
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReductive splitting of hemicellulose with stable ruthenium-loaded USY zeolites†
Thijs
Ennaert
,
Simon
Feys
,
Don
Hendrikx
,
Pierre A.
Jacobs
and
Bert F.
Sels
*
Centre for Surface Chemistry and Catalysis, Faculty of Bioscience Engineering, KU Leuven, Celestijnenlaan 200f, B-3001 Heverlee, Belgium. E-mail: bert.sels@biw.kuleuven.be
First published on 6th June 2016
Abstract
Reductive catalytic splitting to sugar alcohols is a promising technology to valorize (hemi)cellulosic feedstock. This contribution focuses on the conversion of arabinoxylan (AX), a common hemicellulose polymer, to pentitols like xylitol and arabitol in the presence of ruthenium-loaded H-USY zeolites. Both acid and metal sites on the catalyst play a crucial role in the bifunctional catalytic mechanism. Overall, the reaction mechanism involves hydrolysis of AX into shorter (less reactive) xylan oligomer intermediates (XOs), which are in turn hydrolysed into sugar monomers. The first step occurs fast in hot liquid water, but the second step which is rate limiting, requires acid catalysis. Literature has reported successful XO hydrolysis with soluble acids. However, USY zeolites, being non-corrosive instead of the former, are able to hydrolyse XOs more efficiently, likely due to their strong mesopore adsorption capacity. Once formed, the monomeric sugars should be hydrogenated on the metal sites as fast as possible, as otherwise undesired competitive acid catalysed side-reactions will occur. While another catalyst like Ru on carbon can also be used in the one-pot approach close proximity of the two sites, e.g. in the pores of the USY zeolite, is beneficial for the pentitol selectivity, as long as they are well harmonised. After searching for the ideal dual site balance, exceptionally high pentitol yields up to 90 mol% were achieved after only 5 h of reaction. Comparison with earlier reported cellulose reactions shows a narrowing of the ideal acid-to-metal range, besides a shift to lower ratios. Initial regeneration studies show a stable Ru/USY catalytic system able to perform multiple reaction runs with retention of activity and selectivity.
Introduction
Renewable biomass, like carbohydrates, is a sustainable alternative feedstock for the production of highly oxygenated chemical compounds.1–3 Lignocellulose, containing cellulose (40–50 wt%) and hemicellulose (25–40 wt%) next to the aromatic polymer lignin (10–25 wt%), is the most abundant biomass feedstock. Fractionation, usually involving solvent-based extractions in the presence of an acid or a base, isolates the sugar part, which may be converted into valuable chemicals and fuels with bio- and chemocatalytic processes.3–16 Integrated concepts, in which metal catalysis is applied during lignocellulose solvolysis, such as in the ‘lignin-first’ approach, are also emerging.17–24 Hereby, raw lignocellulose is processed in a polar organic solvent at elevated temperatures in the presence of a metal catalyst into a stable lignin oil and a solid pulp. While the lignin oil, containing monomeric and dimeric o-methoxyphenols, can be upgraded to a wide variety of chemicals, including for example phenol, cyclohexanone derivatives, and adipic acid,25–27 processing of the pulp into fuel compounds, like light naphtha, and chemicals like polyols, anhydropolyols and smaller sugars, has also been demonstrated.28–36The essential step in pulp valorisation to chemicals is the hydrolytic breakdown of the carbohydrate polymer into the monomeric sugar units. The recalcitrant structure of the macromolecules, especially of cellulose, demands relatively harsh hydrolysis conditions, viz. the presence of substantial acid quantities and/or temperatures above 150 °C.7 Alternatively, a mechanical or plasma pretreatment can be applied before or integrated in the catalytic conversion step.37–41 The monosaccharides are very reactive and prone to degradation under such conditions into furanic products, like 5-hydroxymethylfurfural and furfural, and levulinic acid. These products are very sensitive to further degradation into insoluble humin products. The fast reaction of the sugar monomers, for instance into stable polyols under reduced conditions in the presence of a metal catalyst, is an elegant solution to avoid such undesired side-reactions.42 The various polyol products are well-integrated in the current chemical industry, and they may serve as a platform for other chemicals.43
Cellulose to polyols, like sorbitol, sorbitans and isosorbide, using a combination of acid and metal catalysis has been researched extensively.11,42,44–64 Similar valorisation of hemicellulose seems somewhat overlooked, though its amorphous nature should predict a much easier reductive hydrolysis. Yet, only a few reports describe considerable C5 sugar alcohol yields from concentrated hemicellulose streams. Yields between 10 and 25% were obtained for instance after 4–24 hours of reaction using heterogeneous catalysis (see Table 1, entries 1–4).65–68 Few studies reported higher sugar alcohol yields in the presence of sulfuric acid (see Table 1, entries 8 and 9).69–71 Finally, some reports dealing with the formation of sugar alcohols from real biomass are also published (Table 1, entries 5–7 and 10).45,72–74 Herein, the obtained pentitol yields show a large variety: from 20–25%72,73 to nearly quantitative yields.74 Note that the latter are exclusively obtained for substrates with a very low C5 sugar content (<6%), as clearly illustrated by entries 6 and 7. So, the need for an efficient heterogeneous system for converting concentrated hemicellulose streams still exists. The reasons for the low pentitol yields with solid catalysts, whereas quantitative yields were reported for cellulose, are barely understood and demand a detailed study. Better knowledge probably should lead to a better definition of a heterogeneous catalytic system for hemicellulose upgrading, if possible.
| Entry | Catalyst | Substratea | T (°C) | t (h) | Metal loading (wt%) | Acidityb (mM) | Y (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a C5 sugar content given in parentheses for the reactions with lignocellulose samples. b Values in parentheses are the amount of Brønsted acidity (in μmol) per gram of catalyst. c Sugar alcohol yield. d AG = arabinogalactan. e Sulfonated mesoporous carbon. f n.g. = not given. g 3% Ru and 1% Pt. h Acidity in mM is the sum of the HCl concentration and zeolite acidity, while acidity in μmol g−1 only comprises the zeolite acidity. | ||||||||
| Heterogeneous acid and hydrogenation catalyst | ||||||||
| 1 | Ru/beta | AGd | 185 | 4 | 4.5 | 0.4 (198) | 10 | 75 |
| 2 | Ru/MCM-48 | AGd | 185 | 24 | 5 | 0.12 (58) | 25 | 66 |
| 3 | Sulf. mesoporous carbone + Ru/C | AGd | 185 | 4 | 4.6 | 5.8 (2900) | 25 | 68 |
| 4 | Ru/USY | AGd | 185 | 4 | 2.5 | 0.46 (231) | 25 | 67 |
| 5 | Pt/BP2000 | Silver grass (24) | 190 | 24 | 1.3 | n.g.f | 20 | 72 |
| 6 | Ru(N)Pt/C | Bagasse (24) | 190 | 16 | 3 and 1g | — | 24 | 73 |
| 7 | Pt/C | Cedar (6) | 190 | 16 | 4 | — | 97 | 74 |
| Homogeneous acid + heterogeneous hydrogenation catalyst | ||||||||
| 8 | H2SO4 + Ir-ReOx/SiO2 | Xylan | 140 | 12 | 4 | 32 | 80 | 69 |
| 9 | H2SO4 + Ru/C | Xylan | 140 | 3 | 5 | 16 | 83 | 70, 71 |
| 10 | HCl + Ru/USY | Birch (20) | 190 | 24 | 0.2 | 6 (519)h | 53 | 45 |
This contribution therefore studies the one-pot conversion of arabinoxylan (AX), a common copolymer of xylose and arabinose found in the primary and secondary cell walls of plants, into their C5 sugar alcohol analogues, xylitol and arabitol (in this paper referred to as ‘pentitols’), in the presence of Ru-loaded H-USY zeolites (Ru/USY). These pentitols are valuable chemicals with high demand in industry as platform chemicals and low-calorie sweeteners, next to their use as anti-cariogenic agents.43 Similar Ru-loaded zeolites have been used successfully in the conversion of cellulose into polyols, yielding hexitols quantitatively.44 Best catalytic requirements with regard to active site accessibility and acid-to-metal balance will be searched in order to obtain complete conversion into pentitols.
Scheme 1 summarizes the main critical steps of such a reductive splitting process, emphasizing the role of acid and metal catalysis. Acidity is required to hydrolyse the insoluble AX into soluble xylo-oligomers (XO) and further into xylose and arabinose. Hydrogenation is foreseen in this study at the Ru sites in the zeolite. This study will demonstrate the importance of different sites and attempts to quantify the acid-to-metal balance to achieve high pentitol yields. While some acidity in solution may be helpful to increase the reaction rate, the acidity in the zeolite is demonstrated to be most effective in hydrolysing the (more persistent) soluble oligomers, likely due to strong adsorption phenomena. Reaction selectivity is largely determined by the spatial proximity of the acid and metal sites and their molar site balance. Designing metal-loaded acidic mesoporous zeolites ultimately proves successful, yielding close to quantitative amounts of pentitols from commercial AX hemicellulose.
 | ||
| Scheme 1 Reaction scheme of the reductive splitting of arabinoxylan (AX) to xylitol and arabitol (pentitols). | ||
Experimental
The USY zeolites are commercially available from Zeolyst International. Detailed structural and compositional information of the different zeolite catalysts can be found in Table 2 and in the ESI.† Ru5/C and arabinoxylan from beech wood (AX) were purchased and used as received from Sigma-Aldrich.| Sample codea | USY zeolite | Bulk Si/Alb | Post-synthetic modification | Brønsted acidity (μmol g−1) | Pore volume (mL g−1) | |||
|---|---|---|---|---|---|---|---|---|
| Steaming | Acid washing | Micropore volume | Mesopore volume | Total pore volume | ||||
| a The sample code is based on the framework T atom fraction of each zeolite as calculated and used in ref. 44. b The framework Si/Al ratios can be found in Table S1 (together with the distribution of framework and extra-framework aluminium). | ||||||||
| USY19 | CBV500 | 2.6 | 1× (mild) | — | 519 | 0.27 | 0.02 | 0.30 |
| USY9 | CBV712 | 6.0 | 1× | Mild | 413 | 0.29 | 0.15 | 0.44 |
| USY6 | CBV720 | 15.0 | 1× | Extensive | 200 | 0.30 | 0.13 | 0.44 |
| USY3 | CBV760 | 30.0 | 2× | Extensive | 127 | 0.31 | 0.15 | 0.46 |
The Ru metal-loaded USY zeolites (Ru/USY) were prepared via ion-exchange with an appropriate amount of aqueous 0.1 mM hexaammineruthenium(III) chloride (Sigma-Aldrich). Ru-loaded zeolites were consequently activated at 400 °C under a flow of H2via an earlier reported procedure.47 In a typical reaction, a 100 mL stainless steel autoclave (Parr Instruments) was loaded with 1 g of xylan, 0.5 g Ru/H-USY catalyst and 50 mL of water, unless otherwise stated. This mixture was stirred at 750 rpm. A control reaction at 500 rpm showed no external mass transfer issues under the reaction circumstances. The reactor was flushed with N2 prior to reaction to remove air and heated to the desired reaction temperature (mostly 160 °C). The reactor was then pressurized at 5 MPa H2. This moment was considered as the start of the reaction. During the reaction, various samples were taken from the reactor at appropriate time intervals and immediately cooled in an ice bath after sampling. Myo-inositol was added as an external standard to 0.5 mL of each sample followed by the evaporation of the water. Afterwards, the obtained solid fraction was dissolved in pyridine and silylated using N-methyl-N-(trimethylsilyl) trifluoroacetamide (Sigma-Aldrich), prior to GC analysis on a Hewlett-Packard 5890 GC equipped with a 50 m CP-Sil-5CB column and a FID detector. Next to GC analysis, the (non silylated) aqueous samples were also analysed on an Agilent 1200 series HPLC equipped with a Varian Metacarb 67C column (300 × 6.5 mm) and a RI detector to determine and quantify the main by-products. Due to the multiplicity of the signals in the obtained chromatogram, it was not possible to make a distinction between non-reduced and reduced XO,76 therefore, the term ‘XO’ includes all xylo-oligomers. All product yields are expressed in C mol% and are based on the total amount of C moles, originating from the sugars in the AX substrate. The standard error of the whole system (including catalyst preparation, catalysis and analysis) is 2%.
The zeolite Brønsted acid density (in μmol g−1) was determined by FTIR-monitored adsorption of pyridine (Acros Organics) vapour. FTIR spectra with a resolution of 2 cm−1 were recorded on a Nicolet 6700 spectrometer with a DTGS detector, requiring accumulation of 128 scans per spectrum. Self-supported zeolite wafers of 5–10 mg cm−2 positioned in a temperature-controlled vacuum cell with ZnSe windows were dried at 400 °C for 1 h. After cooling, a FTIR spectrum was scanned at 150 °C as the blank. Pyridine was adsorbed at 50 °C in pulses until saturation was achieved, after which the sample was heated to 150 °C and evacuated for 20 min. Finally, the pyridine-probed spectrum was recorded at this temperature. The latter spectrum was corrected by subtracting the blank spectrum of the degassed and pyridine-free zeolite sample. The band intensities around 1545 cm−1, normalized for sample mass differences, were used to quantify Brønsted acid density, using the integrated molar extinction coefficient proposed by Emeis.77
Before measuring the porosity, samples were pretreated in N2 at 250 °C for 6 h. The porosity was derived from a nitrogen adsorption–desorption isotherm at −196 °C using a Micromeritics TriStar 3000. Usually, 30 nitrogen uptake points were needed before the nitrogen saturation pressure was reached. Desorption occurred stepwise until saturation before proceeding to the next step. While the micropore volume was calculated from the t-plot, the mesopore volume was taken as the difference between the total pore volume and micropore volume.
Ruthenium dispersion of Ru/USY catalysts was determined with CO-chemisorption. After pelletisation between 250 and 500 μm, catalyst particles were loaded into a tubular quartz reactor. After activation at 400 °C under H2, they were cooled under He to room temperature. For titration of the Ru particle surface, pulses of 5 μL of pure CO were added to a He flow of 10 mL min−1 at an interval of 2 min. The CO concentration in the outlet was continuously monitored at m/z = 28 with a Pfeiffer Omnistar quadrupole mass spectrometer. For quantification, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CO to Ru surface ratio was assumed.
:1 CO to Ru surface ratio was assumed.
Results and discussion
Catalytic evaluation of Ru-based catalytic systems
Metal-loaded acidic zeolites are in principle well suited to facilitate bifunctional reaction mechanisms,9 such as those presented in Scheme 1. A mildly steamed Y zeolite (USY9, faujasite topology) with an intermediate Si/Al ratio and Brønsted acidity (see Tables 2 and S1† for compositional information and acidity quantification) was therefore loaded with 0.6 wt% Ru, whereby its ability to convert AX hemicellulose into pentitols was tested for different catalyst weight loadings (Table 3, entries 1–6). To anticipate a hindered interaction of the zeolite acidic sites with large (insoluble) sugar polymers, reactions were also carried out in the presence of minute amounts (ppm level) of HCl (Table 3, entries 2, 4 and 6), which in accord with earlier reports44,47 is expected to facilitate the solubilisation of AX into XO (see Scheme 1), if required. To demonstrate the importance of proximity between the acid and metal sites, a metal-free acidic USY9 in combination with commercial Ru5/C, was used as reference (Table 3, entry 7). The importance of zeolitic acidity (in contrast to that of soluble acidity) is clarified by comparing the abovementioned zeolite catalysed reactions with reactions catalysed by Ru5/C in the presence of various HCl concentrations (Table 3, entries 8–11).| Entry | Catalysta | Total amount of zeolite acidity (μmol) | Added amount of HCl (μmol) | Time (h) | Yield (mol% C) | |||
|---|---|---|---|---|---|---|---|---|
| Pentitols | XOb | Degradation productsc | Humins | |||||
| a The amount of Ru on every support/catalyst is denoted as wt%. b XO = xylo-oligomers. c The term ‘degradation products’ contain the side-products formed via thermal or acid catalysed dehydration of sugars, like furfural, furfuryl alcohol, levulinic acid, etc. d 0.25 g zeolite catalyst. e 0.5 g zeolite catalyst. f 1 g zeolite catalyst. g 0.030 g Ru/C. | ||||||||
| 1 | Ru0.6/USY9d | 103 | — | 24 | 35 | 49 | 1 | 8 |
| 2 | Ru0.6/USY9d | 103 | 41 | 24 | 49 | 25 | 8 | 13 |
| 3 | Ru0.6/USY9e | 207 | — | 24 | 74 | 8 | 5 | 8 |
| 4 | Ru0.6/USY9e | 207 | 41 | 24 | 79 | 1 | 7 | 10 |
| 5 | Ru0.6/USY9f | 413 | — | 5 | 90 | 0 | 4 | 2 |
| 6 | Ru0.6/USY9f | 413 | 41 | 5 | 89 | 0 | 2 | 3 |
| 7 | Ru5/Cg + USY9 | 207 | — | 24 | 58 | 6 | 18 | 17 |
| 8 | Ru5/Cg | — | — | 24 | 4 | 74 | 9 | 4 |
| 9 | Ru5/Cg | — | 41 | 24 | 5 | 66 | 6 | 24 |
| 10 | Ru5/Cg | — | 203 | 24 | 32 | 30 | 11 | 27 |
| 11 | Ru5/Cg | — | 405 | 24 | 49 | 0 | 14 | 37 |
The catalytic reactions were carried out with an initial 2 wt% AX loading at 160 °C and under 50 bar H2 atmosphere. The conversion of AX is defined as the transformation of insoluble AX into soluble organic carbon (like XO, pentitols and others), as it is also reported for cellulose conversion.46 As expected, AX hemicellulose is rapidly dissolved, viz. already 100% within 1 h, forming XO as the main product. Since fast dissolution also appears in the absence of an acidic catalyst, it is fair to assume that AX to XO dissolution is mainly assisted by hydronium ions produced via water autoprotolysis at an elevated temperature.
Ru/C is known to efficiently hydrogenate monomeric sugars into their corresponding polyols. A test reaction with commercial Ru5/C indeed shows high yield of and selectivity towards xylitol upon xylose hydrogenation (Fig. 1). In contrast, the reaction of commercial AX hemicellulose in the presence of active Ru5/C for 24 hours shows a very low pentitol yield (<5%, Table 3, entry 8). Since soluble XOs are the dominant products, their hydrolysis to monosaccharides seems more difficult than expected. Addition of substantial amounts of mineral acids is helpful to depolymerize XO. The pentitol yield thus increases gradually with the acid concentration at the expense of XO. For instance, a reaction in the presence of 8 mM HCl gives a 49% pentitol yield, whereas it is 5% in the presence of 0.8 mM HCl (compare entries 11 and 9). Obtaining higher pentitol yields by further increasing the HCl concentration is likely not easy under the present conditions as all XO intermediates are already completely converted in the presence of 8 mM HCl. Moreover, detailed inspection of the product distribution shows that huge amounts of dehydration products and humins are formed.
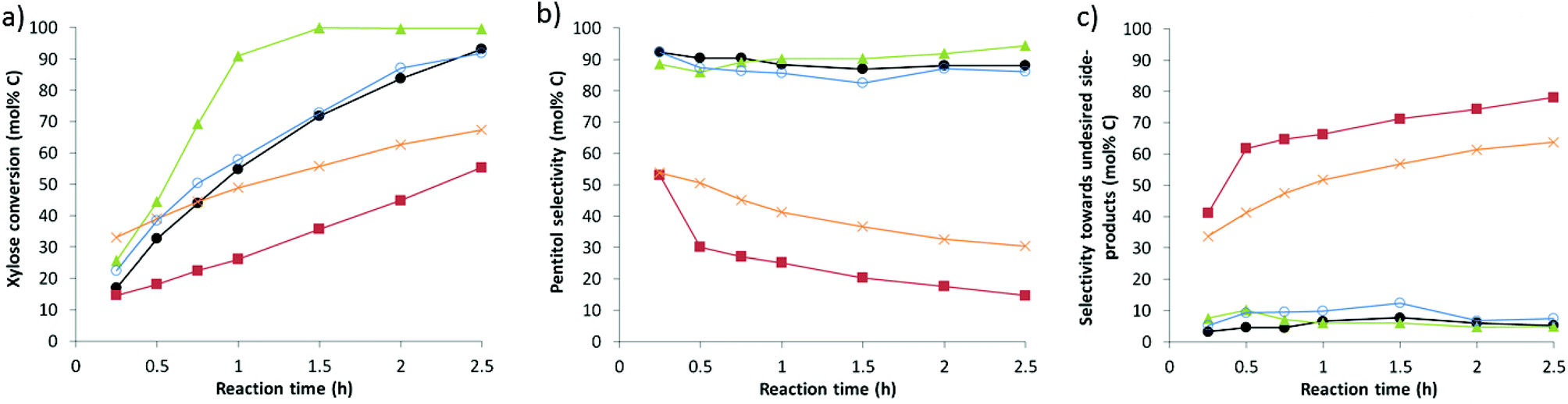 | ||
| Fig. 1 Xylose hydrogenation reactions with Ru/C (black filled circles), Ru0.6/USY9 (red squares), Ru0.6/USY3 (green triangles), Ru/C + HCl (orange crosses) and HCl treated Ru/C (blue open circles): (a) xylose conversion, (b) pentitol selectivity and (c) selectivity towards undesired side-products (xylose dehydration products and humins). | ||
Instead of using soluble acids, an acidic USY9 zeolite was added (entry 7) in the standard Ru5/C reaction. Clearly, a much higher pentitol yield, viz. 58%, was observed despite the lower absolute content of acidic sites and the non-corrosive nature of the reaction solution. Main by-products are strong acid catalysed dehydration, rearrangement and condensation products, alike those appearing in reactions with a high HCl concentration. Yet, USY9 converts more XO at an equimolar proton concentration (Table 3, entries 7 and 10) and is thus capable of further dissolving XO into monosaccharides, which were largely hydrogenated by Ru5/C into pentitols.
Even better results were obtained by locating Ru on the zeolite USY9, instead of using Ru5/C. Indeed, for the same acidity (zeolite loading, see Table 3, entries 3 and 7), a much higher pentitol yield of 74% was observed in the presence of Ru0.6/USY9. Addition of minute amounts of HCl only slightly improved the pentitol yield up to 79% (Table 3, entry 4), pointing that deep polymer solubilisation by using a soluble acid only minorly affects the reaction outcome. The absolute amount of acidity (zeolite loading) has a tremendous effect on the pentitol yield. Increasing the zeolite loading shows a gradual increase of the pentitol yields. For instance, the pentitol yield increases from 35 to 90%, mainly at the cost of intermediate XOs, by increasing the zeolite loading (added acidity in μmol) from 0.5 wt% (103 μmol) to 2 wt% (413 μmol) (compare entries 1, 3 and 5). No soluble HCl is required here to obtain the highest pentitol yields (see Table 3, entries 5 and 6).
Role of zeolite acidity versus soluble acidity
The previous part reveals that proton acidity of a zeolite is more effective than that of soluble protons and that the effectivity gain is largely found in the hydrolysis of soluble XO oligomers.It is known that soluble sugar oligomers strongly adsorb (rate and capacity) in mesoporous materials like USY zeolites.78,79 Such a strong adsorption leads to a strong interaction with the local zeolite protons in a restricted volume. The high local substrate and acid concentrations result in a fast hydrolysis in the zeolite pores, explaining the strongly reduced XO levels after a reaction with Ru5/C + USY9 compared to a reaction with Ru5/C + HCl, despite the equimolar proton concentration (compare entries 7 and 10 in Table 3). Note that both reactions show a large formation of acid catalysed side-reactions, as reflected in the amount of degradation and humin products.
Degradation and humin products are largely secondary (or later) reactions from the monosaccharides, and may therefore be expected if their release is faster than their hydrogenation. A first series of experiments were carried out to prove that Ru supported on both USY and carbon is capable of rapidly hydrogenating xylose into xylitol. In order to circumvent (minimize) a possible role of acidity, both reactions were carried out in the absence of (or presence of low) acidity. In the case of Ru on zeolite, Ru/USY3 was selected for its significantly lower acidity content (see Table 2). The kinetic profiles and selectivity patterns are displayed in Fig. 1. Reactions were carried out at 160 °C for 2.5 hours.
Both catalyst samples show a fast and selective hydrogenation of xylose, the higher rate of Ru0.6/USY3 being in line with the larger Ru dispersion in the zeolite compared to that on the carbon support (as reflected in the number of surface Ru sites measured by CO chemisorption, 5.6 mmol and 2.7 mmol, respectively). The presence of acidity, either by adding HCl to Ru5/C or by using the more acidic Ru0.6/USY9, affects both the hydrogenation rate as well as the selectivity. Conversion rates (per Ru site) are less stable (in the case of carbon) and slower (in the case of zeolites). Catalyst deactivation due to the presence of HCl is excluded, since HCl pretreated Ru/C shows exactly the same high hydrogenation rate. The presence of acidity had also an enormous detrimental impact on the selectivity, in accordance with the highly reactive nature of xylose towards acids. As long as no (little) acidity is present, high pentitol selectivities around 90% were observed. These values indeed dropped below 40% and 20% for Ru5/C + HCl and Ru0.6/USY9, respectively (Fig. 1b), at incomplete conversion. Instead, mainly products originating from acid catalysed side-reactions are observed. These experiments explain the low pentitol yields from AX with the Ru5/C + HCl system: at the high HCl concentrations needed to efficiently depolymerize XO, competitive acid catalysed reactions prevail resulting in less selective xylose hydrogenation and consequently lower pentitol yields.
Though zeolite acidity is required (and more effectively than soluble acidity) to hydrolyse XO, it is also detrimental to the pentitol selectivity. Therefore, close proximity of the metal site to the acid site should beneficially influence the reaction selectivity. Indeed, the addition of an acidic zeolite to Ru5/C showed moderate pentitol selectivity, whereas the pentitol selectivity was significantly higher in the presence of Ru0.6/USY9 for the same zeolite (acid) content (compare entries 3 and 7 in Table 3).
Effect of the acid-to-metal balance on the pentitol yield
The above part suggests that the zeolite acidity determines the reaction rate, XO hydrolysis being the slowest step, but it also suggests that the balance of acid-to-metal could affect the reaction selectivity. Even when the acid and metal sites are in close proximity in the zeolite, a misbalance of both will largely influence the pentitol selectivity. A too high acid-to-metal ratio is expected to lead to acid degradation products instead of selective pentitol formation.Closer inspection of such balances was attempted by synthesizing various Ru-loaded zeolites with different Ru and acid contents. A series of USY zeolites, different in the Si/Al ratio and thus the acidity content, were loaded with different Ru amounts. Table 4 summarizes the investigated Ru-loaded zeolites, together with their total number of acid and metal sites. The molar acid-to-metal site ratio and the reaction outcome are also reported in Table 4.
| Entry | Zeolite catalysta | Acidity (μmol g−1) | Available Ru sites (μmol g−1) | n acid/nmetal | Yield (mol% C) | |||
|---|---|---|---|---|---|---|---|---|
| Pentitols | XOb | Degradation productsc | Humins | |||||
| a The amount of Ru on every support/catalyst is denoted as wt%. b XO = xylo-oligomers. c The term ‘degradation products’ contain the side-products formed via thermal or acid catalysed dehydration of sugars, like furfural, furfuryl alcohol, levulinic acid, etc. | ||||||||
| 1 | Ru0.3/USY19 | 497 | 20 | 25 | 46 | 5 | 25 | 23 |
| 2 | Ru0.2/USY9 | 398 | 14 | 28 | 38 | 0 | 23 | 33 |
| 3 | Ru0.3/USY9 | 393 | 19 | 20 | 75 | 5 | 12 | 1 |
| 4 | Ru0.45/USY9 | 386 | 24 | 16 | 70 | 7 | 10 | 4 |
| 5 | Ru0.6/USY9 | 379 | 28 | 14 | 74 | 8 | 5 | 8 |
| 6 | Ru0.9/USY9 | 362 | 34 | 11 | 61 | 17 | 5 | 13 |
| 7 | Ru1.2/USY9 | 355 | 40 | 9 | 50 | 32 | 7 | 6 |
| 8 | Ru1.5/USY9 | 320 | 50 | 6 | 44 | 37 | 5 | 9 |
| 9 | Ru0.3/USY6 | 196 | 26 | 8 | 41 | 29 | 10 | 6 |
| 10 | Ru0.3/USY3 | 122 | 28 | 4 | 26 | 41 | 9 | 6 |
Plots of the data in Fig. 2 nicely illustrate the impact of zeolite acidity on the conversion of XO (a), and the effect of the balance on the pentitol selectivity (b). Higher zeolite acidity roughly leads to a lower XO content (and thus more polymer hydrolysis), whereas an optimal balance clearly exists to achieve high pentitol selectivity (Fig. 2 and S2†). The highest selectivity here, being 80%, is obtained for a Ru-loaded zeolite with an acid-to-metal site ratio in the range of 15 to 20, valid under the applied reaction conditions, viz. 2 wt% AX hemicellulose, 160 °C and 50 bar hydrogen pressure. Note that this ratio strongly depends on the used reaction conditions, as illustrated for the temperature in the ESI (Fig. S2b†).
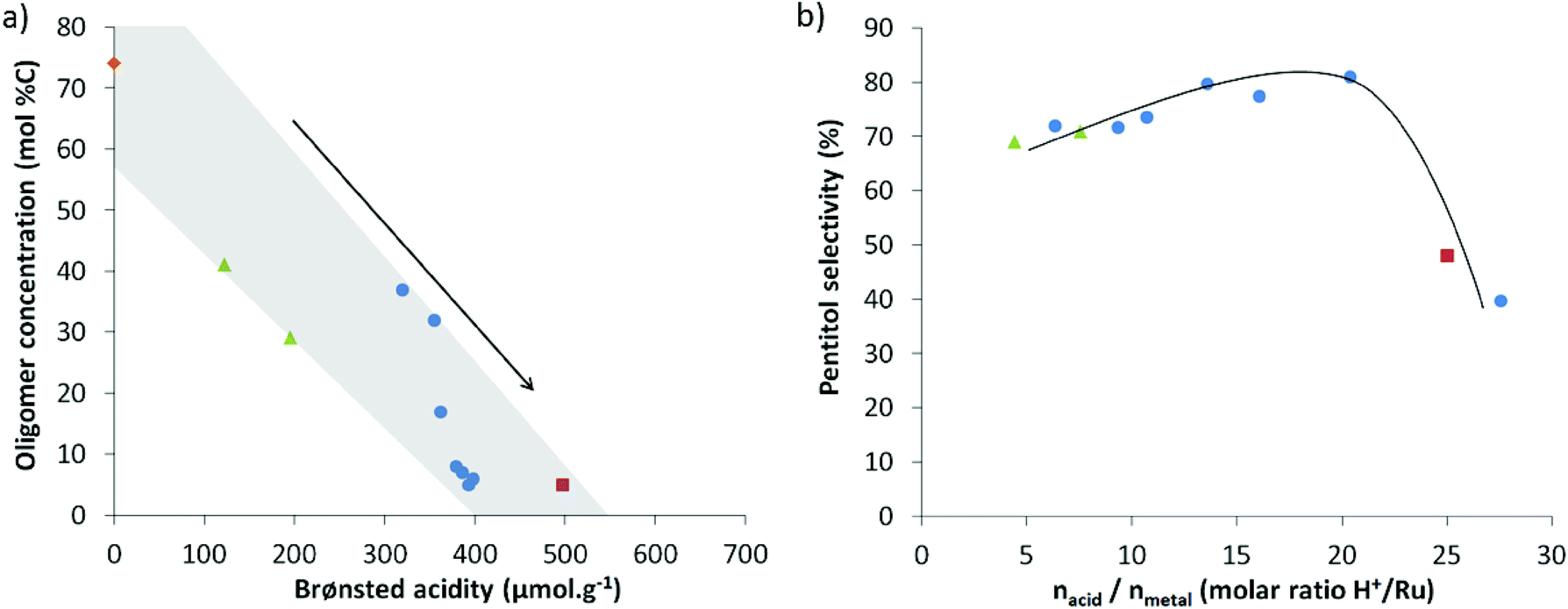 | ||
| Fig. 2 (a) Influence of the Brønsted acidity on the XO conversion and (b) impact of the molar acid/metal ratio on the pentitol selectivity (which is defined as (Ypentitol)/(Ypentitol + Ydegradation products + Yhumins) × 100): USY9 catalysts (filled blue circles), highly acidic USY19 (red square), low acid USY zeolites (green triangles) and Ru/C (orange rhombus). | ||
These trends may be well illustrated with examples in Table 4. Catalysts with low acid-to-metal ratios, like Ru0.3/USY6 and Ru0.3/USY3 (entries 9 and 10), indeed show slow conversion of XO, whereas catalysts with the optimal acid-to-metal site balance secure a high pentitol selectivity (largely irrespective of the XO content) (entries 3, 4 and 5). A too high acidity in the catalytic system forms acid catalysed dehydration products, as exemplified by the data in entries 1 and 2. A slight decrease in selectivity for catalytic systems with a high metal site content is observed based on the data in entries 6 to 8, and the data in Fig. 2b and S1.† Hydrogenolysis of pentitols to shorter polyols clarifies the small drop in selectivity here.80,81
Catalytic activity after regeneration
One of the advantages of heterogeneous catalysts over the homogeneous ones is the easy recyclability of the catalyst, provided a preservation of the activity of the spent catalysts in consecutive runs. However, there are several imaginable deactivation pathways. Fouling of the sites by humins is one possibility, but also the structural integrity of the zeolite (acidity)44,82–84 as well as sintering of Ru under the hot water conditions9,85 could impact the reaction rate (acidity) and the reaction selectivity (through deviation from the ideal acid-to-metal balance). Structural zeolite stability has been recently studied in detail showing that USY zeolites rich in Al, such as applied here, are surprisingly stable in hot liquid water, even during days at elevated temperatures.44The recyclability of the catalytic system is evaluated using the best catalyst in this study, Ru0.6/USY9 (Table 3, entry 5) in 4 consecutive runs. The previous studies indicate that in particular USY9 is stable against structural degradation, certainly at 160 °C after 24 hours. The results, illustrated in Fig. 3 for reactions after 3 and 5 hours, confirm this hypothesis as both full activity and selectivity regeneration were achieved for each run.
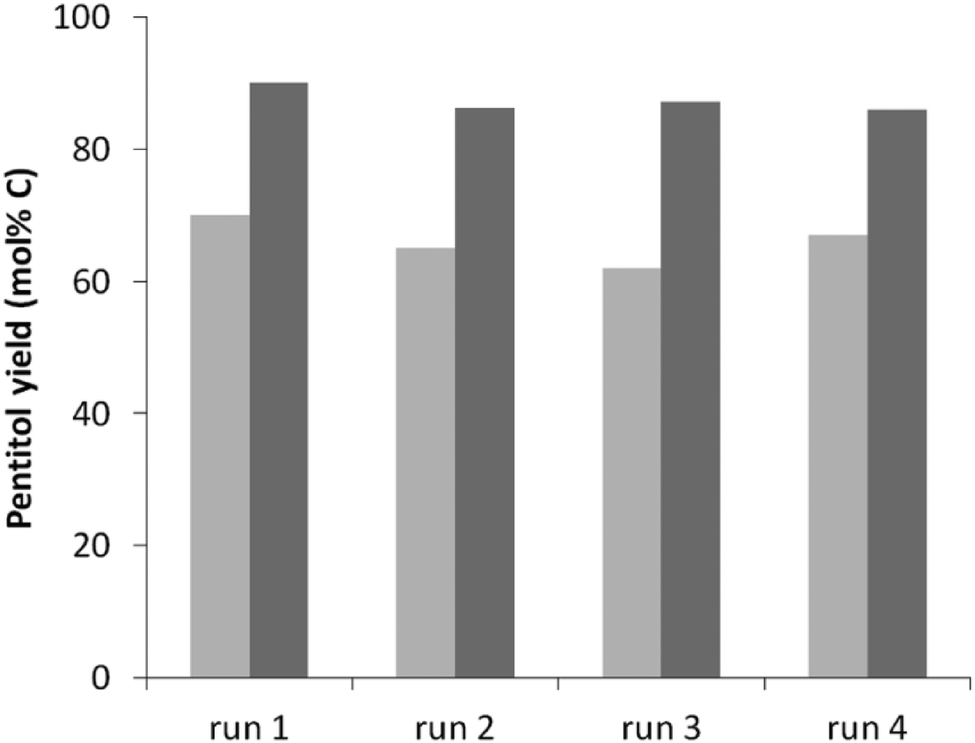 | ||
| Fig. 3 Pentitol yield after 3 h (light grey) and 5 h (dark grey) for multiple regenerations. Reaction conditions: see Table 3, entry 5. | ||
Rationalization and conclusion
A rationalization of the concept of bifunctional catalysis for cellulose versus hemicellulose is reported here. The above data show now that high pentitol yields in the presence of Ru-loaded acidic USY zeolites are possible, alike the high hexitol yields that were earlier observed from cellulose (Fig. 4), but that site balance is of utmost importance. Indeed, both systems bear a similar concept of bifunctional catalysis, in which the acidity is required to solubilize and hydrolyse the carbohydrate substrate, whereas the metal site is used to produce the corresponding (more stable) polyols, avoiding acid catalysed side-reactions with the monosaccharides. Despite the mechanistic similarity, there are significant differences, which are largely due to the more recalcitrant structure of cellulose.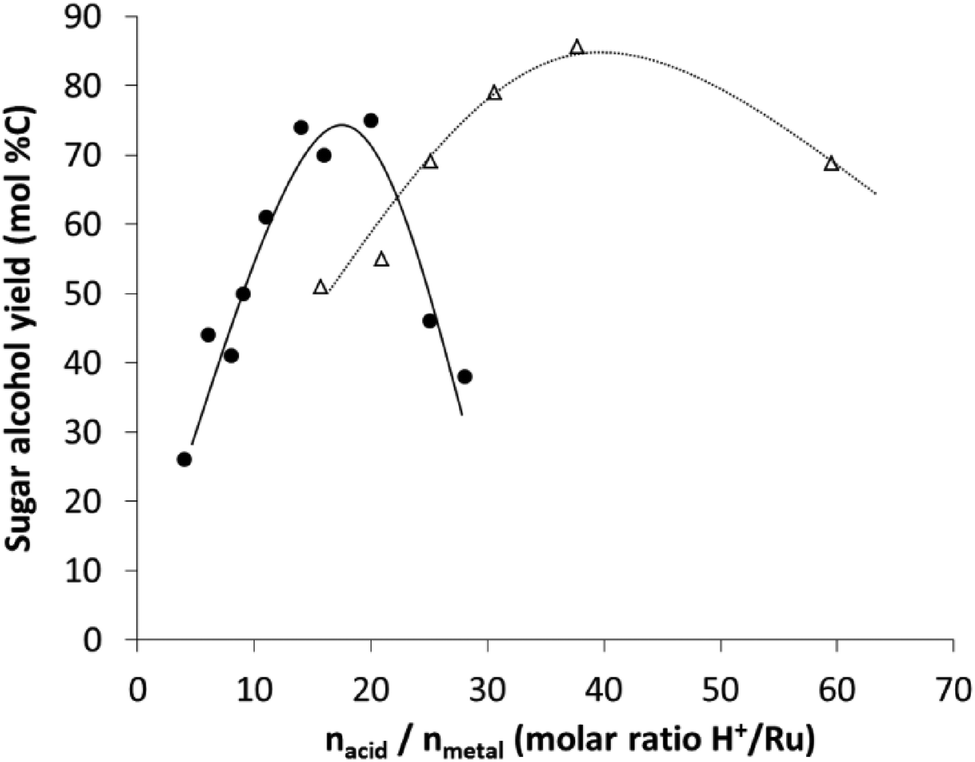 | ||
| Fig. 4 Comparison between the sugar alcohol yield obtained with Ru/USY catalysts in the reductive splitting of cellulose44,47 (open triangles and dashed line) and hemicellulose (filled circles and continuous line). | ||
The first one involves the presence of soluble acidity, like HCl, during cellulose processing. Soluble acidity is highly recommended, if not necessary, with cellulose to fasten cellulose dissolution even though higher reaction temperatures may also be applied to assist in disassembling of cellulose. Temperatures above 200 °C are usually applied to convert cellulose, whereas 160 °C seems sufficient to fully convert AX hemicellulose, even in the absence of an additional mineral acid (160–175 °C being optimal). The lower temperature used in hemicellulose valorisation has the advantage of Ru/USY catalyst stability, especially against metal sintering, as has been reported for reactions at higher temperatures during cellulose processing.44
Once the sugar polymers are dissolved, soluble oligomers are strongly adsorbed into the mesoporous structure of the zeolite, where they react rapidly with the local acidity in the zeolite to ultimately form the sugar monomers. The presence of a metal site in close proximity is essential here in order to circumvent acid catalysed reactions with the reactive sugar molecule, forming furan-like derivatives. This paper clearly emphasizes the important role of the acid-to-metal site balance, preferably in close proximity, for hemicellulose, as it was earlier suggested for other bifunctional catalysts like Ni on oxidized nanofibers in the case of cellulose conversion.86 Since hemicellulose is more easily hydrolysed, relatively more hydrogenation capacity is required to balance the acid catalysis. This is indeed apparent in Fig. 4, plotting hexitol and pentitol yields in the presence of similar Ru-loaded zeolites vs. the molar acid/metal balance. The expected shift to lower acid-to-metal site ratios is clear, as well as a narrower range of the ratio (Fig. 4). The narrowing is in line with the higher sensitivity of pentoses towards unwanted side-product formation (when compared to hexoses), but also the difference in the rate determining step between cellulose and hemicellulose hydrolysis under the applied reaction conditions. Note that the optimal balance is strongly dependent not only on the reaction conditions, but also on the used catalyst (strength of the acid catalyst and hydrogenation capacity of the metal) as for instance an optimal acid-to-metal ratio of 1 was determined for cellulose processing with Ni on oxidized nanofibers.86
The final difference, which is largely a consequence of the different reaction temperature, involves the hydrogenolysis activity. Hydrogenolysis will occur with the polyol products, forming shorter polyols,80 when reactive sugar monomers are not available (and at long reaction times in the batch reactor). As such reactions are energy demanding (with high apparent activation energy), they will especially happen at a large reaction temperature, like in the reactions with cellulose. Contrarily, hydrogenolysis is thus less pronounced with hemicellulose at a lower reaction temperature. Therefore, too many metal sites will decrease the hexitol product yield, whereas the impact on pentitol selectivity will be less significant.
In future, we believe that the understanding of the differences in the reaction behaviour between cellulose and hemicellulose processing could possibly be exploited to valorise real pulp streams, containing both hemicellulose and cellulose, as for example obtained after the ‘first-lignin’ biorefinery process.17,18,23 Due to the different reaction circumstances, one could imagine selective removal of hemicellulose into polyols under mild temperature and acid conditions, leaving an ideal cellulosic biomass for enzymatic hydrolysis, since polyols (in contrast to furanics) are less hazardous for cellulase activity.87
Acknowledgements
T. E. thanks IWT-Vlaanderen for his doctoral fellowship. The Belgian Government is acknowledged for financial support through IAP funding (Belspo). The authors are grateful to Walter Vermandel for the CO chemisorption analyses.Notes and references
- J. Q. Bond, A. A. Upadhye, H. Olcay, G. A. Tompsett, J. Jae, R. Xing, D. M. Alonso, D. Wang, T. Zhang, R. Kumar, A. Foster, S. M. Sen, C. T. Maravelias, R. Malina, S. R. H. Barrett, R. Lobo, C. E. Wyman, J. A. Dumesic and G. W. Huber, Energy Environ. Sci., 2014, 7, 1500–1523 CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- R. Luque, Pure Appl. Chem., 2014, 86, 843–857 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- Y.-C. Lin and G. W. Huber, Energy Environ. Sci., 2009, 2, 68–80 CAS.
- R. Rinaldi and F. Schuth, Energy Environ. Sci., 2009, 2, 610–626 CAS.
- R. Rinaldi and F. Schüth, ChemSusChem, 2009, 2, 1096–1107 CrossRef CAS PubMed.
- G. A. Tompsett, N. Li and G. W. Huber, in Thermochemical Processing of Biomass, ed. R. C. Brown, John Wiley & Sons, Ltd, 2011, pp. 232–279 Search PubMed.
- T. Ennaert, J. Van Aelst, J. Dijkmans, R. De Clercq, W. Schutyser, M. Dusselier, D. Verboekend and B. F. Sels, Chem. Soc. Rev., 2016, 45, 584–611 RSC.
- B. Op de Beeck, M. Dusselier, J. Geboers, J. Holsbeek, E. Morre, S. Oswald, L. Giebeler and B. F. Sels, Energy Environ. Sci., 2015, 8, 230–240 CAS.
- B. Op de Beeck, J. Geboers, S. Van de Vyver, J. Van Lishout, J. Snelders, W. J. J. Huijgen, C. M. Courtin, P. A. Jacobs and B. F. Sels, ChemSusChem, 2013, 6, 199–208 CrossRef CAS PubMed.
- R. de Souza, L. Miranda and R. Luque, Green Chem., 2014, 16, 2386–2405 RSC.
- M. A. Mellmer, C. Sener, J. M. R. Gallo, J. S. Luterbacher, D. M. Alonso and J. A. Dumesic, Angew. Chem., Int. Ed., 2014, 53, 11872–11875 CrossRef CAS PubMed.
- J. S. Luterbacher, D. Martin Alonso and J. A. Dumesic, Green Chem., 2014, 16, 4816–4838 RSC.
- J. S. Luterbacher, J. M. Rand, D. M. Alonso, J. Han, J. T. Youngquist, C. T. Maravelias, B. F. Pfleger and J. A. Dumesic, Science, 2014, 343, 277–280 CrossRef CAS PubMed.
- J.-P. Lange, I. Lewandowski and P. M. Ayoub, in Sustainable Development in the Process Industries, John Wiley & Sons, Inc., 2010, pp. 171–198 Search PubMed.
- P. Ferrini and R. Rinaldi, Angew. Chem., Int. Ed., 2014, 53, 8634–8639 CrossRef CAS PubMed.
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2014, 7, 2154–2158 CrossRef CAS PubMed.
- C. Li, M. Zheng, A. Wang and T. Zhang, Energy Environ. Sci., 2012, 5, 6383–6390 CAS.
- T. Parsell, S. Yohe, J. Degenstein, T. Jarrell, I. Klein, E. Gencer, B. Hewetson, M. Hurt, J. I. Kim, H. Choudhari, B. Saha, R. Meilan, N. Mosier, F. Ribeiro, W. N. Delgass, C. Chapple, H. I. Kenttamaa, R. Agrawal and M. M. Abu-Omar, Green Chem., 2015, 17, 1492–1499 RSC.
- W. Schutyser, S. Van den Bosch, T. Renders, T. De Boe, S.-F. Koelewijn, A. Dewaele, T. Ennaert, O. Verkinderen, B. Goderis, C. M. Courtin and B. F. Sels, Green Chem., 2015, 17, 5035–5045 RSC.
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 CAS.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S. F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 CAS.
- N. Yan, C. Zhao, P. J. Dyson, C. Wang, L.-t. Liu and Y. Kou, ChemSusChem, 2008, 1, 626–629 CrossRef CAS PubMed.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2015, 116, 1540–1599 CrossRef PubMed.
- W. Schutyser, S. Van den Bosch, J. Dijkmans, S. Turner, M. Meledina, G. Van Tendeloo, D. P. Debecker and B. F. Sels, ChemSusChem, 2015, 8, 1805–1818 CrossRef CAS PubMed.
- D. R. Vardon, M. A. Franden, C. W. Johnson, E. M. Karp, M. T. Guarnieri, J. G. Linger, M. J. Salm, T. J. Strathmann and G. T. Beckham, Energy Environ. Sci., 2015, 8, 617–628 CAS.
- C. Chatterjee, F. Pong and A. Sen, Green Chem., 2015, 17, 40–71 RSC.
- M. Dusselier, M. Mascal and B. Sels, in Selective Catalysis for Renewable Feedstocks and Chemicals, ed. K. M. Nicholas, Springer International Publishing, 2014, vol. 353, ch. 544, pp. 1–40 Search PubMed.
- H. Kobayashi and A. Fukuoka, Green Chem., 2013, 15, 1740–1763 RSC.
- X. Liu, X. Wang, S. Yao, Y. Jiang, J. Guan and X. Mu, RSC Adv., 2014, 4, 49501–49520 RSC.
- F. Schüth, R. Rinaldi, N. Meine, M. Käldström, J. Hilgert and M. D. K. Rechulski, Catal. Today, 2014, 234, 24–30 CrossRef.
- M. Yabushita, H. Kobayashi and A. Fukuoka, Appl. Catal., B, 2014, 145, 1–9 CrossRef CAS.
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS PubMed.
- M. Rose and R. Palkovits, Macromol. Rapid Commun., 2011, 32, 1299–1311 CrossRef CAS PubMed.
- I. Delidovich, K. Leonhard and R. Palkovits, Energy Environ. Sci., 2014, 7, 2803–2830 CAS.
- M. Benoit, A. Rodrigues, K. De Oliveira Vigier, E. Fourre, J. Barrault, J.-M. Tatibouet and F. Jerome, Green Chem., 2012, 14, 2212–2215 RSC.
- M. Benoit, A. Rodrigues, Q. Zhang, E. Fourré, K. De Oliveira Vigier, J.-M. Tatibouët and F. Jérôme, Angew. Chem., Int. Ed., 2011, 50, 8964–8967 CrossRef CAS PubMed.
- R. Carrasquillo-Flores, M. Käldström, F. Schüth, J. A. Dumesic and R. Rinaldi, ACS Catal., 2013, 3, 993–997 CrossRef CAS.
- N. Meine, R. Rinaldi and F. Schüth, ChemSusChem, 2012, 5, 1449–1454 CrossRef CAS PubMed.
- A. Shrotri, L. K. Lambert, A. Tanksale and J. Beltramini, Green Chem., 2013, 15, 2761–2768 RSC.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS PubMed.
- H. Schiweck, A. Bär, R. Vogel, E. Schwarz, M. Kunz, C. Dusautois, A. Clement, C. Lefranc, B. Lüssem, M. Moser and S. Peters, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- T. Ennaert, J. Geboers, E. Gobechiya, C. M. Courtin, M. Kurttepeli, K. Houthoofd, C. E. A. Kirschhock, P. C. M. M. Magusin, S. Bals, P. A. Jacobs and B. F. Sels, ACS Catal., 2015, 5, 754–768 CrossRef CAS.
- T. Ennaert, B. Op de Beeck, J. Vanneste, A. T. Smit, W. J. J. Huijgen, A. Vanhulsel, P. A. Jacobs and B. F. Sels, Green Chem., 2015, 18, 2095–2105 RSC.
- J. Geboers, S. Van de Vyver, K. Carpentier, K. de Blochouse, P. Jacobs and B. Sels, Chem. Commun., 2010, 46, 3577–3579 RSC.
- J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Chem. Commun., 2011, 47, 5590–5592 RSC.
- G. Liang, H. Cheng, W. Li, L. He, Y. Yu and F. Zhao, Green Chem., 2012, 14, 2146–2149 RSC.
- G. Liang, L. He, H. Cheng, W. Li, X. Li, C. Zhang, Y. Yu and F. Zhao, J. Catal., 2014, 309, 468–476 CrossRef CAS.
- A. Negoi, K. Triantafyllidis, V. I. Parvulescu and S. M. Coman, Catal. Today, 2014, 223, 122–128 CrossRef CAS.
- A. Shrotri, A. Tanksale, J. N. Beltramini, H. Gurav and S. V. Chilukuri, Catal. Sci. Technol., 2012, 2, 1852–1858 CAS.
- R. Palkovits, K. Tajvidi, J. Procelewska, R. Rinaldi and A. Ruppert, Green Chem., 2010, 12, 972–978 RSC.
- R. Palkovits, K. Tajvidi, A. M. Ruppert and J. Procelewska, Chem. Commun., 2011, 47, 576–578 RSC.
- P. A. Jacobs and H. Hinnekens, EP0329923, 1989 Search PubMed.
- B. Zhang, X. Li, Q. Wu, C. Zhang, Y. Yu, M. Lan, X. Wei, Z. Ying, T. Liu, G. Liang and F. Zhao, Green Chem., 2016, 18, 3315–3323 RSC.
- K. Fabicovicova, M. Lucas and P. Claus, Green Chem., 2015, 17, 3075–3083 RSC.
- L. S. Ribeiro, J. J. M. Orfao and M. F. R. Pereira, Green Chem., 2015, 17, 2973–2980 RSC.
- H. Kobayashi, Y. Hosaka, K. Hara, B. Feng, Y. Hirosaki and A. Fukuoka, Green Chem., 2014, 16, 637–644 RSC.
- W. Zhu, H. Yang, J. Chen, C. Chen, L. Guo, H. Gan, X. Zhao and Z. Hou, Green Chem., 2014, 16, 1534–1542 RSC.
- Y. Liao, Q. Liu, T. Wang, J. Long, L. Ma and Q. Zhang, Green Chem., 2014, 16, 3305–3312 RSC.
- H. Kobayashi, Y. Ito, T. Komanoya, Y. Hosaka, P. L. Dhepe, K. Kasai, K. Hara and A. Fukuoka, Green Chem., 2011, 13, 326–333 RSC.
- H. Kobayashi, H. Matsuhashi, T. Komanoya, K. Hara and A. Fukuoka, Chem. Commun., 2011, 47, 2366–2368 RSC.
- H. Kobayashi, H. Ohta and A. Fukuoka, Catal. Sci. Technol., 2012, 2, 869–883 CAS.
- H. Kobayashi, H. Yokoyama, B. Feng and A. Fukuoka, Green Chem., 2015, 17, 2732–2735 RSC.
- L. Faba, B. T. Kusema, E. V. Murzina, A. Tokarev, N. Kumar, A. Smeds, E. Díaz, S. Ordóñez, P. Mäki-Arvela, S. Willför, T. Salmi and D. Y. Murzin, Microporous Mesoporous Mater., 2014, 189, 189–199 CrossRef CAS.
- B. T. Kusema, L. Faba, N. Kumar, P. Mäki-Arvela, E. Díaz, S. Ordóñez, T. Salmi and D. Y. Murzin, Catal. Today, 2012, 196, 26–33 CrossRef CAS.
- D. Y. Murzin, B. Kusema, E. V. Murzina, A. Aho, A. Tokarev, A. S. Boymirzaev, J. Wärnå, P. Y. Dapsens, C. Mondelli, J. Pérez-Ramírez and T. Salmi, J. Catal., 2015, 330, 93–105 CrossRef CAS.
- D. Y. Murzin, E. V. Murzina, A. Tokarev, N. D. Shcherban, J. Wärnå and T. Salmi, Catal. Today, 2015, 257(Part 2), 169–176 CAS.
- S. Liu, Y. Okuyama, M. Tamura, Y. Nakagawa, A. Imai and K. Tomishige, Green Chem., 2016, 18, 165–175 RSC.
- G. Yi and Y. Zhang, ChemSusChem, 2012, 5, 1383–1387 CrossRef CAS PubMed.
- G. Yi and Y. Zhang, WO201315150A1, 2013 Search PubMed.
- H. Kobayashi, Y. Yamakoshi, Y. Hosaka, M. Yabushita and A. Fukuoka, Catal. Today, 2014, 226, 204–209 CrossRef CAS.
- A. Yamaguchi, O. Sato, N. Mimura and M. Shirai, Catal. Today, 2016, 265, 199–202 CrossRef CAS.
- A. Yamaguchi, O. Sato, N. Mimura, Y. Hirosaki, H. Kobayashi, A. Fukuoka and M. Shirai, Catal. Commun., 2014, 54, 22–26 CrossRef CAS.
- L. Faba, B. T. Kusema, E. V. Murzina, A. Tokarev, N. Kumar, A. Smeds, E. Díaz, S. Ordóñez, P. Mäki-Arvela, S. Willför, T. Salmi and D. Y. Murzin, Microporous Mesoporous Mater., 2014, 189, 189–199 CrossRef CAS.
- L. Negahdar, P. J. C. Hausoul, S. Palkovits and R. Palkovits, Appl. Catal., B, 2015, 166–167, 460–464 CrossRef CAS.
- C. A. Emeis, J. Catal., 1993, 141, 347–354 CrossRef CAS.
- P.-W. Chung, A. Charmot, O. A. Olatunji-Ojo, K. A. Durkin and A. Katz, ACS Catal., 2013, 4, 302–310 CrossRef.
- P.-W. Chung, M. Yabushita, A. T. To, Y. Bae, J. Jankolovits, H. Kobayashi, A. Fukuoka and A. Katz, ACS Catal., 2015, 5, 6422–6425 CrossRef CAS.
- P. J. C. Hausoul, L. Negahdar, K. Schute and R. Palkovits, ChemSusChem, 2015, 8, 3323–3330 CrossRef CAS PubMed.
- J. Sun and H. Liu, Green Chem., 2011, 13, 135–142 RSC.
- F. Héroguel, B. Rozmysłowicz and J. S. Luterbacher, Chimia, 2015, 69, 582–591 CrossRef PubMed.
- W. Lutz, H. Toufar, R. Kurzhals and M. Suckow, Adsorption, 2005, 11, 405–413 CrossRef CAS.
- R. M. Ravenelle, F. Schüβler, A. D'Amico, N. Danilina, J. A. van Bokhoven, J. A. Lercher, C. W. Jones and C. Sievers, J. Phys. Chem. C, 2010, 114, 19582–19595 CAS.
- J.-P. Lange, Angew. Chem., Int. Ed., 2015, 54, 2–14 CrossRef PubMed.
- S. Van de Vyver, J. Geboers, W. Schutyser, M. Dusselier, P. Eloy, E. Dornez, J. W. Seo, C. M. Courtin, E. M. Gaigneaux, P. A. Jacobs and B. F. Sels, ChemSusChem, 2012, 5, 1549–1558 CrossRef CAS PubMed.
- K.-K. Cheng, J.-A. Zhang, E. Chavez and J.-P. Li, Appl. Microbiol. Biotechnol., 2010, 87, 411–417 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ESI contains more details and information about the used zeolites, one additional figure and a consideration about the influence of the reaction temperature on the bifunctional reaction and its optimal acid/metal balance. See DOI: 10.1039/c6gc01439a |
| This journal is © The Royal Society of Chemistry 2016 |