
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Liquid structure of the choline chloride-urea deep eutectic solvent (reline) from neutron diffraction and atomistic modelling†
Oliver S.
Hammond
a,
Daniel T.
Bowron
b and
Karen J.
Edler
*a
aCentre for Sustainable Chemical Technologies, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: k.edler@bath.ac.uk
bISIS Neutron and Muon Source, Science and Technology Facilities Council, Rutherford Appleton Laboratory, Harwell Oxford, Didcot OX11 0QX, UK
First published on 19th January 2016
Abstract
The liquid structure of the archetypal Deep Eutectic Solvent (DES) reline, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar mixture of choline chloride and urea, has been determined at 303 K. This is the first reported liquid-phase neutron diffraction experiment on a cholinium DES. H/D isotopic substitution is used to obtain differential neutron scattering cross sections, and an Empirical Potential Structure Refinement (EPSR) model is fitted to the experimental data. Radial distribution functions (RDFs) derived from EPSR reveal the presence of the anticipated hydrogen bonding network within the liquid, with significant ordering interactions not only between urea and chloride, but between all DES components. Spatial density functions (SDFs) are used to map the 3D structure of the solvent. Interestingly, choline is found to contribute strongly to this bonding network via the hydroxyl group, giving rise to a radially layered structure with ordering between all species. The void size distribution function calculated for reline suggests that the holes present within DESs are far smaller than previously suggested by hole theory. These observations have important implications in the future development of these ‘designer solvents’.
:2 molar mixture of choline chloride and urea, has been determined at 303 K. This is the first reported liquid-phase neutron diffraction experiment on a cholinium DES. H/D isotopic substitution is used to obtain differential neutron scattering cross sections, and an Empirical Potential Structure Refinement (EPSR) model is fitted to the experimental data. Radial distribution functions (RDFs) derived from EPSR reveal the presence of the anticipated hydrogen bonding network within the liquid, with significant ordering interactions not only between urea and chloride, but between all DES components. Spatial density functions (SDFs) are used to map the 3D structure of the solvent. Interestingly, choline is found to contribute strongly to this bonding network via the hydroxyl group, giving rise to a radially layered structure with ordering between all species. The void size distribution function calculated for reline suggests that the holes present within DESs are far smaller than previously suggested by hole theory. These observations have important implications in the future development of these ‘designer solvents’.
Introduction
Deep Eutectic Solvents (DESs) are a sub-category of ionic liquids (ILs), first reported in 2001 by Abbott et al.1 Unlike the binary ion pairing defining classic IL structure, DESs are made by the complexation of a hydrogen bond-capable salt (frequently ammonium halides) with a neutral hydrogen bond donor species (HBD).2 The term ‘Deep Eutectic Solvent’ hails from the low glass transition temperature at a specific molar ratio of salt to HBD.3,4 ILs and DESs share common benefits such as low vapour pressure and a tuneable, designer nature;5 like ILs, over 106 DESs are hypothesized to exist.6 The additional component in DES formulations permits fine-tuning of physicochemical properties. For example, the solvent hydrophobicity can be varied,7 and the compatibility of many DESs with H2O can make systems more tractable whilst preserving the hypothesized supramolecular structure (≤50 wt% H2O).8 The most widely-studied DES to date is the 1:2 choline chloride:urea Type III DES, also known as reline and amongst the first to be reported.9 Reline is a tractable room temperature ionic mixture with facile preparation from cheap, naturally-occurring and readily-available precursors,10 making reline a biodegradable, bactericidal, non-cytotoxic designer solvent.11,12
The primary application of DESs thus far has been as metal electrodeposition agents.13 DESs have also been applied as environmentally-friendly alternatives to molecular solvents in synthesis,14 particularly metal-catalysed reactions,15 and the synthesis of functional nanomaterials.16 DESs have also found green applications in extraction and separation, such as in the purification of biodiesel,17 and CO2 sequestration.18 DESs are one of the few nonaqueous solvent systems capable of supporting spontaneous self-assembly of anionic,19 cationic,20 and phospholipid-based amphiphiles.21 It has also been hypothesised that glass-forming natural DESs (NADESs) act as plant cryoprotectants.22
The key physical driving force for the formation of DESs is generally attributed to charge delocalization between the anion and HBD upon mixing as a result of the formation of hydrogen bonds.23 This assertion is mainly experimentally supported spectroscopically; HOESY NMR experiments by Abbott et al. revealed a correlation between urea and fluoride in a choline fluoride DES.4 The presence of hydrogen bonds in various alcoholic and carboxylic NADESs was shown in NOESY NMR and FT-IR experiments by Dai et al.8,24 Trends observed in DES conductivity and viscosity by Abbott et al. for choline chloride-glycerol support the existence of a 3D intermolecular H-bonding network.25 PFG-NMR spectroscopy experiments by D'Agostino et al. hint at extensive hydrogen-bonded chains of malonic acid molecules in a malonic acid-choline chloride DES,26 and later inferred that there exists a complex set of interactions between different species in DESs, by analysis of the respective diffusion coefficients.27 Solvatochromic probe behaviour was studied spectroscopically in a variety of DESs by the group of Pandey et al., revealing the polarity of DESs and the presence of solvent–solvent interactions attributed to interspecies hydrogen bonding.28,29 Quasi-elastic neutron scattering (QENS) was applied to a choline chloride-glycerol DES by Wagle et al., finding that the DES components have varying differential localized mobilities because of their different interaction strengths.30
To date and despite significant interest, no experimental technique has therefore been applied to DESs that is able to fully probe the phenomena driving DES formation and structuring, as has been extensively studied previously in ILs.31 Methods applied so far are either indirect (such as NMR) with substantial interpretation of dynamic intermolecular interactions, or involve doping with an additive that impacts upon the solvent structure, or are solely computational approaches.32–35 Wide Q-range neutron diffraction is a well-established method for directly measuring the structure of a disordered material.36 In this paper, we present experimental neutron diffraction data of four H/D isotopic contrasts of the DES reline. The structure of the DES is resolved using a reverse modelling protocol, generating 3D configurations commensurate with the measured diffractograms and known physical parameters. We therefore report a complete experimental account of the interactions governing the unique properties of the reline DES, including their geometry and length scale.
Experimental
Preparation of isotopically substituted DES
Hydrogenated choline chloride (≥98%) and urea (≥99%) were purchased from Sigma-Aldrich and used without further purification. d4-urea (CO(ND2)2, 99.6% pure, 99.8 atom% D) and d9-choline chloride ((CD3)3N(CH2)2OHCl), 99% pure, 99.6 atom% D) were purchased from QMX laboratories and used without further purification. Reline samples with isotopic substitution were prepared by mixing protonated and deuterated choline chloride and urea in the eutectic molar ratio at 60 °C to form four DESs with respective choline chloride:urea deuterations of H:H, H:D, D:H and D:D. Samples were dried under vacuum at 60 °C and were determined, using a Mettler-Toledo DL32 Karl Fischer titrator, to have a mean water content of 2252 ± 519 ppm (0.2% H2O) at the point of measurement.
Neutron diffraction
Diffraction data were collected using the SANDALS diffractometer, located in Target Station 1 in the ISIS Neutron and Muon Facility, Rutherford Appleton Laboratories, Harwell Campus, UK. SANDALS is a time-of-flight diffractometer optimized for the study of structurally disordered light element systems, operating using forward scattering detector geometry and a neutron wavelength range of 0.05 to 4.5 Å. Data were collected over the full Q-range of 0.1 to 50 Å−1 using a circularly-collimated neutron beam of diameter 30 mm. Approximately 2 g of each DES sample was added to null-scattering, vacuum-sealed Ti0.68Zr0.32 alloy flat plate cells. The internal dimensions of the cell are 1 × 35 × 35 mm with a wall thickness of 1 mm, giving a sample thickness of 1 mm. A 3 mm thick vanadium standard, the empty instrument, and empty sample cells were measured in addition to the samples for data normalization and instrument calibration. The samples were placed in an auto-changer and the temperature maintained at 303 K using a Julabo circulating heater to preserve the DES liquid phase over the ∼8 h of counting time per sample.Experimental data analysis is achieved using GudrunN, an analysis suite based on the ATLAS software package that is designed to correct raw neutron total scattering data.37 The sample environment background is subtracted and the data normalized to yield differential scattering cross sections that are consistent with the sample isotopic compositions. The inelastic scattering of hydrogen is then subtracted to form merged interference differential scattering cross section datasets that are amenable to analysis using empirical potential structure refinement (EPSR).38 Details on the EPSR procedure are provided in the ESI,† and the assigned bond lengths and atom types are described in Table 1.
|
|
|||
|---|---|---|---|
| Atom A | Atom B | Bond length/Å | ±Bond length/Å |
| 1 | 2 | 1.49 | 0.097 |
| 1 | 3, 4, 5 | 1.48 | 0.096 |
| 2 | 6, 8 | 1.11 | 0.072 |
| 2 | 7 | 1.54 | 0.100 |
| 3, 4, 5 | 9–17 | 1.11 | 0.072 |
| 7 | 20 | 1.40 | 0.091 |
| 7 | 18, 19 | 1.11 | 0.072 |
| 20 | 21 | 0.99 | 0.064 |
| 22 | 23 | 1.22 | 0.079 |
| 22 | 24, 25 | 1.39 | 0.025 |
| 24, 25 | 26, 28 | 1.01 | 0.066 |
| 24, 25 | 27, 29 | 1.01 | 0.066 |
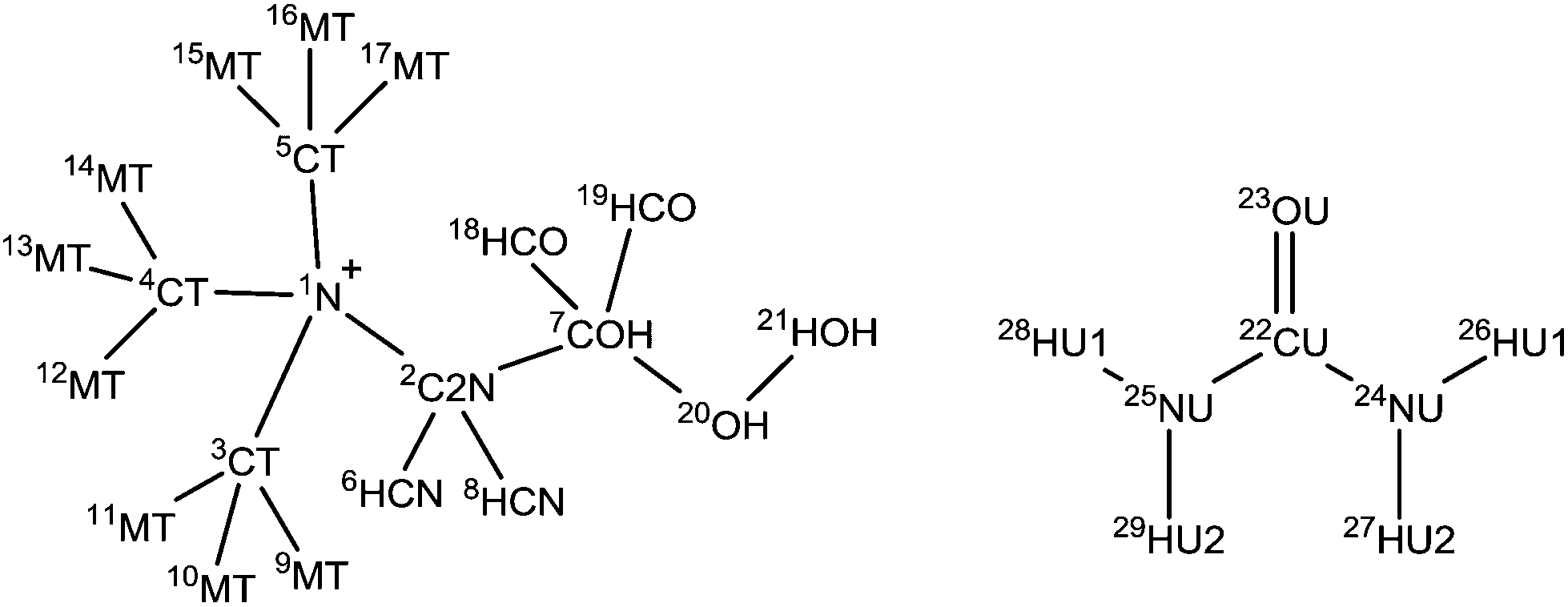
Results and discussion
Fits to the data
Inspection of the experimental diffraction patterns shown in Fig. 1 alongside their EPSR fits reveals that the EPSR model has been able to equilibrate to the experimental data very closely, and transforming the data into r-space further demonstrates the quality of this fit. EPSR calculates a value indicating the quality of fit called the R-factor, where a low R-factor means a better fit. The mean R-factor value over the approximately 4000 iterations accumulated over the refinement procedure was 0.029, a comparatively low value indicating that the EPSR model is as objectively representative of the experimental data as it is possible for it to be.39 It is noted that the major source of discrepancy occurs at Q ≤ 2 Å−1, which is the region affected by the inelastic scattering of light hydrogen. It was not possible to obtain perdeuterated choline chloride, and so the disagreement here between model and experimental data for all isotopic contrasts can be attributed to slight over- or under-subtraction of the substantial wavevector-dependent inelastic scattering background that light hydrogen produces. This is a known issue in the analysis of data from neutron scattering experiments.40,41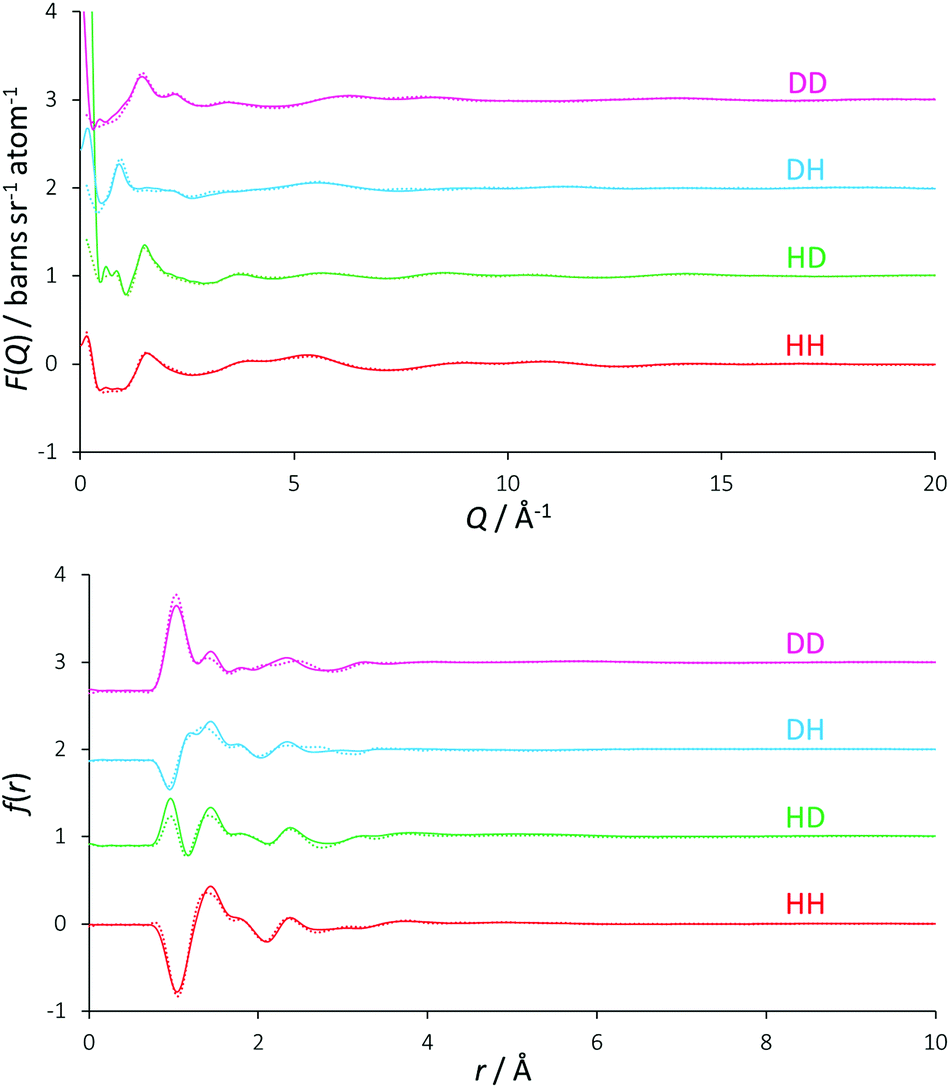 | ||
| Fig. 1 EPSR fits (solid coloured lines) to the total diffraction profiles (coloured circles), shown as a function of Q (upper figure) and r (lower figure) space. | ||
Reline radial distribution functions
Fig. 2 displays the molecularly centred radial distribution functions (or pair correlation functions) derived from EPSR simulation for all of the molecules that are present in reline. These RDFs were computed up to 20 Å, the approximate reliable size resolution of the simulation box. However, the RDFs converge and above approximately 10 Å, there is no evident long-range structural order in reline in the liquid phase. The RDFs are therefore truncated to aid viewing.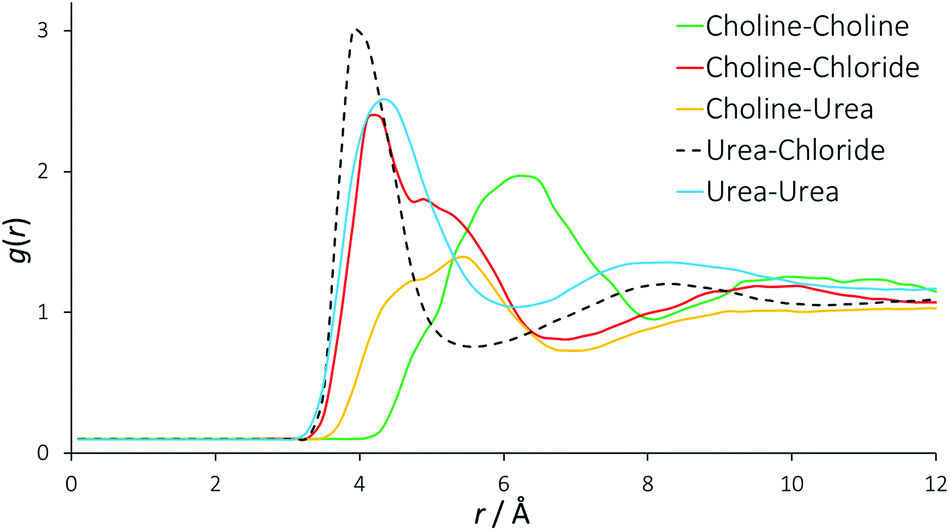 | ||
| Fig. 2 Radial distribution functions between all different species present in the DES mixture. For these RDFs, the molecular centres are defined as the centre of mass. | ||
In previous, similar studies on the structure of conventional 1,3-dialkylimidazolium ILs, the generalized structure comprises each individual ion being solvated by a shell of 7 gegenions.42,43 For reline, we find each choline cation solvated by a shell of approximately 7 other choline cations (intermolecular coordination numbers are shown in Table 2). In the RDF for a typical binary imidazolium-based IL, one would therefore expect to see progressive out-of-phase ion distributions around one another. In the DES system this feature is not seen quite so sharply. There is a clear first feature in the RDF between urea and chloride at 4 Å, which also shows a second peak at 8 Å correlating with a second solvation shell. However, the RDFs for urea–urea and choline–chloride also contribute to the close-range ordering at 4 Å, and the urea–urea RDF has a similar second shell to the urea-chloride RDF at 8 Å. Because these RDFs are derived from the molecular centre of mass, this falls on a length scale commensurate with hydrogen bonds between all of these species. The determined RDF between urea and chloride tends to agree with prior MD work on reline, but due to the manner in which the authors present their data it is almost impossible to draw further meaningful comparisons with other RDFs, other than the observation that the pure MD data appear relatively over-structured.35
| Molecule A | Molecule B | Mean radius/Å | N coord |
|---|---|---|---|
| Urea | Chloride | 3.9 | 2.08 ± 1.01 |
| Choline | Chloride | 4.2 | 4.35 ± 1.30 |
| Choline | Urea | 5.5 | 5.91 ± 2.84 |
| Choline | Choline | 6.3 | 6.74 ± 2.16 |
| Urea | Urea | 4.4 | 6.77 ± 3.05 |
The RDF for choline–chloride has a distinct shoulder at 5 Å, which implies a second major interaction between choline and chloride that is at a slightly greater length scale than the hydrogen bonding interactions. The alternating oscillation in the structuring between RDFs can be observed in the case of the choline–choline RDF, which shows a major correlation at 6 Å. A broad correlation is also observed between choline and urea, at 4 Å to 6 Å. The length scale of interactions in DESs are generally 1 Å shorter than those found in imidazolium-based ILs31 and mixtures of glucose with imidazolium ILs, which show glucose-ion correlations at 5 Å rather than 4 Å in the DES.44 The close-range interactions in the DES suggests that at the high 2:1 ratio of HBD:salt, the structure of reline is driven and dominated by the HBD, rather than the HBD being accommodated with only minor effects, as is the case for ILs at lower HBD:salt ratios.
The insight from these molecular RDFs must therefore be that DESs and ILs share a similar structure of concentric solvation shells, but in the DES this structure is more convoluted than ILs, having a strong close-range interaction with contributions from both choline and urea with chloride, and between separate urea molecules. This implies that rather than the radial, differentially charged solvation shells found in ILs, in this DES a complex structure is formed, driven by hydrogen bonding interactions. The existence of such a structure would allow reline to be stoichiometric and charge-balanced at a localized level due to the presence of the neutral urea molecules, and is complementary to charge delocalization being the main driving force behind DES formation,45,46 whilst also in agreement with prior DFT results for reline.32 This also explains the unusually high stability of proteins in DESs.47
Partial radial distribution functions
Partial (site-site) RDFs between all different atom types that are used in the EPSR simulation give more insight into specific structuring. In the case of reline there are 120 such partial RDFs, many of which provide limited information. The RDFs best describing the structuring within reline are plotted in Fig. 3, and a complete set of RDFs is supplied in the ESI.†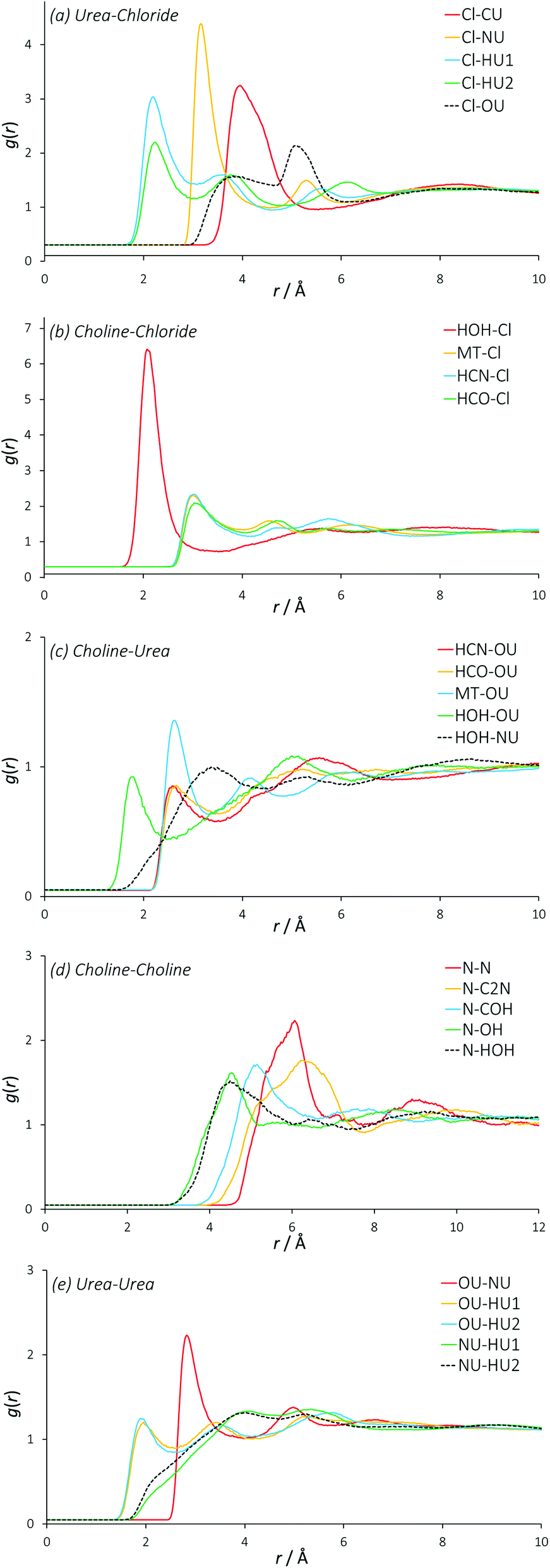 | ||
| Fig. 3 Partial (site-site) radial distribution functions between all different species present in the DES mixture. These RDFs are centred on specific atom types most strongly demonstrating the structuring within liquid reline for simplicity. | ||
The partial RDFs between the two different hydrogen environments of the urea molecule with chloride (Fig. 3a) show a significant first correlation at approximately 2.2 Å. Interestingly, the magnitude of this interaction is significantly stronger for the HU1 hydrogens, which are the two hydrogen atoms furthest from the central axis of the urea molecule. This indicates a preference for urea to form hydrogen bonds with chloride in this direction, perhaps allowing the molecule to orient itself in a configuration that maximizes the interaction energy of its components with other atom types. The second coordination peak of HU1 and HU2 with chloride at ∼4 Å shows a slight preference for HU2, which is a secondary feature from a hydrogen bond that has been formed by HU1 with a chloride. Indeed, the mean coordination number of HU1 (1.73, shown in Table 3) is significantly higher than that of HU2 (1.25) about chloride, demonstrating this preferential orientation. On average, each chloride is coordinated by 4 urea nitrogen atoms at a radius of 3.15 Å. The length scale of the interactions of chloride with urea, particularly HU1 and HU2, prove that urea forms relatively strong hydrogen bonds with chloride as previously hypothesized, at an average coordination of two urea molecules per chloride.
| RDF | Atom A | Atom B | r coord/Å | N coord |
|---|---|---|---|---|
| U–Cl | Cl | CU | 3.3–5.2 | 3.76 ± 2.27 |
| Cl | NU | 2.8–4.3 | 4.10 ± 2.63 | |
| Cl | HU1 | 1.7–3.1 | 1.73 ± 1.58 | |
| Cl | HU2 | 1.7–3.1 | 1.25 ± 1.15 | |
| Ch–Cl | HOH | Cl | 1.6–3.4 | 0.66 ± 0.50 |
| MT | Cl | 2.6–4.0 | 0.70 ± 0.66 | |
| HCN | Cl | 2.6–4.2 | 0.73 ± 0.67 | |
| HCO | Cl | 2.6–4.0 | 0.70 ± 0.66 | |
| Ch–U | HCN | OU | 2.2–3.5 | 0.41 ± 0.60 |
| MT | OU | 2.2–3.4 | 0.48 ± 0.66 | |
| HCO | OU | 2.2–3.6 | 0.45 ± 0.64 | |
| HOH | OU | 1.3–2.6 | 0.16 ± 0.38 | |
| HOH | NU | 1.8–4.2 | 2.08 ± 1.68 | |
| Ch–Ch | N | N | 4.6–6.7 | 3.48 ± 1.72 |
| N | C2N | 4.2–7.5 | 5.34 ± 2.03 | |
| N | COH | 4.0–5.9 | 2.05 ± 1.34 | |
| N | OH | 3.2–5.2 | 2.31 ± 1.11 | |
| N | HOH | 3.2–5.5 | 2.73 ± 1.31 | |
| U–U | OU | NU | 2.5–3.7 | 2.25 ± 1.50 |
| OU | HU1 | 1.4–2.6 | 0.62 ± 0.84 | |
| OU | HU2 | 1.4–2.7 | 2.33 ± 0.95 | |
| NU | HU1 | 1.8–4.6 | 5.62 ± 2.65 | |
| NU | HU2 | 1.8–4.6 | 5.77 ± 2.50 |
The RDFs between choline and chloride demonstrate a significant level of ordering (Fig. 3b). Particularly, the hydroxyl hydrogen of choline has an intense correlation peak at ∼2.1 Å, which shows that choline is participating in strong hydrogen bonding with chloride as the most preferential interaction with 0.7 chlorides coordinated on average. The RDFs of chloride around the other hydrogen atoms of choline show lower hydrogen-bonding with a first correlation at approximately 3 Å, which is the likely cause of the 5 Å shoulder in the choline–chloride RDF. Subsequent features in these other hydrogen RDFs between 4 Å–7 Å show the less-preferred configurations where the chloride is not bound to the hydroxyl group. These RDFs all show very similar structuring, and there is no clear difference between any particular hydrogen sites in terms of chloride coordination number, indicating little preference for chloride to interact with any hydrogen atom other than the strongly hydrogen-bonding hydroxyl group. The free rotation of the hydroxyl group permits many strongly preferred configurations of chloride around choline.
The RDFs describing the orientation of urea around choline show a generally sparse set of interactions (Fig. 3c), with the strongest interaction between the hydroxyl choline hydrogen with the nitrogen of urea at ∼2 Å, and a mean coordination number of 2 over this radius, giving one urea that is associated with the hydroxyl group. This can be rationalized by strong hydrogen bonding, with exchange occurring between the hydroxyl moiety of choline and NU atoms of urea, in addition to secondary structuring effects from the more favourable interactions of urea with atom types such as chloride. The small correlation at 2.4 Å between the urea oxygen atom and the various non-hydroxyl hydrogens of choline shows that the hydrogen bonding for the hydrocarbon chain and methyl hydrogen atoms is relatively weak. This is supported by the relatively low coordination numbers of urea around the moieties of choline other than the hydroxyl group; the second urea molecule around choline is associated weakly between these groups.
The interactions seen between like choline molecules are generally relatively weak and over a longer length scale (Fig. 3d). The most distinct interaction is between like N atoms, with a peak centred at 6 Å and a mean coordination number of 3.4 at this distance. The RDFs between N–OH and N–HOH are similar, but with a slight weighting towards the N–OH correlation. There is therefore no specific preference towards the N correlation with the hydroxyl hydrogen over the hydroxyl oxygen, which rules out inter-choline bridging by chloride bonding. Interestingly, the RDF between N and C2N is approximately 1.3 Å further than the N–COH RDF. Because the C2N carbon atom is closer to the N atom than COH, this means that the ∼7 choline molecules in the solvation shell at 6 Å radius from the central choline are significantly more likely to be oriented with their hydroxyl group pointing in the opposite direction to that of the central choline molecule, which is further confirmed by the hydroxyl partial RDFs main correlation occurring even closer to the N group, and higher coordination numbers for the closer atom types. It is possible that this structural solution most effectively distributes the balance of charge.
There is clear structuring occurring between separate urea molecules (Fig. 3e). Particularly, it seems that a strong hydrogen bond is formed between urea hydrogen atoms and urea oxygen atoms, as may be expected from their electronegativity difference. The HU2 proton shows much stronger structuring with urea at closer range, coordinating 2.3 OU atoms at 2.2 Å. The RDF between the OU and NU atom types has a sharp feature at 2.8 Å, showing the compound of urea hydrogen bonding with urea oxygen atoms. The urea self-correlation function in reline is of the same length scale as that which is observed for urea in water at high concentrations.48 Some inter-urea exchange can be inferred from the high coordination numbers of HU1 and HU2 around NU, demonstrating that the solvent structure also permits urea clusters as a secondary structure, again perhaps with a charge balancing function. Clustering of the HBD species in this manner was observed in previous similar experiments of eutectic mixtures of glucose with imidazolium ionic liquids.44
Spatial density functions
Each chloride, therefore, is solvated by an average of two urea molecules, hydrogen bonded at a distance of 2.2 Å. Every urea molecule is associated with several other urea molecules via urea–urea hydrogen bonding at between 2 Å–3 Å. Each chloride is also strongly associated with one choline molecule mostly via hydrogen bonding with the hydroxyl group, at a distance of 2.2 Å. Finally, each choline is solvated by a shell of 7 other choline molecules, each 6 Å from the origin and likely oriented in opposite directions. A spatial density function (SDF) tracks the distribution of molecular centres in 3D. A set of SDFs for reline may be seen in Fig. 4. To determine these SDFs, the molecular centre of urea was defined as the CU atom, and the centre of choline was taken as the midpoint of atom types N, C2N and COH.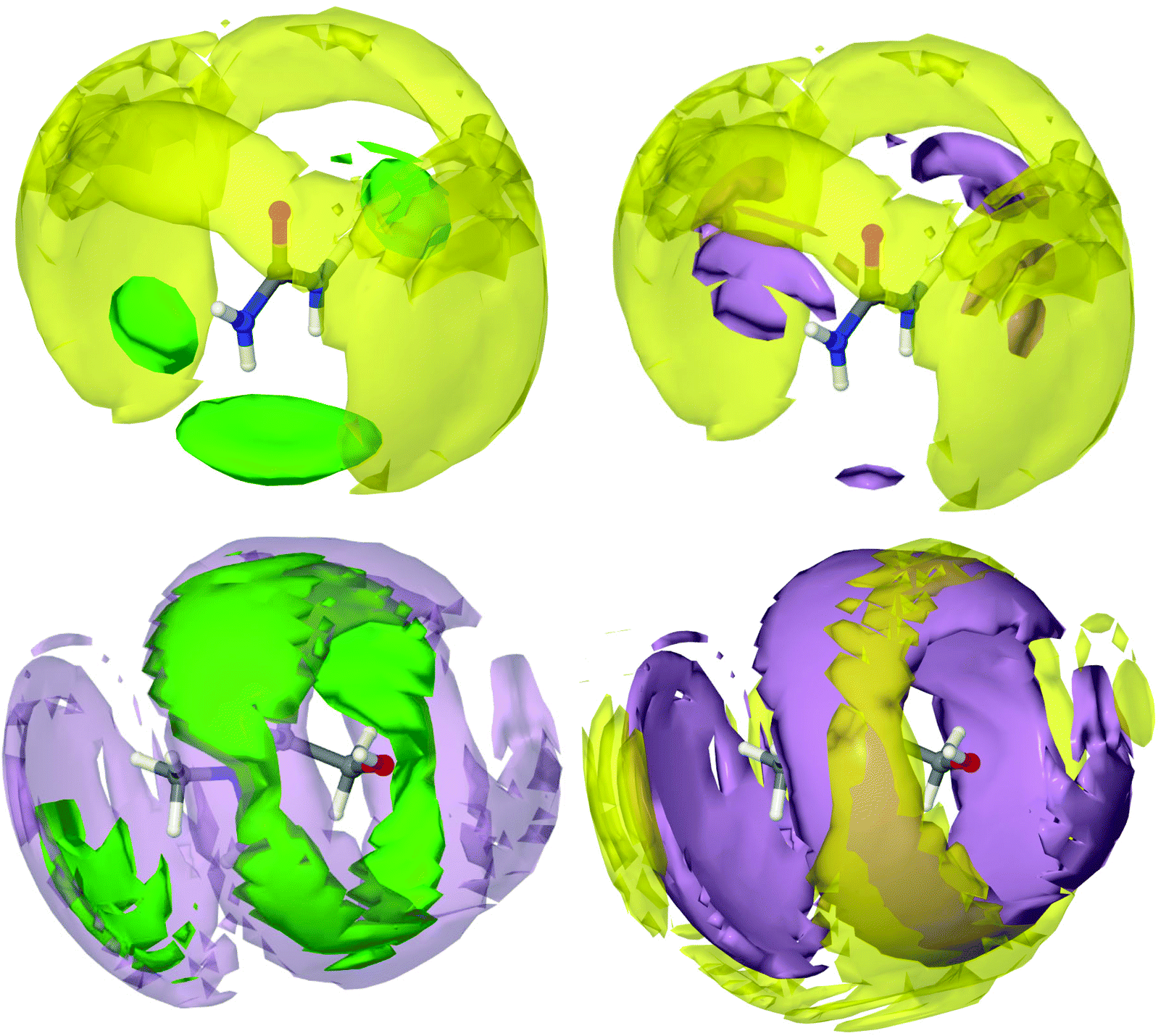 | ||
| Fig. 4 Spatial density functions (SDFs) showing probabilistic 3D structures of the components of reline. Isosurfaces are drawn of the 7.5% most likely configurations of molecular centres. Yellow surfaces depict choline cations, purple surfaces represent urea molecules and green show chloride anions. Each plot represents the SDF for a particular molecule. Two isosurfaces are plotted per SDF to provide a visual reference to aid 3D interpretation. | ||
The SDF of chloride around urea shows very clearly a tightly focused distribution of chloride around the hydrogen bond-donating hydrogen atoms of urea, confirming the strong hydrogen bonding between these species, as was demonstrated previously. The SDF plot of choline around urea shows that the interaction between these two species is not predominantly hydrogen bond driven, with choline molecules more likely to be found radially around the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O axis of urea. Adopting this configuration allows choline to form very strong hydrogen bonds with chloride via its hydroxyl proton, whilst maximizing its interaction energy by also forming weak hydrogen bond interactions with electronegative urea atoms. At the same time, urea is able to coordinate chloride with strong hydrogen bonds. This forms a complementary shell of choline around urea and chloride. A second urea molecule is also able to participate in further hydrogen bonding with the chloride ion that is centred on a first urea, whilst itself interacting with the same choline. This second urea molecule is small enough to be able to occupy a space that cannot be occupied by the bulky choline cation.
O axis of urea. Adopting this configuration allows choline to form very strong hydrogen bonds with chloride via its hydroxyl proton, whilst maximizing its interaction energy by also forming weak hydrogen bond interactions with electronegative urea atoms. At the same time, urea is able to coordinate chloride with strong hydrogen bonds. This forms a complementary shell of choline around urea and chloride. A second urea molecule is also able to participate in further hydrogen bonding with the chloride ion that is centred on a first urea, whilst itself interacting with the same choline. This second urea molecule is small enough to be able to occupy a space that cannot be occupied by the bulky choline cation.
The chloride SDFs centred about choline show that there is a high level of order around choline. However, even at the high probability level selected, plotting the most likely 7.5% portion of the spatial density function, the isosurfaces are relatively large, indicating that there are many available structural conformations that are preferable. It can be seen that chloride is associated most strongly in a circular band around the free rotor hydroxyl group, with which it forms strong hydrogen bonds. In addition to this, the ‘shoulder’ in the choline–chloride RDF at 5 Å can be seen as a second chloride surface at the ammonium moiety of choline. Chloride is the closest species around choline, and is strongly associated with urea. Again, urea is ordered around choline but with many possible conformations. It can be observed that the urea and choline molecules work synergistically to sandwich the chloride ion with hydrogen bonding forces whilst maximizing their interaction energy with one another. Radially further still can be seen the choline solvation shell about choline, which is most strongly associated with the urea shell. This demonstrates how the separate components of DESs form a radially layered sandwich structure, allowing for the best distribution of charge between each component.
It appears that at the eutectic point, the molar ratio of urea and choline is such that the intermolecular hydrogen bonding forces between these two species and chloride becomes balanced, and chloride is therefore strongly affiliated with both species in a sandwich structure. This ability to form strong hydrogen bonds and therefore generate significant intermolecular order, whilst maintaining a sufficient quantity of separate favourable interactions such that the mixture is frustrated from crystallization, can therefore be viewed as the geometric driving force for the formation of the deep eutectic mixture. This is complementary to the argument that charge delocalization causes DES formation, as clearly the ability of urea to delocalize charge drives the formation of the interspecies hydrogen bonds that give rise to the sandwich structure where each chloride is strongly associated with one choline and two urea molecules, as has previously been predicted by DFT.32
It is interesting that the ordering observed around choline is so strong. Although a few studies to date have hinted at a set of complex interactions between all of the components of the DES,27,30 most attribute the effect solely to the HBD. However, choline appears to be a relatively special case in forming DESs, with its ability to participate strongly in hydrogen bonding whilst also frustrating crystallization with its bulky, anisotropic structure.
Hole theory
Traditionally, the differential ionic mobilities in DESs have been rationalized using hole theory,25 and this model has been applied successfully in the prediction of physicochemical properties of DESs such as conductivity.30 This is achieved by considering the liquid as an ionic lattice with a given number of vacancies, where this void concentration and size is determined from the properties of the liquid including its surface tension. Diffusivity is obtained by comparing the radius of an available void space with the hydrodynamic radius of a diffusing species, which then performs a diffusional ‘jump’ into this void space.49,50Using surface tension data, it was determined that the DES reline has a Gaussian distribution of hole sizes centred at a radius of 2 Å, with 9.1% free space in the liquid and a mean activation barrier for diffusion of 58 kJ mol−1.51 EPSR was used to determine the void radial distribution function (VDF) within the DES, which can be seen in Fig. 5. The VOIDS routine translates the simulation box into pixels, and either assigns these pixels as occupied or empty, depending on whether or not there are any atoms within the specified radius. VDFs were calculated for representative radii of 1.8, 2.0 and 2.2 Å.
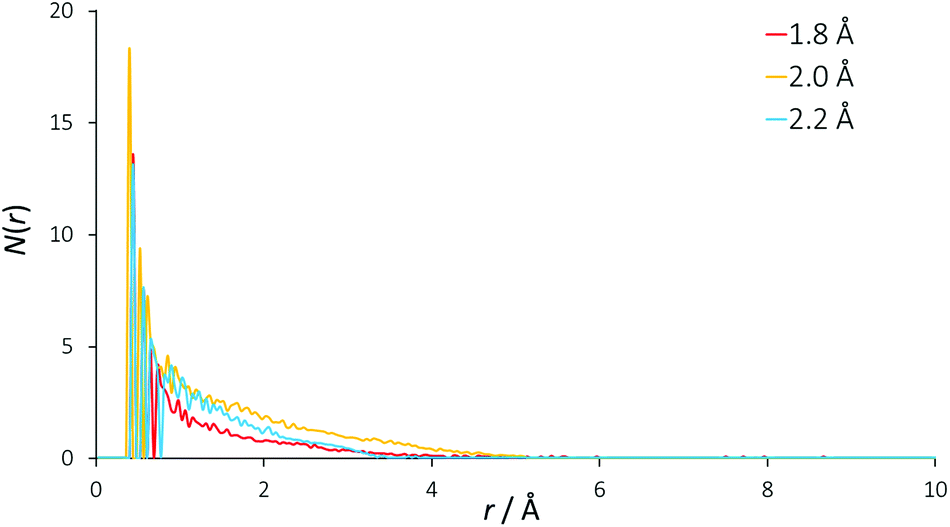 | ||
| Fig. 5 Radial distribution functions of void space in the EPSR model. Separate curves show the result of using different radii from each void origin to define empty spaces. | ||
An exponentially decaying void distribution is observed with the VDFs beginning at a radius of 0.3 Å. The rise in void density at short radii is due to void pixels being more likely to be adjacent to a high density of void pixels than at longer distances. Voids of radius 2 Å are found with a mean occurrence of 1.3 per simulation box, giving a 2 Å void fraction of 0.061%, which is an order of magnitude smaller than the 0.763% previously calculated. This may explain the predicted reline viscosity being low by a similar magnitude (11 cP calculated, 169 cP experimental at 40 °C) from hole theory.51 If there are density fluctuations in the model, as would be observed for spherical voids of 4 Å diameter, the VDF would show oscillating structures, but this is not observed for reline, showing that the liquid mixture is homogeneous even at molecular length scales. Despite being a useful tool for prediction of DES properties, our EPSR model suggests that the void size distribution from hole theory may therefore not be true in the case of reline, and we suggest that the existence of spherical 4 Å diameter holes in this DES is unlikely when considering the strength of the intermolecular interactions.
Conclusions
Wide Q-range neutron diffraction measurements of isotopically-substituted samples of the deep eutectic solvent reline have been interpreted using EPSR modelling to generate, for the first time, an experimentally-validated atomistic configuration of this increasingly popular liquid.We have found that the DES reline does indeed have a strong and complex hydrogen-bonding network between species, but this study builds upon previous works by showing the precise nature of this previously-hypothesized structuring. A significant correlation between the hydrogen bond donor molecule urea and the chloride anion, as had previously been shown by NMR experiments, is shown to be a strong hydrogen bonding interaction. Importantly, we also find that choline interacts very strongly with chloride by hydrogen bonding. This leads to the formation of a complex ion as a most likely 3D configuration, involving one choline, one chloride and two urea molecules. This structure is stabilized by the complementary hydrogen bond formation of choline and urea with chloride, and further by other favourable weak hydrogen bonds that are formed between the different molecules. This set of interactions causes the formation of a radially layered sandwich structure where choline and urea work synergistically to bond with chloride whilst maximizing their own weaker interaction. This sandwich structure can also be visualized as a charge-delocalized, locally stoichiometric cage centred on chloride. The delicate balance of strong forces between all species is sufficient to prevent the crystallization of the mixture at room temperature, thereby accounting for the deep eutectic behaviour of reline.
The information regarding DES structure that has been elucidated in this study will no doubt be able to aid in the informed design of DESs in the future as well as promoting understanding of the properties of the current selection of DESs, hopefully enabling a generation of designer solvents that are ‘green, and finally green enough’.
Acknowledgements
O. S. H. is thankful to STFC and EPSRC for co-funding a PhD studentship via the EPSRC Centre for Doctoral Training in Sustainable Chemical Technologies (Studentship 3578). We thank the ISIS Pulsed Neutron and Muon Source for allocation of experimental beamtime on SANDALS under project RB1510465.Notes and references
- A. P. Abbott, G. Capper, D. L. Davies, H. L. Munro, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2001, 2010–2011 RSC.
- M. Francisco, A. Van Den Bruinhorst and M. C. Kroon, Angew. Chem., Int. Ed., 2013, 52, 3074–3085 CrossRef CAS PubMed.
- H. G. Morrison, C. C. Sun and S. Neervannan, Int. J. Pharm., 2009, 378, 136–139 CrossRef CAS PubMed.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- N. V. Plechkova and K. R. Seddon, in Methods and Reagents for Green Chemistry: An Introduction, 2007, pp. 103–130 Search PubMed.
- D. J. G. P. van Osch, L. F. Zubeir, A. van den Bruinhorst, M. A. A. Rocha and M. C. Kroon, Green Chem., 2015, 4518–4521 RSC.
- Y. Dai, G.-J. Witkamp, R. Verpoorte and Y. H. Choi, Food Chem., 2015, 187, 14–19 CrossRef CAS PubMed.
- B. Tang and K. H. Row, Monatsh. Chem., 2013, 144, 1427–1454 CrossRef CAS.
- Q. Zhang, K. De Oliveira Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- Q. Wen, J.-X. Chen, Y.-L. Tang, J. Wang and Z. Yang, Chemosphere, 2015, 132, 63–69 CrossRef CAS PubMed.
- M. Hayyan, M. A. Hashim, M. A. Al-Saadi, A. Hayyan, I. M. AlNashef and M. E. S. Mirghani, Chemosphere, 2013, 93, 455–459 CrossRef CAS PubMed.
- Q. Zhang, Q. Wang, S. Zhang, X. Lu and X. Zhang, ChemPhysChem, 2015 DOI:10.1002/cphc.201500713 , Ahead of Print.
- P. Liu, J.-W. Hao, L.-P. Mo and Z.-H. Zhang, RSC Adv., 2015, 5, 48675–48704 RSC.
- J. García-Álvarez, Eur. J. Inorg. Chem., 2015, 5147–5157 CrossRef.
- D. V. Wagle, H. Zhao and G. A. Baker, Acc. Chem. Res., 2014, 47, 2299–2308 CrossRef CAS PubMed.
- B. Tang, H. Zhang and K. H. Row, J. Sep. Sci., 2015, 38, 1053–1064 CrossRef CAS PubMed.
- G. Garcia, S. Aparicio, R. Ullah and M. Atilhan, Energy Fuels, 2015, 29, 2616–2644 CrossRef CAS.
- T. Arnold, A. J. Jackson, A. Sanchez-Fernandez, D. Magnone, A. E. Terry and K. J. Edler, Langmuir, 2015, 31, 12894–12902 CrossRef CAS PubMed.
- M. Pal, R. K. Singh and S. Pandey, ChemPhysChem, 2015, 16, 2538–2542 CrossRef CAS PubMed.
- S. J. Bryant, R. Atkin and G. G. Warr, Soft Matter, 2015 10.1039/c5sm02660a , Ahead of Print.
- Y. H. Choi, J. van Spronsen, Y. Dai, M. Verberne, F. Hollmann, I. W. C. E. Arends, G.-J. Witkamp and R. Verpoorte, Plant Physiol., 2011, 156, 1701–1705 CrossRef CAS PubMed.
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef CAS.
- Y. Dai, J. van Spronsen, G. J. Witkamp, R. Verpoorte and Y. H. Choi, Anal. Chim. Acta, 2013, 766, 61–68 CrossRef CAS PubMed.
- A. P. Abbott, R. C. Harris, K. S. Ryder, C. D'Agostino, L. F. Gladden and M. D. Mantle, Green Chem., 2011, 13, 82 RSC.
- C. D'Agostino, R. C. Harris, A. P. Abbott, L. F. Gladden and M. D. Mantle, Phys. Chem. Chem. Phys., 2011, 13, 21383–21391 RSC.
- C. D'Agostino, L. F. Gladden, M. D. Mantle, A. P. Abbott, E. I. Ahmed, A. Y. M. Al-Murshedi and R. C. Harris, Phys. Chem. Chem. Phys., 2015, 15297–15304 RSC.
- A. Pandey, R. Rai, M. Pal and S. Pandey, Phys. Chem. Chem. Phys., 2014, 16, 1559–1568 RSC.
- A. Pandey and S. Pandey, J. Phys. Chem. B, 2014, 118, 14652–14661 CrossRef CAS PubMed.
- D. V. Wagle, G. A. Baker and E. Mamontov, J. Phys. Chem. Lett., 2015, 2924–2928 CrossRef CAS PubMed.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 6358–6405 Search PubMed.
- G. García, M. Atilhan and S. Aparicio, Chem. Phys. Lett., 2015, 634, 151–155 CrossRef.
- S. L. Perkins, P. Painter and C. M. Colina, J. Chem. Eng. Data, 2014, 3652–3662 CrossRef CAS.
- D. Shah and F. S. Mjalli, Phys. Chem. Chem. Phys., 2014, 16, 23900–23907 RSC.
- H. Sun, Y. Li, X. Wu and G. Li, J. Mol. Model., 2013, 19, 2433–2441 CrossRef CAS PubMed.
- K. J. Edler and D. T. Bowron, Curr. Opin. Colloid Interface Sci., 2015, 20, 227–234 CrossRef CAS.
- A. K. Soper, GudrunN and GudrunX: Programs for Correcting Raw Neutron and X-ray Diffraction Data to Differential Scattering Cross Section, Rutherford Appleton Laboratory Technical, RAL-TR-201, 2011 Search PubMed.
- A. K. Soper, Phys. Rev. B: Condens. Matter, 2005, 72, 104204 CrossRef.
- A. K. Soper, Mol. Phys., 2001, 99, 1503–1516 CrossRef CAS.
- A. K. Soper, Mol. Phys., 2009, 107, 1667–1684 CrossRef CAS.
- A. K. Soper, ISRN Phys. Chem., 2013, 2013, 279463 Search PubMed.
- M. Deetlefs, C. Hardacre, M. Nieuwenhuyzen, A. A. H. Padua, O. Sheppard and A. K. Soper, J. Phys. Chem. B, 2006, 110, 12055–12061 CrossRef CAS PubMed.
- C. Hardacre, J. D. Holbrey, S. E. J. McMath, D. T. Bowron and A. K. Soper, J. Chem. Phys., 2003, 118, 273–278 CrossRef CAS.
- T. G. A. Youngs, J. D. Holbrey, C. L. Mullan, S. E. Norman, M. C. Lagunas, C. D'Agostino, M. D. Mantle, L. F. Gladden, D. T. Bowron and C. Hardacre, Chem. Sci., 2011, 2, 1594 RSC.
- D. Carriazo, M. C. Serrano, M. C. Gutiérrez, M. L. Ferrer and F. del Monte, Chem. Soc. Rev., 2012, 41, 4996 RSC.
- K. Haerens, S. Van Deuren, E. Matthijs and B. Van der Bruggen, Green Chem., 2010, 12, 2182 RSC.
- H. Monhemi, M. R. Housaindokht, A. A. Moosavi-Movahedi and M. R. Bozorgmehr, Phys. Chem. Chem. Phys., 2014, 16, 14882–14893 RSC.
- A. Soper, A. K. Castner and E. W. Luzar, Biophys. Chem., 2003, 105, 649–666 CrossRef CAS PubMed.
- A. P. Abbott, ChemPhysChem, 2004, 5, 1242–1246 CrossRef CAS PubMed.
- A. P. Abbott, R. C. Harris and K. S. Ryder, J. Phys. Chem. B, 2007, 111, 4910–4913 CrossRef CAS PubMed.
- A. P. Abbott, G. Capper and S. Gray, ChemPhysChem, 2006, 7, 803–806 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Theory & simulation details for wide angle scattering. Determined molecular coordination numbers and the complete set of partial RDFs. See DOI: 10.1039/c5gc02914g |
| This journal is © The Royal Society of Chemistry 2016 |