
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom trash to resource: recovered-Pd from spent three-way catalysts as a precursor of an effective photo-catalyst for H2 production†
V.
Gombac
ab,
T.
Montini
b,
A.
Falqui
c,
D.
Loche
d,
M.
Prato
e,
A.
Genovese
c,
M. L.
Mercuri
ad,
A.
Serpe
*ad,
P.
Fornasiero
*b and
P.
Deplano
adf
a3R Metals Ltd, Spin-off of the University of Cagliari, at the Dipartimento di Scienze Chimiche e Geologiche, University of Cagliari, S.S. 554 Bivio per Sestu, 09042 Monserrato, Cagliari, Italy. E-mail: serpe@unica.it
bDipartimento di Scienze Chimiche e Farmaceutiche, INSTM, ICCOM-CNR, University of Trieste, Via L. Giorgieri 1, 34127 Trieste, Italy. E-mail: pfornasiero@units.it
cBiological and Environmental Sciences and Engineering Division, King Abdullah University for Science and Technology (KAUST), 23955-6900 Thuwal, Kingdom of Saudi Arabia
dDipartimento di Scienze Chimiche e Geologiche, University of Cagliari, and INSTM research unit, S.S. 554 Bivio per Sestu, 09042 Monserrato, Cagliari, Italy
eNanochemistry Department, Istituto Italiano di Tecnologia (IIT), via Morego 30, 16163 Genova, Italy
fDipartimento di Fisica, University of Cagliari, S.S. 554 Bivio per Sestu, 09042 Monserrato, Cagliari, Italy
First published on 6th January 2016
Abstract
The successful production of a nanostructured and highly dispersed Pd-TiO2 photo-catalyst, using [Pd(Me2dazdt)2](I3)2 (Me2dazdt = N,N′-dimethyl-perhydrodiazepine-2,3-dithione) salt, obtained through the selective and safe recovery of palladium from model exhaust three-way catalysts (TWCs), is reported here. The photo-catalyst prepared by the impregnation/photo-reduction of palladium on the support showed improved performance in H2 production from methanol and in glycerol photo-reforming compared to reference photo-catalysts obtained from conventional Pd-salts. The reported results represent a case of successful palladium “recovery and re-employment” and thus constitute an example of green chemistry by providing, in one route, the environmentally friendly recovery of a critical metal and its employment in the renewable energy field.
Introduction
Significant environmental benefits have been achieved by the introduction of three-way catalysts, which convert pollutants emitted by the car engine into not-harmful gases. Palladium, which has been recognized as a critical material,1 is a crucial, expensive catalytic component of TWCs2 and has limited and geographically localized natural availability. Data from Johnson Matthey show that the use of palladium for autocatalysts represents about 70% of the total demand for the metal.3 Due to the lifetime of TWCs (approximately 160![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 km, corresponding to an average of 8–10 years of use), an accumulation of noble-metals’ (NMs) rich wastes has been growing over the last few years. Palladium recovery has therefore economic and environmental relevance, in particular recovery from exhaust TWCs, especially given the introduction in the 2000s of the so-called Pd-only technology.‡ Despite this interest in NMs recovery, also encouraged by recent EU directives on end-of-life vehicles management,4 currently they are recycled at only a low extent (Pd: 26%; Pt: 25%; Rh: 31%, in 2014),3 primarily by non-selective and unattractive methods, such as pyrometallurgical chlorination5 or dissolution with strong oxidising acids.6,7 As an alternative to these polluting and/or harmful methods, over the last decade we have thoroughly investigated the leaching properties of a new class of reagents – namely dihalogen mixtures with donors bearing two vicinal thionic groups, such as cyclic-dithioxamides [e.g. N,N′-dimethylperhydrodiazepine-2,3-dithione (Me2dazdt) and N,N′-dimethylpiperazine-2,3-dithione (Me2pipdt)] – that show a peculiar capability to dissolve noble and toxic metals effectively and in very mild conditions, as well as their applicability in different fileds.8 In particular, a safe methodology for the effective and selective Pd dissolution and extraction from model spent TWCs, based on the use of the Me2dazdt·2I2 molecular adduct (Chart 1a), have been pointed out and patented (Scheme 1).9
000 km, corresponding to an average of 8–10 years of use), an accumulation of noble-metals’ (NMs) rich wastes has been growing over the last few years. Palladium recovery has therefore economic and environmental relevance, in particular recovery from exhaust TWCs, especially given the introduction in the 2000s of the so-called Pd-only technology.‡ Despite this interest in NMs recovery, also encouraged by recent EU directives on end-of-life vehicles management,4 currently they are recycled at only a low extent (Pd: 26%; Pt: 25%; Rh: 31%, in 2014),3 primarily by non-selective and unattractive methods, such as pyrometallurgical chlorination5 or dissolution with strong oxidising acids.6,7 As an alternative to these polluting and/or harmful methods, over the last decade we have thoroughly investigated the leaching properties of a new class of reagents – namely dihalogen mixtures with donors bearing two vicinal thionic groups, such as cyclic-dithioxamides [e.g. N,N′-dimethylperhydrodiazepine-2,3-dithione (Me2dazdt) and N,N′-dimethylpiperazine-2,3-dithione (Me2pipdt)] – that show a peculiar capability to dissolve noble and toxic metals effectively and in very mild conditions, as well as their applicability in different fileds.8 In particular, a safe methodology for the effective and selective Pd dissolution and extraction from model spent TWCs, based on the use of the Me2dazdt·2I2 molecular adduct (Chart 1a), have been pointed out and patented (Scheme 1).9
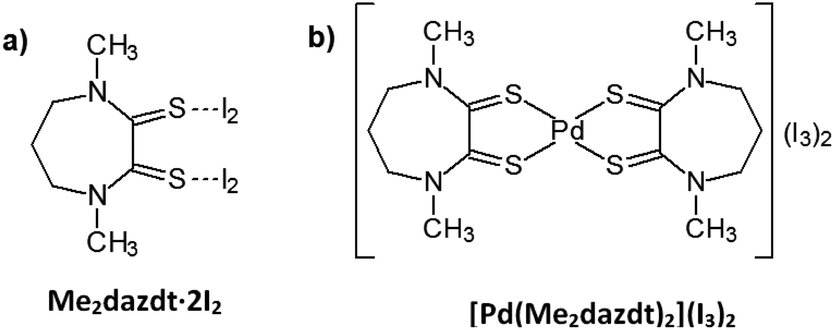 | ||
| Chart 1 Leaching reagent (a) and product (b). | ||
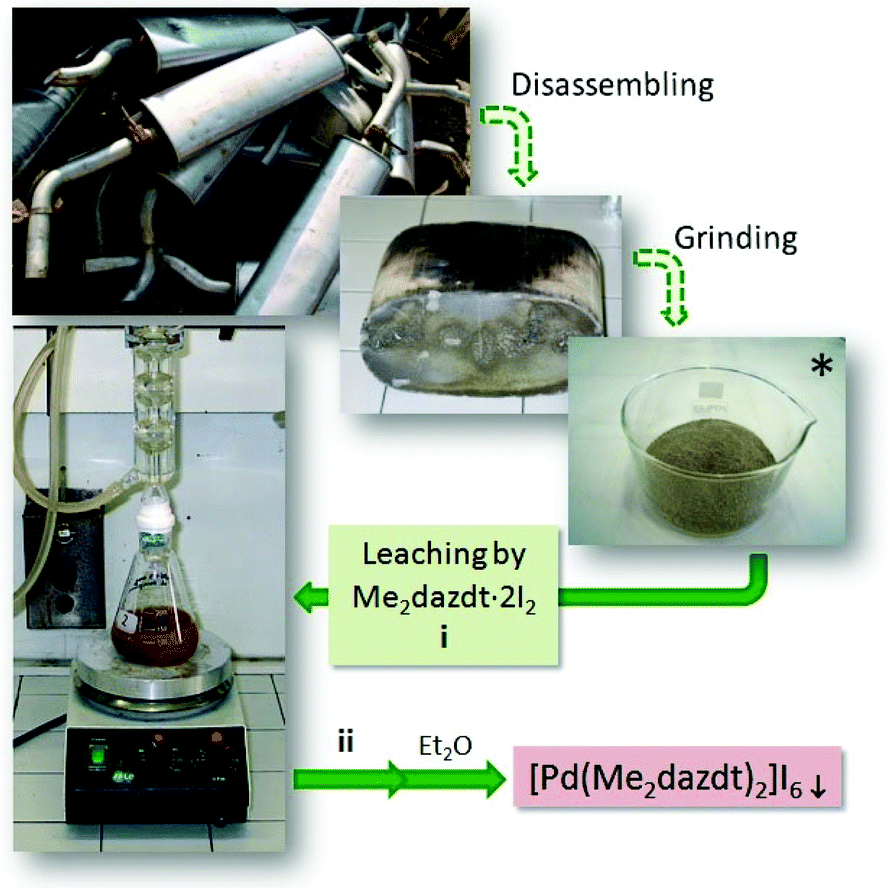 | ||
| Scheme 1 Pd leaching process from spent TWCs. *Pd(0.5–2.8%)-CeO2-ZrO2/Al2O3; ageing protocol: 1050 °C/200 h to simulate the thermal deactivation of TWCs. i: methyl ethyl ketone (MEK), reflux, 168 h, quantitative yield; ii: separation and washing through a glass microfibre thimble (0.8 μm particle retention) in a Soxhlet apparatus. | ||
The use of this non-cytotoxic leaching reagent, capable of combining complexing with oxidizing properties in the same molecule, allows almost quantitative palladium recovery under mild conditions (temperature: 80 °C and at atmospheric pressure without protection from air or moisture) and provides the [Pd(Me2dazdt)2](I3)2 complex (Chart 1b) as the leaching product.
This product, which should be destroyed in order to recover Pd-metal, has instead shown itself to be a very valuable catalyst precursor and useful for designing a sustainable palladium “recovery and re-employment” method, as described herein. Specifically, the growing interest in the photo-catalytic activity of palladium-based materials in the reforming of alcohols and carbohydrates to produce H2 prompted us to study the potential of this complex as a metal precursor for the preparation of photo-catalysts. The key feature of a photo-catalytic process is the presence of a semiconductor material (among these, TiO2 is a representative and commonly used semiconductor) with a 3.2 eV band gap (Eg),10 in which, upon illumination with radiation having an energy higher than Eg, the promotion of an electron (e−) from the valence band (VB) to the conduction band (CB) takes place. Concomitantly, the formation of a positive hole (h+) in the VB occurs. Photo-generated electrons and holes can then either undergo undesired recombination or migrate to the surface of the photo-catalist, where they can initiate reactions with adsorbed species. Photo-generated electrons in the CB can be directly involved in H2 production if the semiconductor CB edge is more negative than the H+ → H2 reduction potential [E(H+/H2) = 0.00 V with respect to the normal hydrogen electrode (NHE) at pH = 0]. NMs (e.g. Pt, Pd, Au), transition metal oxides (e.g. RuO2, IrO2, NiO) or sulphides (e.g. MoS2) can act as electron or hole traps, preventing e−/h+ recombination and enhancing photo-catalytic H2 production.11 The addition of sacrificial agents, which can irreversibly react with photo-generated holes (or OH˙ radicals derived from the h+ reaction with H2O) and undergo an easier oxidation than water, has also been proven to enhance the performance.12 In this respect, the use of largely available organic species in the so-called photo-reforming processes has been increasingly considered. Many oxygenate molecules, mostly derived from biomasses (e.g. sugar, starch, vegetable oils, lignocellulosic crops, algae fuels), have been tested for more sustainable H2 production by these routes.13
In this context, we investigated the production of a Pd-TiO2-based photo-catalyst by a Pd photo-deposition process using [Pd(Me2dazdt)2](I3)2, obtained through a one-step palladium oxidation/extraction from a model spent TWC, and the commercial reference TiO2 Degussa P25 as the supporting semiconductor.
The application of this photo-catalyst for H2 production by the photo-reforming of methanol and glycerol is described here, and compared with reference materials of similar composition but prepared using conventional Pd-salts as metal precursors.
While methanol is a commonly used sacrificial agent for testing the efficiency of photo-catalysts in hydrogen evolution,14 glycerol is a more challenging15 and sustainable substrate as it represent a 10 wt% by-product of biodiesel synthesis.16 Furthermore, the oxidative dehydrogenation of the glycerol and the subsequent catalytic and photo-catalytic reactions might lead also to the simultaneous and sustainable production of hydrogen and valuable products, such as 1,3-dihydroxypropanone or hydroxyacetaldehyde. In particular, 1,3-dihydroxypropanone is a promising compound for the preparation of skincare products and also a building block for the synthesis of biodegradable polymers.17
Results and discussion
Preparation of the precursor
The [Pd(Me2dazdt)2](I3)2 complex was obtained in the form of a crystalline product from the extraction solution recovered at the end of the palladium recovering process from model exhausted TWCs, as summarized in Scheme 1 and in the Experimental section.9 In particular, a model Pd(2.8%)-CeO2-ZrO2/Al2O3 catalyst was submitted to a severe ageing protocol (1050 °C/200 h) in order to simulate the thermal deactivation of what occurs on a real TWC. Subsequently, palladium was leached by reacting the catalyst (250 mg) with 50 mL of a 2.5 × 10−3 M methyl ethyl ketone (MEK) solution of Me2dazdt·2I2 for 168 h under reflux. The Pd-containing solution, separated from the remaining ceramic material by means of a glass microfibre thimble in a Soxhlet apparatus, was concentrated and crystallized by the slow diffusion of Et2O to form [Pd(Me2dazdt)2](I3)2 complex in a quantitative way.Palladium photo-deposition on TiO2
In order to investigate the capability of [Pd(Me2dazdt)2](I3)2 to work as a suitable and sustainable precursor for the preparation of photo-catalysts and to evaluate its activity in the photo-reforming of methanol and glycerol, a series of Pd-TiO2 photo-catalysts were prepared by impregnation of metal precursors followed by metal photo-reduction in the presence of methanol as an additional reductant,18 as summarized in Chart 2.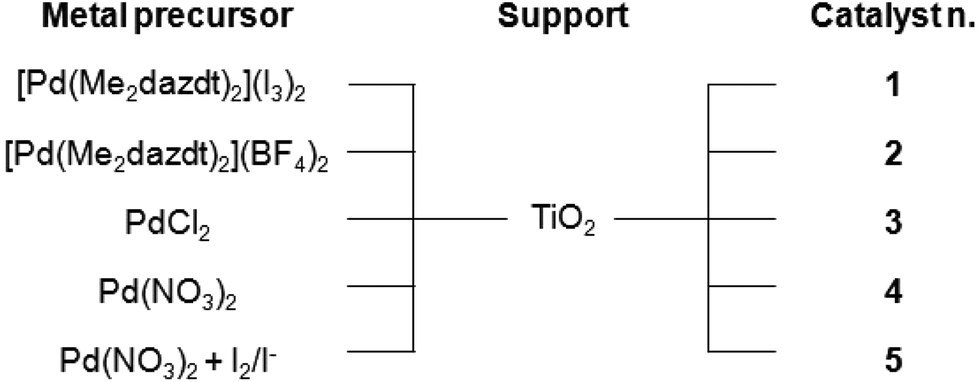 | ||
| Chart 2 Nomenclature of the photo-catalysts used in this work. | ||
The catalysts were prepared by the impregnation of Degussa P25 TiO2 (54 m2 g−1, average pore size distribution of about 50 nm, and total pore volume 0.23 ml g−1) with a proper amount of the selected metal precursor solution to obtain a 0.5% w/w Pd-loading, followed by irradiation in the UV range of the suspension obtained by dispersion of the dry material in a 1:1 methanol/water solution in Ar flow (see details in the Experimental section). After metal deposition, only marginal decreases of surface area, average pore size distribution and total pore volume were measured consistently under the mild reaction conditions employed and with the low metal loadings. Fig. 1 shows the hydrogen evolution during the Pd photo-reduction process on varying the metal precursor. During this step, the concomitant presence of UV electrons and methanol as reducing agents leads to a progressive formation of Pd-metal nanoparticles onto the TiO2 surface, which usually takes place in the first 30–60 min.19 Consistently, the hydrogen production in the preparation of 4 increases during the metal deposition period (first 30 min), and then remains constant for the following 6 h. However, 3 shows, instead, a transient good initial activity, which we also typically observe in the case of other metal halide precursors. This behaviour is present only in the first photo-catalytic process, suggesting that it may be linked to the beneficial but transient presence of some halides associated with palladium. The photo-catalytic production of hydrogen, with the formation of palladium hydrides, favours an efficient removal of the halides (chlorides in this case) leading to a reactivity similar to that of 4. This final performance is consistent with the use of an identical support (TiO2) and comparable metal loading. In fact, it is known that chlorination enhances both UV and visible photo-activity, possibly by involving chlorinated radicals in the photo-reaction. This means that the reactivity depends on the effectiveness of the surface Cl incorporation.20
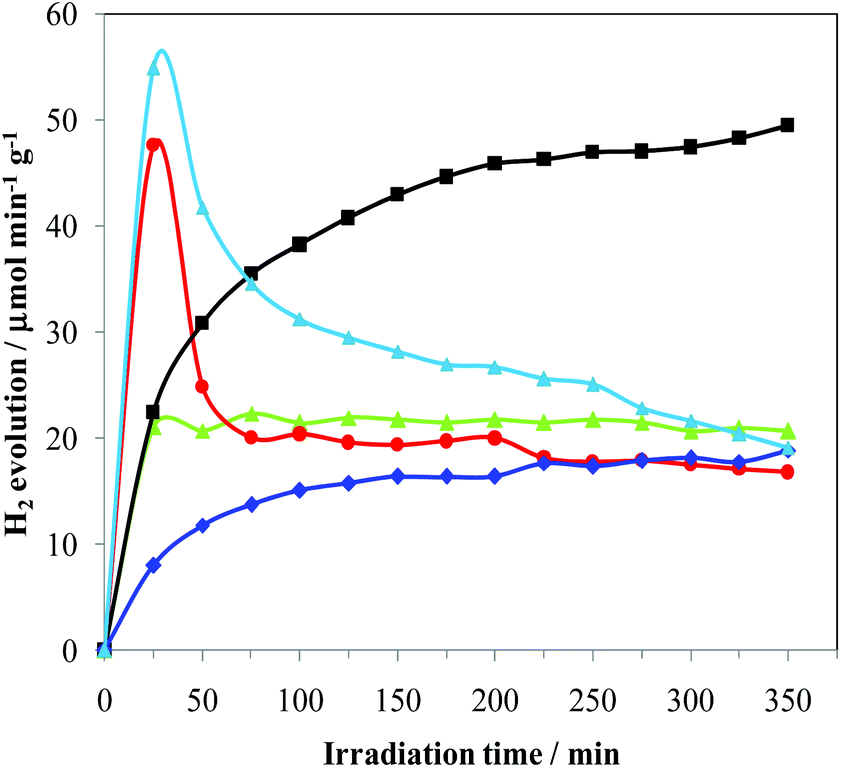 | ||
Fig. 1 H2 production rates during Pd photo-deposition in methanol aqueous solutions by irradiation in the UV-vis region. -■- Pd(0.5%)-complex-I3/TiO2 (1);  Pd(0.5%)-complex-BF4/TiO2 (2); Pd(0.5%)-complex-BF4/TiO2 (2);  Pd(0.5%)-chloride/TiO2 (3); Pd(0.5%)-chloride/TiO2 (3);  Pd(0.5%)-nitrate/TiO2 (4); Pd(0.5%)-nitrate/TiO2 (4);  Pd(0.5%)-nitrate-I2/I−/TiO2 (5). Pd(0.5%)-nitrate-I2/I−/TiO2 (5). | ||
The performances of 2 were investigated in order to assess the role of I2/I−. In fact, the improved photo-catalytic activity of TiO2 upon sensitization with iodine is documented in the literature21 and is attributed to an extended absorption edge up to around 800 nm, resulting in visible light photo-catalytic activity beyond 600 nm and/or beneficial effects for enhancing e−–h+ lifetime. The hydrogen evolution from 2 slowly increases with time, reaching after 6 h a constant rate similar to that of 3 and only slightly lower than that of 4. By using Pd(NO3)2 and I2/I− solution, producing 5, a transient hydrogen evolution behaviour very similar to that observed in the case of 3 was obtained. This may suggest the formation of similar metal-halogenides in solution. Vice versa, 1 shows a progressive increase of photo-catalytic activity, leading to a H2 production that is 80% higher than that of all the other samples. All these findings clearly indicate that not only is [Pd(Me2dazdt)2](I3)2 suitable for the preparation of an effective photo-catalyst but it is also able to be incorporated into the TiO2 I2/I− and/or other species from the complex as sensitizers or dopants in a more effective manner. Consistently, the diffuse reflectance spectra of 1 and 1-used show an underlying broad band in the UV-Vis region (Fig. S1, ESI†) which may be the result of this incorporation, as observed for N- or I-doped TiO2 samples.22 ICP-AES analysis of the samples confirmed that the real metal loading was closed to the nominal one for all the photo-catalysts (Table 1). This highlights that the deposition process was almost quantitative.
| Sample | Metal loading (wt%) | CO/Pd | Pd Surface area (m2 g−1) | Pd NPsa mean particle size (nm) |
|---|---|---|---|---|
|
a Mean particle size calculated from CO chemisorption assuming a spherical geometry and CO:Pd = 1:1 stoichiometry.
b Not determined. This sample was used only to evaluate the effect of counter ion (I3−) on the reactivity.
|
||||
| 1 | 0.47 ± 0.01 | 0.620 | 1.38 | 1.81 |
| 2 | 0.41 ± 0.03 | n.d.b | n.d.b | n.d.b |
| 3 | 0.38 ± 0.01 | 0.875 | 1.72 | 1.28 |
| 4 | 0.50 ± 0.04 | 0.583 | 1.30 | 1.92 |
CO chemisorption experiments performed on 1, 3 and 4 show high metal dispersion, from which it was possible to estimate an average metal particle size of about 1.3 nm for 3 and slightly higher for 1 and 4 (Table 1).
Photo-catalytic activity
Catalysts 1–4 were then applied to the glycerol photo-reforming process through the same procedure followed in the photo-deposition process. Fig. 2 shows the hydrogen evolution from a 1 M aqueous solution of glycerol using UV-Vis light irradiation over each Pd/TiO2 photo-catalyst as a function of reaction time. All the Pd/TiO2 photo-catalysts showed relatively high hydrogen formation rates. Photo-activation of the relatively more complex glycerol molecule with respect to commonly used sacrificial alcohols, such as methanol and ethanol, is difficult and leads to lower H2 yields.23 Notably, under the same experimental conditions, the reference bare TiO2 Degussa P25 is practically inactive. As shown, significant differences in the photo-catalytic H2 production depending on the used metal precursor were observed.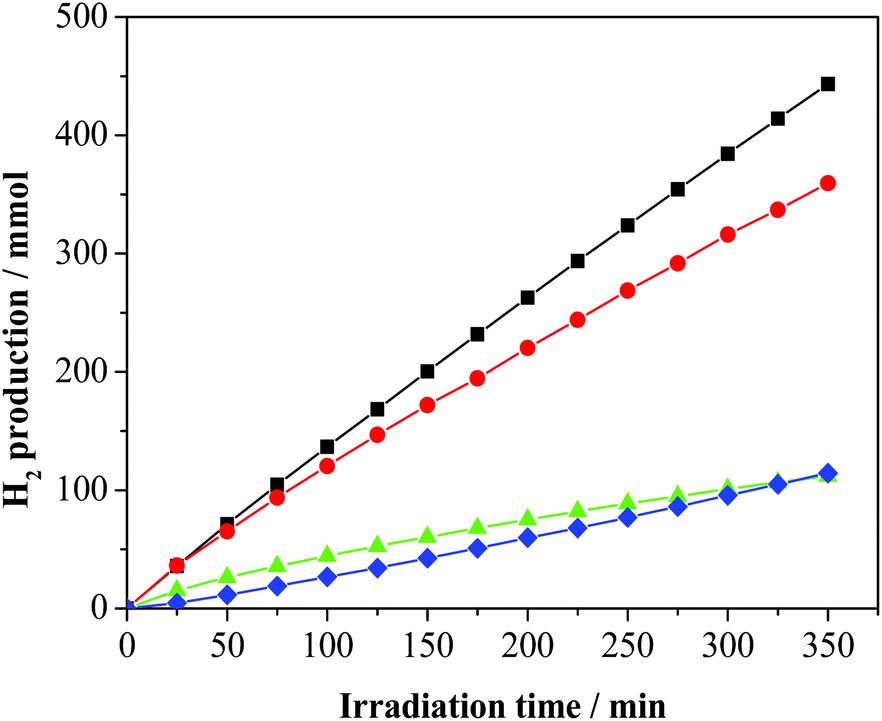 | ||
Fig. 2 H2 production on photo-deposited Pd/TiO2 nanocomposites from glycerol aqueous solutions by irradiation in the UV-vis region: -■- Pd(0.5%)-complex-I3/TiO2 (1);  Pd(0.5%)-complex-BF4/TiO2 (2); Pd(0.5%)-complex-BF4/TiO2 (2);  Pd(0.5%)-chloride/TiO2 (3); Pd(0.5%)-chloride/TiO2 (3);  Pd(0.5%)-nitrate/TiO2 (4). Pd(0.5%)-nitrate/TiO2 (4). | ||
In particular, 2 and 4 are significantly less active than 3 and 1. For all the studied photo-catalysts, the analyses of the gaseous products indicate the formation of H2 and CO2 only. In addition, 1,3-dihydroxypropanone and hydroxyacetaldehyde were detected in the liquid phase as the main by-products of the photo-oxidation of glycerol, as previously observed.17 A small amount of acetaldehyde, 1,2-ethanediol and 1,3-propandiol as well as traces of formic acid, acetic acid and 2,3-dihydroxypropanal (see Fig. S2, ESI†) were detected. These latter compounds derive from subsequent dehydrogenation occurring on the main products.19 The photo-activity of the present metalized TiO2 samples is related to the generation and utilization of photo-carriers (electrons/holes). To reach a high photo-catalytic activity, the Pd-TiO2 photo-catalyst must have a high light absorption capacity and/or a low electron/hole recombination.
Using the same TiO2 semiconductor as the metal support for all the samples, it is reasonable to consider that most of the differences observed in the samples are related to the palladium dispersion, metal crystallinity or surface morphology and/or to the presence of residuals of the synthesis (chlorides, iodine, sulphur, etc.) either as sensitizers or poisons. Therefore, in order to understand the reason for the improved photo-catalytic performances of the material obtained from [Pd(Me2dazdt)2](I3)2, we carefully investigated its metal dispersion before (sample 1) and after (sample 1-used) use.
It is interesting to note that the presence of hydrogen atoms in the alpha position to the alcoholic function has been indicated as an important pre-requirement for efficient hydrogen evolution, with tertiary alcohols showing worse H2 productivity.24,19b
However, these investigations were performed under very diluted solutions. Vice versa, as in the present case, when the concentration of the sacrificial agent significantly increases, a different reactivity order is reported in the literature,25 due to the establishment of complex adsorption equilibria. In fact, the number of reaction by-products increases significantly with the increase in the sacrificial agent complexity.
The XRD analyses (Fig. S3, ESI†) of 1 and 1-used samples show similar patterns. Peaks related to a rutile/anatase mix are present but there is no evidence of Pd, due to its low loading in the catalyst. The similarity between the pre- and post-catalysis sample patterns indicates that the rutile/anatase ratio and the crystalline grain dimensions are preserved. This confirms that the TiO2 support is stable under the reaction conditions.§ Conventional TEM images of samples 1, 3 and 4 were collected in order to detect the presence and size of the Pd nanoparticles. TEM investigation highlighted the presence of Pd nanoparticles with an average size of ∼3 nm on sample 4 (Fig. S4, ESI†), while no TEM evidence of Pd particles was observed on samples 1 and 3. The comparison between 1 and 4 suggests that the higher activity of 1 could be at least partially explained by invoking its higher metal dispersion and reduced nanoparticle size. To detect metal palladium on 1 and to compare it with 3, more sensitive high angle annular dark field (HAADF) STEM measurements were performed. Fig. 3a shows the HAADF STEM imaging and STEM-EDX analysis of 1. The presence of the Ti-phase composition (∼40 nm) was assessed by the chemical analysis carried out on the single nanoparticles and was consistent with the XRD analysis. Pd particles were barely observable due to the low metal loading and likely due to the high dispersion of the metal, which, during the photo-deposition process, forms sub-nanometre particles with the amorphous/paracrystalline structure. On the other hand, HAADF STEM imaging and STEM-EDX characterizations of the 1-used (Fig. 3b) show indeed small (∼3 nm) Pd particles grown over the Ti-particles, as confirmed by the chemical analysis.
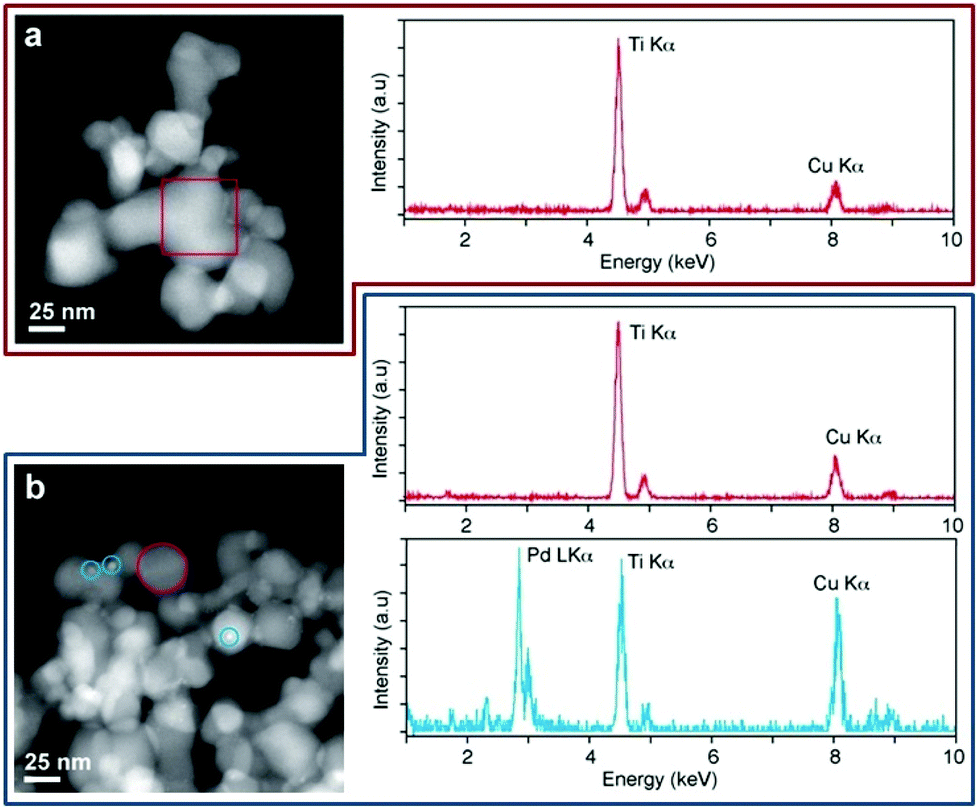 | ||
| Fig. 3 HAADF STEM imaging (left panels) and STEM-EDX analysis (right panels) of 1 and 1-used nanoparticles. (a) 1 cluster showing no Pd signal; (b) 1-used clusters displaying very small Pd nanoparticles, as confirmed by STEM-EDX chemical analysis. Colour code of STEM-EDX: Ti particles in red, Pd particles in light blue. | ||
For comparison, 3 shows no TEM evidences of the presence of Pd particles in the pre- and post-catalysis samples, likely due to higher metal dispersion. As a consequence, the highest activity of 1 may be relatable both to the metal particles sizes and/or the presence of sensitizers or dopants as residues of the complex.
XPS analysis of 1 reveals a complex situation, with the presence of multiple contributions arising from the partial photo-decomposition of the complex. Indeed, as shown in Fig. 4, traces of I, S and N are present in the sample, together with Pd. The Pd:S:N:I atomic ratio is 1:4:2:0.2, as obtained via quantitative analysis of the XPS spectra. The obtained values clearly differ from the stoichiometry of the complex, where a 1:4:4:6 atomic ratio is expected. Also, while the position of the I 3d peaks (with I 3d5/2 peak at 619.3 eV) is consistent with the presence of tri-iodide moieties,26 the S 2p and N 1s spectra are symptomatic of chemical states different from those expected for the [Pd(Me2dazdt)2](I3)2 complex. Indeed, the S 2p profile can be deconvoluted into at least 3 different components, centred at positions typical for thiols (S 2p3/2 at 162.5 eV), disulfides (S 2p3/2 at 164.5 eV) and sulfates (S 2p3/2 at 169 eV), while the N 1s profile can be deconvoluted into two components, centred at 400 and 402 eV and respectively ascribable to N coordinated in an organic ring26 and to an oxidized state of N as NO or NO2 resulting from the decomposition of the molecule chemically adsorbed on the surface of TiO2.27 The data clearly highlight that while a significant fraction of the complex is eliminated during the Pd reduction/deposition process, some species interact with the TiO2 and remain attached to the surface, modifying its properties. Obviously, this process strongly depends on the ageing time, and is influenced by the vicinity of the various species in the complex. As cited above, literature data indicate that the TiO2 photo-catalytic activity may be improved by encapsulated I221a or also by the presence of sulphur and nitrogen as sensitizers or dopants. In catalyst 1 under discussion, it is not so easy to discriminate the different contribution of each component that may furthermore work individually or combined with enhanced activity.
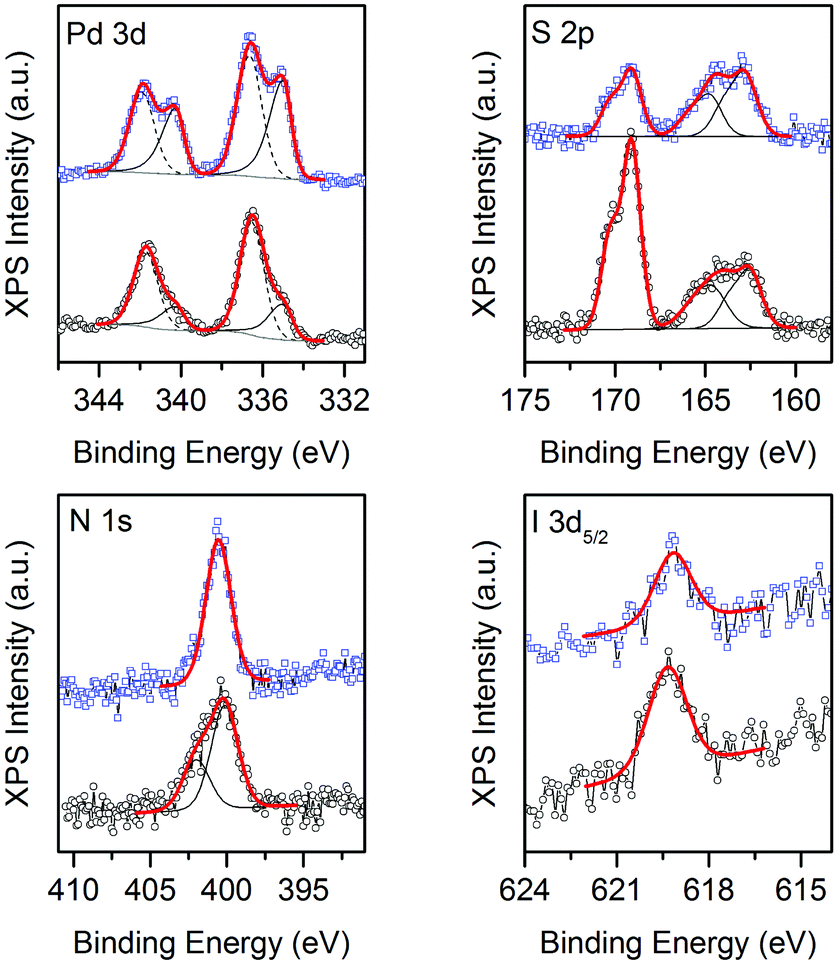 | ||
Fig. 4 XPS of Pd(0.5%)-complex-I3/TiO2 before (○, 1) and after ( , 1-used) H2 photo-reforming from glycerol solution. , 1-used) H2 photo-reforming from glycerol solution. | ||
Concerning the Pd 3d spectrum, it clearly indicates the presence of two different oxidation states. Indeed, the best fitting of the experimental profile was obtained with two components, ascribable to Pd(0) (Pd 3d5/2 centred at 335 eV) and Pd(II) (Pd 3d5/2 centred at 336.5 eV).26 For comparison, the XPS analysis of 1-used reveals a similar scenario, the main differences being a decrease in the intensity of the sulfate component, the disappearance of the oxidized N and an increase in the intensity of the Pd(0) component with respect to the intensity of the Pd(II) one, as shown in Fig. 4.
We estimated moreover the Pd(0) to Pd(II) fraction for all the considered photo-catalysts before and after H2 photo-reforming from glycerol solution (Table 2 and Fig. S5, ESI†).
| Sample | Fresh | Used | ||
|---|---|---|---|---|
| Pd(0) | Pd(II) | Pd(0) | Pd(II) | |
| 1 | 25.0% | 75.0% | 43.8% | 56.2% |
| 2 | 6.7% | 93.3% | 57.5% | 42.5% |
| 3 | 17.5% | 82.5% | 73.3% | 26.7% |
| 4 | 90.3% | 9.7% | 91.8% | 8.2% |
In order to avoid misunderstanding of the data, it is essential to recall the fact that the XPS were recorded ex situ, after exposing the photo-catalysts to air. Consequently, the information cannot be immediately translated into consideration on the status of the photo-catalyst during operational conditions, which are indeed reducing (as indicated by the UV irradiation and the presence of alcohols). With these remarks in mind, the presence of some oxidized Pd species is not surprising and can be related to both an only partial reduction of the metal precursors in the reducing operational conditions and to the exposure of the photo-catalyst to air before XPS analysis. The consequent extent of the partial oxidation/passivation of Pd should be related to the different redox stability of the starting metal precursors and to the final metal particle size, as small Pd nanoparticles are more easily passivated with respect to larger ones. From the data of the fresh samples (Table 2) it appears that palladium nitrate is easily reduced to metal Pd, which eventually is subjected to a minor passivation. Vice versa, the other precursors (both complexes and chloride) appear partially more stable, maintaining a higher fraction of Pd(II). However, as suggested by the TEM and chemisorption studies, the particle sizes of 1 and 3 are significantly smaller than those of 4. This fact suggest that the major contribution of Pd(II) in 1 and 3 could arise from the easy and fast Pd reoxidation of their small nanoparticles by exposition to air before XPS.
Experimental
Preparation of [Pd(Me2dazdt)2](I3)2 and [Pd(Me2dazdt)2](BF4)2
The [Pd(Me2dazdt)2](I3)2 complex was obtained as reported in ref. 9 and led to crystallization through the slow diffusion of Et2O.The [Pd(Me2dazdt)2](BF4)2 complex was obtained as reported in ref. 28.
Preparation of Pd-TiO2-based photo-catalysts for the photo-catalysis experiments
Titanium oxide (TiO2, 97%) was purchased from Degussa, while palladium chloride (P99.9%), palladium nitrate dihydrate (40% Pd basis), acetone (P99.5%), potassium iodide (P99.5%) and iodine (P99.8%) were obtained from Aldrich. All the chemicals were used as received. Water was distilled twice before use.Pd was loaded on to the support by an impregnation/photo-reduction technique. Usually 1 g of TiO2 (P25 Degussa) was impregnated with a solution (7.0 mg mL−1) of Pd complex in acetone for [Pd(Me2dazdt)2](I3)2 and [Pd(Me2dazdt)2](BF4)2 or in water when PdCl2 (solution acidified with an excess of HCl to fully dissolve PdCl2) or Pd(NO3)2 were used as the metal precursors in order to prepare 1–4 palladium photo-catalysts respectively, then stirred for 2 hours in the dark and finally dried in an evaporator. For sample 5, an amount of I2/I− 0.1 M aqueous solution was added in order to obtain the same ratio of Pd/I present in [Pd(Me2dazdt)2](I3)2. The amount of Pd precursor was changed in order to obtain the desired Pd-loading (0.5 wt%). The sample was dispersed in 120 mL of bidistilled water under vigorous stirring and de-aerated with argon flow (50 mL min−1). After stirring for 30 min, the flow of argon was reduced to 15 mL min−1 and maintained during all the reaction time, while methanol was added in order to obtain a 1:1 by volume solution with water. Immediately after methanol addition, the suspension was irradiated for 6 h with a 125 W medium pressure Hg lamp (Helios Italquartz). The photon flux was measured by using a DeltaOHM radiometer HD2302.0. Accordingly to Lianos et al.,29 the approximate average incident radiation power on the photo-catalyst was 129 mW. The samples were finally filtered, washed with acetone and dried. All the samples showed a stable evolution of H2 during the catalysts preparation.
The photo-catalytic activity was evaluated for hydrogen production from aqueous solutions of 1 M glycerol. For the UV-Vis experiments, 500 mg of catalyst were suspended in 240 mL of solution and the suspension was irradiated with the same 125 W medium pressure Hg lamp described above. Ar flow (15 mL min−1) was employed to remove air from the reactor and as a carrier for the reaction products. As the hydrogen production can depend on the sacrificial agent concentration,30 the duration of the long term experiments was optimized to prevent significant alteration of the concentration of the glycerol due to the conversion.
Gaseous products were analyzed by GC analysis using a TCD for the quantification of H2 and a FID, coupled with a methanator, for the quantification of the C-containing compounds (CO, CO2, CH4, etc.).
Characterization of the catalysts
Inductively coupled plasma atomic emission spectroscopy (ICP-AES) measurements were performed on fresh catalysts for palladium quantitative determination. The palladium contents for samples 1–4 are summarized in Table 1. Experimental details on the samples’ digestion and analysis are described in the ESI.†Fresh and used catalysts were characterized by XRD and TEM, as detailed in the ESI.†
X-ray diffraction (XRD) patterns were recorded on a Panalytical Empyrean diffractometer equipped with a graphite monochromator on the diffracted beam and a X'Celerator linear detector. The scans were collected within the range of 10–90° (2θ) using Cu Kα radiation. The average size of the crystallite domains was calculated using the Scherrer equation corrected for instrumental broadening using a standard LaB6 sample.
The materials were characterized by N2 physisorption at liquid nitrogen temperature to obtain the Brunauer–Emmet–Teller (BET) surface area, average pore size distribution (adsorption) and cumulative pore volume using a Micromeritic ASAP2020 after degassing the samples overnight at 350 °C.
CO chemisorption experiments were performed at 35 °C on a Micromeritics ASAP 2020. Typically, 0.2 g of dried samples after photo-reduction of the metal precursor were used. Since photo-reduction leads to deposition of metal nanoparticles in the zero-valent state, only a degas at 150 °C for 6 h was applied as a pre-treatment of the samples. The adsorbed volumes were determined by extrapolation to zero pressure of the linear part of the adsorption isotherm (100–400 torr) after elimination of the so-called reversible adsorption by subtracting the contribution of the bare support.
Conventional TEM images of 1, 3 and 4 samples were recorded on a Hitachi H-7000 Microscope, operating at 125 kV, equipped with a W thermionic electron source. Prior to observation, the samples were deposited on a carbon-coated copper grid.
Crystal structure determination and chemical analysis of the selected 1, 1-used and 3 samples were analyzed using a JEOL JEM 2200-FS transmission electron microscope (TEM) operating at 200 kV, equipped with a Schottky field emission electron source and a CEOS spherical aberration corrector of the objective lens. The same microscope was used in Scanning TEM (STEM) mode and high angle annular dark field (HAADF) geometry, providing an electron probe of 0.7 nm to perform energy dispersive spectroscopy (EDS) analysis. The latter was performed using a Bruker Quantax 400 XFlash 6 T silicon drift detector (SDD) with an area of 60 mm2. The copper X-ray peaks found in the EDS spectra were the unavoidable but very well-known consequence of the use of the copper TEM grid.
X-ray photoelectron spectroscopy (XPS) measurements were performed on a Kratos Axis Ultra DLD spectrometer, equipped with a monochromatic Al Kα source operated at 15 kV and 20 mA. High resolution narrow scans were acquired at a constant pass energy of 10 eV and steps of 0.1 eV. The photoelectrons were detected at a take-off angle of 0° with respect to the surface normal. The pressure in the analysis chamber was kept below 7 × 10−9 torr during data acquisition. The binding energy scale was internally referenced to the C 1s peak at 284.8 eV. Data were converted to the VAMAS format and processed using CasaXPS 2.3.16 software. Data fitting was performed using Shirley-type background and Gauss-Lorentz profiles. For the fitting of S 2p doublets, a spin orbit splitting of 1.2 eV and a branching ratio of 1/2 were assumed. Similarly, for the Pd 3d doublets, a spin orbit splitting of 5.2 eV and a branching ratio of 2/3 were used. For convenience, the position of S 2p and Pd 3d doublets were identified by the position of the S 2p3/2 and Pd 3d5/2 components, respectively.
Conclusions
In this study, the capability of [Pd(Me2dazdt)2](I3)2, the leaching product of a sustainable Pd-recovering method from TWCs, to act as a suitable metal precursor for a photo-reforming catalyst was demonstrated. The prepared photo-catalyst showed an 80% enhanced activity in H2-production from methanol and glycerol with respect to other catalysts prepared starting from commercially available Pd-salts. This amazing application of [Pd(Me2dazdt)2](I3)2 allowed us to design a virtuous “recovery and re-employment” process based on the green leaching of palladium from TWCs and allowed further valorization of the obtained product in the challenging renewable energy field.Acknowledgements
3R Metals Ltd, Sardegna Ricerche (Project POR Sardegna 2000-06 Mis. 3.13 Az. Creazione Imprese Innovative), University of Cagliari and Trieste (Project FRA2013), MIUR – Italy (project HI-PHUTURE protocol 2010N3T9M4), Regione Sardegna (Project P.O. FSE 2007-2013 Asse IV Capitale umano – Linea di attività I.3.1, 2011) and FBS (Project “Metodi di recupero eco-compatibili di metalli nobili da rifiuti Hi-Tech: valorizzazione e trasferimento tecnologico, 2013”) are greatly acknowledged for financing the research activity on this topic. The authors acknowledge Filippo Drago of Nanochemistry Department, IIT and Americo Rigoldi of Dipartimento di Scienze Chimiche e Geologiche, University of Cagliari, for their support in the ICP-AES analysis.Notes and references
- Report on Critical Raw Materials for the EU, May 2014, http://ec.europa.eu/DocsRoom/documents/10010/attachments/1/translations/en/renditions/native.
- J. Kaspar, P. Fornasiero and N. Hickey, Catal. Today, 2003, 77, 419 CrossRef CAS.
- http://www.platinum.matthey.com/services/market-research/may-2015 .
- 2000/53/EC Council Directive.
- M. W. Ojeda, M. D. C. Ruiz, M. E. Godoy and J. B. Rivarola, Trans. Instn. Min. Metall., Sect. C: Mineral Process. Extr. Metall., 1999, 108, C33 CAS , and refs cited therein.
- J.-P. Cuif, US6455018, 2002 Search PubMed.
- P. Yong, N. A. Rowson, J. P. G. Farr, I. R. Harris and L. E. Macaskie, Environ. Technol., 2003, 24(3), 289 CrossRef CAS PubMed.
- (a) A. Serpe, F. Artizzu, M. L. Mercuri, L. Pilia and P. Deplano, Coord. Chem. Rev., 2008, 252, 1200 CrossRef CAS, and references cited therein; (b) A. Serpe, F. Artizzu, L. Marchiò, M. L. Mercuri, L. Pilia and P. Deplano, Cryst. Growth Des., 2011, 11, 1278 CrossRef CAS; (c) A. Serpe, F. Artizzu, D. Espa, A. Rigoldi, M. L. Mercuri and P. Deplano, Green Process. Synth., 2014, 3, 141 CAS; (d) P. Deplano, M. L. Mercuri, L. Pilia, A. Serpe and M. Vanzi, EP1964936, 2005 Search PubMed.
- (a) A. Serpe, F. Bigoli, M. C. Cabras, P. Deplano, P. Fornasiero, M. Graziani, M. L. Mercuri, T. Montini, L. Pilia and E. F. Trogu, Chem. Commun., 2005, 1040 RSC; (b) P. Deplano, P. Fornasiero, M. Graziani, M. L. Mercuri, A. Serpe and E. F. Trogu, EP1743044B1, 2008 Search PubMed.
- O. Carp, C. L. Huisman and A. Reller, Prog. Solid State Chem., 2004, 32, 33 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503 CrossRef CAS PubMed.
- D. Barreca, G. Carraro, V. Gombac, A. Gasparotto, C. Maccato, P. Fornasiero and E. Tondello, Adv. Funct. Mater., 2011, 21, 2611 CrossRef CAS; M. Kitano, K. Tsujimaru and M. Anpo, Top. Catal., 2008, 49, 4 CrossRef.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- L. S. Al-Mazroai, M. Bowker, P. Davies, A. Dickinson, J. Greaves, D. James and L. Millard, Catal. Today, 2007, 122, 46 CrossRef CAS; M. Bowker, D. James, P. Stone, R. Bennett, N. Perkins, L. Millard, J. Greaves and A. Dickinson, J. Catal., 2003, 217, 427 CrossRef; M. Bowker, Green Chem., 2011, 13, 2235 RSC.
- R. A. Sheldon, Green Chem., 2014, 16, 950 RSC.
- M. Gupta and N. Kumar, Renewable Sustainable Energy Rev., 2012, 16, 4551 CrossRef CAS.
- C. H. Zhou, J. N. Beltramini, Y. X. Fan and G. Q. Lu, Chem. Soc. Rev., 2008, 37, 527 RSC.
- (a) M. I. Litter, Appl. Catal., B, 1999, 23, 89 CrossRef CAS; (b) T. Montini, V. Gombac, L. Sordelli, J. J. Delgado, X. Chen, G. Adami and P. Fornasiero, ChemCatChem, 2011, 3, 574 CrossRef CAS; (c) S. Meyer, S. Saborowski and B. Schäferl, ChemPhysChem, 2006, 7, 572 CrossRef CAS PubMed.
- (a) T. Montini, V. Gombac, L. Sordelli, J. J. Delgado, X. Chen, G. Adami and P. Fornasiero, ChemCatChem, 2011, 3, 574 CrossRef CAS; (b) I. Rossetti, ISRN Chem. Eng., 2012, 2012, 1 CrossRef; (c) N. H. Tran and G. S. K. Kannangara, Chem. Soc. Rev., 2013, 42, 9454 RSC.
- R. Yuan, T. Chen, E. Fei, J. Lin, Z. Ding, J. Long, Z. Zhang, X. Fu, P. Liu, L. Wu and X. Wang, ACS Catal., 2011, 1, 200 CrossRef CAS.
- (a) S. Usseglio, A. Damin, D. Scarano, S. Bordiga, A. Zecchina and C. Lamberti, J. Am. Chem. Soc., 2007, 129, 2822 CrossRef CAS PubMed; (b) X. Hong, Z. Wang, W. Cai, F. Lu, J. Zhang, Y. Yang, N. Ma and Y. Liu, Chem. Mater., 2005, 17, 1548 CrossRef CAS; (c) S. Tojo, T. Tachikawa, M. Fujitsuka and T. Majima, J. Phys. Chem. C, 2008, 112, 14948 CrossRef CAS; (d) G. Liu, C. Sun, X. Yan, L. Cheng, Z. Chen, X. Wang, L. Wang, S. C. Smith, G. Q. Lu and H.-M. Cheng, J. Mater. Chem., 2009, 19, 2822 RSC.
- L. G. Devi and R. Kavitha, Appl. Catal., B, 2013, 140–141, 559 CrossRef CAS.
- M. Cargnello, A. Gasparotto, V. Gombac, T. Montini, D. Barreca and P. Fornasiero, Eur. J. Inorg. Chem., 2011, 28, 4309 CrossRef.
- H. Bahruji, M. Bowker, P. R. Davies and F. Pedrono, Appl. Catal., B, 2011, 107, 205 CrossRef CAS.
- See for instance: C. R. López, E. Pulido Melián, J. A. Ortega Méndez, D. E. Santiago, J. M. Doña Rodríguez and O. González Díaz, J. Photochem. Photobiol., A, 2015, 312, 45 CrossRef.
- NIST X-ray Photoelectron Spectroscopy Database, Version 4.1, National Institute of Standards and Technology, Gaithersburg, 2012, http://srdata.nist.gov/xps/ Search PubMed.
- M. Sathish, B. Viswanathan and R. P. Viswanath, Appl. Catal., B, 2007, 74, 307 CrossRef CAS.
- F. Bigoli, P. Deplano, M. L. Mercuri, M. A. Pellinghelli, L. Pilia, G. Pintus, A. Serpe and E. F. Trogu, Inorg. Chem., 2002, 41, 5241 CrossRef CAS PubMed.
- N. Strataki, V. Bekiari, D. I. Kondarides and P. Lianos, Appl. Catal., B, 2007, 77, 184 CrossRef CAS.
- D. I. Kondarides, A. Patsoura and X. E. Verykios, J. Adv. Oxid. Technol., 2010, 13, 116 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: ICP-AES measurements, XRD patterns and DR-spectra of samples 1 and 1-used, GC-MS analysis of liquid phase recovered from glycerol photo-reforming, TEM of sample 4, XPS spectra of samples 2–4. See DOI: 10.1039/c5gc02908b |
| ‡ Pd, Pt and Rh were in a tunable molar ratio: 2:4:1 in 1997, 28:1:1 in 2000. The NMs content of catalysts depends closely from the market price of these metals. In the last years, an increased use of Pt in car catalysts has been observed, due to the jump of the Pd price in 2001. |
| § Similar results have been obtained for samples 3 and 4. |
| This journal is © The Royal Society of Chemistry 2016 |