
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sustainable synthesis of enantiopure fluorolactam derivatives by a selective direct fluorination – amidase strategy†
Nicky J.
Willis
a,
Craig A.
Fisher
b,
Catherine M.
Alder
a,
Antal
Harsanyi
b,
Lena
Shukla
*a,
Joseph P.
Adams
a and
Graham
Sandford
*b
aGlaxoSmithKline R&D Ltd, Medicines Research Centre, Gunnels Wood Road, Stevenage, Hertfordshire SG1 2NY, UK. E-mail: Lena.2.Shukla@gsk.com
bChemistry Department, Durham University, South Road, Durham DH1 3LE, UK. E-mail: Graham.Sandford@durham.ac.uk
First published on 13th October 2015
Abstract
Pharmaceutically important chiral fluorolactam derivatives bearing a fluorine atom at a stereogenic centre were synthesized by a route involving copper catalyzed selective direct fluorination using fluorine gas for the construction of the key C–F bond and a biochemical amidase process for the crucial asymmetric cyclisation stage. A comparison of process green metrics with reported palladium catalyzed enantioselective fluorination methodology shows the fluorination-amidase route to be very efficient and more suitable for scale-up.
Introduction
Enzyme catalysed reaction of functional fluoromalonate building blocks, prepared using fluorine gas, has been used for the first time for the enantioselective synthesis of a pharmaceutically important chiral fluorolactam derivative. An inexpensive, highly economically competitive, lower waste stream process that does not rely on precious metal catalysis and has been quantified by green metric analysis is described.The synthesis of chemical intermediates bearing a fluorine atom at a stereogenic centre is becoming increasingly important for applications across the materials and life-science sectors.1 While fluoroaromatic derivatives appear as sub-units in many commercially valuable pharmaceutical products,2 there are far fewer fluorinated drugs on the market where a single fluorine atom is attached to an sp3 carbon, apart from several anti-inflammatory fluorosteroid derivatives.3 One reason for the relative lack of commercial products that bear fluorine at a stereogenic centre is the often very difficult synthesis, but much progress in the field of enantioselective chemical fluorination has been made in recent years.4 Fluorination of positions α to a carbonyl group by an electrophilic fluorination process is a common approach to the synthesis of enantiopure fluorinated systems and various Selectfluor™-cinchona alkaloid combinations,5 palladium or zinc catalysed processes using N-fluorobenzenesulfonamide (NFSI),6 organocatalyst-fluorinating agent combinations7 and chiral fluorinating agents based upon Selectfluor™-type derivatives8 have been devised and successfully implemented to give a range of enantiopure fluorinated building blocks (Scheme 1). Whilst these chemical approaches can be very valuable at the discovery stage of a medicinal chemistry process, the application of chemical enantioselective fluorination strategies at larger scale is severely hampered by the usually prohibitive expense of the reagent-ligand combinations and the large waste streams generated.
 | ||
| Scheme 1 Examples of reagent combinations used for the synthesis of enantiopure systems with fluorine located at a stereogenic centre. | ||
Pharmaceutical companies are increasingly concerned about the environmental impact of their commercial products and, for example, GSK recently announced an environmental strategy with the objective that the company's operations become carbon neutral by 2050.9a Additionally, the European Federation of Pharmaceutical Industries and Associations (EFPIA) continues to develop the Eco-Pharmaco-Stewardship (EPS) proposal to develop methods to minimise the effect of pharmaceuticals within the environment including in the development and manufacturing stages.9b
Consequently, highly efficient low-waste synthetic processes for pharmaceutical manufacture are required to meet the industry's ambitious environmental goals. Therefore, methods for assessing the efficiency and amount of waste generated by a synthetic strategy are used, in part, to identify a suitable final process for pharmaceutical manufacture. Green metrics packages allow a holistic comparison between potential synthetic reaction pathways using a mixture of quantitative and qualitiative assessment criteria.10 Calculations of total process mass intensity (PMI) enables the synthetic chemist to simply compare the environmental effect of competing synthetic strategies from common starting materials, thus aiding the selection of the final preparative route.11
A series of pre-clinical candidate spleen tyrosine kinase (Syk) inhibitors121 have been synthesised from chiral fluorolactam building blocks 2 (Scheme 2). General synthetic procedures for the preparation of enantiopure 2-fluoro-1,3-amidoesters are relatively rare13 and are limited to enantioselective fluorination of malonate esters using NFSI with Zn(OAc)2/DBFOX-Ph catalyst followed by amide formation,6c ligand catalysed chiral alkylation of fluoromalonate derivatives followed by amide formation14 or fluorination using NFSI with chiral palladium catalysis.12,15
 | ||
| Scheme 2 Retrosynthetic approach to Syk inhibitors 1.12 | ||
The NFSI–palladium catalysis protocol reported by Sodeoka15 was adopted for scale up and 2a was synthesised on 100 g scale12 (Scheme 3). However, the route12a,15 requires a structurally complex palladium catalyst prepared by multi-step procedures and purification of the desired enantiomer by time-consuming chiral HPLC due to the relatively low 44% ee obtained for the fluorination stage when performed on the large scale.
 | ||
| Scheme 3 Process mass intensity (PMI), mass intensity (MI), atom economy (AE) and reaction mass efficiency (RME) calculations for the literature synthesis of 2a. | ||
Results and discussion
Our assessment of the reported synthesis of 2 (Scheme 3) using green metric analysis (SI-2†), shows that the single-step enantioselective fluorination reaction has an estimated calculated process mass intensity (PMI) value of 925 (SI-2†). Inspection of each stage of the synthetic route shows that most waste is generated in the key enantioselective fluorination stage because, of course, NFSI is synthesised by reaction of the corresponding sulfonamide with fluorine gas,16 which must be taken into account when calculating PMI measurements, and loss of material due to the low ee and subsequent resolution. We assumed that all solvent used in the HPLC resolution was recycled and the waste generated in the multi-step synthesis of the palladium catalyst was not included in the PMI calculation. Consequently, the PMI 925 is a low estimate and offers a reasonable benchmark for process development.As an alternative synthetic strategy, initially we investigated the synthesis of related fluorolactam derivative 2b (R = Me) using a combined chemical and biochemical synthetic approach from fluoromalonate ester starting materials (Scheme 2). While enzyme catalysed asymmetric hydrolysis of various fluoromalonate derivatives have been developed,17 no asymmetric amidase reactions of fluoromalonate derivatives have been reported.
Fluoromalonate ester 4a is synthesised in the high yield direct fluorination reaction of dimethyl malonate ester using fluorine gas catalysed by copper nitrate in acetonitrile solution.18a Recently, we described the optimisation of this process which is routinely carried out on the 50 g scale and assessed to have a mass intensity MI = 9 (Scheme 4).18b
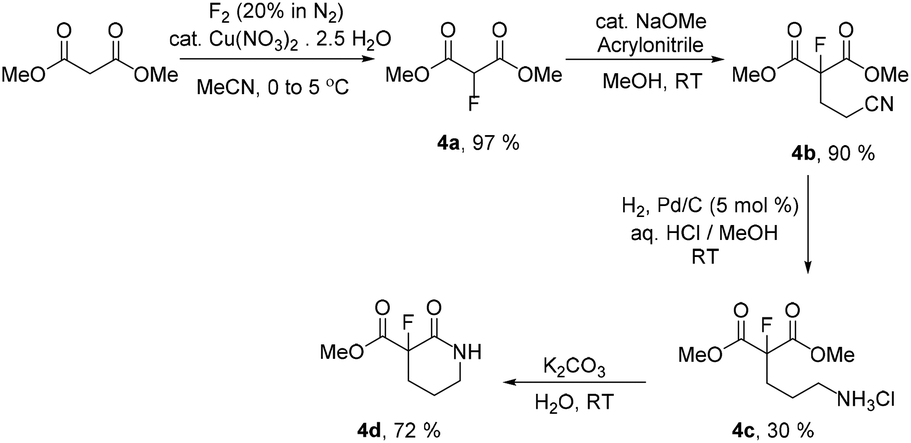 | ||
| Scheme 4 Initial unoptimised synthesis of racemic 4a–d. | ||
Initial unoptimised synthesis of a range of racemic monofluorinated functional precursors 4 for subsequent enzymatic transformation reactions were carried out. Michael addition of acrylonitrile19 to fluoromalonate 4a gave the desired nitrile 4b in 90% yield and subsequent reduction of the nitrile group of 4b by hydrogen over palladium enabled the isolation of salt 4c. Base catalysed ring closure gave racemic fluorolactam 4d (Scheme 4, SI-1.2†). With products 4b–d in hand we began attempts to resolve each fluorinated intermediate by appropriate enzymatic methods to identify the most effective synthetic sequence for the large scale synthesis of the desired enantiopure chiral fluorolactam 2b.
Initially, hydrolase catalysed resolution of 4b was attempted adapting literature protocols.20 However, nitrile 4b was unstable in mildly basic aqueous media (pH 7.0–7.1) and so this approach was discounted as a viable starting material for desymmetrisation (SI-1.3†).
Attempted hydrolase promoted amidation in anhydrous tertiary amyl alcohol as the solvent21 gave only racemic product 4d from salt 4c using various enzyme catalysts (SI-1.4†). After determining that 4d does not hydrolyse in aqueous phosphate buffer (pH 7.3) to the corresponding acid at 20–25 °C over 16 hours (SI-1.4†), enzymatic transformations of 4c were explored in this aqueous buffered medium and, indeed, 4d could be resolved by a variety of hydrolases. Following an initial screening process of 56 enzymes (SI-1.5†), 25 promising hydrolases that afforded 10–60% hydrolysis of 4d in 8 hours were analysed further (SI-1.5†). A number of highly enantioselective processes were observed (Table 1) giving both acids 5a,b by hydrolysis (SI-1.6†) and the corresponding esters 2b,c as reaction products. Both 2b and 2c were isolated by preparative scale HPLC (SI-3†) and their structures and absolute stereochemistries confirmed by X-ray crystallography (Fig. 1, SI-4†).
 | ||
| Fig. 1 Molecular structures of (S)-2b (above) and (R)-2c (below). | ||

CAL-B 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 is a recombinant Candida Antartica Lipase B that is commercially available from Fermase and used to catalyse a range of biotransformations on the large scale.22 Since inexpensive CAL-B 10000 affords the desired fluorolactam (S)-2b (entry 4, Table 1), and is available for purchase on the multi-kilogram and tonne scale, this hydrolase was selected for further reaction optimisation. The possibility of telescoping the formation and resolution of 2b from salt 4c was explored to reduce the work-up process. Initially when 4c was added to buffer solution at room temperature to form a 25 mM solution, we observed that the pH reduced from 7.3 to 6.7 after 15 minutes and that no side-reactions or degradation could be detected. However, when the pH of the solution was readjusted to 7.3 by addition of 2 N NaOH (0.92 equiv.), 19F NMR spectroscopic and chiral HPLC (SI-3†) analysis of the crude reaction mixture indicated full conversion to the desired enantiopure lactam 2b in 47% yield and 98% ee (Scheme 5, SI-1.7†).
000 is a recombinant Candida Antartica Lipase B that is commercially available from Fermase and used to catalyse a range of biotransformations on the large scale.22 Since inexpensive CAL-B 10000 affords the desired fluorolactam (S)-2b (entry 4, Table 1), and is available for purchase on the multi-kilogram and tonne scale, this hydrolase was selected for further reaction optimisation. The possibility of telescoping the formation and resolution of 2b from salt 4c was explored to reduce the work-up process. Initially when 4c was added to buffer solution at room temperature to form a 25 mM solution, we observed that the pH reduced from 7.3 to 6.7 after 15 minutes and that no side-reactions or degradation could be detected. However, when the pH of the solution was readjusted to 7.3 by addition of 2 N NaOH (0.92 equiv.), 19F NMR spectroscopic and chiral HPLC (SI-3†) analysis of the crude reaction mixture indicated full conversion to the desired enantiopure lactam 2b in 47% yield and 98% ee (Scheme 5, SI-1.7†).
 | ||
| Scheme 5 Synthesis of fluorolactam 6 from 4c. | ||
The most operationally simple experimental protocol for the transformation of 4c to 2b would be to add 4c in one portion to the reaction mixture and then adjust the pH to 7.3. Unfortunately, at 250 mM concentrations, the solution became too acidic (pH 4.9) and hydrolysis by-products were formed. This issue was, however, resolved by slow addition of the salt and base, such that the pH was maintained between 6.8 and 7.3. Consequently, the desymmetrization reaction could be telescoped very successfully at 257 mM concentration and the desired fluorolactam 2b was separated efficiently by solid phase extraction. CAL-B enzyme was recovered quantitatively and recycled three times without any observed loss of reactivity profile in subsequent cyclisation processes.
With basic operational parameters for the synthesis of enantiopure 2b using inexpensive reagents and solvents in place, we carried out studies to optimise the multistep synthesis in order to assess the green metrics of the chemo-enzymatic process in comparison to the published enantioselective fluorination strategy (Scheme 6).
 | ||
| Scheme 6 Optimised synthesis of 2b. | ||
In order to reduce the solvent use in reaction work-up, the possibility of carrying out the subsequent Michael addition reaction of 4a with acrylonitrile in a one-pot process without any work-up after the fluorination stage was explored. Firstly, the Michael addition reaction between the crude direct fluorination product mixture and acrylonitrile was assessed but no alkylation reaction occurred due to problems associated with the presence of copper nitrate and HF in the reaction mixture. Consequently, reactions in which a short series of environmentally benign bases including DBU, potassium phosphate and 2-methyl pyridine, were added to the crude direct fluorination product mixture were screened. Addition of 0.5 equivalents of potassium phosphate to the reaction mixture allowed the Michael reaction to proceed to full conversion at room temperature. Scale-up of the one-pot process on 100 mmol scale, where the acrylonitrile was added to the crude direct fluorination reaction mixture via syringe pump over 30 minutes, gave 4b in 60% yield after 1.5 hours.
Reduction of the nitrile group of 4b was carried out in a Parr hydrogenator with palladium/carbon in methanol and conc. hydrochloric acid. Upon completion of the hydrogenation, a white precipitate formed upon washing the crude reaction mixture with ethanol which allowed simple filtration of the ammonium hydrochloride salt 4c. After process optimisation, the solvent volume used for the reduction could be reduced significantly, providing 4c in 84% yield after recrystallisation. The telescoped cyclisation and resolution process was carried out on 10 g scale to obtain realistic metrics data, generating 2b in 43% isolated yield, 99% ee from 4c (Scheme 6, SI-1.9–11†).
The three stage, enhanced synthesis of (S)-2b from dimethyl malonate ester gave a calculated PMI = 201, over four times lower than the corresponding enantioselective chemical synthesis strategy used previously.15
Experimental
Optimised synthesis of 2b (Scheme 6)
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 117.88 (s, CN), 92.73 (d, 1JCF 201.0, C–F), 53.86 (s, CH3O), 30.16 (d, 2JCF 21.5, CH2), 11.48 (d, 3JCF 5.5, CH2); m/z (ASAP) 204.1 (100%, [MH]+), 162.1 (25).
O), 95.57 (d, 1JCF 197.4, C–F), 54.10 (s, CH3O), 40.21 (s, CH2NH2), 32.18 (d, 2JCF 21.6, CH2CF), 22.38 (d, 3JCF 3.2, CH2); m/z (ASAP) 208.1 (100%, [M − Cl]+), 191 (14), 176 (8).
:1, pH 7.3) followed by 4c (10.00 g, 41.0 mmol) in small portions using 0.5 M NaOH to buffer the solution to pH 7.3. The solution was filled to 328 mL total volume with further 0.06 M Na2HPO4: 0.06 M KH2PO4 buffer (3:1, pH 7.3) to give a 257 mM solution Fermase immobilised CAL-B 10000 (7.2 g) was added to the reaction mixture which was stirred at 100 rpm at 20 °C for 8 h. The reaction mixture was poured onto a Waters Oasis HLB 12cc 5 g LP extraction cartridge under reduced pressure and water (30 mL) was added to the cartridge such that the acid was fully eluted (ca. 1 mL per min). The washed enzyme was removed and the ester was eluted with 20% formic acid (30 mL). The solution was concentrated under reduced pressure at RT to give a solid, which was recrystallised from acetone (10 mL) to give (S)-methyl 3-fluoro-2-oxopiperidine-3-carboxylate2b (3.15 g, 44%, >99% ee) as white crystals; mp 115–116 °C; [α]D +14.393° (c 1.00, MeCN); ([MH]+, 176.0718. C7H11FNO3 requires: [MH]+, 176.0723); IR (neat, cm−1) 3200, 3074, 2968, 2888, 1760, 1668, 1435; 1H NMR (400 MHz, CDCl3) δ 7.49 (1H, s, NH), 3.85 (3H, s, OCH3), 3.45–3.37 (2H, m, CH2), 2.42–2.20 (2H, m, CH2), 2.04–1.89 (2H, m, CH2); 19F NMR (376 MHz, CDCl3) δ −156.07 (dd, 3JHF 28.2, 3JHF 20.0); 13C NMR (101 MHz, CDCl3) δ 168.68 (d, 2JCF 26.2, NH–CO), 165.13 (d, 2JCF 22.4, CO2), 90.87 (d, 1JCF 190.8, C–F), 53.29 (s, CH3O), 42.22 (s, CH2–NH), 31.25 (d, 2JCF 22.4, CH2–CF), 18.26 (d, 3JCF 2.6, CH2); m/z (ASAP) 176 (100%, [MH]+), 162 (9).
O), 117.88 (s, CN), 92.73 (d, 1JCF 201.0, C–F), 53.86 (s, CH3O), 30.16 (d, 2JCF 21.5, CH2), 11.48 (d, 3JCF 5.5, CH2); m/z (ASAP) 204.1 (100%, [MH]+), 162.1 (25).
O), 95.57 (d, 1JCF 197.4, C–F), 54.10 (s, CH3O), 40.21 (s, CH2NH2), 32.18 (d, 2JCF 21.6, CH2CF), 22.38 (d, 3JCF 3.2, CH2); m/z (ASAP) 208.1 (100%, [M − Cl]+), 191 (14), 176 (8).
:1, pH 7.3) followed by 4c (10.00 g, 41.0 mmol) in small portions using 0.5 M NaOH to buffer the solution to pH 7.3. The solution was filled to 328 mL total volume with further 0.06 M Na2HPO4: 0.06 M KH2PO4 buffer (3:1, pH 7.3) to give a 257 mM solution Fermase immobilised CAL-B 10000 (7.2 g) was added to the reaction mixture which was stirred at 100 rpm at 20 °C for 8 h. The reaction mixture was poured onto a Waters Oasis HLB 12cc 5 g LP extraction cartridge under reduced pressure and water (30 mL) was added to the cartridge such that the acid was fully eluted (ca. 1 mL per min). The washed enzyme was removed and the ester was eluted with 20% formic acid (30 mL). The solution was concentrated under reduced pressure at RT to give a solid, which was recrystallised from acetone (10 mL) to give (S)-methyl 3-fluoro-2-oxopiperidine-3-carboxylate2b (3.15 g, 44%, >99% ee) as white crystals; mp 115–116 °C; [α]D +14.393° (c 1.00, MeCN); ([MH]+, 176.0718. C7H11FNO3 requires: [MH]+, 176.0723); IR (neat, cm−1) 3200, 3074, 2968, 2888, 1760, 1668, 1435; 1H NMR (400 MHz, CDCl3) δ 7.49 (1H, s, NH), 3.85 (3H, s, OCH3), 3.45–3.37 (2H, m, CH2), 2.42–2.20 (2H, m, CH2), 2.04–1.89 (2H, m, CH2); 19F NMR (376 MHz, CDCl3) δ −156.07 (dd, 3JHF 28.2, 3JHF 20.0); 13C NMR (101 MHz, CDCl3) δ 168.68 (d, 2JCF 26.2, NH–CO), 165.13 (d, 2JCF 22.4, CO2), 90.87 (d, 1JCF 190.8, C–F), 53.29 (s, CH3O), 42.22 (s, CH2–NH), 31.25 (d, 2JCF 22.4, CH2–CF), 18.26 (d, 3JCF 2.6, CH2); m/z (ASAP) 176 (100%, [MH]+), 162 (9).
Conclusions
In conclusion, the combined three stage chemo-enzymatic synthesis of enantiopure fluorolactam 2b using fluorine gas for the construction of the C–F bond and amidase CAL-B 10000 for the key desymmetrization step has been optimised on a reasonable scale and is suitable for scale-up. The PMI of the fluorination-amidase route is PMI = 201 compared to PMI = 925 for the enantioselective fluorination literature synthesis. Clearly, the fluorination-amidase route established here has a PMI that is highly competitive with the corresponding chemical synthesis and demonstrates the very effective use of amidase enzymes for larger scale synthesis of challenging pharmaceutically relevant enantiopure fluorinated systems. However, the still relatively high PMI for the three step synthetic process is largely due to the final resolution step which, by definition, leads to the loss of half the product material.
Perhaps of more importance for synthesis on the large scale is that the cost of the overall fluorination-amidase process is several orders of magnitude less expensive than the use of enantioselective fluorination strategies that require the utilisation of relatively expensive N–F electrophilic fluorinating agents prepared from fluorine gas and the use of structurally complex precious metal catalysts. Simple recycling of the enzyme catalyst by filtration, recycling of solvents and a high yielding inexpensive, copper catalysed fluorination step make the strategy very attractive for scale-up.
The use of the fluorine-enzyme multi-step approach complements existing chemical enantioselective fluorination procedures that are more applicable to discovery scale synthesis. Here, we have demonstrated that the development of new fluorinated sub-units within drug structures bearing fluorine at a chiral sp3 centre assessed in the discovery phase by chemical enantioselective fluorination on the small scale can, when required, be scaled up by a combined inexpensive fluorination-enzyme catalysed approach thus extending the chemical space for fluorinated aliphatic units within the structures of drug candidates.
Acknowledgements
The research leading to these results has received funding from the Innovative Medicines Initiative Joint Undertaking project CHEM21 under grant agreement no 115360, resources of which are composed of financial contribution from the European Union's Seventh Framework Programme (FP7/2007–2013) and EFPIA companies’ in kind contribution. We thank Dr D.S. Yufit for X-ray crystallography.Notes and references
- Fluorine at stereogenic centres: (a) Asymmetric fluoroorganic chemistry. Synthesis, applications, and future directions, ed. P.V. Ramachandran, ACS Symposium Series 746, ACS, Washington, DC, 2000 Search PubMed; (b) V. A. Soloshonok, Enantiocontrolled synthesis of fluoro-organic compounds, Wiley, Chichester, 1999 Search PubMed.
- For reviews on fluorinated pharmaceuticals, see: (a) K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (b) Fluorine in Medicinal Chemistry and Chemical Biology, ed. I. Ojima, Wiley-Blackwell, Oxford, 2009 Search PubMed; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 Search PubMed; (d) C. Isanbor and D. O'Hagan, J. Fluorine Chem., 2006, 127, 303–319 CrossRef CAS; (e) K. L. Kirk, J. Fluorine Chem., 2006, 127, 1013–1029 CrossRef CAS; (f) E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS PubMed; (g) B. R. Smith, C. M. Eastman and E. J. Njardarson, J. Med. Chem., 2014, 57, 9764–9773 CrossRef CAS PubMed; (h) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed.
- For a recent review see: J.-P. Begue and D. Bonnet-Delpon, J. Fluorine Chem., 2006, 127, 992–1012 CrossRef CAS.
- For reviews on enantioselective fluorination, see: (a) N. Shibata, T. Ishimaru, S. Nakamura and T. i. Toru, J. Fluorine Chem., 2007, 128, 469–483 CrossRef CAS; (b) C. Bobbio and V. Gouverneur, Org. Biomol. Chem., 2006, 4, 2065–2075 RSC; (c) S. Lectard, Y. Hamashima and M. Sodeoka, Adv. Synth. Catal., 2010, 352, 2708–2732 CrossRef CAS; (d) J. Ma and D. Cahard, Chem. Rev., 2004, 104, 6119–6146 CrossRef CAS PubMed; (e) J. Ma and D. Cahard, Chem. Rev., 2008, 108, PR1–PR43 CrossRef CAS PubMed; (f) S. V. Brunet and D. O'Hagan, Angew. Chem., Int. Ed., 2008, 47, 1179–1182 Search PubMed; (g) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826–870 CrossRef CAS PubMed.
- (a) B. Mohar, J. Baudoux, J.-C. Plaquevent and D. Cahard, Angew. Chem., Int. Ed., 2001, 40, 4214–4216 CrossRef CAS; (b) N. Shibata, E. Suzuki, T. Asahi and M. Shiro, J. Am. Chem. Soc., 2001, 123, 7001–7009 CrossRef CAS PubMed; (c) N. Shibata, E. Suzuki and Y. Takeuchi, J. Am. Chem. Soc., 2000, 122, 10728–10729 CrossRef CAS.
- (a) Y. Hamashima, K. Yagi, H. Takano, L. Tamas and M. Sodeoka, J. Am. Chem. Soc., 2002, 124, 14530–14531 CrossRef CAS PubMed; (b) Y. Hamashima, T. Suzuki, H. Takano, Y. Shimura and M. Sodeoka, J. Am. Chem. Soc., 2005, 127, 10164–10165 CrossRef CAS PubMed; (c) D. S. Reddy, N. Shibata, J. Nagai, S. Nakamura, T. Toru and S. Kanemasa, Angew. Chem., Int. Ed., 2008, 47, 164–168 CrossRef CAS PubMed.
- (a) D. D. Steiner, N. Mase and C. F. Barbas, Angew. Chem., Int. Ed., 2005, 44, 3706–3710 CrossRef CAS PubMed; (b) T. D. Beeson and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 8826–8828 CrossRef CAS PubMed; (c) Y. Huang, A. M. Walji, C. H. Larsen and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 15051–15053 CrossRef CAS PubMed.
- (a) V. Rauniyar, A. D. Lackner, G. L. Hamilton and F. D. Toste, Science, 2011, 334, 1681–1684 CrossRef CAS PubMed; (b) R. J. Phipps, K. Hiramatsu and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 8376–8379 CrossRef CAS PubMed; (c) T. Honjo, R. J. Phipps, V. Rauniyar and F. D. Toste, Angew. Chem., Int. Ed., 2012, 124, 9822–9826 CrossRef; (d) R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 1268–1271 CrossRef CAS PubMed; (e) J. R. Wolstenhulme, J. Rosenqvist, O. Lozano, J. Ilupeju, N. Wurz, K. M. Engle, G. W. Pidgeon, P. R. Moore, G. Sandford and V. Gouverneur, Angew. Chem., Int. Ed., 2013, 52, 9796–9800 CrossRef CAS PubMed.
- (a) See GSK press release: http://www.gsk.com/en-gb/media/press-releases/2011/gsk-publishes-2010-corporate-responsibility-report/; (b) See related press releases on the EFPIA website: http://www.efpia.eu/mediaroom/226/43/Collaboration-is-the-key-to-managing-pharmaceuticals-in-the-environment.
- General green chemistry and process metrics reviews: (a) D. J. C. Constable, A. D. Curzons and V. L. Cunningham, Green Chem., 2002, 4, 521–527 RSC; (b) J. Andraos, Org. Process Res. Dev., 2005, 9, 149–163 CrossRef CAS; (c) J. Augé, Green Chem., 2008, 10, 225–231 RSC; (d) C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef CAS; (e) Green Chemistry in the Pharmaceutical Industry, ed. P. J. Dunn, A. S. Wells and M. T. Williams, Wiley-VCH, Weinheim, 2010 Search PubMed; (f) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- Metrics packages: (a) ACS Tools for Green Chemistry, http://www.acs.org/content/acs/en/greenchemistry/research-innovation/tools-for-green-chemistry.html; (b) C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121 RSC; (c) T. V. T. Phan, C. Gallardo and J. Mane, Green Chem., 2015, 17, 2846–2852 RSC.
- (a) F. L. Atkinson, M. D. Barker, C. Douault, N. S. Garton, J. Liddle, V. K. Patel, A. G. Preston and D. M. Wilson, U.S. Pat. Appl, 40984A1, 2013 Search PubMed; (b) N. R. Curtis, S. Davies, M. Gray, S. G. Leach, R. A. McKie, L. E. Vernon and A. Walkington, Org. Process Res. Dev., 2015, 19, 865–871 CrossRef CAS.
- (a) C. A. Fisher, A. Harsanyi, G. Sandford, D. S. Yufit and J. A. K. Howard, Chimia, 2014, 68, 425–429 CrossRef CAS PubMed; (b) G. P. Jadhav, S. Chhabra, G. Telford, D. S. W. Hooi, K. Righetti, P. Williams, B. Kellam, D. I. Pritchard and P. M. Fischer, J. Med. Chem., 2011, 54, 3348–3359 CrossRef CAS PubMed.
- S. Hong, M. Kim, M. Jung, M. W. Ha, M. Lee, Y. Park, M. Kim, T. Kim, J. Lee and H. Park, Org. Biomol. Chem., 2014, 12, 1510–1517 CAS.
- T. Suzuki, T. Goto, Y. Hamashima and M. Sodeoka, J. Org. Chem., 2007, 72, 246–250 CrossRef CAS PubMed.
- W. J. Wagner, G. H. Shia and A. J. Poss, US Pat. Appl, 5403957, 1992 Search PubMed.
- T. Kitazume and T. Yamazaki, Top. Curr. Chem., 1997, 193, 91–129 CrossRef CAS.
- (a) R. D. Chambers and J. Hutchinson, J. Fluorine Chem., 1998, 92, 45–52 CrossRef CAS; (b) A. Harsanyi and G. Sandford, Green Chem., 2015, 17, 3000–3009 RSC.
- N. F. Albertson and J. L. Fillman, J. Am. Chem. Soc., 1949, 71, 2818–2820 CrossRef CAS.
- S. Banerjee, W. J. Wiggins, J. L. Geoghegan, C. T. Anthony, E. A. Woltering and D. S. Masterson, Org. Biomol. Chem., 2013, 11, 6307–6319 CAS.
- A. L. Gutman, E. Meyer, X. Yue and C. Abell, Tetrahedron Lett., 1992, 33, 3943–3946 CrossRef CAS.
- (a) V. Gotor-Fernandez, E. Busto and V. Gotor, Adv. Synth. Catal., 2006, 348, 797–812 CrossRef CAS; (b) S. van Pelt, R. L. M. Teeuwen, M. H. A. Janssen, R. A. Sheldon, P. J. Dunn, R. M. Howard, R. Kumar, I. Martinez and J. W. Wong, Green Chem., 2011, 13, 1791–1798 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: For experimental details (SI-1), metrics calculations (SI-2), HPLC analysis (SI-3), X-ray crystallography (SI-4) and NMR spectra (SI-5). CCDC 1401917-1401922. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5gc02209f |
| This journal is © The Royal Society of Chemistry 2016 |