
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterogeneous catalytic approaches in C–H activation reactions
Stefano
Santoro
a,
Sergei I.
Kozhushkov
b,
Lutz
Ackermann
*b and
Luigi
Vaccaro
 *a
*a
aLaboratory of Green Synthetic Organic Chemistry, Dipartimento di Chimica Biologia e Biotecnologie, Università di Perugia, Via Elce di Sotto, 8 – 06123 Perugia, Italy. E-mail: luigi.vaccaro@unipg.it; Web: http://www.dcbb.unipg.it/greensoc
bInstitut für Organische und Biomolekulare Chemie, Georg-August-Universität Göttingen, Tammannstrasse 2, 37077 Göttingen, Germany. E-mail: Lutz.Ackermann@chemie.uni-goettingen.de; Web: http://www.ackermann.chemie.uni-goettingen.de
First published on 10th March 2016
Abstract
Despite the undisputed advances and progress in metal-catalyzed C–H functionalizations, this atom-economical approach had thus far largely been developed with the aid of various metal catalysts that were operative in a homogeneous fashion. While thereby major progress was accomplished, these catalytic systems featured notable disadvantages, such as low catalyst recyclability. This review summarizes the development of user-friendly, recyclable and easily separable heterogeneous catalysts for C–H activation. This strategy was characterized by a remarkably broad substrate scope, considerable levels of chemo- and site-selectivities and proved applicable to C–C as well as C–heteroatom formation processes. Thus, recyclable catalysts were established for arylations, hydroarylations, alkenylations, acylations, nitrogenations, oxygenations, or halogenations, among others. The rapid recent progress in selective heterogeneous C–H functionalizations during the last decade until December 2015 is reviewed.
 Stefano Santoro | Stefano Santoro received his Ph.D. degree from the University of Perugia in 2009 (with Prof. C. Santi and Prof. M. Tiecco). Between 2006 and 2007 he was a visiting Ph.D. student at Aarhus University, in the group of Prof. K. A. Jørgensen. In 2010 he moved to Stockholm University as a postdoctoral researcher with Prof. F. Himo, where he stayed until 2014. Since 2014 he has been an assistant professor at the University of Perugia. His main research interests are in the areas of homogeneous and heterogeneous catalysis, C–H functionalizations and mechanistic investigations by means of DFT calculations. |
 Sergei I. Kozhushkov | Sergei I. Kozhushkov (1956) was born in Kharkov, USSR. He studied chemistry at Lomonosov Moscow State University, where he obtained his doctoral degree in 1983 and performed his “Habilitation” in 1998. In 1991, he joined the research group of Professor A. de Meijere (Georg-August-Universität Göttingen) as an Alexander von Humboldt Research Fellow. Since 2007, he has been working in the research group of Prof. Dr. L. Ackermann (Georg-August-Universität Göttingen). |
 Lutz Ackermann | Lutz Ackermann (1972) studied Chemistry at the Christian-Albrechts-University Kiel, Germany, and received his Ph.D. in 2001 for research under the supervision of Prof. Dr. Alois Fürstner at the Max-Plank-Institut für Kohlenforschung in Mülheim/Ruhr. He was a postdoctoral coworker in the laboratory of Prof. Dr. Robert G. Bergman at the University of California at Berkeley before initiating his independent research career in 2003 at the Ludwig-Maximilians-University München supported by the Emmy Noether-programme of the DFG. In 2007, he was appointed as a Full (W3) Professor at the Georg-August-University Göttingen. His recent awards and distinctions include a JSPS visiting professor fellowship (2009), an AstraZeneca Excellence in Chemistry Award (2011) and an ERC Grant (2012) as well as visiting professorships at the Università degli Studi di Milano, the University of Wisconsin at Madison, the Università di Pavia, and the Università degli Studi di Perugia. The development and application of novel concepts for sustainable catalysis constitutes his current research interests, with a major focus on C–H activation. |
 Luigi Vaccaro | Luigi Vaccaro obtained his Laurea in Chemistry from the University of Naples – Federico II; then he earned a PhD in Chemical Sciences from the University of Perugia where currently he is an Associate Professor leading the Green S.O.C. group. He was habilitated as a Full Professor in 2013. He is currently appointed as an Associate Editor of RSC Advances and Beilstein Journal of Organic Chemistry. He received the Europa Medal from the Society of Chemical Industry – London (2001), the ADP Award from Merck's Chemistry Council for “Creative work in organic chemistry” (2006 and 2007), the G. Ciamician Medal of the Società Chimica Italiana (2007) and the Vigevani Visiting Professorship (2014). His research is currently focused on the development of heterogeneous catalysis, safer media and flow chemistry towards the definition of novel green/sustainable synthetic tools. |
1. Introduction
During the last decade, the direct functionalization of otherwise inert C–H bonds as latent functional groups has received considerable attention.1 Compared with the traditional cross-coupling methods (Fig. 1a), this step-economical approach avoids the use of prefunctionalized substrates, and thereby provides means for environmentally-benign and economically-attractive organic syntheses. In fact, while well-established stoichiometric and catalytic methods for cross-coupling reactions (Fig. 1a) have allowed access to key molecular structures, they also require specific pre-functionalization with the corresponding inevitable formation of undesired by-products such as halides, salts, borates etc. Therefore, C–H activation/functionalization strategies represent a more step-economical efficient alternative. | ||
| Fig. 1 Evolution of environmentally-friendly and technologically-sound syntheses (presented for arylation). | ||
As a consequence, this strategy has found a growing number of applications in the preparation of pharmaceuticals, naturally occurring compounds, agrochemicals and other practically useful products.2 In this context, powerful transition-metal catalysts for C–H activation have been developed, which have enabled a streamlining of organic synthesis (Fig. 1b).
A major challenge of this approach is related to the possibility to achieve site-selectivity, and impressive advances have already been made in this direction.3
Despite these considerable advances, the vast majority of C–H activations were achieved with homogeneous catalysts, which leaves ample room for optimization for the practitioner. Thus, these usually rather expensive catalysts possess low recyclability, and purification of the desired products from traces of a metal contaminant commonly calls for special reagents or chromatographic methods, particularly when considering the preparation of pharmaceuticals in an industrial setting. Still, industrial production often prefers to utilize well-established homogeneous methods.4 Obviously, decisions in the industries are mainly based on economical reasons, which also heavily depend on regulations and the price/availability of materials. Since the latter change with time, for instance with environmental regulations becoming increasingly stringent and the depletion of natural resources, a chemical approach that is not economically sustainable at present can become a reasonable alternative in the future. In this perspective heterogeneous catalytic approaches, which inherently allow an easier recovery of the catalyst, are certainly promising, provided that the recovered catalyst can be reused with no loss of activity.
Another interesting aspect is the possibility to use heterogeneous catalysis to achieve a specific site-selectivity, or a switch in selectivity compared to related homogeneous processes, and possibly an increase in the catalytic activity. This could in principle be accomplished through the design of hybrid molecular structures.
This aspect is of key interest in the context of green chemistry, where chemical efficiency has to be combined with economic and environmental needs.
Therefore, the development of recyclable heterogeneous catalysts is highly desirable not only because these systems allow for a facile separation by filtration and hence can be recyclable (Fig. 1c),5 but also for devising novel chemical processes.
Herein we summarize the rapid recent progress in selective C–H functionalizations through heterogeneous catalysis until December 2015.
2. C–C bond formation
2.1. C–H arylation
Arguably, the first example of heterogeneous palladium catalyzed C–H arylation was reported by Nakamura et al. in 1982.6 In this contribution, palladium on charcoal was used to introduce aryl groups into isoxazole, allowing slightly lower yields compared to the reaction promoted by Pd(OAc)2.A seminal example of C–H arylation by a heterogeneous catalyst was disclosed by Fagnou and coworkers in 2005, when they reported about the use of palladium hydroxide on carbon – Pearlman's catalyst – to promote the direct C–H arylation with aryl iodides or bromides (Scheme 1).7 The catalytic system worked efficiently both for intramolecular arylation (leading to benzo[c]chromenes and dihydrophenanthridine derivatives 1) and intermolecular processes, although in the latter case only heteroaromatics were used as the substrates. The nature of the active catalytic species was investigated by means of two experiments. In the first one, a three-phase test, the aryl halide was supported on a Wang resin and allowed to react under normal reaction conditions. After 4.5 hours, a complete conversion to the product was observed, thus suggesting a catalytic role of the homogeneous leached palladium species. This was confirmed by the use of a thiol-based scavenger resin, which leads to less than 5% of the product formation after 16 hours, presumably due to sequestration of the homogeneous species.
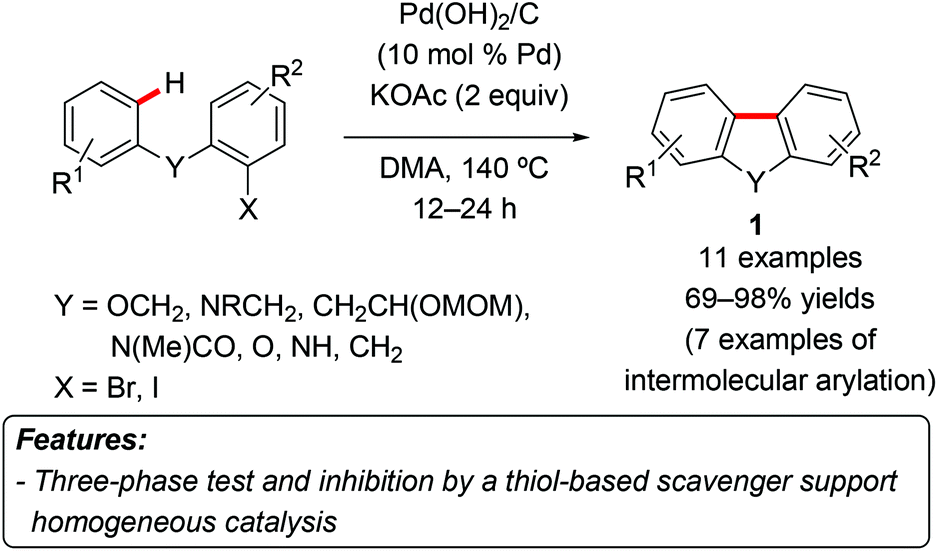 | ||
| Scheme 1 Intramolecular Pd(OH)2/C-catalyzed direct arylation. | ||
Djakovitch et al. showed that two heterogeneous palladium catalysts were able to promote the C3-arylation of 2-substituted indoles 2 when reacting with aryl bromides 3.8 The catalysts represented either a microporous [Pd(NH3)4]2+/NaY system obtained by ion exchange from NaY zeolite, or a mesoporous [Pd]/SBA-15 material prepared by immobilizing a phosphine ligand on a calcined SBA-15 mesoporous silica. Only two representative substrates have been tested and revealed very poor reactivity. The [Pd]/SBA-15 catalytic system was more efficient than [Pd(NH3)4]2+/NaY, affording the products in 35–36% conversions after 12 days, as determined by GC analyses. It should be also emphasized that the related homogeneous process employing Pd(OAc)2 occurs very slowly and with poor yields as well (48–55% after 12 days). Interestingly, the use of aryl iodides led to selective N-arylation, instead of the desired C3-arylation products 4. In a follow-up report the authors extended the scope of the C3-arylation still using the [Pd(NH3)4]2+/NaY catalyst and aryl bromides 3 as the arylating reagent, but changing the base from NaOAc to K2CO3 and the solvent from NMP to 1,4-dioxane (Scheme 2).9
 | ||
| Scheme 2 Heterogeneous palladium-catalyzed approach to 2- and 2,3-substituted indoles 4. | ||
Almost all the tested substrates gave moderate to good conversions, with some exceptions mainly depending on the indole substitution pattern. When isolated yields were tested, they were in line with the conversions measured by GC analyses. Neither the recyclability of the catalyst nor the nature of the catalytically active species was tested.
Alami and coworkers used Pearlman's catalyst to promote the regioselective arylation of adenine derivatives using aryl iodides, bromides or chlorides as the arylating agents with either conventional or microwave heating.10 The protocol proved effective for NH2-free adenines and selectively afforded the products of C8-arylation in yields ranging from moderate to good. The authors did not investigate the recyclability of the catalyst or the mechanism of the process.
SanMartin, Domínguez and coworkers synthesized a series of pyrazolophenanthridines 6 by intramolecular direct arylation of diarylpyrazoles 5 (Scheme 3).11
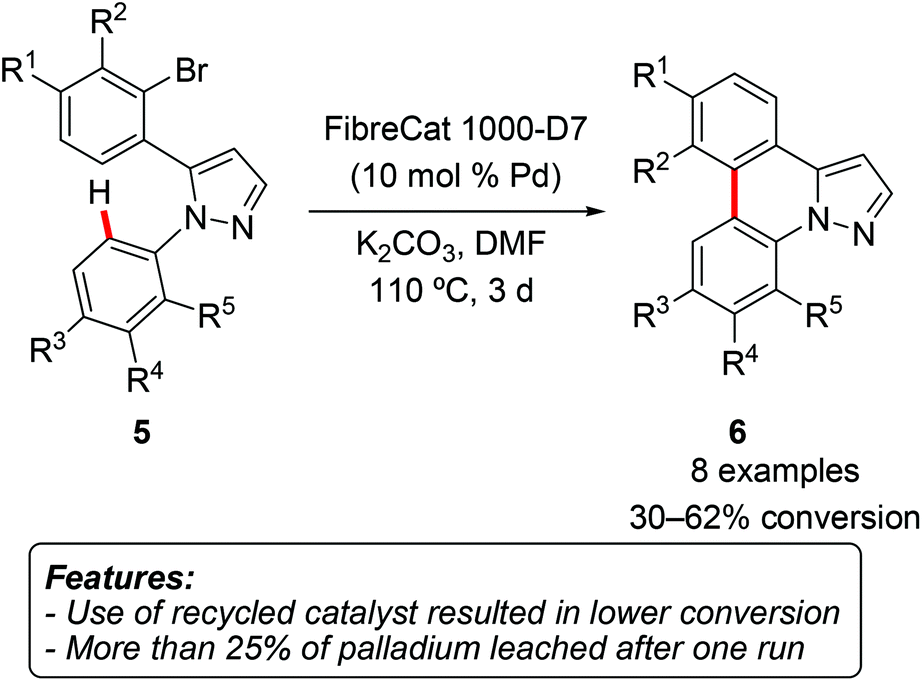 | ||
| Scheme 3 Direct C–H arylation in the synthesis of pyrazolophenanthridines 6. | ||
Among the other palladium sources, the reaction could be catalyzed by commercially available heterogeneous polymer-supported palladium catalysts FibreCat 1001 and FibreCat 1000-D7. The results obtained with heterogeneous catalysts were somewhat disappointing in comparison with those obtained, for instance, with simple Pd(OAc)2. The recovered catalyst was used in further reaction runs, but a significantly lower conversion of only 21–25% was observed. Moreover, ICP (Inductively Coupled Plasma) analysis of the filtrate revealed the presence of a considerable (>25%) amount of leached palladium.
Jafarpour et al. used Pearlman's catalyst to promote the C2-site-selective C–H arylation of pyrroles 7 with aryl iodides 8 (Scheme 4).12 The reaction was performed in triethylamine at 100 °C and proceeded best with NH-free pyrroles 7 (Scheme 4, R1 = H), while N-methylpyrrole and N-phenylpyrrole 7 (Scheme 4, R = Me or Ph) afforded somewhat lower yields. Both electron-rich and electron-poor aryl iodide could be used as arylating agents. Unfortunately, neither the recyclability of the catalyst nor the nature of the actual catalytic species was investigated.
 | ||
| Scheme 4 Palladium-catalyzed C2-selective arylation of NH-free pyrroles 7. | ||
Ranu and coworkers reported on the palladium catalyzed C–H arylation of benzothiazoles claiming that the reaction is actually catalyzed by palladium nanoparticles formed in situ from Pd(OAc)2.13 Although the authors were able to observe the nanoparticles by TEM (transmission electron microscopy) analyses after 4 hours of reaction, no proof has been obtained that the catalytically active species are the nanoparticles. Indeed, the employment of preformed nanoparticles led to a much lower yield, suggesting a predominant role of the homogeneous palladium species.
Cai and coworkers prepared fluorous silica gel-supported perfluoro-tagged palladium nanoparticles (Pdnps/FSG) and tested their application as catalysts for the C–H arylation of indoles 9.14 The catalyst design relied on the stabilization of metal nanoparticles by compounds possessing long perfluorinated alkyl chains, and on the possibility to use these groups also to support the nanoparticles on fluoric silica gel by noncovalent fluorous interactions. The catalyst proved to be very efficient in catalyzing the regioselective C2-arylation of heteroarenes 9 with a catalyst loading as low as 0.1 mol% (Scheme 5a). The highest yields were obtained with N-methylindoles 9 (Scheme 5, R2 = Me) and N-benzylindole 9 (R2 = Bn), whereas NH-free substrate 9 (Scheme 5, R2 = H) gave only moderate results. Aryl iodides 10a (Scheme 5, X = I) were the arylating reagents of choice, while the use of aryl bromides or chlorides (Scheme 5, X = Cl or Br) resulted in lower yields. The catalyst could be recovered by simple centrifugation and decantation, and was reused for seven consecutive runs with a slight decrease in activity. ICP-AAS (Inductively Coupled Plasma Atomic Absorption Spectrometry) analysis showed a negligible amount of palladium in the crude reaction mixtures. TEM analysis of the recovered particles showed a slight increase in the size of the palladium particles.
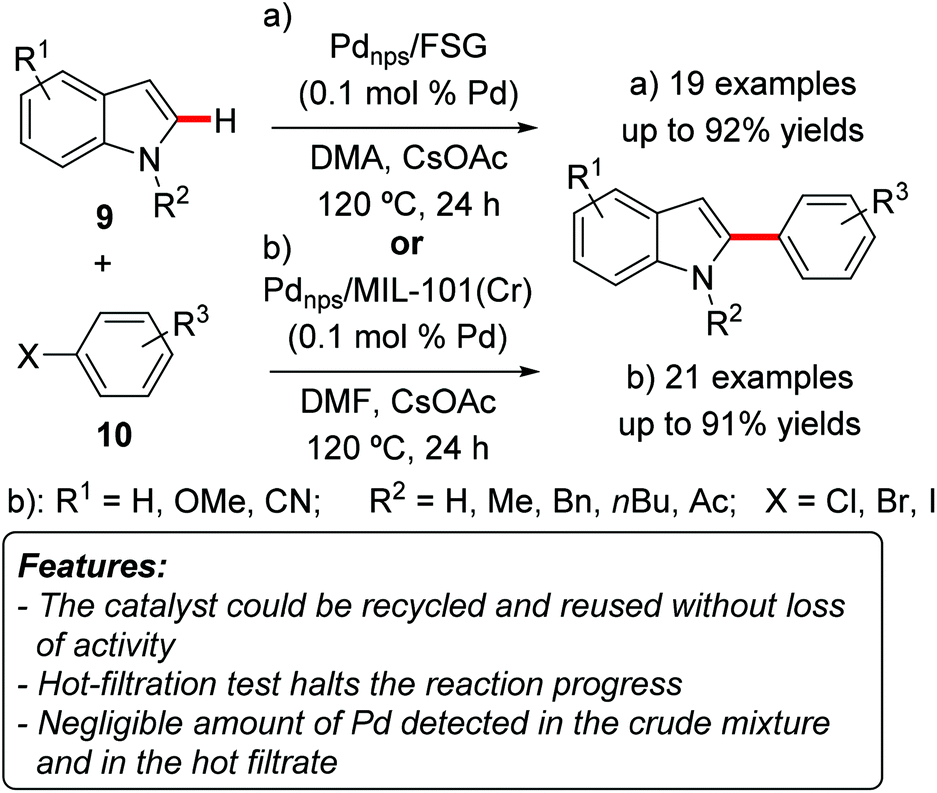 | ||
| Scheme 5 Nanoparticles as reusable catalysts for indole C2-arylation. | ||
A hot-filtration test evidenced that the removal of the solid catalyst completely stopped the reaction. ICP-AAS analysis was also performed on the filtrate, showing again a negligible amount of palladium. All these findings strongly supported a genuine heterogeneous catalysis.
The C2-selective C–H arylation of indoles 9 was also realized by Cao and coworkers using palladium nanoparticles encapsulated in a MOF MIL-101(Cr) (Cr3(F,OH)(H2O)2O[(O2C)–C6H4–(CO2)]3·nH2O; n ≈ 25) as catalysts (Scheme 5b).15 Also in this case only 0.1 mol% of the catalyst could be used, and the best arylating reagents were aryl iodides, whereas aryl bromides and chlorides furnished lower product yields. N-Methyl-9a (R2 = Me) and N-butylindoles 9b (R2 = nBu) gave higher yields than the parent NH-free compound 9c (R2 = H), similarly to the results reported by Cai and coworkers.14 ICP-AES (Inductively Coupled Plasma Atomic Emission Spectrometry) analysis revealed a very low amount of palladium in the crude mixture after the workup (0.4 ppm). A hot-filtration test showed that removal of the heterogeneous catalyst completely stopped the reaction. Moreover, the catalyst could be recovered and reused for at least five reaction runs, with the yield decreasing by only 4% at the fifth run. TEM analysis of the recovered catalyst showed that the size of the palladium particles is very similar to those in the freshly prepared catalyst, which was attributed to the fact that the particles are confined in the mesoporous cages and their growth is therefore restricted.
The same research group later prepared palladium nanocrystals stabilized by cucurbit[6]uril and used this system (CB[6]-PdNPs) as a heterogeneous catalyst for the direct arylation of polyfluoroarenes with aryl iodides and bromides 10 (Scheme 6).16 A variety a polyfluorinated arenes 11 were found to be suitable for this transformation, generally affording the product in good yields. When multiple possible arylation sites were present, the reaction preferentially afforded the product of monoarylation at the kinetically most acidic C–H site. Electron-rich and electron-poor aryl halides can be used, and the process is tolerant to functional groups, such as esters, ketones, cyano and trifluoromethyl substituents. The heterogeneous catalyst was recycled for five reaction cycles, retaining 90% of its original activity at the fifth run.
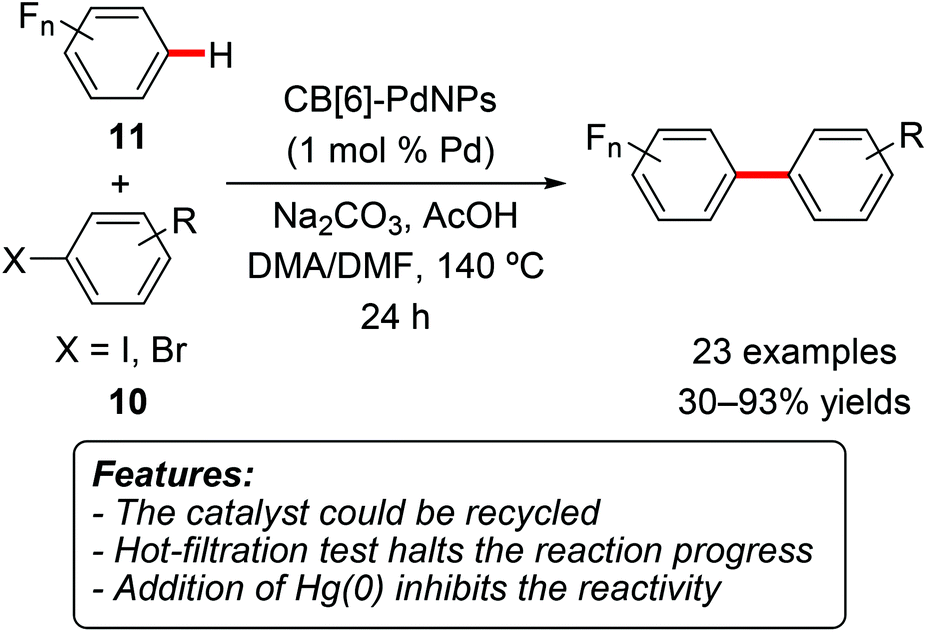 | ||
| Scheme 6 Cucurbit[6]uril-stabilized palladium nanoparticles for direct C–H functionalization of oligofluoroarenes 11. | ||
Both the addition of mercury(0) and the removal of the catalyst by hot filtration completely stopped the reactivity, suggesting that the active catalytic species are heterogeneous in nature.
Very recently, the same research group reported on the use of a perfluoroalkane-functionalized mesoporous MOF as a platform to encapsulate palladium nanoparticles. The MOF was prepared by reacting preformed NU-1000 MOF [Zr6(μ3-OH)8(OH)8(TBAPy)2; TBAPy = 1,3,6,8-tetrakis(p-benzoate)pyrene] with fluoroalkane carboxylic acids. The catalyst (Pd@F15-NU-1000) was used to promote the C2-regioselective C–H arylation of indoles in water.17 The catalyst could be reused for several consecutive reaction runs without loss of activity and metal leaching.
Vorotyntsev, Hierso and coworkers prepared dispersed palladium–polypyrrole nanocomposites (Pd@PPy) via redox polymerization of pyrrole with [Pd(NH3)4Cl2], and tested their efficiency in the catalytic C–H arylation of 2-butylfuran 12a (X = O) and 2-butylthiophene 12b (X = S) with aryl bromides 13 (Scheme 7).18 The reaction occurred generally with good conversion and positioned selectively at the C5-position. The recyclability of the catalyst was investigated but diverse results were found on the basis of the substrates of the reaction. In the best case, a second catalytic run was possible, with GC conversion decreasing from 100 to 85%. TEM analysis showed a general growth of the particle size in the recovered material. The authors hypothesized that the reaction proceeded through the release of highly active molecular palladium followed by redeposition of these soluble species on the existing metallic sites.
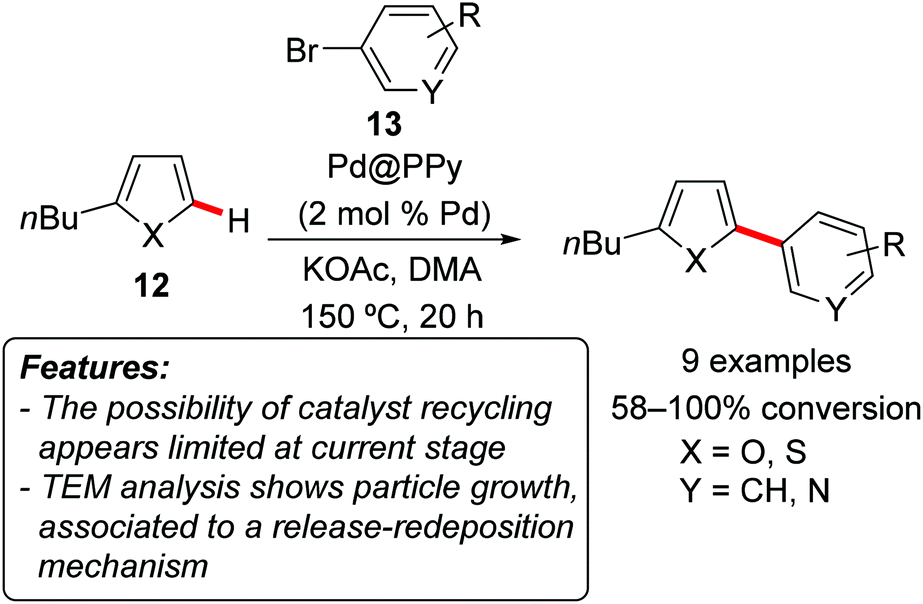 | ||
| Scheme 7 Palladium–polypyrrole nanocomposites as catalysts for the arylation of heteroaromatics 12. | ||
Sanford, Matzger and coworkers reported on the use of a novel crystalline material based on a defective metal–organic framework [MOF-5(Oh), obtained by reacting 1,4-benzenedicarboxylic acid with 1,3,5-tris(4-carboxyphenyl)benzene in the presence of Zn(NO3)2] impregnated with Pd(OAc)2 in the direct C–H arylation of naphthalene.19 Diphenyliodonium tetrafluoroborate (Ph2IBF4) was employed as an arylating reagent. The catalyst afforded a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of α- and β-phenylnaphthalenes in higher yield than soluble Pd(OAc)2 did (64 vs. 17%). Two distinct hot-filtration tests were conducted. In the first one, the catalyst was removed after 3 hours, showing no further progress of the reaction. In the second test, the catalyst was heated in the reaction solvent at the reaction temperature without substrates for 3–16 hours, followed by hot filtration. Next, the filtrate was added to the substrates and kept at the reaction temperature for 12 hours, giving only 2–8% of the phenylated product. These tests suggested that the catalytic role of the released homogeneous palladium species should be negligible. XRD (X-ray powder diffraction) analysis of the recovered catalyst showed its gradual degradation at the reaction temperature, resulting in a complete loss of crystallinity after 12 hours.
:1 mixture of α- and β-phenylnaphthalenes in higher yield than soluble Pd(OAc)2 did (64 vs. 17%). Two distinct hot-filtration tests were conducted. In the first one, the catalyst was removed after 3 hours, showing no further progress of the reaction. In the second test, the catalyst was heated in the reaction solvent at the reaction temperature without substrates for 3–16 hours, followed by hot filtration. Next, the filtrate was added to the substrates and kept at the reaction temperature for 12 hours, giving only 2–8% of the phenylated product. These tests suggested that the catalytic role of the released homogeneous palladium species should be negligible. XRD (X-ray powder diffraction) analysis of the recovered catalyst showed its gradual degradation at the reaction temperature, resulting in a complete loss of crystallinity after 12 hours.
Kim, Lee and coworkers applied nanocrystals of bimetallic Pd–Fe3O4 as a catalyst in the direct C–H arylation of imidazo[1,2-a]pyridine (14) with a series of aryl bromides 3 (Scheme 8).20 The reaction selectively afforded the products of C3-functionalization and tolerates a variety of functional groups such as nitrile, chloride, ketone, aldehyde and ether. Aryl bromides ranging from electron-rich to electron-poor could be used, with the latter giving in general better results in terms of yield. The catalyst could be magnetically recovered after completion of the arylation and successfully reused for ten consecutive reaction runs without loss of activity.
 | ||
| Scheme 8 C–H bond arylation of imidazo[1,2-a]pyridine (14) using magnetically recyclable Pd–Fe3O4 nanoparticles. | ||
Glorius and coworkers demonstrated that commercially available Pd/C in combination with catalytic amounts of CuCl could promote the C3-regioselective C–H arylation of benzo[b]thiophenes 15 using aryl chlorides 16 as the arylating agents (Scheme 9).21 It is noteworthy that the vast majority of methodologies reported for C–H arylations within this class of heterocycles favored functionalization at the C2-position. Electron-rich and electron-neutral aryl chlorides 16 gave generally excellent product yields, while the reactions with electron-poor arylating agents afforded the corresponding products in lower yields. Sterically encumbered aryl chlorides could also be used with satisfactory results. Unsubstituted (15a, R = H) and C5-functionalized with various substituents benzo[b]thiophenes 15 were amenable to the reaction conditions. An efficient recycling of the catalyst could not be established. The authors conducted a very accurate investigation in order to determine the nature of the active catalyst, relying on several tests. Four different 3-phase tests showed that supported reagents did not react under the standard reaction conditions. Both the addition of elemental mercury or the removal of the heterogeneous catalyst by hot filtration stopped the reaction. The effect of the addition of PPh3, generally considered as a poison for heterogeneous catalysts, was also checked, resulting in an inhibition of the reactivity. The presence of leached metal in the filtrate was also measured by X-ray fluorescence (XRF) spectroscopy, which showed the presence of less than 4 ppm of palladium (corresponding to 0.01% of the initial palladium loading). Additionally, the catalytic activity of different homogeneous palladium sources was investigated. The vast majority of them exhibited no reactivity, while PdCl2(PPh3)2 and Pd(PPh3)4 catalyzed the reaction, but interestingly afforded exclusively the product of C2-arylation. Thus the collected observations strongly suggested the catalytic species to be heterogeneous in nature.
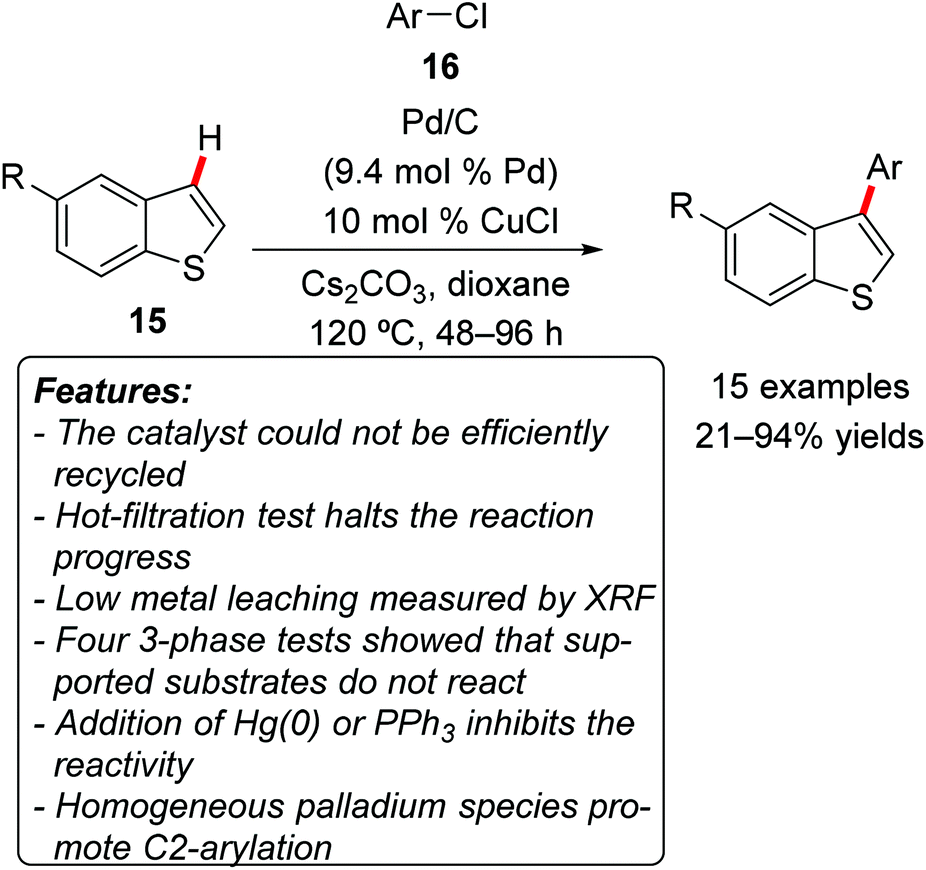 | ||
| Scheme 9 Site-specific C3-functionalization of benzo[b]thiophenes 15. | ||
Subsequently, the same research group developed another methodology, again based on simple Pd/C as a catalyst but relying on iodonium salts as arylating reagents with the purpose to provide the C3-site-selective C–H arylation of benzo[b]thiophenes 15 [R = (CH)4] and thiophenes 17 (R = H) (Scheme 10).22
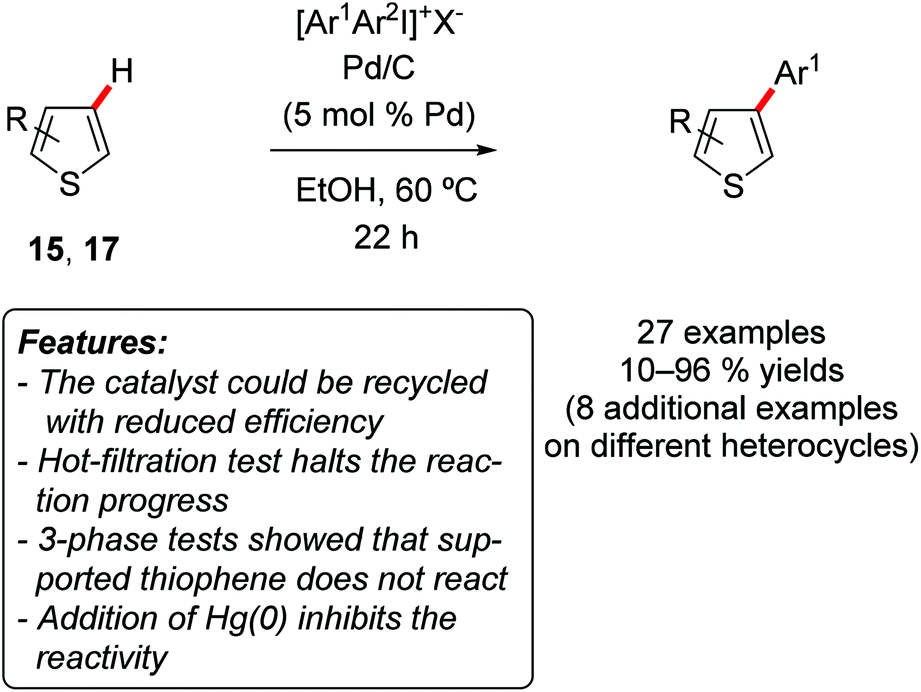 | ||
| Scheme 10 Pd/C-catalyzed C–H arylation of benzo[b]thiophenes 15 and thiophenes 17 under mild conditions. | ||
Simple diphenyliodonium tetrafluoroborate [Ph2I]BF4 was employed for phenylation reactions, while the use of unsymmetrical [ArI(TRIP)]OTf (TRIP = 2,4,6-triisopropylphenyl) resulted in the selective transfer of the less hindered aryl group. Notably, the reaction occurred under rather mild reaction conditions (60 °C), and proved to be insensitive to the presence of water or air. The methodology could also be extended to other heteroarenes, such as benzofuran, 1H-indole (2a, R1 = H), 2-butylfuran (12a), although in these substrates the arylation occurred selectively on the C2-position. Also in this case a detailed mechanistic investigation, based on 3-phase tests, Hg(0) poisoning and hot-filtration test, suggested that a heterogeneous species was responsible for the catalysis. The recycling of the catalyst was investigated, with a decrease in the yield observed for the second and third reaction run (from 89 to 64 and then to 40% yield).
Very recently the scope of this catalytic system based on Pd/C and diaryliodonium salts was expanded towards the direct arylation of polyaromatic hydrocarbons, including triphenylene, naphthalene, anthracene, pyrene and phenanthrene.23 On the basis of mechanistic investigations, Glorius and coworkers suggested that the reaction should proceed through the in situ formation of insoluble catalytically active metal nanoparticles.
Hayashi and coworkers used Pd/C as the catalyst in the direct (hetero)arylative polycondensation toward thiophene-based π-conjugated polymers.24 Using 2-bromo-3-hexylthiophene, they obtained regioregular head-to-tail poly(3-hexylthiophene-2,5-diyl) (HT-P3HT). The regularity in the polymer depended on the site-selectivity of the C–H bond functionalization, which occurred preferentially on the C5-position under the optimized reaction conditions.
Investigating the applicability of the commercially available silica-supported heterogeneous palladium catalyst SiliaCat® DPP-Pd for the synthesis of semiconductors, Welch and coworkers found that this transformation could efficiently be accomplished through direct heteroarylation rather than through traditional Stille and Suzuki–Miyaura cross-couplings.25 In certain cases, the use of a heterogeneous catalyst significantly reduced the formation of side-products derived from the homo-coupling of aryl bromides.
Yamada and coworkers developed a silicon nanowire array-stabilized palladium nanoparticle catalyst (SiNA-Pd) and tested it, among other palladium-catalyzed processes, for the direct C–H arylation of thiophenes and indoles with phenyl iodide.26 The procedure requires a very low catalyst loading (0.3 mol% Pd) and 2 equivalents of CsOAc as a base. The reaction on 2-methyl- and 2-ethylthiophenes afforded selectively the product of C5-phenylation in good yields (80 and 81%, respectively). The functionalization on N-methylindole gave a 4:2:1 mixture of C2-, C3- and diarylated products in 70% overall yield. Finally, the reaction on 1,3-dimethylindole selectively afforded the product of C2-phenylation in 63% yield. The nature of the active catalytic species was studied for other processes, but not with respect to this C–H arylation.
Wang and coworkers prepared a palladium catalyst immobilized on magnetic Fe3O4 nanoparticle (SiO2@Fe3O4-triazole-Pd) and applied it to the direct C2-site-selective C–H arylation of indoles 9 with arylboronic acids 18 (Scheme 11).27 The catalyst was prepared by forming triazoles applying click-chemistry on silica-coated Fe3O4 nanoparticles, followed by reaction of the particles with Pd(OAc)2. Substituted arylboronic acids 18 with both electron-donating and electron-withdrawing functionalities were suitable substrates.
 | ||
| Scheme 11 Palladium-immobilized on Fe3O4 magnetic nanoparticles as a catalytic system for the direct C2-arylation of indoles 9 with arylboronic acids 18. | ||
Electron-rich indoles 9 reacted smoothly, while indoles substituted with electron-withdrawing groups gave slightly lower yields. The catalyst could be reused for eight consecutive reaction runs without significant loss of activity, and ICP-AES analysis revealed that the Pd content in the reaction solution was less than 0.20 ppm.
Bäckvall, Olofsson and coworkers used palladium nanoparticles supported on amino-functionalized mesocellular foam (Pd0-AmP-MCF) to catalyze the C2-regioselective C–H arylation of indoles 9 with diaryliodonium salts 19 as the arylating reagents (Scheme 12).28 The reaction occurred in water at ambient temperature or 40 °C and tolerated a variety of functionalities both on the iodonium salt and on the indole motif. Both unprotected and N-protected indoles proved to be suitable substrates. The catalyst could be employed for three consecutive reaction runs, although with a gradual decrease of activity (yield reduced from 100 to 80 and then to 67%). ICP-OES (Inductively Coupled Optical Emission Spectrometry) analyses showed a very low amount of leached Pd in the filtrate at the end of the reaction (0.6 ppm). In the control experiment the authors also verified that the leached palladium was not catalytically active.
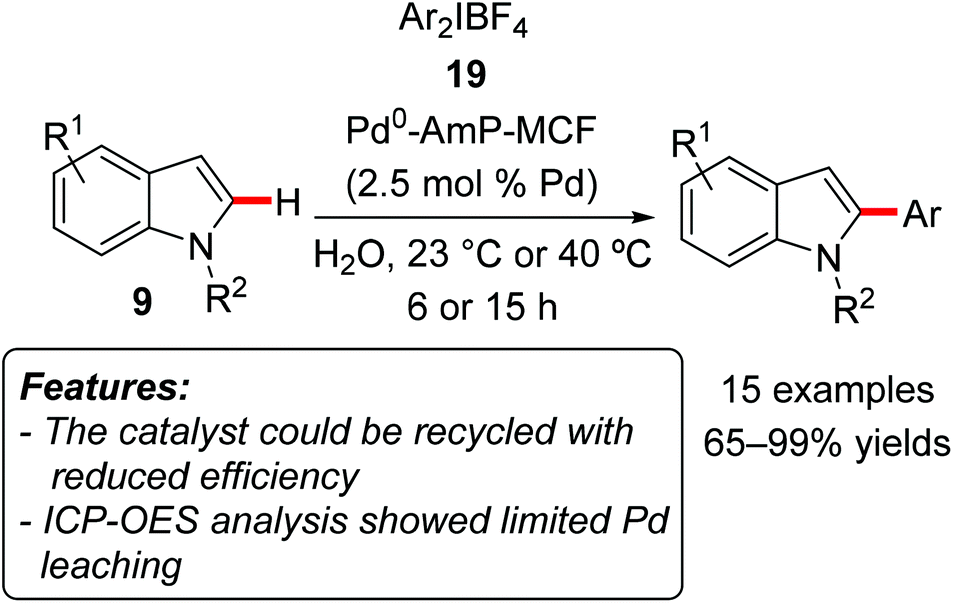 | ||
| Scheme 12 C2-selective arylation of indoles 9 catalyzed by heterogeneous nanopalladium. | ||
Srinivasu and coworkers prepared a mesoporous silica with a highly dispersed palladium nanoparticle composite catalyst (PS-3) and tested it for the direct C–H arylation of benzamides 20 with aryl iodides 8 (Scheme 13).29 The reaction occurred with excellent site-selectivity towards ortho-functionalization and was tolerant to the presence of functional groups, such as halides, ketones and ethers. Both electron-rich and electron-poor benzamides were competent substrates for the process. It is noteworthy that this catalytic system led to the predominant formation of monoarylated products 21, whereas the formation of diarylated benzamides was limited. Palladium was not detected by ICP-AES analysis in the filtrate at the end of the reaction. The catalyst could be recycled for five consecutive reaction runs with consistent activity and selectivity. XRD, XPS (X-ray photoelectron spectroscopy) and TEM (transmission electron microscopy) analyses of the recovered catalyst demonstrated that the catalyst is stable under the reaction conditions.
 | ||
| Scheme 13 ortho-Arylation of benzamides 20 over Pd/mesoporous silica. | ||
Jia, Xi and coworkers immobilized palladium on an amino group-functionalized reduced graphene oxide (PdII-FRGO) and tested the obtained material as a catalyst for a series of Pd-catalyzed reactions.30 Among other processes, the catalyst was found to be efficient in promoting the C5-selective C–H arylation of 2-ethylthiophene with aryl bromides. Four examples were reported, and the products were obtained in good yields (67–85%).
While exploring the use of palladium nanoparticles supported on modified single-walled carbon nanotubes as catalysts for Ullman-type N-arylation of indoles and imidazoles, Veisi and Morakabati found that changing the arylating reagents from aryl iodides to aryl chlorides or bromides led to a notable switch in the chemo-selectivity of the reaction using indoles, with the preferential formation of the C3-functionalized product.31 The authors rationalized this behavior in terms of the different electronic character of the formed Pd(II)-intermediate. The species generated from chlorides and bromides were “harder electrophiles” and thus favored the reaction on C3, while the “softer electrophiles” generated from aryl iodides would prefer N-functionalization. The nature of the active catalytic species was investigated in an Ullman-type reaction by hot filtration and by measurement of the palladium leaching by ICP-AAS. Both experiments supported a genuinely heterogeneous process.
Keshipour and Shaabani immobilized Cu(I) and Pd nanoparticles on ethylenediamine-functionalized cellulose and used the obtained heterogeneous species [Cu(I)/PdNPs@EDAC] as a catalyst for the 1,3-dipolar cycloaddition/direct arylation sequence leading to 1,4,5-trisubstituted-1,2,3-triazoles 24 (Scheme 14).32 In the first step the organic azide formed in situ from the reaction between benzyl bromide (22) and sodium azide reacted with phenylacetylene (23) in a copper-catalyzed 1,3-dipolar cycloaddition to give a 1,4-disubstituted-1,2,3-triazole. The addition of an aryl halide after two hours made the subsequent palladium-catalyzed C–H arylation to occur, affording the final 1,4,5-trisubstituted-1,2,3-triazole 24. Several aryl iodides and bromides, including heteroaryl bromides, were employed in the reaction with good results. The reaction proved to be tolerant to various functional groups, including halides, ethers, esters and ketones. The catalyst could be recycled for five consecutive cycles with only a minor loss of catalytic activity.
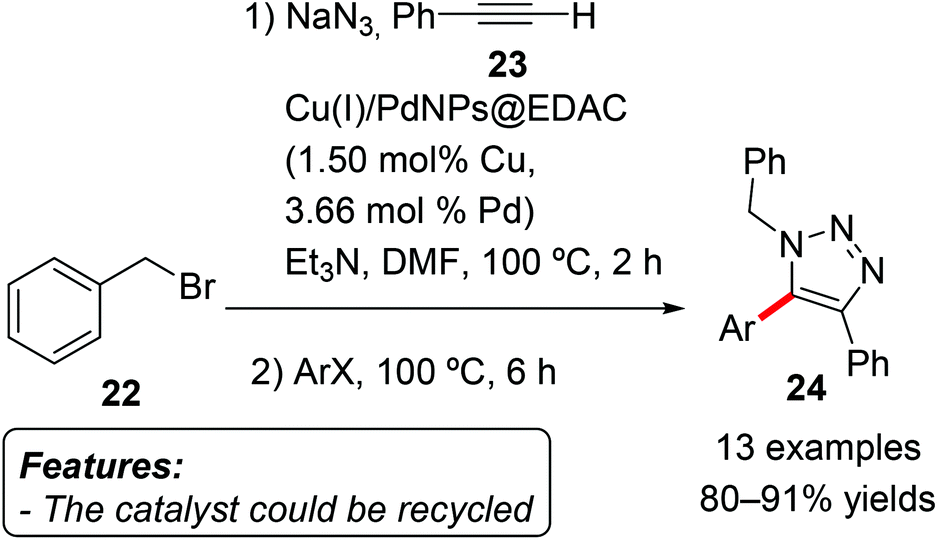 | ||
| Scheme 14 Copper(I) and palladium nanoparticles as an efficient catalyst for the 1,3-dipolar cycloaddition/direct arylation sequence. | ||
Lee and Park realized a meta-selective C–H arylation of anilides using a heterogeneous copper catalyst composed of metal nanoparticles entrapped in aluminum oxyhydroxide nanofibers [Cu/AlO(OH)] and diaryliodonium salts as arylating reagents (Scheme 15).33
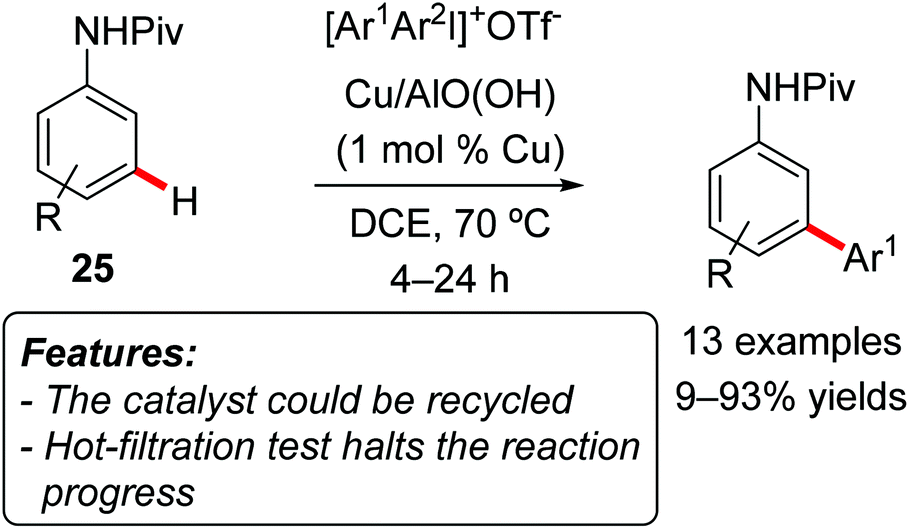 | ||
| Scheme 15 Recyclable copper catalyst for meta-selective C–H arylation. | ||
The reaction was efficient with pivalanilides 25 bearing electron-donating groups, while arylation of 2-fluoropivalanilide 25a (R = 2-F) resulted in a very low conversion. Due to the para-directing effect of its methoxy-group, 3-methoxypivalanilide 25b (R = 3-OMe) furnished the ortho-phenylated derivative as the major product. The choice of iodonium salts is not limited to Ph2IOTf, and the use of unsymmetrical arylmesityliodonium triflates (ArMesIOTf) resulted in the selective transfer of the less bulky aryl group, affording the meta-arylated anilide in generally good yields. A hot-filtration test completely stopped the reactivity, and the catalyst could be reused for five consecutive reaction runs in a sample reaction. In the first three runs the isolated yield remained almost constant (from 93 to 90%), with a more noticeable decay in the fourth run (down to 80%).
Zhang, Wang and coworkers used CuO nanospindles to catalyze the direct C–H arylation of heterocycles 26, such as benzoxazole (26a, X = O), benzothiazole (26b, X = S) and N-methylbenzimidazole (26c, X = NMe), with aryl iodides 8 (Scheme 16).34
 | ||
| Scheme 16 CuO-nanospindle-catalyzed ligand-free arylation of C–H bonds in heterocycles 26. | ||
The reaction worked efficiently and selectively, generally resulting in higher product yields when electron-poor aryl iodides were employed. The protocol could not be extended to aryl bromides. The catalyst could be recycled and reused for at least three reaction runs without significant loss of activity. The removal of the catalyst by centrifugation after one hour stopped the reaction, since no further conversion could be observed after 3 hours. ICP-AAS analysis evidenced a slight leaching of copper during the reaction (<1 ppm). This research group later reported on the CuO nanoparticle-catalyzed alkenylation with alkenyl bromides of C–H bonds with comparable substrates under similar reaction conditions.35 The corresponding products were obtained in moderate to excellent yields. Here, the addition of PPh3 proved to be beneficial. Also in this case the catalyst could be recycled and displayed almost the same activity in at least three reaction runs. The removal of the heterogeneous species by hot filtration stopped the reaction, and ICP-AAS analysis showed that less than 1 ppm of copper leached into the solution.
Phan and coworkers prepared a series of copper-containing crystalline porous MOFs and used them as catalysts for the direct C–H arylation of various heterocycles with aryl halides 10.36
Thus, the reaction catalyzed by Cu2(BPDC)2(BPY) (BPDC = 4,4′-biphenyldicarboxylate; BPY = 4,4′-bipyridine) was tested on benzoxazole (26a, Scheme 17a), benzothiazole (26b), 2-chlorothiophene, 4-methylthiazole, and N-methylbenzimidazole (26c), with results in terms of yields ranging from moderate to excellent.36a Various aryl iodides, bromides and chlorides 10 could be used, with the reaction being tolerant to functional groups such as nitrile, ketone, nitro and ethers. The catalyst could be reused for six reaction runs with a limited decrease in the catalytic efficacy. Analysis of the recovered catalyst by FT-IR and XRD showed limited changes in its structure. However, among other Cu-MOFs, Cu2(OBA)2(BPY) [OBA = 4,4′-oxybis(benzoate); BPY = 4,4′-bipyridine] displayed the highest catalytic activity for direct arylations of benzothiazole (26b, Scheme 17b) with aryl iodides 10a, when tBuOLi was employed as the base in dioxane at 120 °C (Scheme 17b).36b
 | ||
| Scheme 17 Direct C–H arylation of heteroarenes 26 with aryl halides 10 over heterogeneous MOFs Cu2(BPDC)2(BPY) and Cu2(OBA)2(BPY). | ||
The same research group synthesized a nickel-containing crystalline mesoporous metal–organic framework Ni2(BDC)2(DABCO) (BDC = 1,4-benzenedicarboxylate; DABCO = 1,4-diazabicyclo[2.2.2]octane) and tested it in the direct C–H arylation of benzoxazole with simple or para-substituted phenylboronic acids.37 Results were found to be highly dependent on the nature of the boronic acid, with yields ranging from poor to good. The catalyst could be recycled for seven reaction runs with almost constant activity. The removal of the heterogeneous catalyst from the reaction mixture by centrifugation halted the reaction, suggesting that the latter was not catalyzed by leached metallic species.
Wada and coworkers prepared a series of solid ruthenium catalysts and tested them in the chelation-assisted arylation of aromatic C–H bonds in substrates 27 with aryl halides 10 (Scheme 18).38
 | ||
| Scheme 18 Solid ruthenium catalysts for the direct arylation of aromatic C–H bonds. | ||
The most efficient catalytic system was obtained by impregnating CeO2 with a solution of [{RuCl2(CO)3}2] and subsequent calcination at 400 °C. Further, after treating the solid ruthenium-system with three equivalents of PPh3 under a hydrogen atmosphere, a more active catalyst (3PPh3–Ru/CeO2) was also prepared. Both electron-rich and electron-poor aryl chlorides 10 (Scheme 18, X = Cl) reacted with benzo[h]quinoline 27 to afford the arylation products selectively and in good yields. When the reaction was performed on 2-phenylpyridine or on 1-phenyl-2-pyrazol 27 (Scheme 18, not fused aromatic ring), mixtures of mono- and diarylated products were obtained. The recycling of the catalyst was tested in a sample reaction. After being isolated by centrifugation, washing, calcining at 400 °C and treatment again with PPh3, the catalyst could be reused without loss of activity for at least three runs.
Li, Jiang and coworkers reported a transition metal-free direct C–H arylation of aromatics with aryl iodides or bromides, using a MOF catalyst [MOF-253, Al(OH)(BPYDC); BPYDC = 2,2′-bipyridine-5,5′-dicarboxylate] (Scheme 19).39 The reactions were conducted using the arene 28 as a reactant and solvent, with tBuOK as the base and at reaction temperatures ranging from 100 to 140 °C. Product yields for the arylation of simple benzene 28 (Scheme 19, R1 = H) were generally very high, using a wide range of aryl iodides or bromides 10 as well as heteroaryl iodides 29 or 30. The direct arylation of substituted benzene derivatives gave more variable results, both in terms of product yields and regioselectivity. The catalyst could be reused for at least five reaction runs without significant loss in terms of yield. A hot-filtration test showed that the reaction does not proceed after the removal of the solid catalyst.
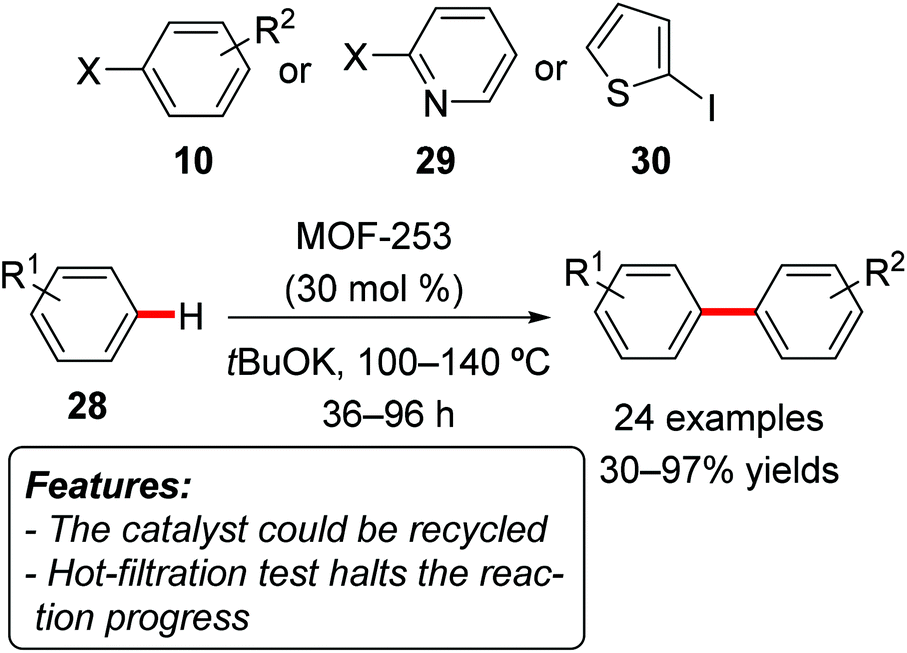 | ||
| Scheme 19 Arylation of unactivated arenes 28 with aryl halides over recyclable heterogeneous catalysts. | ||
2.2. Hydroarylation of alkenes
Only a limited number of C–C bond formation reactions other than direct arylations of (hetero)aromatic substrates have been reported in the context of heterogeneous catalysis. For instance, Wada, Inoue and coworkers demonstrated that ruthenium immobilized on CeO2 efficiently catalyzes the hydroarylation of vinylsilanes with a range of aromatic ketones 31 (Scheme 20).40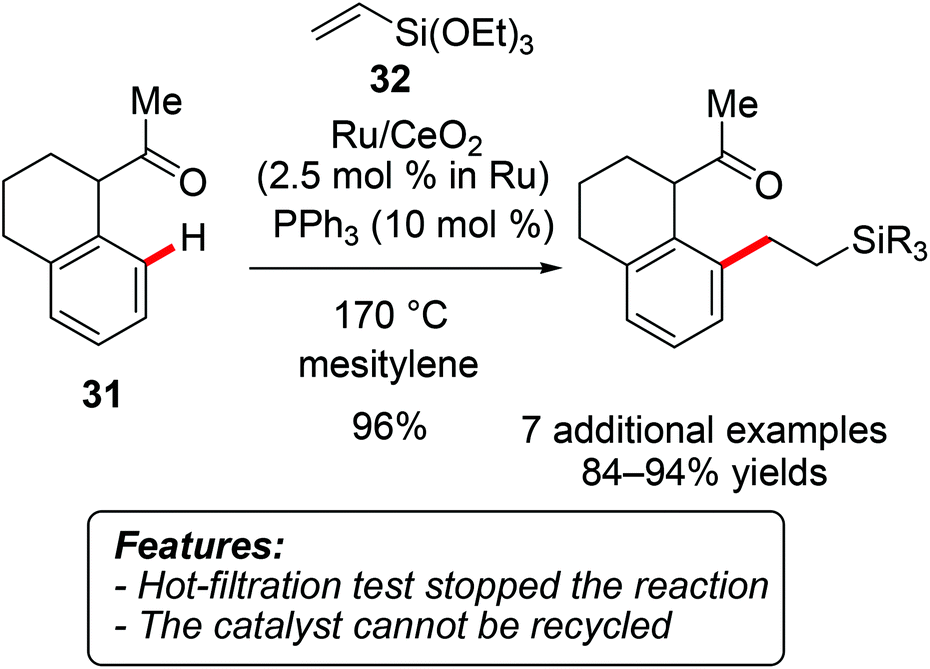 | ||
| Scheme 20 Ru/CeO2-catalyzed hydroarylation with arenes 31. | ||
Triethoxy(vinyl)silane (32) was mainly used as a substrate, although one example of ethoxydimethyl(vinyl)silane has also been reported. The reaction could be conducted without an additional reaction medium on a larger scale of 35 mmol, thereby significantly reducing the catalyst loading to 0.06 mol%, albeit under rather drastic reaction conditions. The hot-filtration test showed that removal of the solid catalyst completely inhibited the reaction. However, ICP-AES analysis of the product illustrated that 3% of the ruthenium leached from the support. Attempts to recycle the catalytic system were thus unsuccessful.
Haldar and Koner prepared the iron-containing mesoporous aluminosilicate (Fe–Al-MCM-41) which proved to be effective in catalyzing the hydroarylation of styrene and its derivatives with phenols in good yields and selectivities.41 The procedure allowed the effective preparation of 10 different 1,1-diarylethanes with GC measured conversions ranging from 81 to 99%. One representative example with a 65% isolated yield after chromatographic purification was reported. The catalyst could be reused for 4 consecutive runs, with only a negligible loss of activity. The genuinely heterogeneous conditions of the process were supported by a hot-filtration test and by proving that no metal leaching was detected in the products by ICP-AAS analysis.
2.3. Fujiwara–Moritani reaction
The heterogeneously catalyzed coupling of arenes with olefins has also been reported through the oxidative Fujiwara–Moritani process.42 Thus, Ying and coworkers developed a palladium-polyoxometalate nanomaterial Pd-H6PV3Mo9O40/C (abbreviated as PdV3Mo9/C below) and tested it in the oxidative olefination of anilides 33 with acrylates 34 (R3 = CO2-alkyl), with molecular oxygen as the terminal oxidant (Scheme 21).43 The reaction proved to be quite sensitive to the electronic nature of the anilide, with the best yields being obtained with unsubstituted or with monomethylated substrates. The catalyst could be recycled after reloading the recovered solid with PV3Mo9 prior to the next run. ICP-MS (Inductively Coupled Plasma-Mass Spectrometry) analysis of the filtrate detected a relatively low amount of leached palladium (7 ppm).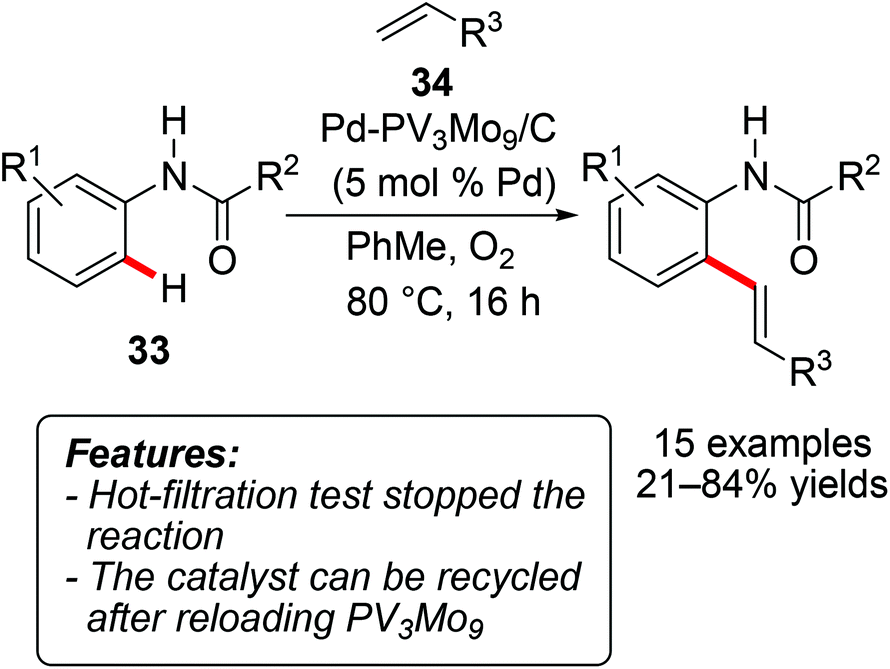 | ||
| Scheme 21 Hydroarylations with anilides 33 catalyzed by heterogeneous Pd. | ||
Lei, Zhao and coworkers prepared a series of 2,2′-bipyridine-grafted periodic mesoporous organosilica materials and used them as a support for Pd(II) species.44 The obtained catalytic systems were tested for the Fujiwara–Moritani reaction obtaining moderate to good results. Interestingly, the catalyst showed higher TOF and selectivity than simple Pd(OAc)2 and could be reused at least three times remaining unchanged, as indicated by XANES (X-ray Absorption Near Edge Structure) and EXAFS (Extended X-Ray Absorption Fine Structure) analyses after the recycling.
Recently, the preparation of a heterogeneous catalyst obtained by deposition of Pd(II) on MoS2 nanosheets has been reported. This catalyst was used, among other processes, for the oxidative C–H alkenylation of heteroaromatics, using Cu(OAc)2 as a terminal oxidant. However, the catalyst could not be recycled.45
2.4. C–H acylation
The heterogeneous palladium-based catalytic system Pd(II)/Mg–La mixed oxide was used for the oxidative acylation of aromatic substrates by Kantam, Venugopal and coworkers.They realized an oxidative direct acylation of 2-arylpyridines 27 with alcohols 35 (R2 = aryl, alkyl), using tert-butyl hydroperoxide (TBHP) as the oxidant (Scheme 22).46 The corresponding ketones 36 were usually obtained in good yields, applying either benzylic or aliphatic alcohols. The reactions tolerated several different functional groups, such as fluoro, chloro, bromo, trifluoromethyl and methoxy substituents. The catalyst could be reused four times without a significant loss of activity, and the palladium(II) content of the original and the recovered catalyst measured by ICP analysis was 9.1 and 8.9%, respectively, suggesting only a minor metal leaching.
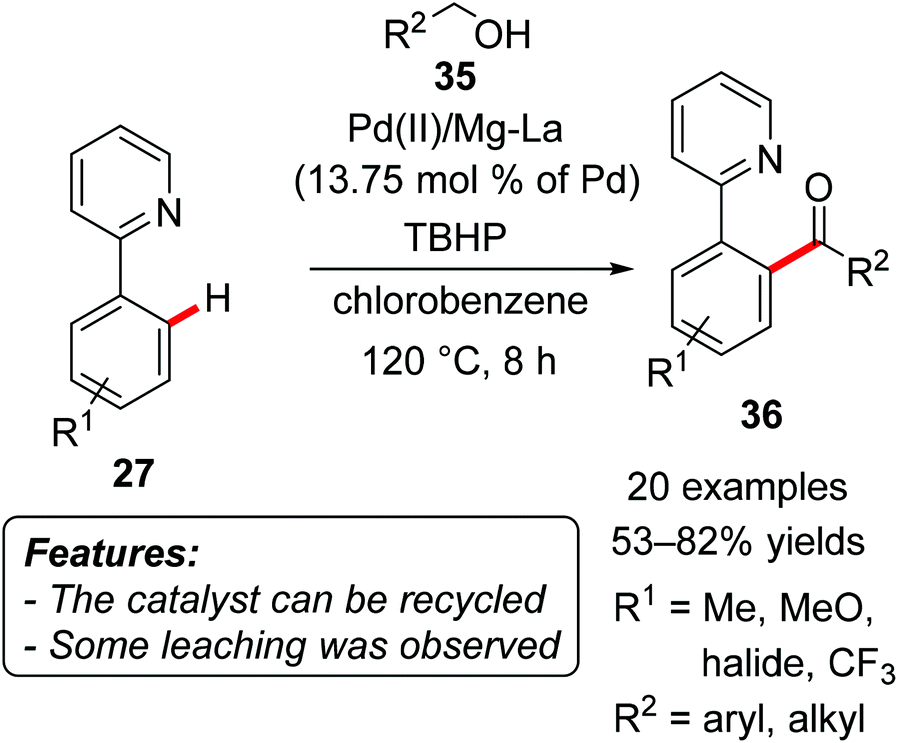 | ||
| Scheme 22 Pd/Mg–La mixed oxide-catalyzed oxidative C(sp2)–H bond acylation using alcohols 35. | ||
Very recently Wu and coworkers reported a Pd/C-catalyzed carbonylative cyclization of N-arylpyridin-2-amine, using simple DMF as the CO source and K2S2O8 as the oxidant. Yet, the catalyst could not be reused.47
2.5. C–H cyanation
Kantam and coworkers employed their Pd(II)/Mg–La mixed oxide catalytic system (see chapter 2.4) also in the C–H cyanation of 2-arylpyridine derivatives 27 using the combination of NH4HCO3 and DMSO as a cyanating source (Scheme 23).48 The reaction proceeded efficiently on a variety of substrates with different functional groups on the aromatic moiety. The recyclability of the catalyst was tested, showing a negligible loss of activity in four consecutive reaction runs. | ||
| Scheme 23 A Pd(II)/Mg–La mixed oxide catalyst for cyanation of aromatic C–H bonds. | ||
The palladium loading of the heterogeneous catalyst was measured by ICP-AAS after each reaction, revealing only a slight decrease (from 9.76% for the fresh catalyst to 9.60% after the fourth cycle). This arguably indicates a limited metal leaching into the products. Two different hot-filtration tests were performed. According to the first test, the reaction was filtered after three hours and the filtrate was left at the reaction temperature for additional 18 hours. The reaction did not proceed further after the filtration. In the second experiment, the catalyst was stirred at the reaction temperature for ten hours before being filtered. The filtrate was used to perform the cyanation reaction, but no product formation was observed. All these findings suggested that the process is actually catalyzed by a heterogeneous catalyst, rather than by leached homogeneous species.
Other examples of C–H cyanation processes catalyzed by heterogeneous species have been reported sporadically. Hence, Khorshidi showed that a ruthenium(III) exchanged zeolite was able to promote the cyanation of indoles using K4[Fe(CN)6] as the cyanide source in the presence of K2CO3 and Cu(OAc)2.49 The reaction afforded the product of C3-functionalization site-selectively and in generally good yields. The catalyst could be recycled, however, a 15% loss of efficiency was observed after five runs.
A few examples of cyanation via C(sp3)–H functionalization have also been reported. Verma et al. used a starch-immobilized ruthenium catalyst for the oxidative cyanation of tertiary amines with NaCN as a cyanide source and H2O2 as a terminal oxidant.50 The catalyst was prepared by reacting silyl-functionalized imidazolium chloride with expanded corn starch, followed by treatment with ruthenium(III) chloride. Different aryl dialkylamines could be converted to the corresponding α-aminonitriles in good yields. The reaction on N,N-dimethyl benzylamine was much slower and furnished the product in a reduced yield. The recyclability of the catalyst was tested for six consecutive reaction runs, and no loss of activity was noted. Moreover, ruthenium could not be detected by ICP-AES analysis of the filtrate obtained at the end of the reaction. It should be considered that from these results it is not possible to rule out a release-and-catch mechanism.51
Mizuno and coworkers used manganese oxide-based molecular sieves (OMS-2, KMn8O16·nH2O) to promote the α-cyanation of tertiary amines 37 with trimethylsilyl cyanide (TMSCN, 38) as a cyano source and oxygen as the terminal oxidant (Scheme 24).52 The reaction proved to be efficient on a variety of alkyl and aryl substituted amines, affording exclusively the product of mono-cyanation 39, usually with good selectivity in those cases where several different α-positions were suitable for the functionalization. A hot-filtration test, the low amount of manganese detected by ICP-AES in the filtrate, and the recyclability of the catalyst (reused at least five times) indicated that the reaction was promoted by a heterogeneous catalyst rather than by leached homogeneous species. Mechanistic investigations revealed that the transformation possibly occurs through an initial single-electron transfer (SET) followed by a deprotonation. A second SET and the final attack of a cyanide ion on the iminium intermediate furnished the final product.
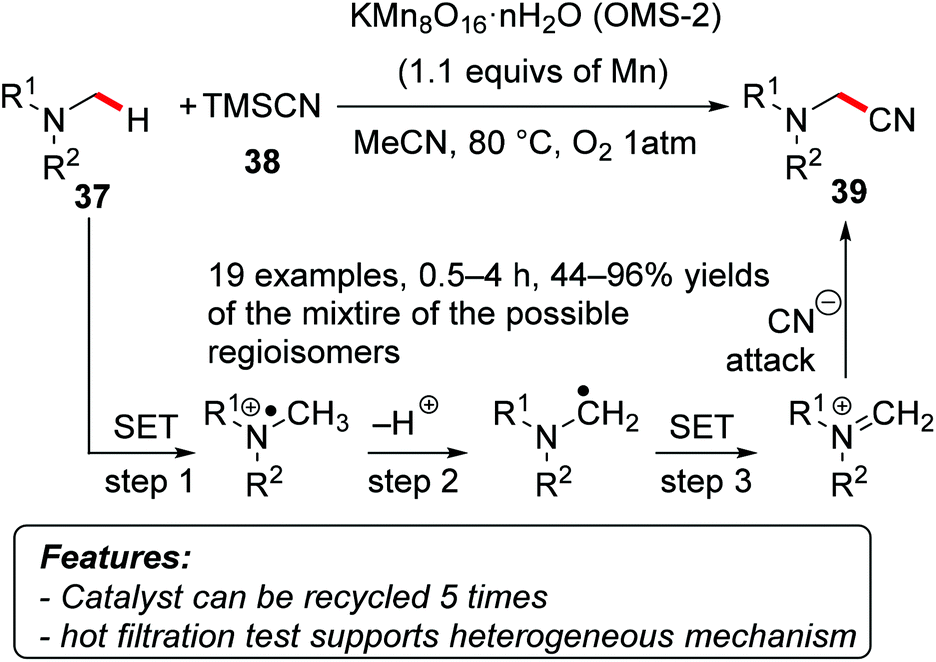 | ||
| Scheme 24 Regioselective α-cyanation of tertiary amines 37 catalyzed by heterogeneous manganese oxides. | ||
Jain and coworkers reported on an alternative approach to the same process, using magnetic Fe3O4-succinic acid-PEG nanoparticles as a catalyst, NaCN as a cyanide source and H2O2 as a terminal oxidant.53 The reaction worked efficiently on aryldialkylamines 40, while benzyldimethylamines demonstrated slower conversions with lower yields. The catalyst, which can be easily recovered at the end of the reaction with an external magnet, could be used for six runs without appreciable loss of catalytic efficacy.
2.6. Miscellaneous
Vila and Rueping elegantly realized a photo-induced titanium dioxide-catalyzed multicomponent reaction between tertiary amines 40, isocyanides 41 and water, affording a variety of α-aminoamides 42 (Scheme 25).54a The reaction tolerated a wide range of functional groups and afforded the products in moderate to good yields (40–82%). The catalyst could be recovered by simple centrifugation and reused in five consecutive runs without a considerable loss of reactivity or selectivity. | ||
| Scheme 25 Visible light-mediated/TiO2-catalyzed three-component reaction. | ||
Under essentially the same conditions, but in EtOH as the solvent, the successful direct arylation of heteroaromatics with diazonium salts was achieved recently by the same research group.54b
Phan and coworkers demonstrated that a copper-based metal–organic framework [MOF-199, Cu3(BTC)2; BTC = 1,3,5-benzenetricarboxylate] was able to catalyze the oxidative coupling of N,N′-dimethylanilines 43 with terminal alkynes 44 using TBHP as a terminal oxidant to afford the corresponding propargylamines 45 (Scheme 26).55 The explored substrate scope was rather limited, with yields ranging from moderate to good (55–81%). The catalyst could be recycled ten times without significant loss in the catalytic activity. Moreover, FT-IR and XRD analyses showed that the catalyst was unchanged after recovery from the reaction mixture. A hot-filtration test was also performed highlighting that the reaction was stopped after removal of the heterogeneous catalyst.
 | ||
| Scheme 26 Metal–organic framework (MOF)-catalyzed oxidative synthesis of propargylamines 45. | ||
Wu and coworkers reported on a Pd/CeO2-catalyzed oxidative synthesis of indoles 48 from anilines 46 and alkynes 47 through a C–H activation process. The reaction was co-catalyzed by copper(II) species and used atmospheric oxygen as the terminal oxidant (Scheme 27).56 Several functional groups were well tolerated under the reaction conditions, and the products were obtained – with few exceptions – in generally good yields. When aryl, alkyl-substituted alkynes 47 were used, the process occurred with good regioselectivity, placing the alkyl moiety in 48 distal to nitrogen, being thus reminiscent of the selectivity in homogeneous catalysis. A mercury test was performed by adding elemental mercury to the reaction mixture after 15 minutes, when the conversion achieved was 15%. After two hours, the reaction reached 20% conversion, while in a control mercury-free reaction the conversion was 88%. The catalyst could not be recycled, since a drastic decrease in the product yield was observed already in the second run.
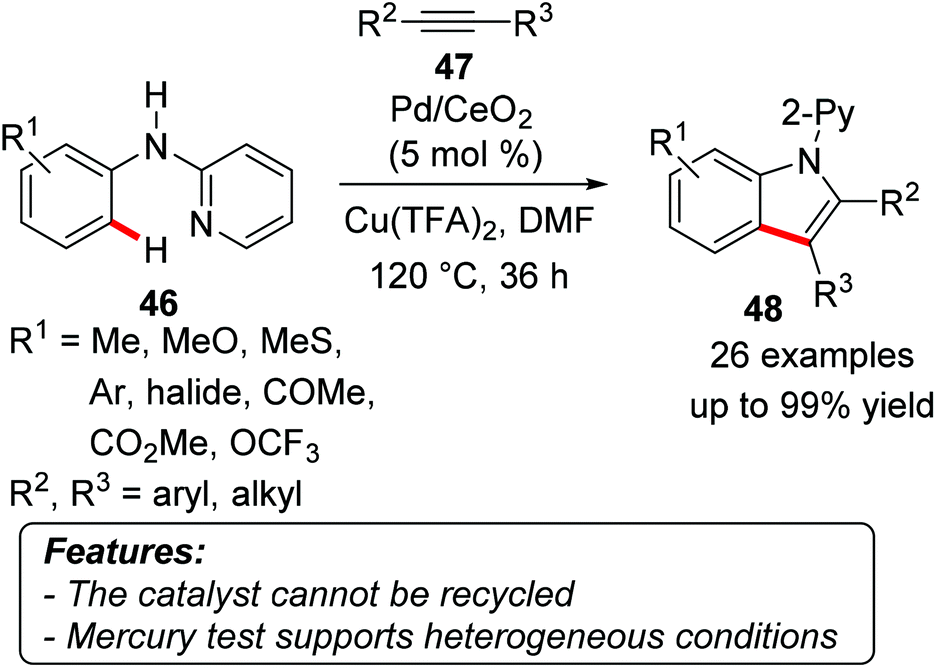 | ||
| Scheme 27 Pd/cerium(IV) oxide-catalyzed oxidative synthesis of indoles 48. | ||
An operationally simple protocol for the direct C–H bond perfluoroalkylation of arenes 28 with perfluoroalkyl iodides and bromides 49 using a supported platinum catalyst has been recently reported (Scheme 28).57 A variety of perfluoroalkyl-substituted aromatic compounds 50 were efficiently synthesized with excellent substrate scope, whereas the catalyst could easily be filtered or centrifuged off and used again.
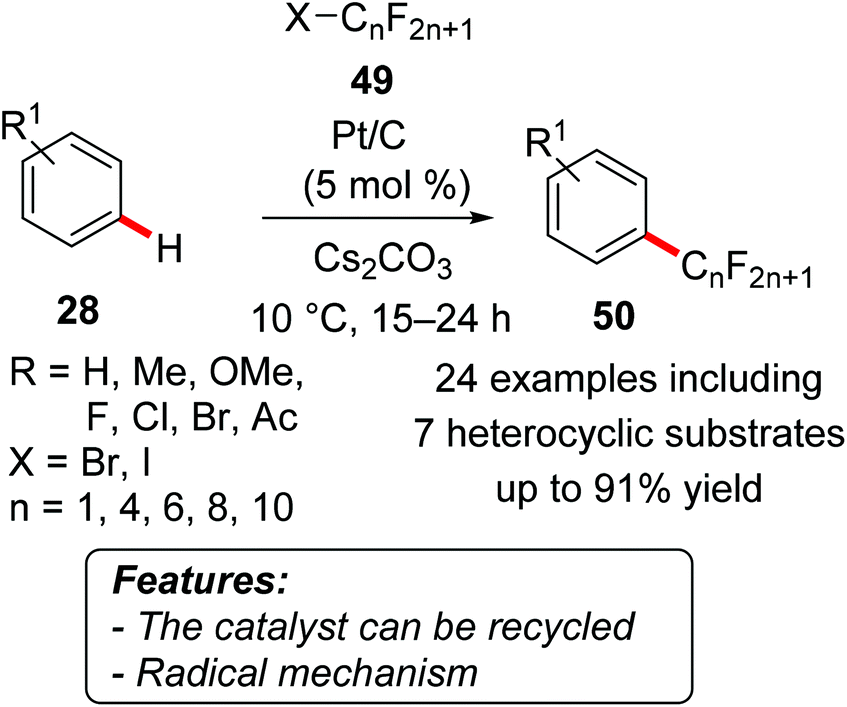 | ||
| Scheme 28 Heterogeneous Pt/C-catalyzed oxidative C–H perfluoroalkylation. | ||
A few other examples of the oxidative activation of tertiary amines have been reported, using different heterogeneous catalysts based on iron,58 ruthenium,59 titanium,60 palladium61 or gold.62 Most of the examples involve the coupling of substituted tetrahydroisoquinolines with pro-nucleophiles, such as nitromethane or other activated carbon nucleophiles.
The first coupling of aryl amines 51via oxidative C–H activation63 using a heterogeneous Rh/C catalyst in the presence of acids was recently reported (Scheme 29).63a Biaryl compounds 52 were selectively prepared in good yields and subsequently converted into carbazoles.
 | ||
| Scheme 29 Rh/C-catalyzed oxidative synthesis of biaryl compounds 52. | ||
Moreover, silica-supported dodecatungstophosphoric acid (DTP/SiO2) allowed C(sp3)–H bond functionalizations of a methyl group in various (azaaryl)methanes to be performed in good to excellent yield.64 The DTP/SiO2 catalyst could be recovered by simple filtration and reused several times without the loss of catalytic activity.
3. C–Heteroatom bond formation
3.1. Nitrogenation
Thus far, only a few examples of heterogeneous catalytic C–H activation processes towards the formation of C–heteroatom bonds have been reported, mainly focusing on the formation of C–N, C–O or C–halogen bonds.Ying and coworkers used their (vide supra) palladium-polyoxometalate nanomaterial Pd-H6PV3Mo9O40/C (below mentioned as PdV3Mo9/C) as a catalyst for the oxidative coupling of diaryl amines 54 (R1, R2 = aryl) with acrylates 53 (Scheme 30).43 The process required an O2 atmosphere and appeared to be quite sensitive to the electronic nature of the substrates. Moreover, the replacement of one of the aryl groups by a butyl group as well as of the acrylate by methyl vinyl ketone resulted in low yields. The catalyst could be recycled after reloading with PV3Mo9, although with a loss of activity (1st cycle: 87; 2nd cycle: 79 and 4th cycle: 67% yield, respectively). A hot-filtration test illustrated that the reaction progressed even after removal of the solid catalyst, thus proving that some homogeneous Pd species can contribute to the catalysis.
 | ||
| Scheme 30 Palladium-polyoxometalate nanomaterials for the C–N bond formation through oxidative amination. | ||
Alternatively, porous γ-MnO2 was employed by Ghosh, Panda and coworkers as an efficient heterogeneous catalyst for the direct oxidative amination of substituted benzoxazoles through C–H activation with secondary amines in the presence of O2.65 The catalyst could be easily separated by filtration and applied several times with essentially the same efficacy.
Li and coworkers developed a methodology for the synthesis of benzamides 56 through an oxidative C(sp2)–H bond functionalization of benzaldehydes 54 (Scheme 31).66
 | ||
| Scheme 31 Heterogeneous cobalt(II)-based catalysts for the C–H bond functionalization of benzaldehydes 54. | ||
The reaction was catalyzed by heterogeneous cobalt-based catalysts obtained from pyrolysis of a cobalt-containing MOF (Co9(BTC)6(TPT)2(H2O)15; BTC = 1,3,5-benzenetricarboxylate; TPT = 2,4,6-tris(4-pyridyl)-1,3,5-triazine). The pyrolysis could be conducted at different temperatures, and the material with the highest catalytic activity was obtained at 600 °C (Co@C-N600). Several aromatic and a few aliphatic aldehydes were reacted with substituted formamides 55 using TBHP as an oxidant. The substrate scope was widely explored and the process proved to be robust and tolerant to different functional groups. At the end of the reaction the catalyst could be recovered using an external magnet and, after being washed and dried, it could be reused for five consecutive cycles in a sample reaction with only a slight loss in the product yield. ICP-AAS analysis detected a limited amount of Co (less than 0.1% of the total) in the solution obtained by hot filtration at the end of the reaction.
Biffis and coworkers reported that copper species dispersed on oxide supports catalyzed the C–H amidation in the α-position of cyclic ethers, using chloramine T as a nitrene source.67 The authors proposed that the catalyst worked as a reservoir of active catalytic species, which are released into the solution. The catalyst could be recovered, but the product yield dropped after each cycle.
Voutchkova and Crabtree showed that commercially available Ir/C was able to promote direct functionalizations of benzylic C–H bonds in simple aromatic substrates to give aldehydes, esters and imines, albeit in low yields and poor selectivity.68 The catalyst could be recycled twice without loss of activity.
Very recently, a Cu-based MOF [Cu2(BPDC)2(DABCO); BPDC = 4,4′-biphenyldicarboxylate; DABCO = 1,4-diazabicyclo[2.2.2]octane] was reported to promote the oxidative coupling between heteroaromatics and amides, using di-tert-butyl peroxide as an oxidant.69 The removal of the catalyst before reaction completion halted the reactivity, and ICP-MS analysis of the filtrate showed very limited metal leaching, suggesting that the reaction is actually catalyzed by the solid catalyst rather than by homogeneous leached metal particles. Moreover, the catalyst could be recovered and reused several times without significant loss of activity.
3.2. C–H oxygenation
Fei and Cohen prepared a palladium-containing MOF by initial incorporation of the thiocatecholate (TCAT) ligand into a UiO-type MOF, and then using it as a support for palladium.70 The obtained material UiO-66-PdTCAT was able to catalyze the regioselective oxidative C–H alkoxylation of 7,8-benzoquinoline 56a (Scheme 32, R1,R2 = –(CH)2–).70 The alkoxylation reaction made use of simple alcohols and (diacetoxyiodo)benzene as the oxidant (Scheme 32). Methanol, ethanol and 2,2,2-trifluoroethanol provided a virtually complete conversion (95–99%), while the degree of transformation with 2-propanol was considerably lower (21%). The methoxylation reaction could be catalyzed by homogeneous Pd(OAc)2 as well, furthermore in a nearly quantitative conversion and in a shorter reaction time. However, the heterogeneous catalyst has the obvious advantage of being recyclable, as it could be reused for five reaction runs without a significant decrease in the yield. A hot-filtration test and the very low amount of leached Pd into the filtrate (<0.1 ppm) supported a heterogeneous catalysis mechanism.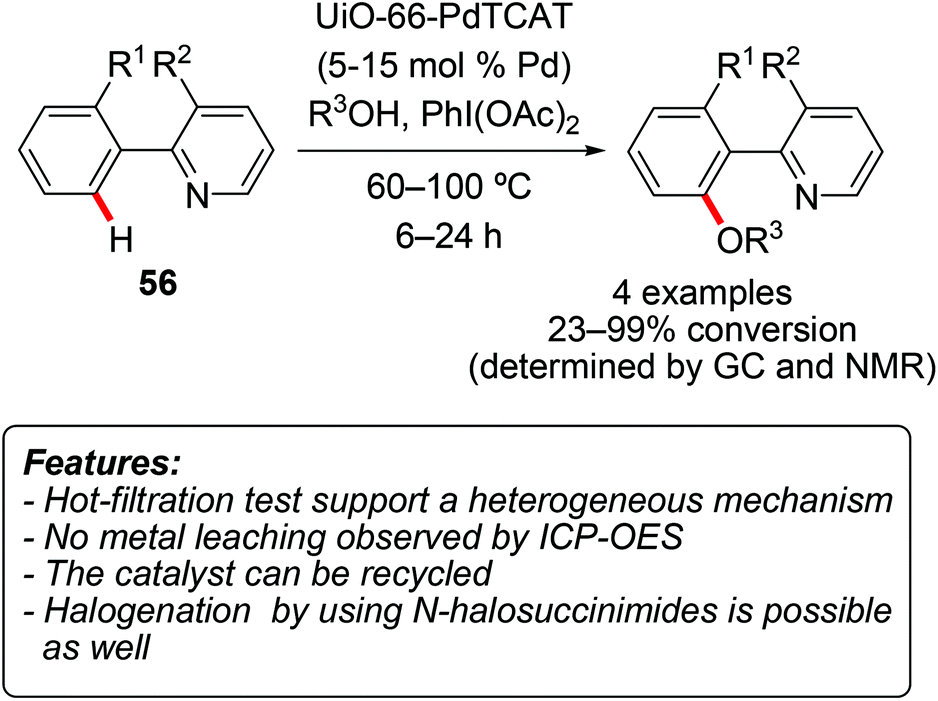 | ||
| Scheme 32 Palladium on Zr(IV)-based MOF for selective C–H functionalization. | ||
Phan et al. synthesized a crystalline porous copper-containing MOF [Cu2(BPDC)2(BPY); BPDC = 4,4′-biphenyldicarboxylate; BPY = 4,4′-bipyridine] and tested it for the oxidative C–O coupling of 1,4-dioxane (57) and phenols 58 (Scheme 33).71 A careful screening of reaction conditions was conducted, with several parameters being optimized. The oxidant of choice was TBHP, and the reaction could be conducted using an excess of ether without an additional solvent. The tested scope of the reaction is rather limited, since good results are only obtained with ortho-substituted phenols and 1,4-dioxane (57) as the ether. The removal of the catalyst by centrifugation in the early stages of the reaction resulted in halting of the reactivity. The catalyst could be recycled for at least eight consecutive cycles without a significant loss in catalytic activity.
 | ||
| Scheme 33 Oxidative C–O coupling by direct C–H activation of ethers 57 over heterogeneous Cu2(BPDC)2(BPY) catalyst. | ||
Li, Luque and coworkers reported on the reusable heterogeneous bimetallic catalyst system consisting of Au–Pd and supported by porous MOF MIL-101 [Au–Pd/MIL-101 material; MIL-101 is a MOF constructed from Cr3+ and terephthalate].72a The latter allowed accomplishing a direct transformation of substituted toluenes into benzoates,72b when reacting with alcohols under an atmosphere of O2 as a benign oxidant, in 32–95% yield.
3.3. Chalco- and halogenation
An example of C–S formation through a C–H activation process catalyzed by a heterogeneous catalyst was recently disclosed. Hence, Glorius and coworkers used Pd/Al2O3 and diaryldisulfides 60 (Y = S) in the presence of CuCl2 to perform the regioselective C–H thiolation of heteroaromatics 9, 15, 17, or 59 (Scheme 34).73 2-Substituted thiophenes 17 were selectively functionalized at the C5-position, while a 2,5-disubstituted thiophene reacted on C3. 3-Substituted thiophenes appeared to be less reactive, affording mixtures of C2- and C5-functionalized products. N-Methylindoles 9 reacted selectively on C3 carbon, furnishing the corresponding thiolated products in good yields. Benzo[b]thiophenes 15 and benzofurans 59 were generally less reactive, requiring harsher reaction conditions. The scope in terms of disulfides was also explored. Both electron-rich and electron-poor disulfides proved to be competent reacting partners, with the electron-rich substrates generally providing higher yields. The reaction proved to be tolerant to various functional groups. For instance, halogens were well accepted on both the heteroarene and the disulfide, thus allowing for further functionalization of the products. Although total reflection X-ray fluorescence (TXRF) showed the presence of palladium in solution, the results of a hot-filtration test and of a three-phase test suggested that these homogeneous palladium species should not be catalytically active. The recyclability of the catalyst was examined, showing a decrease in product yield already in the second reaction run. The protocol could be extended to the selenenylation of heteroarenes by using diselenides 60 (Y = Se) instead of disulfides. | ||
| Scheme 34 Pd/Al2O3-catalyzed C–H bond thiolation of heteroarenes 9, 15, 17 or 59. | ||
Fei and Cohen used their UiO-66-PdTCAT MOF (vide supra) as a catalyst for the halogenation of 7,8-benzoquinoline 56a and of 3-methyl-2-phenylpyridine.70 The reaction was conducted using N-halosuccinimide both as a halogenating reagent and as an oxidant. While chlorination and bromination of 7,8-benzoquinoline occurred very efficiently (95 and 97% conversion, respectively), the iodination of the same substrate was unsuccessful. On the other hand, while chlorination and bromination processes were not very successful on 3-methyl-2-phenylpyridine (50 and 32% conversion, respectively), the iodination of this substrate gave an acceptable 87% conversion. Also in this case, the catalyst could be reused for at least five reaction cycles, with only a minor decrease in product yields.
Martín-Matute and coworkers used a palladium-containing iron-based MOF [Pd@MIL-88B-NH2(Fe)] and a palladium-containing chromium-based MOF [Pd@MIL-101-NH2(Cr)] as catalysts for the C–H halogenation of several aromatic substrate bearing directing groups.74 By adjusting the reaction conditions – particularly the temperature and the solvent – the selective monoiodination or diiodination of 2-arylpyridines 27 could be achieved with good to excellent efficacy (Scheme 35). 2-Arylpyridines could also be efficiently mono-brominated, while the chlorination reaction occurred in low yields. Similarly, the iodination and bromination of N-acetyl-2-methylindoline proceeded very efficiently, while chlorination afforded only traces of the desired product. However, whereas the chlorination and bromination of 7,8-benzoquinoline 56a was efficacious, the iodination of the same substrate did not occur. The selective halogenation of meta-disubstituted substrates was also attempted, but with variable results.
 | ||
| Scheme 35 Selective heterogeneous halogenations catalyzed by Pd@MOF nanocomposites. | ||
Both catalysts could be recycled for at least five consecutive reaction runs, consistently leading to full conversion. Palladium leaching was measured after each run, conducted under different conditions. The amount of leached palladium was always very low, with the exception of the bromination reaction performed with the iron-based MOF in acetic acid, which led to up to 6.7 ppm of Pd-leaching. TEM analysis revealed significant agglomeration of Pd on the surface of the MOF crystals. This was interpreted by the authors as a consequence of a leaching-redepositing mechanism. Hot-filtration tests gave results depending on the nature of the substrate, particularly the coordinating ability of the directing group. This was also in accordance with a leaching-redepositing mechanism.
Alternatively, monochlorination, -bromination and -iodination of aromatic compounds with N-halosuccinimides in the presence of molecular oxygen as an oxidant were achieved in a highly selective process using heterogeneous Cu–MnO catalyst under irradiation with visible light.75 The reactions proceeded without any additive or ligands and proved to be efficient for halogenations of electron-rich as well as electron-poor substrates, including benzoates. The heterogeneous nature of this catalyst was confirmed by a hot-filtration test.
3.4. Borylation
The synthetic utility of C–H borylation reactions resides in the effective preparation of organoboron compounds that can be considered as transient functional groups and, among others, find extensive use in cross-coupling processes.76 The vast majority of the methodologies reported to date rely on homogeneous catalysts employing rhodium or iridium complexes. However, a few examples using heterogeneous or easily recoverable transition metal complexes have also been devised.Yinghuai and coworkers prepared iridium(0) nanoparticles and tested them in the borylation of unsubstituted and monosubstituted benzenes 28 with pinacolborane (HBpin, 61).77 The nanoparticles were stabilized by trihexyltetradecylphosphonium methylsulfonate [THTdP][MS] as an ionic liquid, and the use of microwave (MW) heating in combination with tetra-2-pyridinylpyrazine (TPy) as a ligand proved to be beneficial for the efficiency of the process. Additional studies would be necessary for fully understanding this process, particularly the influence of microwave heating. The reported scope was limited to four examples, with the yields of boronic acids 18 ranging from 39 to 89% after subsequent hydrolysis of the boronic acid ester (Scheme 36). The catalytic system could be recycled at least six times with negligible loss of the catalytic activity. This evidence, in connection with the low amount of the leached metal and with the results of a mercury test, of TEM and of XPS analyses conducted after four reaction runs, provided strong support for the reaction being actually promoted by a heterogeneous species.77
 | ||
| Scheme 36 Ir(0)-NPs catalyzed borylations of arenes 28. | ||
Nishida and coworkers used [IrCl(COD)]2 in combination with [2,2′-bipyridine]-4,4′-dicarboxylic acid (63) as a ligand for the C–H borylation of arenes [benzene derivatives 28, benzofuran (59) and benzo[b]thiophene (15)] with bis(pinacolato)diboron (B2pin2, 62) (Scheme 37).78
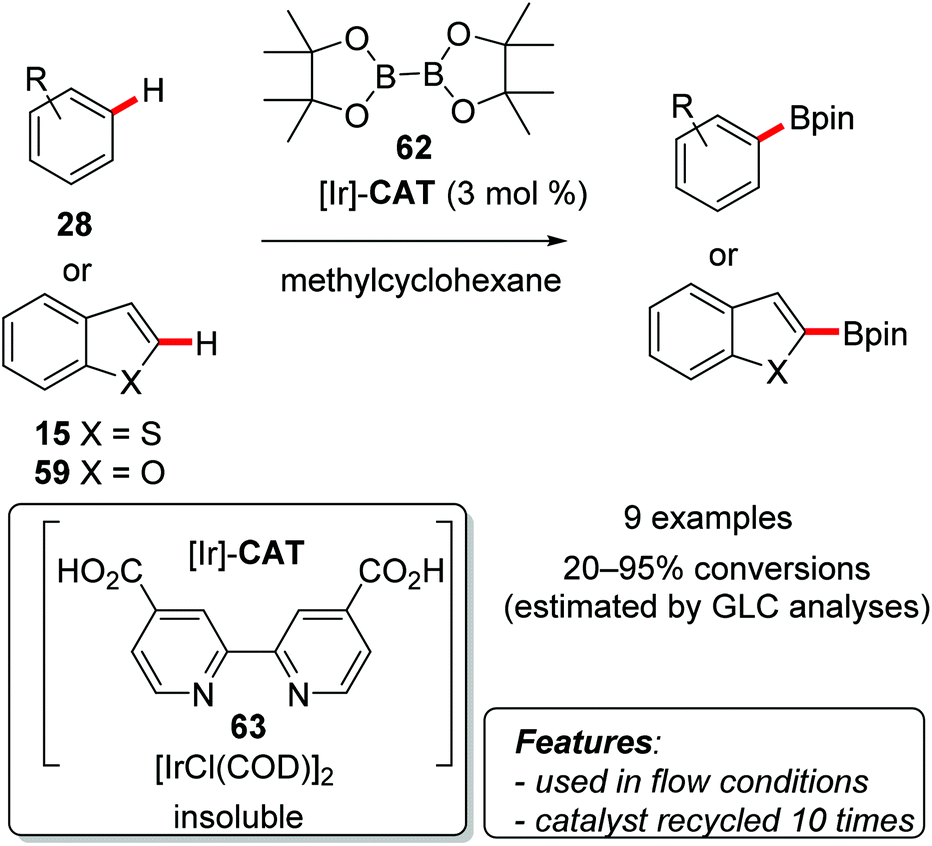 | ||
| Scheme 37 Borylation reactions catalyzed by insoluble/recoverable iridium/bipyridine complex 63. | ||
Monosubstituted arenes afforded mixtures of meta- and para-functionalized products in higher yields for electron-poor substrates. The functionalization of disubstituted aromatics proved to be superior, both in terms of the yield (83–95%) and selectivity, with the formation of a single regioisomer resulting from the substitution occurring on the least sterically hindered position. Benzothiophene (15) and benzofuran (59) were also borylated selectively at the C2-position in good yields of 86 and 78%, respectively. After completion of the reaction the catalytic system could be recovered by filtration under nitrogen in a glove box, and reused more than ten times. The presence of homogeneous iridium species in solution was evaluated by ICP analysis of the filtrate, proving that no metal was leached at all. It is worth mentioning that when filtration was performed under air, the recovered catalytic system showed no catalytic activity. To overcome this limitation, the same authors also elaborated a continuous-flow procedure for the borylation of arenes, which allowed the use of the catalyst outside of the glove box.79 In most cases the amount of the leached metal detected in the products was very low.
Sawamura and coworkers prepared several silica-supported phosphine/iridium or rhodium catalysts and extensively investigated their application in C–H borylation reactions.80 The catalysts were used for both C(sp2)–H and C(sp3)–H borylations, typically employing B2pin2 (62) as the borylating agent. In most cases, a directing group was present on the substrate, increasing the reactivity and dictating the site-selectivity and, in certain cases, also the stereoselectivity80j of the process (Scheme 38). The borylation of heterocycles such as indoles 9, thiophenes 17, pyrroles 7, furans 12 and quinolines was also reported.80b,i Mostly, the exact working mode and the nature of the catalytically active species were not investigated. However, the few attempts at recycling the catalyst were either unsuccessful80d,g or a significant decrease of activity was observed.80a
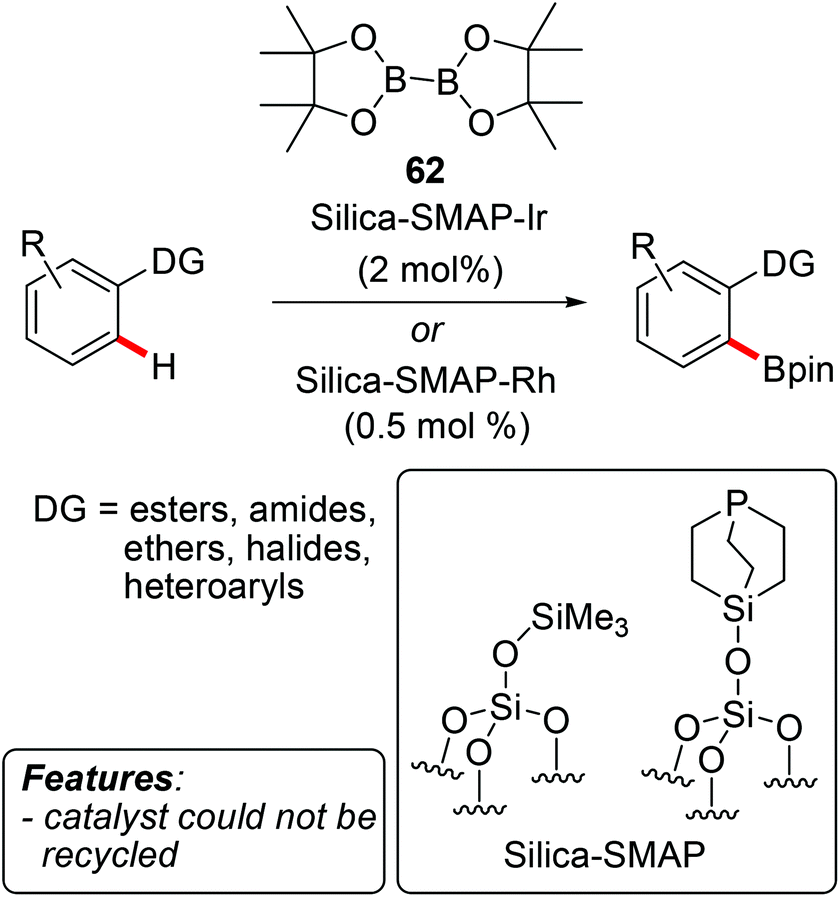 | ||
| Scheme 38 Silica-supported phosphine–iridium catalysts for the borylation reaction. | ||
Jones and coworkers reported on the preparation of an immobilized bipyridine–iridium system and its application in C–H borylation of arenes using bis(pinacolato)diboron (62) (Scheme 39).81 The reaction afforded the borylated products in moderate to good yields, with a selectivity that favored the least sterically hindered position. The catalyst was recycled for three reaction runs with 1,2-dichlorobenzene as the substrate. Although the initial turnover frequency of the catalyst decreased after each run, the product was obtained in 94% yield even in the second and third runs by increasing the reaction time from 24 to 48 hours. A hot-filtration test and the analysis of the filtered catalyst proved that the reaction was actually catalyzed by a heterogeneous species rather than by leached homogeneous particles, and that the catalyst remained stable under the reaction conditions.81
 | ||
| Scheme 39 Reusable silica-supported iridium/bipyridine catalyst for C–H borylation. | ||
Copéret and coworkers prepared a periodic mesoporous organosilica (PMO) containing bipyridylene moieties and functionalized it with chloro(1,5-cyclooctadiene)iridium(I) dimer [{IrCl(COD)}2] (Scheme 40).82 The resulting heterogeneous iridium catalyst was tested for the borylation of simple and monofunctionalized benzenes 28 with B2pin2 (62) as the borylating reagent.
 | ||
| Scheme 40 Ir–bpy/PMO as a solid catalyst for borylation reaction. | ||
While the reaction with benzene, toluene and anisole afforded the corresponding products in good yields, the functionalization of nitrobenzene was much less efficient, and a possible deactivation of the catalyst by the substrate was put forward. The reaction on monosubstituted arenes was not site-selective, affording a nearly statistical mixture of meta- and para-borylated products. The catalyst was recycled three times in the reactions with toluene and anisole with no apparent loss in activity. Furthermore, the catalyst wash-off contained a very little amount of the metal (≤1 ppm), except in the borylation of nitrobenzene, where ca. 5 ppm of the metal was detected.
Independently, Maegawa and coworkers reported on a very similar approach to access a periodic mesoporous organosilica containing bipyridine ligands within the pore walls.83 This was allowed to react with several transition metal complexes, including ruthenium, iridium, rhenium and palladium species. The systems obtained by the reaction of the mesoporous organosilica with [IrCl(COD)]2 proved to efficiently catalyze the borylation of arenes in the presence of B2pin2 (62). 1,2- and 1,3-disubstituted benzene derivatives selectively afforded the product of the functionalization occurring on the least sterically hindered position. However, monosubstituted arenes such as toluene, anisole and methyl benzoate afforded regioisomeric mixtures of the meta- and para-borylated products. The catalyst could be recycled at least three times, with a relatively low decrease in the product yield (1st run: 94, 2nd run: 91, 3rd run: 88 and 4th run: 78%, respectively). A hot-filtration test and ICP-AES analysis of the filtrate suggested that the catalysis is affected by the supported metal, rather than by leached homogeneous species. Later, the same research group showed that the catalytic system is more efficient in combination with HBpin instead of B2pin2. Applying this method, heteroaromatic substrates could also be functionalized in good yields.84
Lin and coworkers prepared several bipyridyl- and phenanthryl-based metal–organic frameworks (MOFs) of the UiO structure.85 Postsynthetic metalation with [Ir(COD)(OMe)]2 provided catalysts which were used for, among other reactions, the C–H borylation of arenes. The borylation of benzene, mono- and disubstituted arenes was achieved in excellent yields. Also in this case, disubstituted benzenes afforded selectively the product derived from functionalization on the least sterically hindered position, while monosubstituted benzenes gave mixtures of meta- and para-borylated compounds. The transformation of heteroarenes was also viable with these catalytic systems. Remarkably, the catalysts were recyclable for at least 15 times without significant loss of activity.
4. Conclusions and outlook
Recent years have witnessed tremendous progress in metal-catalyzed C–H functionalizations. Despite these undisputed advances, the main efforts were as of yet focused on exploring various metal complexes in a homogeneous fashion. From the current scenario it is also easily evident that most of the catalytic protocols employ as a reaction medium a variety of generally non-benign solvents while a clear attention to this aspect is urgently needed.86 Moreover, the efficient reusability of the catalytic system is still a major challenge.Environmentally-friendly and economically-sound heterogeneous catalysts were until recently underappreciated for the direct functionalization of otherwise unreactive C(sp3)–H and C(sp2)–H bonds. However, considerable success has been accomplished in the last decade. Thus, most user-friendly, easily separable heterogeneous materials emerged as recyclable catalysts that featured a broad substrate scope, and displayed high chemo- and site-selectivities in C–C as well as C–heteroatom formatting processes. It is particularly notable that unusual site-selectivities could be achieved with these heterogeneous systems, as for instance in the β-selective arylation of thiophene derivatives. As to future developments, methods for asymmetric C–H bond functionalizations can open a new horizon in this research arena. For instance, bis(oxazoline) ligands supported by electrostatic interactions on laponite clay copper complexes represent partially recoverable hybrid catalysts, which catalyzed the insertion of carbenoids into C(sp3)–H bonds with moderate to good enantioselectivities.87
Moreover, the rational design of materials with confined nano-sized geometries should prove instrumental for controlling confined chemical reactions, and thereby for achieving challenging remote functionalizations. Considering the practical importance of atom- and step-economical C–H functionalization on a large scale, for organic synthesis, materials sciences or medicinal chemistry, significant further progress is expected in this rapidly evolving research area.
Acknowledgements
L. A. sincerely thanks all his former and present co-workers involved in the C–H bond functionalization projects for their experimental work and their intellectual contributions. The cited studies from his laboratories were partly funded by the European Research Council under the European Community 7th Framework Program and the Fonds der Chemischen Industrie. The Univeristà degli Studi di Perugia is thanked for financial support. S. S. gratefully acknowledges the MIUR for “Programma Giovani Ricercatori, Rita Levi Montalcini” the fellowship N. PGR123GHQY.Notes and references
- For selected representative recent reviews on C–H bond functionalizations, see: (a) Z. Huang, H. N. Lim, F. Mo, M. C. Young and G. Dong, Chem. Soc. Rev., 2015, 44, 7764–7786 RSC; (b) J. Yang, Org. Biomol. Chem., 2015, 13, 1930–1941 RSC; (c) B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308–1318 CrossRef CAS PubMed; (d) K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040–1052 CrossRef CAS PubMed; (e) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef CAS PubMed; (f) B. Liu, F. Hu and B.-F. Shi, ACS Catal., 2015, 5, 1863–1881 CrossRef CAS; (g) Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66–81 CrossRef CAS PubMed; (h) M. Zhang, Y. Zhang, X. Jie, H. Zhao, G. Li and W. Su, Org. Chem. Front., 2014, 1, 843–895 RSC; (i) L. Ackermann, J. Org. Chem., 2014, 79, 8948–8954 CrossRef CAS PubMed; (j) T. Mesganaw and J. A. Ellman, Org. Process Res. Dev., 2014, 18, 1097–1104 CrossRef CAS PubMed; (k) S. Tani, T. N. Uehara, J. Yamaguchi and K. Itami, Chem. Sci., 2014, 5, 123–135 RSC; (l) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74–100 CrossRef CAS PubMed; (m) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed; (n) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed; (o) J. Schipper and K. Fagnou, Chem. Mater., 2011, 23, 1594–1600 CrossRef; (p) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed; (q) T. Satoh and M. Miura, Chem. – Eur. J., 2010, 16, 11212–11222 CrossRef CAS PubMed; (r) O. Daugulis, Top. Curr. Chem., 2010, 292, 57–84 CrossRef CAS PubMed; (s) P. Sehnal, R. J. K. Taylor and I. J. S. Fairlamb, Chem. Rev., 2010, 110, 824–889 CrossRef CAS PubMed; (t) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC; (u) L. Ackermann, R. Vicente and A. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed; (v) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (w) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed , and references cited therein.
- For recent reviews on C–H bond functionalizations in the synthesis of biologically active, and practically useful products, see: (a) Y. Kuninobu and S. Sueki, Synthesis, 2015, 3823–3845 CrossRef CAS; (b) F. J. Lombard and M. J. Coster, Org. Biomol. Chem., 2015, 13, 6419–6431 RSC; (c) L. Ackermann, Org. Process Res. Dev., 2015, 18, 260–269 CrossRef; (d) J. Hong, Chem. – Eur. J., 2014, 20, 10204–10212 CrossRef CAS PubMed; (e) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed; (f) Y.-K. Chen and S. W. Youn, Chem. – Eur. J., 2012, 18, 9452–9474 CrossRef PubMed; (g) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC; (h) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC.
- For reviews on remote C–H functionalizations, see ref. 1b and: (a) J. Li, S. D. Sarkar and L. Ackermann, Top. Organomet. Chem., 2016, 55, 217–257 CrossRef. Highlights: (b) J. Schranck, A. Tlili and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 9426–9428 CrossRef CAS PubMed; (c) F. Juliá-Hernández, M. Simonetti and I. Larrosa, Angew. Chem., Int. Ed., 2013, 52, 11458–11460 CrossRef PubMed. For selected examples of remote C–H functionalizations, see: (d) Y. Kuninobu, H. Ida, M. Nishi and M. Kanai, Nat. Chem., 2015, 7, 712–717 CrossRef CAS PubMed; (e) A. J. Paterson, S. St John-Campbell, M. F. Mahon, N. J. Press and C. G. Frost, Chem. Commun., 2015, 51, 12807–12810 RSC; (f) J. Li, S. Warratz, D. Zell, S. D. Sarkar, E. E. Ishikawa and L. Ackermann, J. Am. Chem. Soc., 2015, 137, 13894–13901 CrossRef CAS PubMed; (g) R. Y. Tang, G. Li and J.-Q. Yu, Nature, 2014, 507, 215–220 CrossRef CAS PubMed; (h) S. Lee, H. Lee and K. L. Tan, J. Am. Chem. Soc., 2013, 135, 18778–18781 CrossRef CAS PubMed; (i) N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877–5884 CrossRef CAS PubMed; (j) D. Leow, G. Li, T. S. Mei and J.-Q. Yu, Nature, 2012, 486, 518–522 CrossRef CAS PubMed; (k) O. Saidi, J. Marafie, A. E. Ledger, P. M. Liu, M. F. Mahon, G. Kociok-Kohn, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298–19301 CrossRef CAS PubMed; (l) L. Ackermann, N. Hofmann and R. Vicente, Org. Lett., 2011, 13, 1875–1877 CrossRef CAS PubMed.
- S. Hübner, J. G. de Vries and V. Farina, Adv. Synth. Catal., 2016, 358, 3–25 CrossRef.
- For recent reviews, see: (a) A. J. Reay and I. J. S. Fairlamb, Chem. Commun., 2015, 51, 16289–16307 RSC; (b) R. Cano, A. F. Schmidt and G. P. McGlacken, Chem. Sci., 2015, 6, 5338–5346 RSC; (c) L. Djakovitch and F.-X. Felpin, ChemCatChem, 2014, 6, 2175–2187 CrossRef CAS. For selected early theoretical and experimental reports on heterogeneous catalysis, predominantly focusing on C(sp3)–H oxidation and H/D exchange reactions in hydrocarbons, see: (d) G. Bianchini, M. Crucianelli, C. Crestini and R. Saladino, Top. Catal., 2006, 40, 221–227 CrossRef CAS; (e) G. Bianchini, M. Crucianelli, F. De Angelis, V. Neri and R. Saladino, Tetrahedron Lett., 2005, 46, 2427–2432 CrossRef CAS; (f) H. Sajiki, F. Aoki, H. Esaki, T. Maegawa and K. Hirota, Org. Lett., 2004, 6, 1485–1487 CrossRef CAS PubMed; (g) R. Burch, D. J. Crittle and M. J. Hayes, Catal. Today, 1999, 47, 229–234 CrossRef CAS; (h) T. H. Ballinger and J. T. Yates, Jr., J. Phys. Chem., 1992, 24, 9979–9983 CrossRef; (i) W. F. Maier, Angew. Chem., Int. Ed. Engl., 1989, 28, 135–145 CrossRef.
- N. Nakamura, Y. Tajima and K. Sakai, Heterocycles, 1982, 17, 235–245 CrossRef CAS.
- M. Parisien, D. Valette and K. Fagnou, J. Org. Chem., 2005, 70, 7578–7584 CrossRef CAS PubMed.
- (a) L. Djakovitch, V. Dufaud and R. Zaidi, Adv. Synth. Catal., 2006, 348, 715–724 CrossRef CAS. For a review on homogeneous C–H arylations of indoles, see: (b) L. Joucla and L. Djakovitch, Adv. Synth. Catal., 2009, 351, 673–714 CrossRef CAS.
- G. Cusati and L. Djakovitch, Tetrahedron Lett., 2008, 49, 2499–2502 CrossRef CAS . For the comparison of various methods for the selective arylation of indoles, see: ref. 8b.
- (a) S. Sahnoun, S. Messaoudi, J.-F. Peyrat, J.-D. Brion and M. Alami, Tetrahedron Lett., 2008, 49, 7279–7283 CrossRef CAS; (b) S. Sahnoun, S. Messaoudi, J.-D. Brion and M. Alami, Org. Biomol. Chem., 2009, 7, 4271–4278 RSC.
- S. Hernández, I. Moreno, R. SanMartin, G. Gómez, M. T. Herrero and E. Domínguez, J. Org. Chem., 2010, 75, 434–441 CrossRef PubMed.
- F. Jafarpour, S. Rahiminejadan and H. Hazrati, J. Org. Chem., 2010, 75, 3109–3112 CrossRef CAS PubMed.
- D. Saha, L. Adak and B. C. Ranu, Tetrahedron Lett., 2010, 51, 5624–5627 CrossRef CAS.
- (a) L. Wang, W.-B. Yi and C. Cai, Chem. Commun., 2011, 47, 806–808 RSC . For the recent comparison of efficacy of homogeneous and heterogeneous palladium-catalyzed direct arylation of indoles and of other heterocycles, see: ; (b) A. J. Reay, L. K. Neumann and I. J. S. Fairlamb, Synlett, 2016, 27, 1211–1216 CrossRef CAS.
- Y. Huang, Z. Lin and R. Cao, Chem. – Eur. J., 2011, 17, 12706–12712 CrossRef CAS PubMed.
- (a) M. Cao, D. Wu, W. Su and R. Cao, J. Catal., 2015, 321, 62–69 CrossRef CAS. For early examples of direct arylation of perfluorinated aromatics through homogeneous palladium catalysis, see: (b) M. Lafrance, C. N. Rowley, T. K. Woo and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 8754–8756 CrossRef CAS PubMed; (c) L. Ackermann and S. Fenner, Chem. Commun., 2011, 47, 430–432 RSC.
- Y.-B. Huang, M. Shen, X. Wang, P. Huang, R. Chen, Z.-J. Lin and R. Cao, J. Catal., 2016, 333, 1–7 CrossRef CAS.
- V. A. Zinovyeva, M. A. Vorotyntsev, I. Bezverkhyy, D. Chaumont and J.-C. Hierso, Adv. Funct. Mater., 2011, 21, 1064–1075 CrossRef CAS.
- T.-H. Park, A. J. Hickman, K. Koh, S. Martin, A. G. Wong-Foy, M. S. Sanford and A. J. Matzger, J. Am. Chem. Soc., 2011, 133, 20138–20141 CrossRef CAS PubMed.
- J. Lee, J. Chung, S. M. Byun, B. M. Kim and C. Lee, Tetrahedron, 2013, 69, 5660–5664 CrossRef CAS.
- (a) D.-T. D. Tang, K. D. Collins and F. Glorius, J. Am. Chem. Soc., 2013, 135, 7450–7453 CrossRef CAS PubMed. For a related homogeneous process, see: (b) K. Ueda, S. Yanagisawa, J. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2010, 49, 8946–8949 CrossRef CAS PubMed.
- (a) D.-T. D. Tang, K. D. Collins, J. B. Ernst and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 1809–1813 CrossRef CAS PubMed. For reviews on hypervalent iodine reagents, see: (b) V. V. Zhdankin, Hypervalent Iodine Chemistry: Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds, John Wiley & Sons, N.-Y, 2013, 480 pp.; Search PubMed; (c) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 121, 9052–9070 CrossRef PubMed. For the use of hypervalent iodine reagents in C–H functionalizations, see: (d) R. Narayan, S. Manna and A. P. Antonchick, Synlett, 2015, 1785–1803 CAS; (e) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (f) N. R. Deprez, D. Kalyani, A. Krause and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 4972–4973 CrossRef CAS PubMed; (g) Y. Zhu, M. Bauer, J. Ploog and L. Ackermann, Chem. – Eur. J., 2014, 20, 13099–13102 CrossRef CAS PubMed.
- K. D. Collins, R. Honeker, S. Vásquez-Céspedes, D.-T. D. Tang and F. Glorius, Chem. Sci., 2015, 6, 1816–1824 RSC.
- S. Hayashi, Y. Kojima and T. Koizumi, Polym. Chem., 2015, 6, 881–885 RSC.
- (a) S. M. McAfee, J. S. J. McCahill, C. M. Macaulay, A. D. Hendsbee and G. C. Welch, RSC Adv., 2015, 5, 26097–26106 RSC; (b) J. Areephong, A. D. Hendsbee and G. C. Welch, New J. Chem., 2015, 39, 6714–6717 RSC.
- Y. M. A. Yamada, Y. Yuyama, T. Sato, S. Fujikawa and Y. Uozumi, Angew. Chem., Int. Ed., 2014, 53, 127–131 CrossRef CAS PubMed.
- L. Zhang, P. Li, C. Liu, J. Yang, M. Wang and L. Wang, Catal. Sci. Technol., 2014, 4, 1979–1988 CAS.
- J. Malmgren, A. Nagendiran, C.-W. Tai, J.-E. Bäckvall and B. Olofsson, Chem. – Eur. J., 2014, 20, 13531–13535 CrossRef CAS PubMed.
- T. Parsharamulu, D. Venkanna, M. L. Kantam, S. K. Bhargava and P. Srinivasu, Ind. Eng. Chem. Res., 2014, 53, 20075–20084 CrossRef CAS.
- S. Wang, D. Hu, W. Hua, J. Gu, Q. Zhang, X. Jia and K. Xi, RSC Adv., 2015, 5, 53935–53939 RSC.
- H. Veisi and N. Morakabati, New J. Chem., 2015, 39, 2901–2907 RSC.
- (a) S. Keshipour and A. Shaabani, Appl. Organomet. Chem., 2014, 28, 116–119 CrossRef CAS. For related homogeneous cycloaddition/C–H functionalization sequences, see: (b) L. Ackermann, H. K. Potukuchi, D. Landsberg and R. Vicente, Org. Lett., 2008, 10, 3081–3084 CrossRef CAS PubMed; (c) R. Jeyachandran, H. K. Potukuchi and L. Ackermann, Beilstein J. Org. Chem., 2012, 8, 1771–1777 CrossRef CAS PubMed.
- (a) E. Y. Lee and J. Park, ChemCatChem, 2011, 3, 1127–1129 CrossRef CAS. For a related homogeneous reaction, see: (b) R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593–1597 CrossRef CAS PubMed.
- W. Zhang, Q. Zeng, X. Zhang, Y. Tian, Y. Yue, Y. Guo and Z. Wang, J. Org. Chem., 2011, 76, 4741–4745 CrossRef CAS PubMed.
- W. Zhang, Y. Tian, N. Zhao, Y. Wang, J. Li and Z. Wang, Tetrahedron, 2014, 70, 6120–6126 CrossRef CAS.
- (a) H. T. N. Le, T. T. Nguyen, P. H. L. Vu, T. Truong and N. T. S. Phan, J. Mol. Catal. A: Chem., 2014, 391, 74–82 CrossRef CAS; (b) T. Truong, V. T. Nguyen, H. T. X. Le and N. T. S. Phan, RSC Adv., 2014, 4, 52307–523154 RSC. For examples of homogeneous copper-catalyzed C–H functionalizations, see: (c) A. Kulkarni and O. Daugulis, Synthesis, 2009, 4087–4109 CAS; (d) H.-Q. Do and O. Daugulis, J. Am. Chem. Soc., 2007, 129, 12404–12405 CrossRef CAS PubMed.
- (a) N. T. S. Phan, C. K. Nguyen, T. T. Nguyen and T. Truong, Catal. Sci. Technol., 2014, 4, 369–377 RSC. For examples of homogeneous nickel-catalyzed C–H functionalizations, see: (b) J. Yamaguchi, K. Muto and K. Itami, Eur. J. Org. Chem., 2013, 19–30 CrossRef CAS; (c) Y. Nakao, Chem. Rec., 2011, 11, 242–251 CrossRef CAS PubMed.
- (a) H. Miura, K. Wada, S. Hosokawa and M. Inoue, Chem. – Eur. J., 2010, 16, 4186–4189 CrossRef CAS PubMed; (b) H. Miura, K. Wada, S. Hosokawa and M. Inoue, Patent application JP002010184881 A, 2010 Search PubMed. For selected related homogeneous processes, see: (c) L. Ackermann and R. Vicente, Top. Curr. Chem., 2010, 292, 211–229 CrossRef CAS PubMed; (d) S. Oi, H. Sato, S. Sugawara and Y. Inoue, Org. Lett., 2008, 10, 1823–1826 CrossRef CAS PubMed; (e) L. Ackermann, A. Althammer and R. Born, Angew. Chem., Int. Ed., 2006, 45, 2619–2622 CrossRef CAS PubMed; (f) L. Ackermann, R. Vicente and A. Althammer, Org. Lett., 2008, 10, 2299–2302 CrossRef CAS PubMed.
- H. Liu, B. Yin, Z. Gao, Y. Li and H. Jiang, Chem. Commun., 2012, 48, 2033–2035 RSC.
- (a) H. Miura, K. Wada, S. Hosokawa and M. Inoue, ChemCatChem, 2010, 2, 1223–1225 CrossRef CAS. For homogeneous ruthenium-catalyzed hydroarylations, see: (b) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529–531 CrossRef CAS; (c) L. N. Lewis and J. F. Smith, J. Am. Chem. Soc., 1986, 108, 2728–2735 CrossRef CAS; (d) F. Kakiuchi and S. Murai, Acc. Chem. Res., 2002, 35, 826–834 CrossRef CAS PubMed; (e) F. Kakiuchi and N. Chatani, in Ruthenium Catalysts and Fine Chemistry, ed. C. Bruneau and P. H. Dixneuf, Springer, Berlin, Heidelberg, 2004 Search PubMed; (f) F. Kakiuchi and N. Chatani, in Ruthenium in Organic Synthesis, ed. S.-I. Murahashi, Wiley-VCH, Weinheim, 2004 Search PubMed. For a recent example, see: (g) M. Schinkel, I. Marek and L. Ackermann, Angew. Chem., Int. Ed., 2013, 52, 3977–3980 CrossRef CAS PubMed.
- (a) S. Haldar and S. Koner, J. Org. Chem., 2010, 75, 6005–6008 CrossRef CAS PubMed. For examples of homogeneous hydroarylation reaction, see: (b) L. T. Kaspar, B. Fingerhut and L. Ackermann, Angew. Chem., Int. Ed., 2005, 44, 5972–5974 CrossRef CAS PubMed; (c) L. L. Anderson, J. Arnold and R. G. Bergman, J. Am. Chem. Soc., 2005, 127, 14542–14543 CrossRef CAS PubMed.
- (a) I. Moritani and I. Y. Fujiwara, Tetrahedron Lett., 1967, 8, 1119–1122 CrossRef; (b) T. Kitamura and Y. Fujiwara, in From C–H to C–C Bonds: Cross-Dehydrogenative-Coupling, ed. C.-J. Li, RSC Publishing, Cambridge, 2014, pp. 33–54 Search PubMed; (c) S. I. Kozhushkov and L. Ackermann, Chem. Sci., 2013, 4, 886–896 RSC. For recent reviews involving C–H olefinations, see: (d) L. Zhou and W. Lu, Chem. – Eur. J., 2014, 20, 634–642 CrossRef CAS PubMed; (e) J. Le Bras and J. Muzart, Chem. Rev., 2011, 111, 1170–1214 CrossRef CAS PubMed; (f) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed; (g) T. Satoh and M. Miura, Synthesis, 2010, 3395–3409 CrossRef CAS.
- L. L. Chng, J. Zhang, J. Yang, M. Amoura and J. Y. Ying, Adv. Synth. Catal., 2011, 353, 2988–2998 CrossRef CAS.
- H. Duan, M. Li, G. Zhang, J. R. Gallagher, Z. Huang, Y. Sun, Z. Luo, H. Chen, J. T. Miller, R. Zou, A. Lei and Y. Zhao, ACS Catal., 2015, 5, 3752–3759 CrossRef CAS.
- S. Wang, G. Deng, J. Gu, W. Hua, X. Jia and K. Xi, Appl. Catal., A, 2015, 508, 80–85 CrossRef CAS.
- R. Kishore, M. L. Kantam, J. Yadav, M. Sudhakar, S. Laha and A. Venugopal, J. Mol. Catal. A: Chem., 2013, 379, 213–218 CrossRef CAS . Loading of palladium was calculated based on data from ref. 12.
- J. Chen, K. Natte and X.-F. Wu, Tetrahedron Lett., 2015, 56, 6413–6416 CrossRef CAS.
- (a) R. Kishore, J. Yadav, B. Venu, A. Venugopal and M. L. Kantam, New J. Chem., 2015, 39, 5259–5264 RSC. For examples of homogeneous C–H cyanation reactions, see: (b) A. B. Pawar and S. Chang, Org. Lett., 2015, 17, 660–663 CrossRef CAS PubMed; (c) J. Li and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 3635–3638 CrossRef CAS PubMed; (d) D.-G. Yu, T. Gensch, F. de Azambuja, S. Vásquez-Céspedes and F. Glorius, J. Am. Chem. Soc., 2014, 136, 17722–17725 CrossRef CAS PubMed; (e) W. Liu and L. Ackermann, Chem. Commun., 2014, 50, 1878–1881 RSC.
- (a) A. Khorshidi, Chin. Chem. Lett., 2012, 23, 903–906 CrossRef CAS. For an example of a homogeneous ruthenium-catalyzed C–H cyanation, see: (b) S.-I. Murahashi, N. Komiya and H. Terai, Angew. Chem., Int. Ed., 2005, 44, 6931–6933 CrossRef CAS PubMed.
- S. Verma, S. L. Jain and B. Sain, ChemCatChem, 2011, 3, 1329–1332 CrossRef CAS.
- M. Gruttadauria, F. Giacalone and R. Noto, Green Chem., 2013, 15, 2608–2618 RSC.
- K. Yamaguchi, Y. Wang and N. Mizuno, ChemCatChem, 2013, 5, 2835–2838 CrossRef CAS.
- V. Panwar, P. Kumar, A. Bansal, S. S. Ray and S. L. Jain, Appl. Catal., A, 2015, 498, 25–31 CrossRef CAS.
- (a) C. Vila and M. Rueping, Green Chem., 2013, 15, 2056–2059 RSC; (b) J. Zoller, D. C. Fabry and M. Rueping, ACS Catal., 2015, 5, 3900–3904 CrossRef CAS.
- G. H. Dang, D. T. Nguyen, D. T. Le, T. Truong and N. T. S. Phan, J. Mol. Catal. A: Chem., 2014, 395, 300–306 CrossRef CAS.
- (a) J. Chen, L. He, K. Natte, H. Neumann, M. Beller and X.-F. Wu, Adv. Synth. Catal., 2014, 356, 2955–2959 CrossRef CAS ; For related homogeneous processes, see: ; (b) L. Ackermann, Acc. Chem. Res., 2014, 47, 281–295 CrossRef CAS PubMed; (c) T. Satoh and M. Miura, Chem. – Eur. J., 2010, 16, 11212–11222 CrossRef CAS PubMed; (d) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC; (e) W. Song and L. Ackermann, Chem. Commun., 2013, 49, 6638–6640 RSC; (f) L. Ackermann and A. V. Lygin, Org. Lett., 2012, 14, 764–767 CrossRef CAS PubMed.
- L. He, K. Natte, J. Rabeah, C. Taeschler, H. Neumann, A. Brückner and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 4320–4324 CrossRef CAS PubMed.
- (a) P. Liu, C.-Y. Zhou, S. Xiang and C.-M. Che, Chem. Commun., 2010, 46, 2739–2741 RSC; (b) T. Zeng, G. Song, A. Moores and C.-J. Li, Synlett, 2010, 2002–2008 CAS.
- Q.-Y. Meng, Q. Liu, J.-J. Zhong, H.-H. Zhang, Z.-J. Li, B. Chen, C.-H. Tung and L.-Z. Wu, Org. Lett., 2012, 14, 5992–5995 CrossRef CAS PubMed.
- M. Rueping, J. Zoller, D. C. Fabry, K. Poscharny, R. M. Koenigs, T. E. Weirich and J. Mayer, Chem. – Eur. J., 2012, 18, 3478–3481 CrossRef CAS PubMed.
- L. De Luca and A. Porcheddu, Eur. J. Org. Chem., 2011, 5791–5795 CrossRef CAS.
- H. E. Ho, Y. Ishikawa, N. Asao, Y. Yamamoto and T. Jin, Chem. Commun., 2015, 51, 12764–12767 RSC.
- (a) K. Matsumoto, K. Dougomori, S. Tachikawa, T. Ishii and M. Shindo, Org. Lett., 2014, 16, 4754–4757 CrossRef CAS PubMed; (b) K. Matsumoto, M. Yoshida and M. Shindo, Angew. Chem., Int. Ed., 2016, 55, 5272–5276 CrossRef CAS PubMed . For the recent example of copper-catalyzed cross-dehydrogenative coupling of pyridine N-oxides with cyclic ethers, see: ; (c) E. Kianmehr, N. Faghih, S. Karaji, Y. A. Lomedasht and K. M. Khan, J. Organomet. Chem., 2016, 801, 10–13 CrossRef CAS.
- S. A. R. Mulla, M. Y. Pathan and S. S. Chavan, RSC Adv., 2013, 3, 20281–20286 RSC.
- P. Pal, A. K. Giri, H. Singh, S. C. Ghosh and A. B. Panda, Chem. – Asian J., 2014, 9, 2392–2396 CrossRef CAS PubMed.
- C. Bai, X. Yao and Y. Li, ACS Catal., 2015, 5, 884–891 CrossRef CAS.
- R. Gava, A. Biffis, C. Tubaro, F. Zaccheria and N. Ravasio, Catal. Commun., 2013, 40, 63–65 CrossRef CAS.
- A. M. Voutchkova and R. H. Crabtree, J. Mol. Catal. A: Chem., 2009, 312, 1–6 CrossRef CAS.
- T. Truong, K. D. Nguyen, S. H. Doan and N. T. S. Phan, Appl. Catal. A, 2016, 510, 27–33 CrossRef CAS.
- (a) H. Fei and S. M. Cohen, J. Am. Chem. Soc., 2015, 137, 2191–2194 CrossRef CAS PubMed. For an example of a homogeneous C–H halogenation reaction, see: (b) L. Wang and L. Ackermann, Chem. Commun., 2014, 50, 1083–1085 RSC.
- N. T. S. Phan, P. H. L. Vu and T. T. Nguyen, J. Catal., 2013, 306, 38–46 CrossRef CAS.
- (a) H. Liu, G. Chen, H. Jiang, Y. Li and R. Luque, ChemSusChem, 2012, 5, 1892–1896 CrossRef CAS PubMed. For a review on catalytic transformations of toluenes into benzoic acid derivatives, see: (b) R. Vanjari and K. N. Singh, Chem. Soc. Rev., 2015, 44, 8062–8096 RSC.
- S. Vásquez-Céspedes, A. Ferry, L. Candish and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 5772–5776 CrossRef PubMed.
- V. Pascanu, F. Carson, M. V. Solano, J. Su, X. Zou, M. J. Johansson and B. Martín-Matute, Chem. – Eur. J., 2016, 22, 3729–3737 CrossRef CAS PubMed.
- P. Pal, H. Singh, A. B. Panda and S. C. Ghosh, Asian J. Org. Chem., 2015, 4, 879–883 CrossRef CAS.
- For selected reviews, see: (a) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed; (b) J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992–2002 RSC; (c) A. Ros, R. Fernández and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229–3243 RSC.
- Z. Yinghuai, K. Chenyan, A. T. Peng, A. Emi, W. Monalisa, L. K.-J. Louis, N. S. Hosmane and J. A. Maguire, Inorg. Chem., 2008, 47, 5756–5761 CrossRef PubMed.
- T. Tagata, M. Nishida and A. Nishida, Tetrahedron Lett., 2009, 50, 6176–6179 CrossRef CAS.
- T. Tagata, M. Nishida and A. Nishida, Adv. Synth. Catal., 2010, 352, 1662–1666 CrossRef CAS.
- (a) S. Kawamorita, H. Ohmiya, K. Hara, A. Fukuoka and M. Sawamura, J. Am. Chem. Soc., 2009, 131, 5058–5059 CrossRef CAS PubMed; (b) S. Kawamorita, H. Ohmiya and M. Sawamura, J. Org. Chem., 2010, 75, 3855–3858 CrossRef CAS PubMed; (c) K. Yamazaki, S. Kawamorita, H. Ohmiya and M. Sawamura, Org. Lett., 2010, 12, 3978–3981 CrossRef CAS PubMed; (d) S. Kawamorita, T. Miyazaki, H. Ohmiya, T. Iwai and M. Sawamura, J. Am. Chem. Soc., 2011, 133, 19310–19313 CrossRef CAS PubMed; (e) S. Kawamorita, T. Miyazaki, T. Iwai, H. Ohmiya and M. Sawamura, J. Am. Chem. Soc., 2012, 134, 12924–12927 CrossRef CAS PubMed; (f) T. Iwai, T. Harada, K. Hara and M. Sawamura, Angew. Chem., Int. Ed., 2013, 52, 12322–12326 CrossRef CAS PubMed; (g) S. Kawamorita, R. Murakami, T. Iwai and M. Sawamura, J. Am. Chem. Soc., 2013, 135, 2947–2950 CrossRef CAS PubMed; (h) T. Iwai, R. Murakami, T. Harada, S. Kawamorita and M. Sawamura, Adv. Synth. Catal., 2014, 356, 1563–1570 CrossRef CAS; (i) S. Konishi, S. Kawamorita, T. Iwai, P. G. Steel, T. B. Marder and M. Sawamura, Chem. – Asian J., 2014, 9, 434–438 CrossRef CAS PubMed; (j) R. Murakami, K. Tsunoda, T. Iwai and M. Sawamura, Chem. – Eur. J., 2014, 20, 13127–13131 CrossRef CAS PubMed.
- F. Wu, Y. Feng and C. W. Jones, ACS Catal., 2014, 4, 1365–1375 CrossRef CAS.
- W. R. Grüning, G. Siddiqi, O. V. Safonova and C. Copéret, Adv. Synth. Catal., 2014, 356, 673–679 CrossRef.
- M. Waki, Y. Maegawa, K. Hara, Y. Goto, S. Shirai, Y. Yamada, N. Mizoshita, T. Tani, W.-J. Chun, S. Muratsugu, M. Tada, A. Fukuoka and S. Inagaki, J. Am. Chem. Soc., 2014, 136, 4003–4011 CrossRef CAS PubMed.
- Y. Maegawa and S. Inagaki, Dalton Trans., 2015, 44, 13007–13016 RSC.
- (a) K. Manna, T. Zhang and W. Lin, J. Am. Chem. Soc., 2014, 136, 6566–6569 CrossRef CAS PubMed; (b) K. Manna, T. Zhang, F. X. Greene and W. Lin, J. Am. Chem. Soc., 2015, 137, 2665–2673 CrossRef CAS PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- (a) J. M. Fraile, J. A. Mayoral, A. Muñoz and J. Santafé-Valero, Tetrahedron, 2013, 69, 7360–7364 CrossRef CAS; (b) J. M. Fraile, J. I. García, J. A. Mayoral and M. Roldán, Org. Lett., 2007, 9, 731–733 CrossRef CAS PubMed . For preparation of the catalyst, see: ; (c) J. M. Fraile, P. López-Ram-de-Viu, J. A. Mayoral, M. Roldán and J. Santafé-Valero, Org. Biomol. Chem., 2011, 9, 6075–6081 RSC.
| This journal is © The Royal Society of Chemistry 2016 |