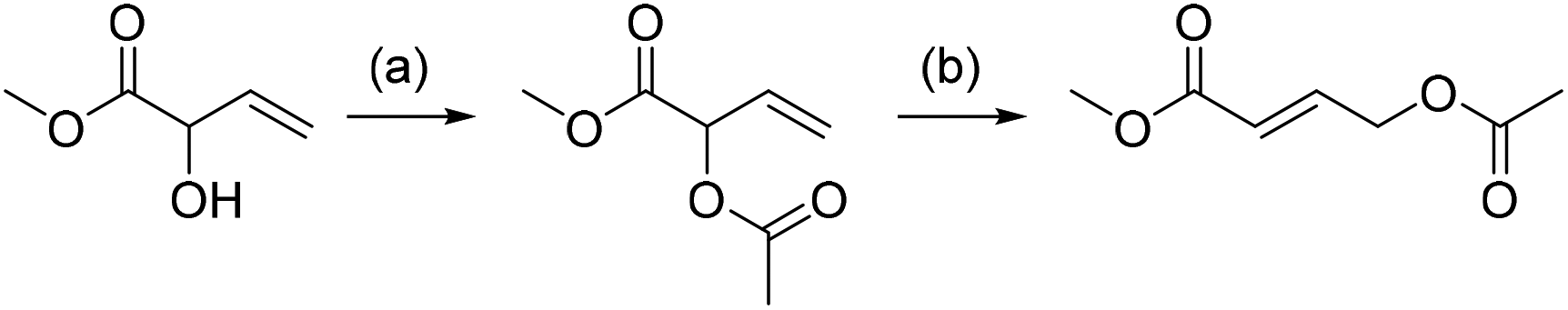 | ||
| Scheme 4 Rearrangement of allylic alcohol. (a) AcCl, pyridine, CH2Cl2, 97% yield. (b) Pd(MeCN)2Cl2, THF, reflux, 59% yield. | ||
While methyl 4-acetoxybut-2-enoate may be an interesting substrate on its own, the most direct application would arise by the removal of the acetyl group to give methyl 4-hydroxybut-2-enoate. The latter may subsequently be reduced completely to 1,4-butanediol or partially reduced and cyclized to yield γ-butyrolactone, both of them valuable large scale chemicals.
Conclusions
We have reported a range of new transformations of MVG into industrially promising compounds utilizing either metathesis or rearrangement reactions. The most successful reaction is the dimerization of MVG to dimethyl (E)-2,5-dihydroxyhex-3-enedioate by a homo metathesis reaction with the Hoveyda–Grubbs 2nd generation catalyst. This reaction proceeds in excellent yield to afford the product as a single stereoisomer. Another metathesis based transformation is the cross metathesis reactions of MVG with long-chain terminal olefins to give unsaturated α-hydroxy FAME's, which may serve as precursors for a variety of surfactants. Furthermore, two Claisen-type rearrangements of MVG have been developed and proven to work well. The products are unsaturated derivatives of adipic acid, which are highly coveted as renewable chemicals. Finally, rearrangement of the allylic alcohol of MVG has been achieved through a palladium catalyzed transposition of the acetate of MVG, yielding a 1,4-dioxygenated motif, which may serve as a precursor for chemicals like 1,4-BDO or GBL. These results show that MVG holds great promise as a novel renewable platform molecule.Experimental section
General remarks
All solvents used were of HPLC grade, all chemicals were bought from commercial suppliers. For dry column vacuum chromatography (DCVC)32 was used Merck Silica Gel 60, 0.015–0.040 mm. Reactions were monitored by GC/MS on a Shimadzu GCMS-QP5000 instrument, which was also used for mass spectrometry (data sets consist of mass and relative intensity). NMR spectra were recorded on a Bruker Ascend 400 MHz instrument. Chemical shifts were measured relative to the signals of residual CHCl3 (δH 7.26 ppm, δC 77.16 ppm) or CD3OH (δH 4.87 ppm, δC 49.00 ppm) and are reported in ppm. HRMS data was obtained by ultrahigh performance liquid chromatography high resolution mass spectrometry (UHPLC-HRMS) on a maXis G3 quadrupole time of flight mass spectrometer (Bruker Daltronics, Bremen) equipped with an electrospray (ESI) source. Melting points were recorded on a Stuart SMP30 melting point apparatus and are uncorrected.1H NMR (400 MHz, CD3OD): δ 6.04 (d, J = 2.4 Hz, 2H), 4.74 (d, J = 2.3 Hz, 2H), 3.76 (s, 6H). 13C NMR (101 MHz, CD3OD): δ 174.3, 130.4, 71.9, 52.7. MS: m/z 205 (0.03) [MH+] 145 (11), 127 (44), 113 (12), 85 (100), 59 (28), 57 (60), 55 (18). M.p. 129.7–131.2 °C (recrystallized from ethyl acetate). Crystal data from single-crystal diffraction studies for dimethyl (2R,5S,E)-2,5-dihydroxyhex-3-enedioate. C8H12O6, space group P21/c, colorless, a = 7.8642(8) Å, b = 4.0675(3) Å, c = 14.5155(13) Å, V = 460.958 Å3, T = 120 K, Z = 2, Rint = 0.0553.
1H NMR (400 MHz, CDCl3): δ 5.81 (dtd, J = 15.1, 6.8, 1.3 Hz, 1H), 5.43 (ddt, J = 15.3, 6.2, 1.4 Hz, 1H), 4.54 (t, J = 5.0 Hz, 1H), 3.72 (s, 3H), 2.94 (d, J = 5.5 Hz, 1H), 1.99 (q, J = 7.0 Hz, 2H), 1.34–1.27 (m, 2H), 1.19 (s, 14H), 0.81 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 174.4, 135.1, 126.0, 71.6, 52.8, 32.2, 32.0, 29.7, 29.7, 29.6, 29.4, 29.2, 28.9, 22.8, 14.2. MS: m/z 256 (0.1) [M+], 197 (30), 109 (34), 95 (61), 81 (42), 67 (25), 57 (100), 55 (43), 43 (31), 41 (31). HRMS calcd for C15H29O3 [MH+]: 257.2111, found: 257.2108. For C15H28O3Na [MNa+]: 279.1931, found: 279.1929.
1H NMR (400 MHz, CDCl3): δ 5.90–5.79 (m, 1H), 5.47 (dd, J = 15.4, 6.1 Hz, 1H), 4.58 (d, J = 6.1 Hz, 1H), 3.76 (s, 3H), 3.00 (s, br, 1H), 2.03 (q, J = 7.0 Hz, 2H), 1.39–1.32 (m, 2H), 1.23 (s, 10H), 0.85 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 174.35, 135.01, 126.01, 71.52, 52.74, 32.22, 31.94, 29.48, 29.33, 29.21, 28.90, 22.73, 14.14. MS: m/z 228 (0.1) [M+], 169 (34), 109 (25), 95 (75), 83 (19), 81 (43), 71 (13), 70 (12), 69 (29), 67 (27), 57 (100), 55 (35), 43 (31), 41 (41). HRMS calcd for C13H24O3Na [MNa+]: 251.1618, found: 251.1624.
1H NMR (400 MHz, CDCl3): δ 5.89–5.77 (m, 1H), 5.46 (dd, J = 15.3, 6.1 Hz, 1H), 4.57 (d, J = 6.0 Hz, 1H), 3.74 (s, 3H), 3.11 (s, br, 1H), 2.01 (q, J = 7.0 Hz, 2H), 1.39–1.30 (m, 2H), 1.22 (s, 18H), 0.84 (t, J = 6.6 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 174.29, 134.85, 126.02, 77.16, 71.48, 52.63, 32.20, 31.97, 29.74, 29.71, 29.70, 29.66, 29.52, 29.41, 29.20, 28.89, 22.73, 14.12. MS: m/z 284 (0.2) [M+], 225 (32), 123 (15), 111 (16), 109 (35), 97 (17), 95 (59), 83 (35), 81 (48), 71 (16), 70 (20), 69 (47), 67 (30), 57 (100), 55 (42), 43 (48), 41 (46). HRMS calcd for C17H32O3Na [MNa+]: 307.2244, found: 307.2253; for C17H32O3K [MK+]: 323.1983, found: 323.1995.
1H NMR (400 MHz, CDCl3): δ 6.87 (dt, J = 15.7, 6.4 Hz, 1H), 5.78 (dt, J = 15.6, 1.5 Hz, 1H), 4.06 (q, J = 7.1 Hz, 2H), 3.64 (s, 3H), 2.49–2.35 (m, 4H), 1.17 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 172.1, 166.7, 147.0, 121.8, 60.6, 51.4, 32.4, 27.2, 14.1. MS: m/z 186 (0.1) [M+], 154 (32), 140 (29), 109 (45), 108 (100), 81 (58), 71 (26), 55 (27), 53 (31). The observed chemical shifts are in accordance with the literature values.33
1H NMR (400 MHz, CDCl3): δ 6.89 (dt, J = 15.7, 6.4 Hz, 1H), 5.80 (dt, J = 15.6, 1.5 Hz, 1H), 3.66 (s, 3H), 3.63 (s, 3H), 2.51–2.39 (m, 4H). 13C NMR (101 MHz, CDCl3): δ 172.7, 166.8, 146.9, 121.9, 51.8, 51.5, 32.2, 27.2. MS: m/z 173 (0.2) [MH+], 141 (34), 140 (84), 113 (26), 112 (24), 111 (16), 109 (41), 108 (100), 97 (16), 85 (11), 82 (20), 81 (87), 80 (24), 71 (68), 68 (10), 59 (52), 55 (24), 54 (14), 53 (59), 43 (11), 42 (12), 41 (36). The observed chemical shifts are in accordance with the literature values.34
1H NMR (400 MHz, CDCl3): δ 5.90 (ddd, J = 16.9, 10.5, 6.3 Hz, 1H), 5.50–5.46 (m, 1H), 5.45–5.41 (m, 1H), 5.36–5.29 (m, 1H), 3.71 (s, 3H), 2.12 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 170.0, 168.9, 130.0, 119.8, 73.0, 52.6, 20.6. MS: m/z 159 (0.1) [MH+], 116 (8), 99 (21), 43 (100). HRMS calcd for C7H11O4 [MH+]: 159.0652, found: 159.0652. For C7H10O4Na [MNa+]: 181.0471, found: 181.0471.
The reaction was allowed to cool down to r.t., then the mixture was treated with 15% (w/w) NaOH (20 mL) and washed with diethyl ether (2 × 20 mL) and the aqueous phase acidified with concentrated HCl while cooling in an ice bath. The solution was evaporated to dryness on a rotary evaporator and the residue extracted with Et2O. The organic phase was dried over Na2SO4, filtered and concentrated in vacuo to yield the crude product as a yellow solid. Yield: 0.6748 g, 4.7 mmol; 66%. Recrystallized from heptane/ethyl acetate. M.p. 162.2–163.2 °C (lit. 166–167 °C).35
1H NMR (400 MHz, CD3OD): δ 6.96 (dt, J = 15.5, 6.4 Hz, 1H), 5.88–5.79 (m, 1H), 2.53–2.44 (m, 4H). 13C NMR (101 MHz, CD3OD): δ 176.1, 169.8, 149.1, 123.2, 33.2, 28.3.
1H NMR (400 MHz, CDCl3): δ 6.85 (dt, J = 15.8, 4.6 Hz, 1H), 5.94 (dt, J = 15.8, 2.0 Hz, 1H), 4.65 (dd, J = 4.6, 2.0 Hz, 2H), 3.65 (s, 3H), 2.02 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 170.1, 166.1, 141.4, 121.7, 62.4, 51.6, 20.6. MS: m/z 116 (19) [MH − Ac+], 99 (23), 87 (13), 85 (23), 84 (11), 55 (15), 43 (100). The observed chemical shifts are in accordance with the literature values.36
Acknowledgements
We thank the Innovation Fund Denmark and Haldor Topsøe A/S for financial support.Notes and references
- T. J. Farmer and M. Mascal in Introduction to Chemicals from Biomass, ed. J. Clark and F. Deswarte, John Wiley & Sons Ltd., 2nd edn, 2015, pp. 89–155 Search PubMed.
- P. N. R. Vennestrøm, C. M. Osmundsen, C. H. Christensen and E. Taarning, Angew. Chem., Int. Ed., 2011, 50, 10502–10509 CrossRef PubMed.
- J. Sun and Y. Wang, ACS Catal., 2014, 4, 1078–1090 CrossRef CAS.
- (a) R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. López Granados, Energy Environ. Sci., 2016, 9, 1144–1189 RSC; (b) K. Yan, G. Wu, T. Lafleur and C. Jarvis, Renewable Sustainable Energy Rev., 2014, 38, 663–676 CrossRef CAS.
- (a) A. J. Kumalaputri, G. Bottari, P. M. Erne, H. J. Heeres and K. Barta, ChemSusChem, 2014, 7, 2266–2275 CrossRef CAS PubMed; (b) X. Hu, R. J. M. Westerhof, L. Wu, D. Dong and C.-Z. Li, Green Chem., 2015, 17, 219–224 RSC; (c) J. J. Pachecho, J. A. Labinger, A. L. Sessions and M. E. Davis, ACS Catal., 2015, 5, 5904–5913 CrossRef.
- (a) M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602–605 CrossRef CAS PubMed; (b) M. S. Holm, Y. J. Pagán-Torres, S. Saravanamurugan, A. Riisager, J. A. Dumesic and E. Taarning, Green Chem., 2012, 14, 702–706 RSC.
- M. Dusselier, P. Van Wouwe, F. de Clippel, J. Dijkmans, D. W. Gammon and B. F. Sels, ChemCatChem, 2013, 5, 569–575 CrossRef CAS.
- R. De Clercq, M. Dusselier, C. Christiaens, J. Dijkmans, R. I. Iacobescu, Y. Pontikes and B. F. Sels, ACS Catal., 2015, 5, 5803–5811 CrossRef CAS.
- S. Tolborg, S. Meier, I. Sádaba, S. G. Elliot, S. K. Kristensen, S. Shunmugavel, A. Riisager, P. Fristrup, T. Skrydstrup and E. Taarning, Green Chem., 2016, 18, 3360–3369 RSC.
- Y. D. Hang and E. E. Woodams, Lebensm. – Wiss. Technol., 2001, 34, 140–142 CrossRef CAS.
- See ESI† for full calculation.
- H. Stach, W. Huggenberg and M. Hesse, Helv. Chim. Acta, 1987, 70, 369–374 CrossRef CAS.
- K. E. Koenig, WO9832735A1, 1998 Search PubMed.
- (a) M. Freiría, A. J. Whitehead and W. B. Motherwell, Synlett, 2003, 805–808 Search PubMed; (b) M. Freiría, A. J. Whitehead, D. A. Tocher and W. B. Motherwell, Tetrahedron, 2004, 60, 2673–2692 CrossRef.
- M. Dusselier, P. Van Wouwe, A. Dewaele, P. A. Jacobs and B. F. Sels, Science, 2015, 349, 78–80 CrossRef CAS PubMed.
- M. Dusselier, P. Van Wouwe, S. De Smet, R. De Clercq, L. Verbelen, P. Van Puyvelde, F. E. Du Prez and B. F. Sels, ACS Catal., 2013, 3, 1786–1800 CrossRef CAS.
- M. T. Stone, Org. Lett., 2011, 13, 2326–2329 CrossRef CAS PubMed.
- (a) T. Terasaka, K. Nakamura, N. Seki, M. Kuno, S. Tsujimoto, A. Sato, I. Nakanishi, T. Kinoshita, N. Nishio, H. Okumura and K. Tsuji, WO00/05217, 2000 Search PubMed; (b) F. Sakai, N. Seki, Y. Tenda, H. Yamazaki, C. Miyamoto, M. Kuno, H. Okumura and K. Nakamura, WO01/26605A2, 2001 Search PubMed.
- V. I. Ognyanov and M. Hesse, Synthesis, 1985, 645–647 CrossRef CAS.
- (a) M. A. Boudreau and J. C. Vederas, Org. Biomol. Chem., 2007, 5, 627–635 RSC; (b) W. P. Unsworth, K. Stevens, S. G. Lamont and J. Robertson, Chem. Commun., 2011, 47, 7659–7661 RSC.
- J. Chanet-Ray, M. O. Charmier-Januario, R. Vessière and M. Zucarelli, J. Heterocycl. Chem., 1994, 31, 1667–1671 CrossRef CAS.
- A. J. Pierik, T. Graf, L. Pemberton, B. T. Golding and J. Rétey, ChemBioChem, 2008, 9, 2268–2275 CrossRef CAS PubMed.
- N. Lebrasseur, G.-J. Fan, M. Oxoby, M. A. Looney and S. Quideau, Tetrahedron, 2005, 61, 1551–1562 CrossRef CAS.
- For a recent review on industrial applications of olefin metathesis, see: C. S. Higman, J. A. M. Lummiss and D. E. Fogg, Angew. Chem., Int. Ed., 2016, 55, 3552–3565 CrossRef CAS PubMed.
- (a) R. Pettigrew and P. Tissington, US3822222A, 1974 Search PubMed; (b) T. Koizumi and H. Matsuzawa, US4802999A, 1989 Search PubMed; (c) A. A. El-Sawy, S. A. Essawy, M. M. El-Sukkary and A. M. F. Eissa, Hung. J. Ind. Chem., 1992, 20, 25–28 CAS; (d) R. J. Yu and E. J. Van Scott, WO96/40047, 1996 Search PubMed.
- A. Dewaele, L. Meerten, L. Verbelen, S. Eyley, W. Thielemans, P. Van Puyvelde, M. Dusselier and B. Sels, ACS Sustainable Chem. Eng., 2016, 4 DOI:10.1021/acssuschemeng.6b00807.
- T. W. Abraham, H. Kaido, C. W. Lee, R. L. Pederson, Y. Schrodi and M. J. Tupy, US20090264672A1, 2009 Search PubMed.
- A. K. Chatterjee, T.-L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360–11370 CrossRef CAS PubMed.
- W. S. Johnson, L. Werthemann, W. R. Bartlett, T. J. Brocksom, T. Li, J. Faulkner and M. R. Petersen, J. Am. Chem. Soc., 1970, 92, 741–743 CrossRef CAS.
- R. E. Ireland and R. H. Mueller, J. Am. Chem. Soc., 1972, 94, 5897–5898 CrossRef CAS.
- L. E. Overman and F. M. Knoll, Tetrahedron Lett., 1979, 20, 321–324 CrossRef.
- D. S. Pedersen and C. Rosenbohm, Synthesis, 2001, 2431–2434 CAS.
- J. Y. Lee and S. Kim, Synlett, 2008, 49–54 Search PubMed.
- E. Hauptman, S. Sabo-Etienne, P. S. White, M. Brookhart, J. M. Garner, P. J. Fagan and J. C. Calabrese, J. Am. Chem. Soc., 1994, 116, 8038–8060 CrossRef CAS.
- M. Viscontini and H. Köhler, Helv. Chim. Acta, 1954, 37, 41–45 CrossRef CAS.
- R. V. Hoffman and B. S. Severns, J. Org. Chem., 1996, 61, 5567–5573 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra with assignments for all compounds. CCDC 1481046 (crystal structure in Fig. 2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6gc01556e |
| This journal is © The Royal Society of Chemistry 2016 |