
Alkylphenols to phenol and olefins by zeolite catalysis: a pathway to valorize raw and fossilized lignocellulose†
Danny
Verboekend
*,
Yuhe
Liao
,
Wouter
Schutyser
and
Bert F.
Sels
*
Department M2S, K.U. Leuven, Kasteelpark Arenberg 23, 3001 Heverlee, Belgium. E-mail: danny.verboekend@biw.kuleuven.be; bert.sels@biw.kuleuven.be
First published on 17th August 2015
Abstract
Selective conversion of alkylphenols to phenol and olefins is presented as a challenging key step in upgrading raw and fossilized lignocellulose. An exceptional and stable dealkylation performance is achieved by application of an acidic ZSM-5 zeolite, in which co-feeding of water is crucial to maintain catalytic activity. The role of water is attributed to competitive adsorption of water and phenol. The lignin-first pathway towards phenol yields a tenfold improvement of phenol compared to the state-of-the-art single-step lignocellullosic depolymerization techniques.
Introduction
The contemporary large-scale consumption of fossil fuels will eventually lead to the depletion of the world's (cheap and clean) oil reserves. The energy supplies should on the long term be replaced by alternative energy sources as wind, water, and solar. However, as the oil runs out, also the main source of everyday base-chemicals required in the production of polymers, pharmaceuticals, and other specialty chemicals may be lost. Of the different types of base chemicals, aromatics represent an important building block for the chemical industry in the manufacture of plastics, rubbers, and other synthetic fibers.1 Ultimately, these aromatics should be derived from a sustainable source, such as woody biomass. On the middle-long term the shortage of oil may, at least partially, be compensated by the increased valorization of solid fossilized lignocellulosic resources, such as peat, lignite, and predominately coals. Hence, the most likely important future sources of aromatics will be derived from solid lignocellulose in its raw unprocessed form (wood) or its fossilized form (coal).The main constituents of raw lignocellulose are cellulose, hemi-cellulose, and lignin. While today many useful chemicals can be derived from (hemi-)cellulose,2–11 the valorization of lignin is of high relevance as it is the only of the three types that comprises a substantial amount of aromatics (ca. 40 wt% benzene units).12 In addition, lignin is a common by-product of the paper industry and of the second generation bioethanol manufacture. In these processes, the surplus lignin is often simply burnt off for heat and/or electricity recovery.13,14 The extraction of aromatics from lignin therefore represents a challenge of importance for today's and future industry.15–19 Moreover, in order to ensure the economic feasibility of future wood-fueled biorefineries, the valorisation of lignin is prerequisite.17–21
Several pathways to depolymerize lignin have been reported.16–19 A particularly interesting lignin-first technology simultaneously extracts and dissembles protolignin from the lignocellulose matrix.22–26 Of the latter, a highly efficient process was devised that manages to, without exhaustive preparative steps, highly selectively convert protolignin into methoxylated and alkylated phenols and oligomers thereof (conversion B, Table 1, Scheme 1). About 50% of the wood protolignin is for instance converted into phenolic monomers,7 approaching the theoretical maximum. The state of the art also provides the knowledge to demethoxylate such methoxylated (alkyl)phenols, yielding selectively the alkylphenol at high conversions (and methane or methanol, conversion C).28–38
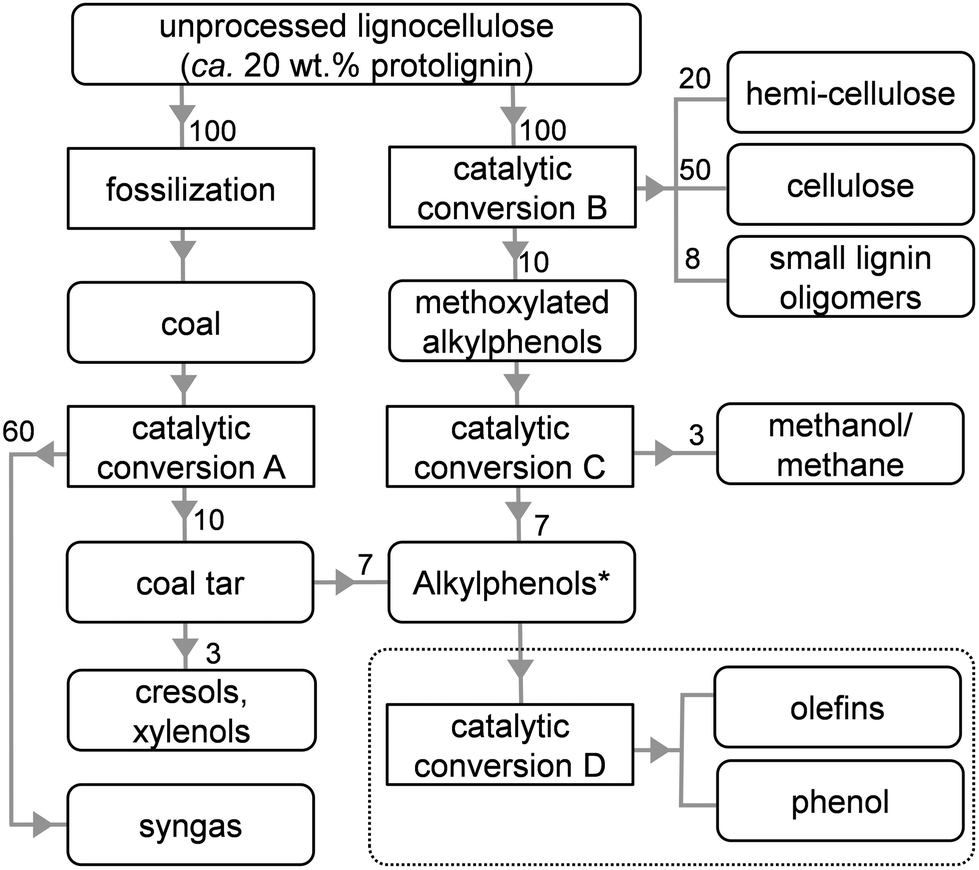 | ||
| Scheme 1 Illustration highlighting the relatively large streams of alkylphenols to be derived from fossilized (coal) or raw (wood) lignocellulose. The values represent streams in wt%. *Cresols and xylenols are not included. The dashed box highlights the desired products in the dealkylation of alkylphenols (catalytic conversion D). The details regarding the conversions A–D are summarized in Table 1. | ||
| Phenol yielda | ||||||
|---|---|---|---|---|---|---|
| Conversion | Reagents | Catalyst/process | Products | Step (wt%) | Overall (wt%) | Source |
| a Maximum phenol yields with respect to phenol present in protolignin. b Close to the expected theoretical maximum yield of birch wood lignin.24,27 | ||||||
| A | Fossilized lignocellulose: coal (+O2 and H2O) | Lurgi phenoraffin | Syngas, coal tar oil (rich in alkylphenols) | — | — | 45–47 |
| B | Raw lignocellulose: birch (+methanol and H2) | e.g. Ru/C, Ni/C | Cellulose, hemicellulose, small lignin oligomers and lignin monomers (methoxylated alkylphenols) | 50b | 50 | 27, 48 |
| C | Methoxylated alkylphenols (+H2) | e.g. NiMo/Al2O3, Pd/C, Ru/TiO2 | Alkylphenols, (methanol/methane not mentioned) | 95 | 48 | 28–38 |
| D | Alkylphenols (+H2O) | H-ZSM-5 | Phenol and olefins | 98 | 47 | This work |
The high selectivity to lignin monomers using the lignin-first strategy stands in great contrast to many other state-of-the-art depolymerization techniques, such as pyrolysis,39,40 base-catalyzed depolymerisation,41,42 and hydrogenolysis.43,44 The latter works yield, besides often substantial unwanted side products as char and light gases, a wide variety of up to 20 or more products, and a phenol yield limited to ca. 5%, with respect to the phenol present in protolignin (Table S1†). Such a broad product slate not only requires dedicated separation efforts, the upgrading of all the products into useful chemicals requires a wide variety of catalytic reforming. In contrast, the lignin-first strategy (Scheme 2) yields essentially three products, facilitating their separation and catalytic upgrading.
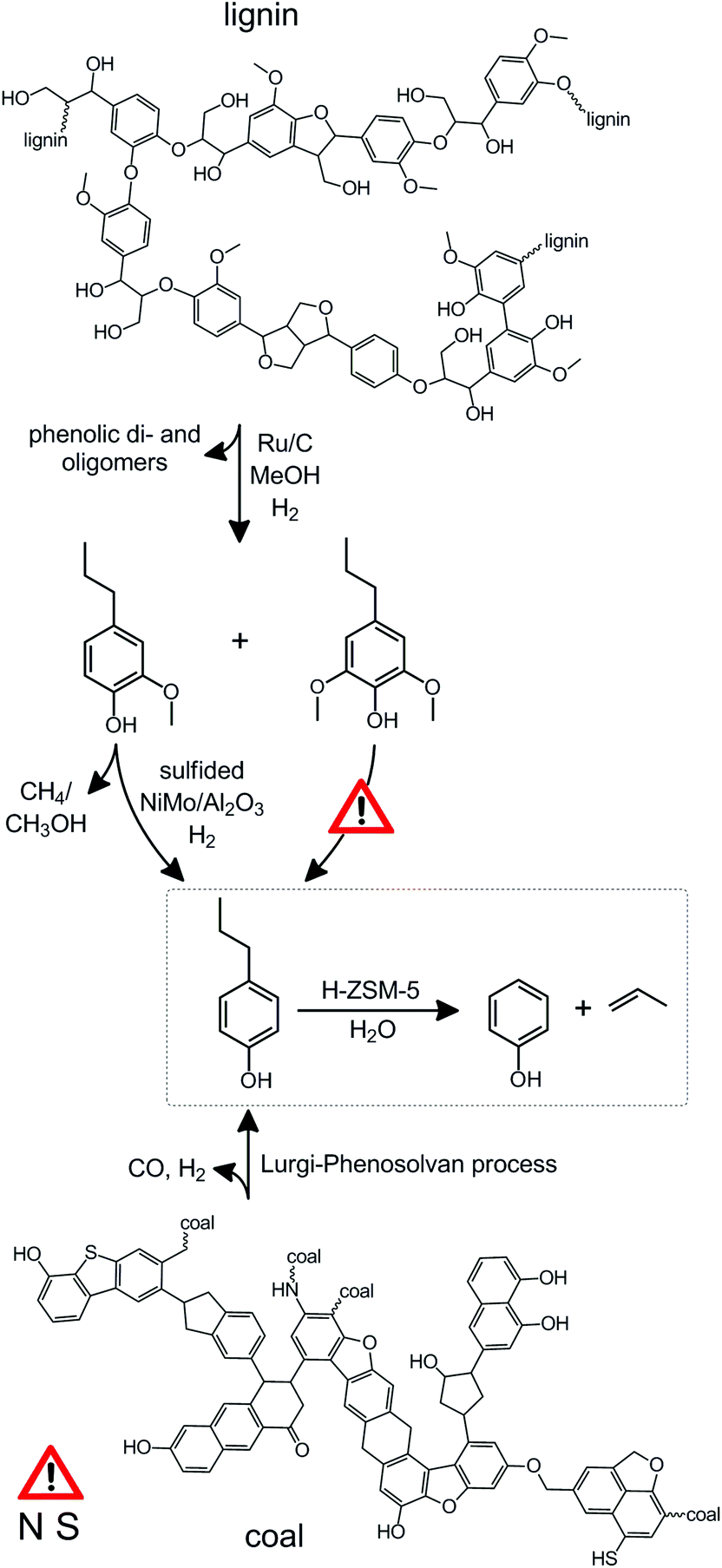 | ||
| Scheme 2 The catalytic upgrading of alkylphenols derived from coal and lignin. The details of the reaction are summarized in Table 1. The structure of lignin and coal were based on ref. 18 and 41, respectively. The dashed box highlights the challenging reaction of interest within this contribution. The overall phenol yield (47%) on lignin basis (steps B to D, Table 1) exceeds the yields of state-of-the-art single step lignin conversion processes which do not typically attain phenolic monomer yields exceeding 5% (Table S1†). The exclamation signs highlight that the selective demethoxylation of propylsyringol remains challenging and that, like is the case for oil, the presence of sulphur and nitrogen could cause catalyst poisoning, which needs to be addressed, for example, by hydrotreating.53 | ||
Alternatively, the technology to isolate aromatics from coal was developed decades ago.45–47 A good example is the Lurgi process, which results primarily into syngas, but also yields a residue (coal tar), which is rich in ethyl and propyl phenols (conversion A, Table 1, Scheme 1).45–48 Hence, using the pathway from either coal or wood, for every ton of (dry) lignocellulosic matter processed, roughly 100 kg of alkylphenols are made available (Scheme 1). Hence, to fuel tomorrow's chemical industry, the valorization of novel intermediates, such as alkylphenols is highly relevant. However, in contrast to the commonly known opposite reaction, viz. the alkylation of phenols,49–52 the knowledge on the dealkylation of alkylphenols is not very well established.
Herein, we stress the need to process alkylated phenols in future wood- or coal-fuelled biorefineries. A highly efficient zeolite-based process that enables to convert (mixtures of) ethyl and propylphenols into high-value products, that is, phenol and the associated olefins, ethylene and propylene (Scheme 2), is pioneered. The developed pathway represents an elegant example of the appeal of harvesting the functionality present within biomass. Combined with the lignin-first depolymerization pathway, the overall process enables to obtain a tenfold higher yield of phenol compared to the state-of the art single-step processes. Whereas large pore zeolites (H-USY) are less selective and deactivate rapidly, a medium pore H-ZSM-5 is established as a superior catalyst, enabling a high selectivity and long catalysts life-time. Additionally, the results open the door to a unique type of zeolite-based catalysis, in which water plays a pivotal role.
Experimental
Materials
The catalyst used were commercial ZSM-5 zeolites (code ‘ZSM5’) of distinct nominal Si/Al ratios (Si/Al = 15: CBV 3024E, Si/Al = 40: CBV 8014, Si/Al = 140: CBV 28014) and a USY zeolite (code ‘USY’, Si/Al = 40: CBV 780), all supplied by Zeolyst. The NH4-ZSM-5 zeolites were calcined in static air (550 °C, 5.5 h, ramp rate 5 °C min−1) to obtain the protonic form. The number following the zeolite code indicates the Si/Al ratio of the sample.Characterization
Thermo-gravimetric analysis (TGA) was performed by using a TGA-Q500 analyzer. The measurements were conducted in air (30 cm3 STP min−1) ramping the temperature from 30 to 800 °C at 10 °C min−1. Carbon content was determined by combustion analysis using a Thermo Finnigan Flash EA 1112. 27Al MAS NMR was performed using Bruker Avance 400 spectrometer (B0 = 9.4 T) on fully hydrated samples, packed into 4 mm zirconia rotors. Scans (18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) were accumulated with a recycle delay of 200 ms. Powder XRD measurements were performed on a STOE Stadi P instrument in transmission mode by using CuKα radiation. Nitrogen-sorption measurements were executed at −196 °C with a Micromeritics TriStar 3000 instrument.
000) were accumulated with a recycle delay of 200 ms. Powder XRD measurements were performed on a STOE Stadi P instrument in transmission mode by using CuKα radiation. Nitrogen-sorption measurements were executed at −196 °C with a Micromeritics TriStar 3000 instrument.
Catalytic testing
Dealkylation experiments were performed in a custom-built plug-flow fixed-bed reactor equipped with 4 parallel quartz reactors (inner diameter of 3 mm). Typically, the quartz reactors were filled with in total 120 mg of catalysts (sieve fraction: 0.125–0.250 mm), yielding a catalyst bed height of ca. 13 mm. The alkylphenols (Sigma-Aldrich, >97%) and water were brought to the gas phase using a nitrogen flow (20 mL min−1) passed through a saturator. Afterwards these flows were mixed, yielding a gas mixture of molar composition: 0.86/0.12/0.02 (N2/H2O/alkylphenol), and passed through the reactors. In the absence of water (only in Fig. 1 and 2) the molar flow composition was 0.98/0/0.02 (N2/H2O/alkylphenol). Effluent gasses were characterized using an in-line gas chromatograph equipped with a HP1 column and a FID detector. The WHSV was varied by reducing the (total) catalyst amount from 120 mg to 7.5 mg, while maintaining the catalysts bed height by diluting the catalyst bed with quartz of the same sieve fraction (0.125–0.25 mm). The dealkylation of a mixture of ethylphenol and propylphenol was achieved using an additional saturator and a molar flow composition of 0.81/0.11/0.06/0.02 (N2/H2O/ethylphenol/propylphenol).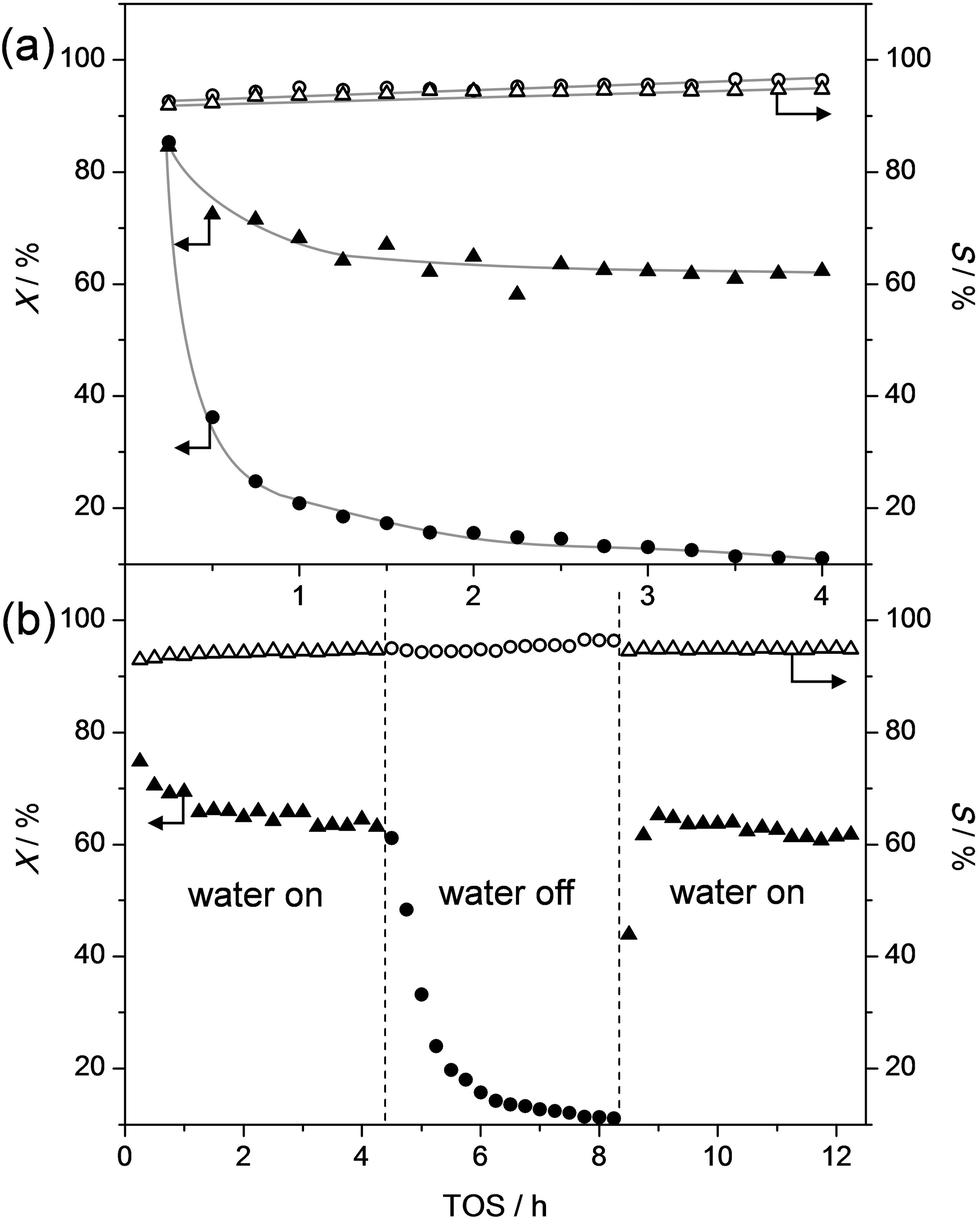 | ||
| Fig. 1 (a) Catalytic conversion (X) of propylphenol and selectivity (S) to phenol and propylene over ZSM5-40 in the presence (triangles) and absence of water (circles). (b) Influence of the temporal removal of water during the dealkylation of propylphenol performed over ZSM5-40. The absence of water was compensated with nitrogen to maintain a constant flow of alkylphenol. T = 305 °C and WHSV = 3.7 h−1. | ||
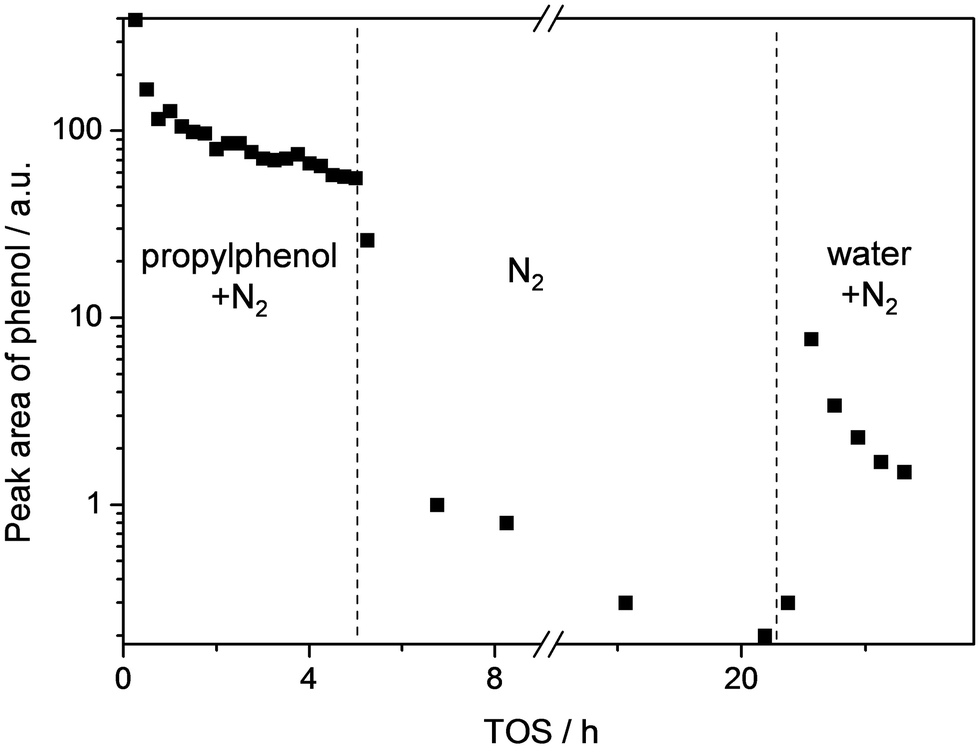 | ||
| Fig. 2 Peak area of phenol as analysed by GC over a ZSM5-40 catalyst as a function of TOS in different conditions (the labels indicate the flows present). The absence of propylphenol or water was compensated with nitrogen to maintain a constant flow. T = 305 °C and (for TOS 0–5 h) WHSV = 3.7 h−1. | ||
Results and discussion
Alkylbenzene versus alkylphenol
Solutions to challenges of future bio-refineries may often be inspired by the currently-available technologies.54 For example, an obvious approach to successfully dealkylate alkylphenols to valuable chemicals may be the evaluation and utilization of the knowledge on similar existing processes in the petrochemical industry. A relevant comparison to the dealkylation of alkylphenols may be the dealkylation of alkylbenzenes. However, phenols behave markedly different compared benzenes,55,56 and parallels of their chemistry are mostly based on the similar carbon skeletons. The dealkylation of alkylbenzenes is not heavily studied as oil-derived aromatics are usually not strongly alkylated,53 and dealkylation is merely performed to valorize side products from petrochemical processes, such as in the catalytic reforming of naphta.57,58 The dealkylation of ethyl and propylbenzenes is most commonly performed in the gas phase using catalysts based on acidic zeolites, like H-ZSM-5.57,58 This reaction was termed shape-selective as a very high selectivity to benzene was obtained. However, the reaction is only selective with a zeolite modified with rare-earth cations, MgO, or most commonly with noble metals, like Pt. In addition, the reaction is performed under a (costly) H2 atmosphere, yielding instead of alkenes, lower-value alkanes in addition to benzene. These adaptions are required as the unmodified H-ZSM-5 originates severe (deactivation by) coking.59–624-n-Propylphenol is the most abundant alkylphenol to be derived from lignin and is also very abundant in tar oil.16,17,48 This molecule was therefore selected as a prime substrate to study within this contribution. Based on the above-described experiences on alkylbenzenes, H-ZSM-5 was first studied (sample ZSM5-40, Fig. 1a). The result shows an initial highly-selective conversion to phenol and propylene. However, within 1 h the conversion was reduced from 80% to ca. 20%. Inspired by patent literature63 and the need for catalysts, which process lignocellulose, to cope with moist streams,64 the influence of co-feeding water was tested. Remarkably, the addition of water to the stream enabled to maintain the catalytic activity. Moreover, the water-free deactivation was completely reversible (Fig. 1b). Importantly, during the addition or removal of water, no influence on the selectivity was observed. This means that the ZSM-5 catalysts yields, unlike in the dealkylation of alkylbenzenes, highly selectively phenol and olefin without the need for noble metals and associated H2 atmosphere or other modifications. This outstanding performance stems from the unique role that water plays.
On the role of water
In general, the role of water in zeolite-catalysed reactions is only partially understood. Though the role of water has been recognized, the underlying fundamentals were not systematically studied. In a variety of reactions, mostly dealing with condensation reactions of alcohols and aldehydes,49–52,56,65–68 the water effect is even completely neglected or only scarcely mentioned. More specifically for phenols, a study on the zeolite-catalyzed alkylation of phenol with propylene evidenced a clear influence of water.49 It enhanced activity by 25%, but the nature of the enhancement remains unclear. Similarly, the liquid-phase condensation of formaldehyde with phenol performed using beta zeolites requires the use of externally added water.65 Other recent works evidenced that the alkylation of phenol proceeds well in water with beta zeolites.50,51 Alternatively, it was demonstrated that in the alkylation of cresol with propanol the zeolite hydrophobicity plays a key role.52 However, in these reactions the exact role of water was not clarified. The latter may be due to that alkylations are, on an academic scale, often executed in batch reactors, which inhibits the study of deactivation phenomena (as can be observed in Fig. 1). The origin of the water effect in zeolite catalysis has been tentatively attributed to a variety of mostly co-catalytic reasons. For example, it was reported that it might shorten the lifetime of carbenium ion intermediates.50 Similarly, the water could actively participate by moderating the zeolite's acidity by the formation of water clusters.66 On another account, effects have been attributed to confinement effects or nest effects caused by water within the zeolite pores.51 Conversely, water could also act as a sweeping agent, forcing formed products to diffuse away from the zeolite,67 and rendering the role of water to be mostly adsorptive, hence physical, rather than chemical. However, clear evidence of the role of water has thus far remained absent.Although further study may be required to unravel the role of water in closer detail, the results in this contribution enable to conclude that the role of the water is mostly sorptive. First of all, should water be required to generate the active site, no activity at all should be observed in the water-free dealkylation. In contrast, Fig. 1a clearly demonstrates a significant activity during the first 2 h TOS in the absence of water. Moreover, the deactivation in the absence of water (Fig. 1b) is fully reversible and should therefore not be related to the presence of excessive coking. To study this in more detail, characterization of the spent catalysts in Fig. 1a was executed. Thermo-gravimetric analysis (TGA) was performed to assess the deposits on the catalysts after reaction. The results (Fig. SI1†) reveal that in the range where coke species may be expected (350–700 °C)69 both catalysts show exactly the same weight loss centered around 500 °C. However, the sample that was tested in the absence of water displayed an additional weight loss around 150 °C, which could very well be due to the presence of (alkyl)phenol. The carbon content of the spent catalysts was quantified using elemental analysis. These results show that the ZSM-5 catalytically evaluated in the presence of water comprised 4.4 wt% of carbon, whereas the one tested the absence of water comprised 6.0 wt% of carbon. Based on the TGA, the difference 1.6 wt% likely constitutes (alkyl)phenol. The presence of easily removable species on the ZSM-5 zeolite in the absence of water was further supported by the carbon content of the sample tested in Fig. 1b. This ZSM5-40 was treated in the absence of water for over 4 h followed by the (re)introduction of water for another 4 h. The resulting sample, although catalytically evaluated for 3 times longer, showed carbon content of 4.8 wt%. This is substantially lower than that of the sample treated for only 4 h in the absence of water in Fig. 1a (6.0 wt%). The difference in carbon content (1.2 wt%) indicates that during the absence of water, species are deposited on the solid, which are removed in presence of water. The non-chemical role of water is further corroborated by the similar selectivities in Fig. 1. Should the role of water be chemical, then its presence may change the nature of the active site, which likely cause an increase in side products. In contrast, no significant change in selectivity is observed at any stage of the reaction.
For a more direct demonstration of the origin of the water-free deactivation, we analyzed the products that are released by a spent catalysts after exposure to water (Fig. 2). To this end, a ZSM5-40 catalyst was evaluated in the dealkylation of propylphenol in the absence of water. As in Fig. 1a, a rapid deactivation was observed during the first hour. After 5 h the reactor was flushed with nitrogen for 15 h. During this period, the phenol concentration reduced almost to zero confirming that all weakly-adsorbed phenol was removed. After ca. 20 h TOS, water was introduced into the stream, which induced an immediate increase in the concentration of phenol. On the other hand, no such effect was observed for propylphenol (Fig. SI2†). These results demonstrate that water enables to maintain catalytic activity by means of sweeping phenol from the catalyst.
Shape selectivity
In order to assess the nature of the high selectivity of ZSM-5, additional experiments were performed using a USY zeolite with the FAU topology. This zeolite features larger micropores (0.74 nm) compared to ZSM-5 (0.55 nm). Nevertheless, the acidity and the dimensionality (3D) of the micropore network are the same. The USY has as additional advantage that, unlike ZSM-5, its synthesis does not involve costly organic templates.70,71 The catalytic performance of the H-USY is compared to that of H-ZSM-5 in Fig. 3. The H-ZSM-5 displays an exceptional catalytic behavior showing a clear activity lift off around 275 °C and an almost constant selectivity to phenol and olefin of ca. 95%. The latter performance becomes even more impressive when compared to the USY data. The faujasite catalyst displayed a higher activity compared to ZSM-5, as conversion starts to occur around 225 °C. However, in the 225–350 °C-range the formation of byproducts, mostly cresols, ethylphenols, and dipropylphenols, account for almost 70% of the converted molecules. The superiority of the ZSM-5 catalysts is confirmed with stability test at constant temperature (Fig. 4a). These reveal a lower selectivity for the USY and a more pronounced deactivation. Importantly, this deactivation should not be related to any structural collapse as both the ZSM-5 and USY zeolite displayed a preserved crystallinity, porosity, and Al coordination before and after reaction (Fig. SI3–SI5†). This deactivation is more likely related to coking, which is corroborated by the higher C content on USY (5.3 wt%) compared to ZSM-5 (4.4 wt%) after reaction.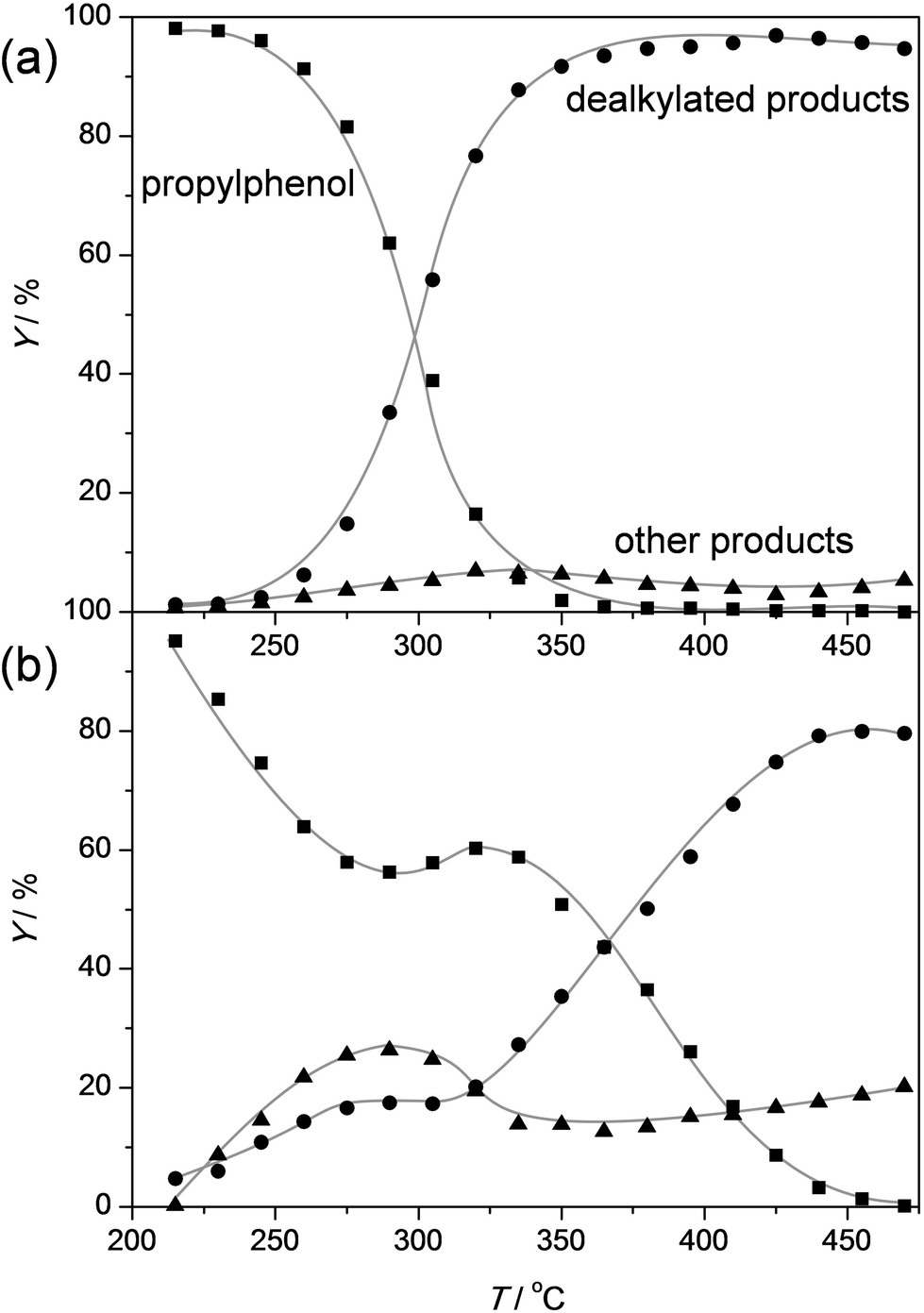 | ||
| Fig. 3 Yields of propylphenol, phenol + olefin (dealkylated products), and other products as a function of temperature over ZSM5-40 (a) and USY-40 (b) zeolite catalysts. The temperature was raised using a ramp rate of 1 °C min−1. WHSV = 3.7 h−1. | ||
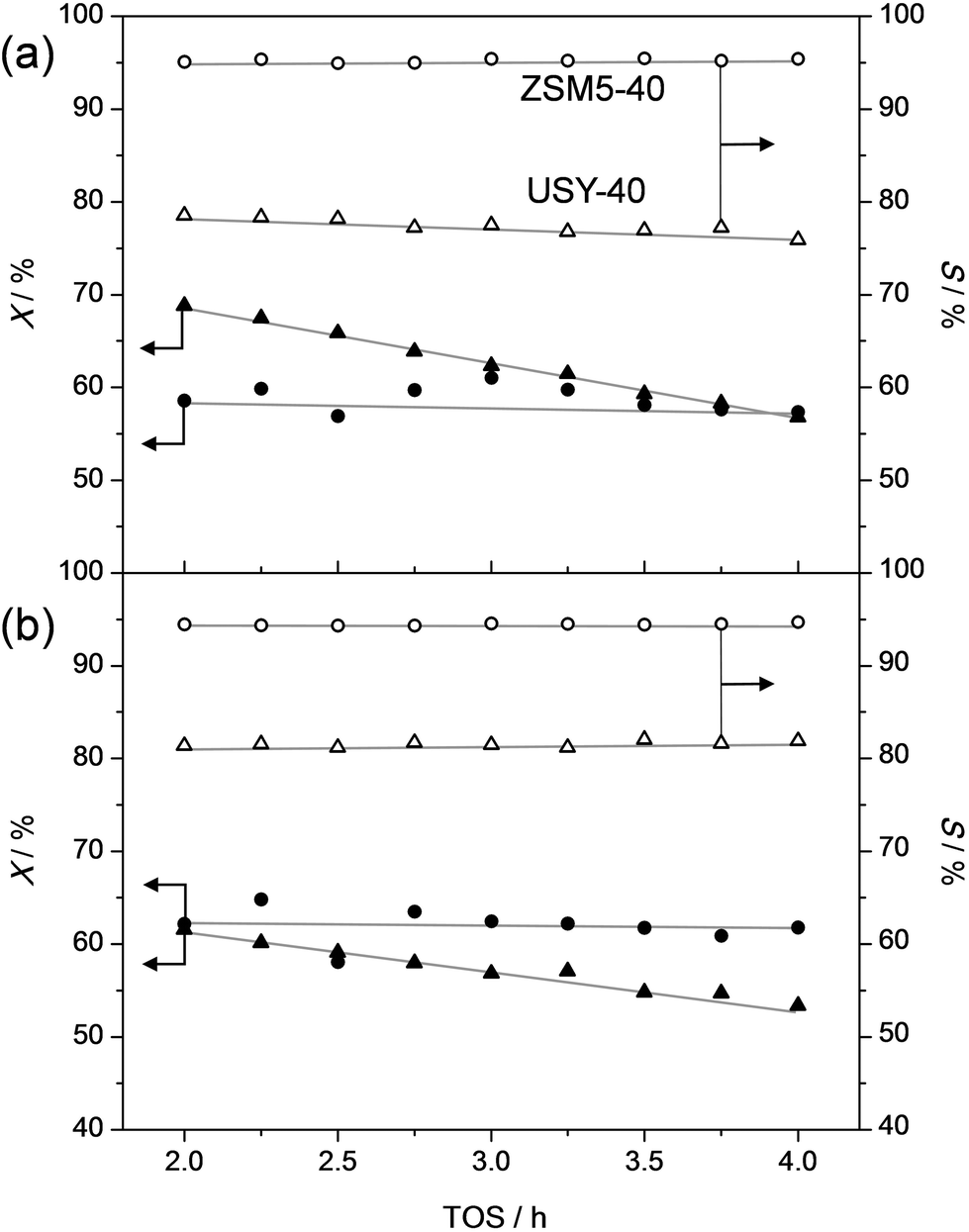 | ||
| Fig. 4 Conversion (X) of propylphenol (a) and ethylphenol (b) and selectivity to dealkylated products (phenol + olefin) (S) as a function of time on stream (TOS) over ZSM5-40 (circles) and USY-40 (triangles). For propylphenol (a), the temperatures were 305 °C (ZSM5-40) and 365 °C (USY-40). In the case of ethylphenol (b), the used temperatures were 385 °C (ZSM5-40) and 445 °C (USY-40). WHSV for ethylphenol and propylphenol were 3.3 h−1 and 3.7 h−1, respectively. | ||
Hence, the faujasite catalyst is noticeably less selective than ZSM-5. Accordingly, it is clear that, in addition to water, the micropore dimensionality is of paramount importance to attain an active and selective performance. Since the n-alkylphenols and some side products (cresols and ethylphenols) fit into the ZSM-5 micropores, the high phenol and olefin yields may be attributed to a transition-state shape selectivity. Herein, the tight fit of the linear alkylphenols, olefins, and phenol inside zeolite micropores inhibits the formation of side products. However, more than in the case of alkylbenzenes, strong chemical effects, based on the unique interaction of phenols in the zeolite micropore, may occur. It should be noted that an accurate determination of the nature of a shape-selectivity requires dedicated study.72–75 The latter lies beyond this contribution.
Acidity and contact time
In order to gain more insight in the influence of the relative abundance of the acid sites, ZSM-5 zeolites of different Si/Al ratios were evaluated in the dealkylation of 4-n-propylphenol (Fig. 5). By variation of the Si/Al ratio, the total number of Brønsted acid sites was varied, which clearly influenced the activity: by increasing the Si/Al from 15 to 140 the conversion lift off shifted from ca. 225 °C to 325 °C. This activity trend could be suitably related by plotting the temperature of 40% conversion (T40) to the total acidity (Fig. 6a). Besides the activity affect, the Si/Al ratio also displayed clear influence on the selectivity and stability of the catalysts. Fig. 5b, shows that the selectivity for dealkylated products and the stability increases with higher Si/Al ratio, hence lower total acidity. In addition, the carbon content of the spent catalysts is lower for the less-acidic zeolites. The latter was related in Fig. 6b, and may be explained by the closer proximity of acid sites at low Si/Al ratio, enhancing the occurrence of undesired secondary reactions.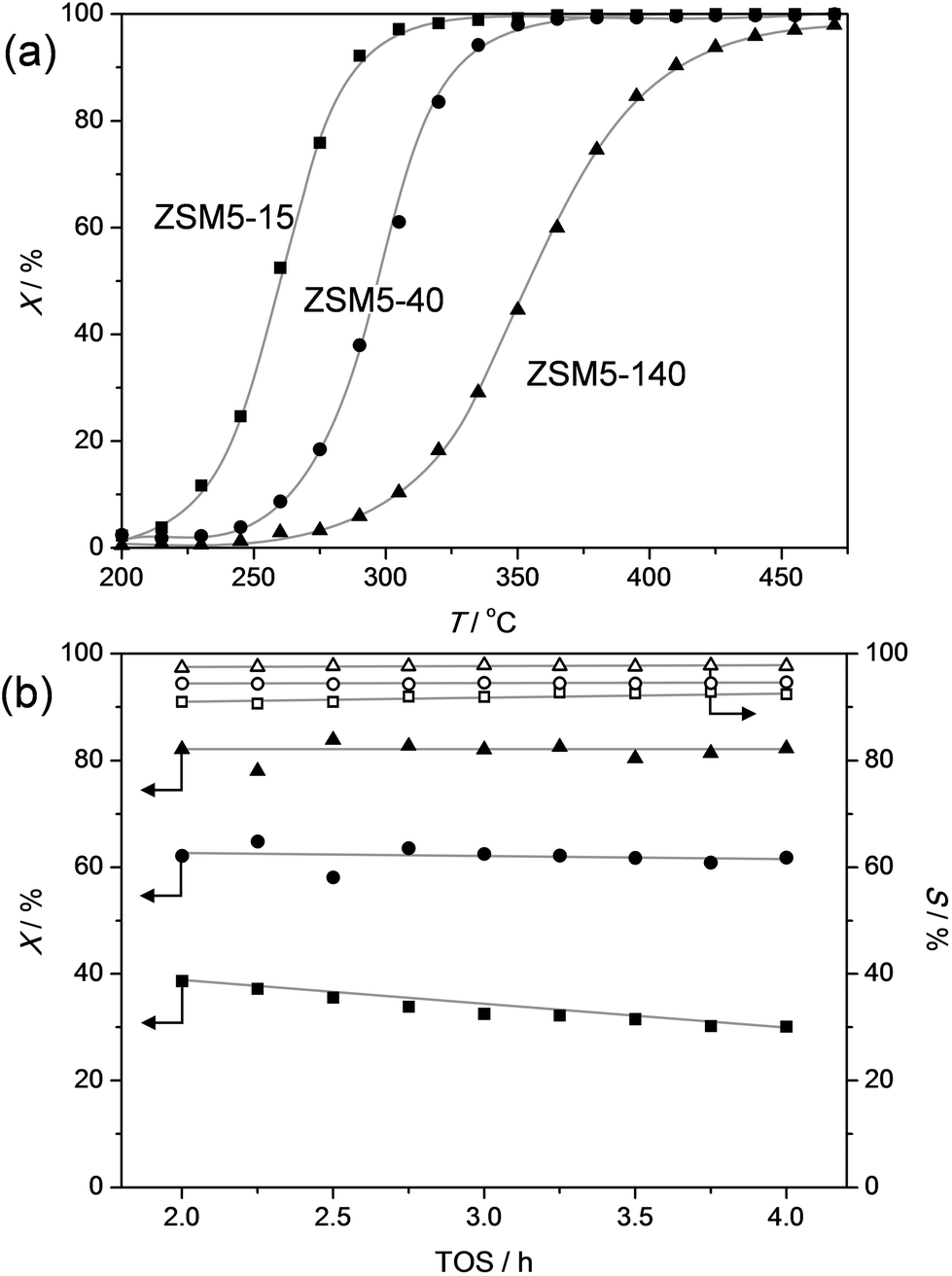 | ||
| Fig. 5 The dealkylation of propylphenol over ZSM-5 zeolites with distinct Si/Al ratios: 15 (ZSM5-15), 40 (ZSM5-40), and 140 (ZSM5-140). (a) Conversion of propylphenol (X) as a function of temperature. The temperature was raised using a ramp rate of 1 °C min−1. (b) Conversion (X) of propylphenol and selectivity to dealkylated products (phenol + olefin) (S) as a function of time. The temperatures in (b) were 270 °C (ZSM5-15), 305 °C (ZSM5-40), and 365 °C (ZSM5-140). WHSV in (a) and (b) was 3.7 h−1. | ||
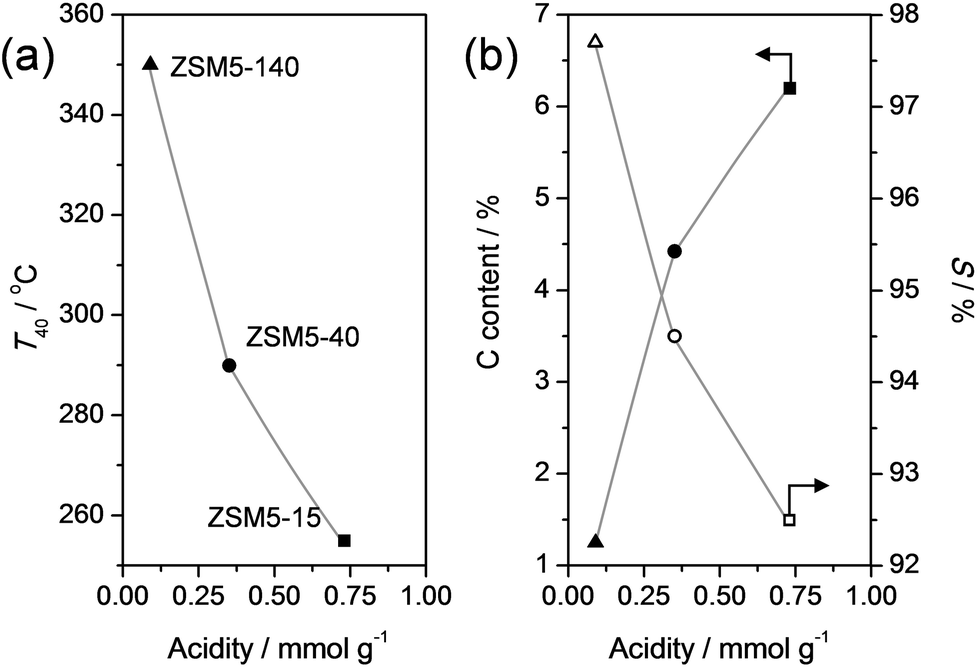 | ||
| Fig. 6 (a) Temperature at 40% conversion of propylphenol (T40) from the experiments in Fig. 5a as a function of the total acidity. (b) Carbon content of spent catalysts (solid symbols) and selectivity to dealkylated products (phenol + olefin) (S, open symbols) as a function of the total acidity. S and the carbon content were determined for catalysts that were exposed to the conditions in Fig. 5b for 4 h. The total acidities were adapted from ref. 76. | ||
In any conversion where secondary reactions are to be suppressed, a crucial parameter is the contact time. Accordingly, we have varied the weight hourly space velocity (WHSV) from ca. 4 to 60 h−1 at 385 °C (Fig. 7). At low WHSV the conversion, as well as the selectivity, are approaching 100%. Only at the WSHV's exceeding ca. 15 h−1, the conversion starts to gradually drop down to 50% at WHSV of 60 h−1. Moreover, an additional attractive feature is that with increasing WHSV the selectivity gradually increases to 98%. This implies that, in line with the results from Fig. 6, the formation of byproducts is the result of secondary reactions. Hence, the highest selectivity can be obtained by coupling a relatively high reaction temperature to low contact times and/or low zeolitic acidity.
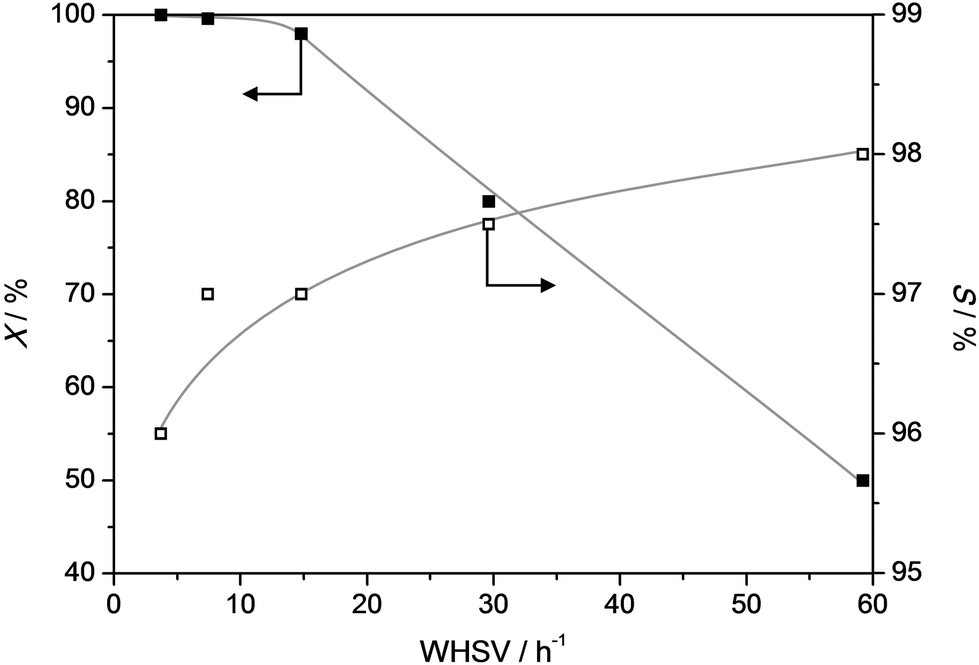 | ||
| Fig. 7 Conversion (X) of propylphenol and selectivity (S) to dealkylated products (phenol + olefin) as a function of WHSV (T = 385 °C) over a ZSM5-40 catalyst. X and S were taken after 4 h TOS. | ||
Versatility and stability
To get a better idea of the potential of the water-assisted dealkylation process, its effectivity was also tested in the conversion of 4-ethylphenol, one of the most abundant alkylphenols in coal tar oils.48Fig. 8 illustrates that, even more clearly like in the case of propylphenol (Fig. 3), ZSM-5 is superior compared to USY based on a higher selectivity to phenol and olefins. As expected, the dealkylation of ethylphenol occurs at a higher temperature (ca. 70 °C higher) compared to propylphenol. Nevertheless, the side reactions, in the case of USY40, occur at the same temperature. In addition, the same deactivation trend was observed in the conversion of 4-ethylphenol (Fig. 4b). Besides activity and selectivity concerns, the life time of the catalyst is of crucial importance. Therefore, ZSM5-140 was tested for the duration of 72 h (Fig. 9). The resulting performance highlights the remarkable stability of the ZSM-5 catalysts, attaining, after the initial deactivation in the first 5 h TOS, a constant conversion of ca. 70% with S > 95% to phenol and olefin. After 72 h, the spent catalysts comprised 1.6 wt% of coke. This is only 0.3 wt% more than the coke content after 5 hours of TOS (1.3 wt%, see Fig. 6b), which suggests that the coking occurs primarily during the early stages of the reaction.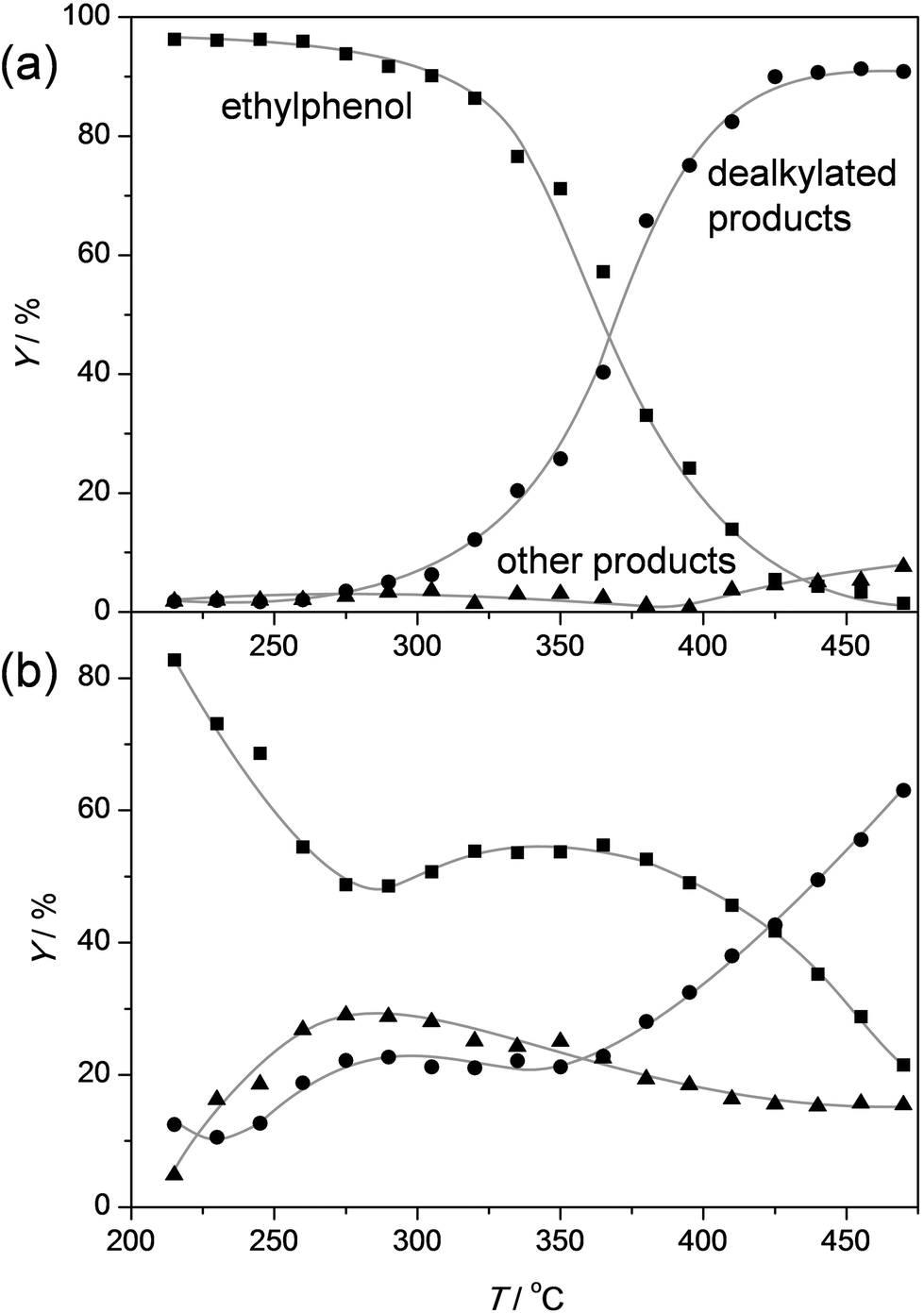 | ||
| Fig. 8 Yields of ethylphenol, phenol + olefin (dealkylated products), and other products as a function of temperature over ZSM5-40 (a) and USY-40 (b) zeolite catalysts. The temperature was raised using a ramp rate of 1 °C min−1. WHSV = 3.3 h−1. | ||
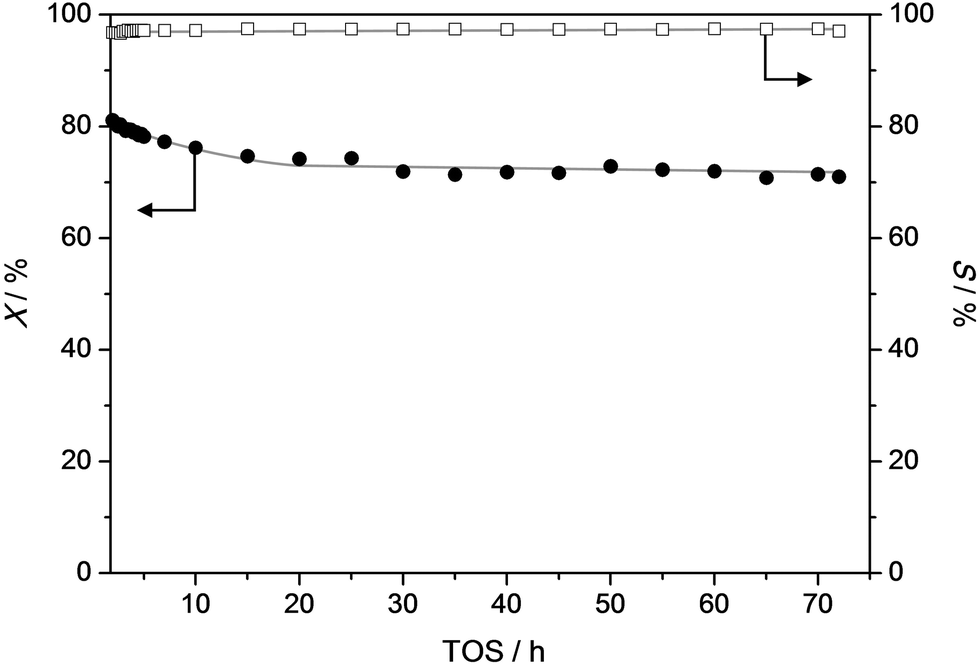 | ||
| Fig. 9 Conversion (X) of propylphenol and selectivity (S) to dealkylated products (phenol + olefin) as a function of time over a ZSM5-140 catalyst. T = 365 °C, WHSV = 3.7 h−1. | ||
As coal tars usually yield a mixture of alkylphenols,48 the capacity of the ZSM-5 catalyst to handle a feed containing both ethylphenol and propylphenol was tested (Fig. 10). The used molar ratio of ethylphenol/propylphenol was equal to three, which is the typical ratio in coal tars.48 The temperature-ramped evaluation of the conversion shows, as can be expected, that the propylphenol is first converted followed by the conversion of ethylphenol around 70 °C higher. Like in the dealkylation of the individual components, the selectivity to phenol and olefins exceeded 95% at all stages of the testing. The similar behavior of the mixture and the individual components was corroborated by the stable performance in the evaluation of the ZSM-5 catalysts as a function of time on stream.
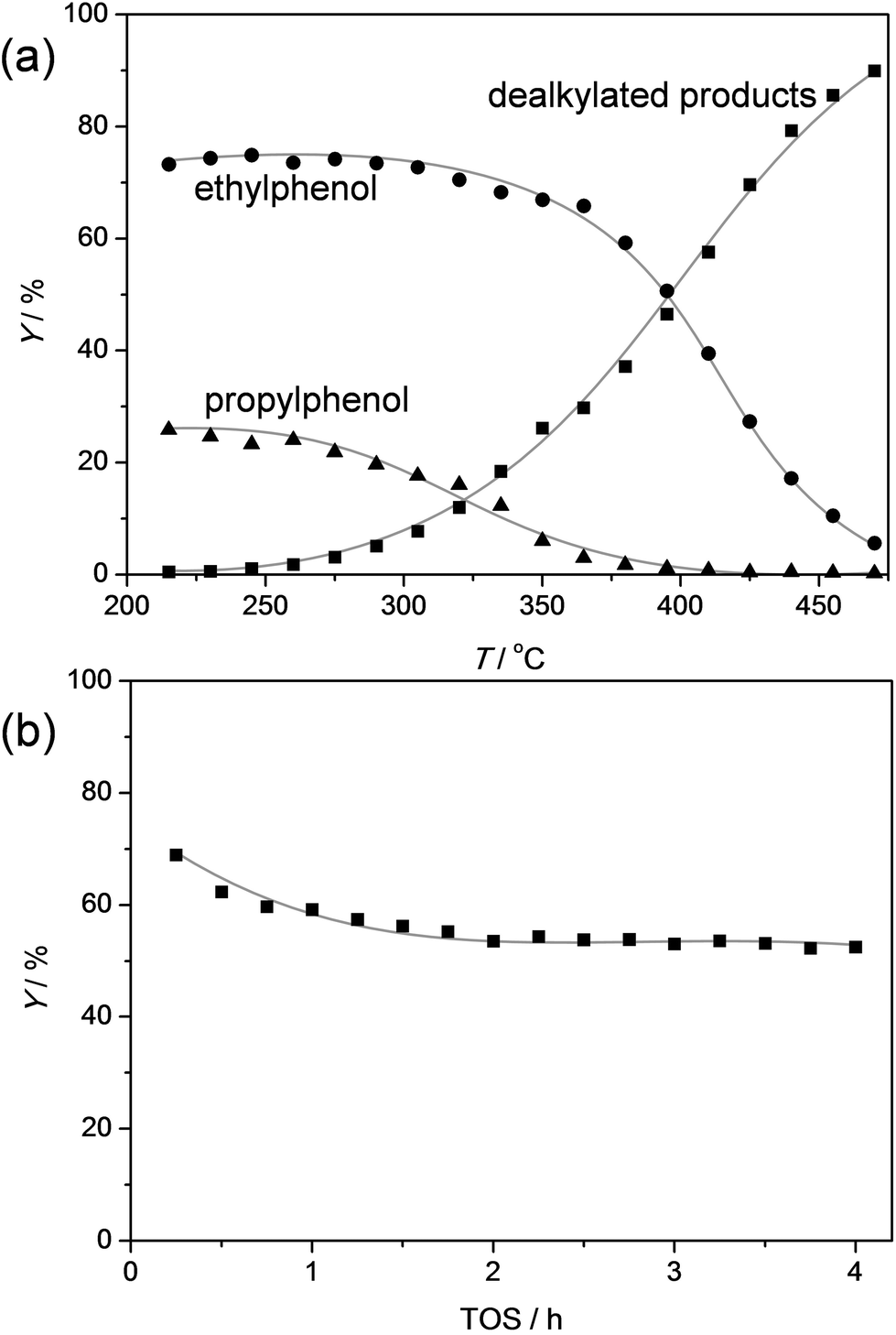 | ||
| Fig. 10 (a) Yields of ethylphenol, propylphenol, phenol + olefin (dealkylated products), and other products as a function of temperature over ZSM5-40. The temperature was raised using a ramp rate of 1 °C min−1. (b) Yield of dealkylated products as function of time over a ZSM5-40 catalyst. T = 415 °C. In (a,b) the WHSV of alkylphenol = 8 h−1, molar ratio of ethylphenol/propylphenol = 3. | ||
It should be stressed that, whereas in deriving phenol from lignin a negligible amount of poisons is to be encountered, coal tar may contain substantial S- and N-containing compounds (Scheme 2). These have a reputation to deactivate catalysts rapidly, which is why they are often removed from refinery streams by hydrotreating.53 However, based on the absence of a metal function on the acidic zeolite catalysts, the deactivation by poisons is limited to N-containing organic bases, such alkyl pyridines.48 However, since the majority of these bulky molecules cannot enter the MFI micropores, their influence should be small.48 Finally, the use of steam in the process should further suppress deactivation by poisoning of the acid sites.
In the petrochemical industry, the production of phenol requires several steps: oil refining, separation of benzene, alkylation of benzene to cumene, oxidation of cumene to cumene hydroperoxide (CHP), and finally the decomposition of CHP to phenol.77 Therefore, the selective conversion of lignin into phenol, with valuable byproducts, and using a limited number of steps may provide an appealing alternative. Moreover, deriving phenol from lignocellulose has as additional advantage over oil and coal that S- and N-containing poisons can be largely avoided.
Conclusions
In future wood or coal-fueled (bio-)refineries, the processing of alkylphenols will constitute a key reaction. It was demonstrated here that (mixtures of) the most abundant alkylphenols to be derived from lignocellulose, that is, ethyl and propylphenol, can be highly-selectively converted to relevant base chemicals phenol and corresponding olefin. The highly efficient and robust dealkylation is achieved using unmodified metal-free ZSM-5 zeolite catalysts and the co-feeding of water. The catalytic results pave the way for further study on the fundamental origin of the shape-selective behaviour and the competitive adsorptive of water and phenols.Acknowledgements
D.V. and W.S. acknowledge funding from the Research Foundation Flanders (FWO) for a post-doctoral fellowship and a doctoral fellowship, respectively. The authors are grateful to W. Vermandel for technical assistance during the catalytic testing, I. Cuppens for elemental analysis, and Dr K. Houthoofd for MAS NMR experiments. FISCH-ARBOREF financial support by the Flemish government is acknowledged.References
- K. Weissermel and H. J. Arpe, in Industrial Organic Chemistry, Wiley-VCH Verlag GmbH, Weinheim, Germany, 4th edn, 2003, pp. 316–336 Search PubMed.
- M. Dusselier, M. Mascal and B. Sels, in Selective Catalysis for Renewable Feedstocks and Chemicals, ed. K. M. Nicholas, Springer International Publishing, Switzerland, 2014, vol. 353, pp. 1–40 Search PubMed.
- S. Van de Vyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82–94 CrossRef CAS.
- J. A. Geboers, S. Van de Vyver, R. Ooms, B. O. de Beeck, P. A. Jacobs and B. F. Sels, Catal. Sci. Technol., 2011, 1, 714–726 Search PubMed.
- J. S. Luterbacher, D. M. Alonso and J. A. Dumesic, Green Chem., 2014, 16, 4816–4838 RSC.
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS PubMed.
- J. Q. Bond, A. A. Upadhye, H. Olcay, G. A. Tompsett, J. Jae, R. Xing, D. M. Alonso, D. Wang, T. Zhang, R. Kumar, A. Foster, S. M. Sen, C. T. Maravelias, R. Malina, S. R. H. Barrett, R. Lobo, C. E. Wyman, J. A. Dumesic and G. W. Huber, Energy Environ. Sci., 2014, 7, 1500–1523 CAS.
- Q. Zhang and F. Jerome, ChemSusChem, 2013, 6, 2042–2044 CrossRef CAS PubMed.
- J. Hilgert, N. Meine, R. Rinaldi and F. Schüth, Energy Environ. Sci., 2013, 6, 92–96 CAS.
- H. Kobayashi, H. Ohta and A. Fukuoka, Catal. Sci. Technol., 2012, 2, 869–883 CAS.
- B. Op de Beeck, M. Dusselier, J. Geboers, J. Holsbeek, E. Morre, S. Oswald, L. Giebeler and B. F. Sels, Energy Environ. Sci., 2015, 8, 230–240 CAS.
- G. Gellerstedt and G. Henriksson, in Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Amsterdam, 2008, pp. 201–224 Search PubMed.
- J. Lora, in Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Amsterdam, 2008, pp. 225–241 Search PubMed.
- F. G. Calvo-Flores and J. A. Dobado, ChemSusChem, 2010, 3, 1227–1235 CrossRef CAS PubMed.
- J. J. Bozell, J. E. Holladay, D. Johnson and J. F. White, Top Value added candidates from biomass, Volume II: Results of screening for potential candidates from biorefinery lignin, PNNL-16983, Pacific Northwest National Laboratory, Richland, Washington, 2007 Search PubMed.
- C. Xu, R. A. D. Arancon, J. Labidi and R. Luque, Chem. Soc. Rev., 2014, 43, 7485–7500 RSC.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- J. Bozell, in Selective Catalysis for Renewable Feedstocks and Chemicals, ed. K. M. Nicholas, Springer International Publishing, Switzerland, 2014, vol. 353, pp. 229–255 Search PubMed.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- D. R. Vardon, M. A. Franden, C. W. Johnson, E. M. Karp, M. T. Guarnieri, J. G. Linger, M. J. Salm, T. J. Strathmann and G. T. Beckham, Energy Environ. Sci., 2015, 8, 617–628 CAS.
- J. R. Bower, L. M. Cooke and H. Hibbert, J. Am. Chem. Soc., 1943, 65, 1192–1195 CrossRef CAS.
- J. M. Pepper and W. Steck, Can. J. Chem., 1963, 41, 2867–2875 CrossRef CAS.
- N. Yan, C. Zhao, P. J. Dyson, C. Wang, L. T. Liu and Y. Kou, ChemSusChem, 2008, 1, 626–629 CrossRef CAS PubMed.
- Q. Song, F. Wang, J. Y. Cai, Y. H. Wang, J. J. Zhang, W. Q. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 CAS.
- T. Parsell, S. Yohe, J. Degenstein, T. Jarrell, I. Klein, E. Gencer, B. Hewetson, M. Hurt, J. I. Kim, H. Choudhari, B. Saha, R. Meilan, N. Mosier, F. Ribeiro, W. N. Delgass, C. Chapple, H. I. Kenttamaa, R. Agrawal and M. M. Abu-Omar, Green Chem., 2015, 17, 1492–1499 RSC.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S. F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 Search PubMed; S. Van den Bosch, W. Schutyser, S. F. Koelewijn, T. Renders, C. M. Courtin and B. F. Sels, Chem. Commun., 2015, 51, 13158–13161 RSC.
- J. Sun, A. M. Karim, H. Zhang, L. Kovarik, X. S. Li, A. J. Hensley, J. S. McEwen and Y. Wang, J. Catal., 2013, 306, 47–57 CrossRef CAS.
- T. Omotoso, S. Boonyasuwat and S. P. Crossley, Green Chem., 2014, 16, 645–652 RSC.
- J. B. Bredenberg, M. Huuska, J. Räty and M. Korpio, J. Catal., 1982, 77, 242–247 CrossRef CAS.
- T. Prasomsri, M. Shetty, K. Murugappan and Y. Román-Leshkov, Energy Environ. Sci., 2014, 7, 2660–2669 CAS.
- L. Jongerius, R. W. Gosselink, J. Dijkstra, J. H. Bitter, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemCatChem, 2013, 5, 2964–2972 CrossRef.
- N. Joshi and A. Lawal, Ind. Eng. Chem. Res., 2013, 52, 4049–4058 CrossRef CAS.
- M. A. Ratcliff, D. K. Johnson, F. L. Posey and H. L. Chum, Appl. Biochem. Biotechnol., 1988, 17, 151–160 CrossRef CAS.
- V. N. Bui, D. Laurenti, P. Delichère and C. Geantet, Appl. Catal., B, 2011, 101, 246–255 CrossRef CAS.
- D. Gao, C. Schweitzer, H. T. Hwang and A. Varma, Ind. Eng. Chem. Res., 2014, 53, 18658–18667 CrossRef CAS.
- H. Y. Zhao, D. Li, P. Bui and S. T. Oyama, Appl. Catal., A, 2011, 391, 305–310 CrossRef CAS.
- J. Chang, T. Danuthai, S. Dewiyanti, C. Wang and A. Borgna, ChemCatChem, 2013, 5, 3041–3049 CrossRef CAS.
- K. Wang, K. H. Kim and R. C. Brown, Green Chem., 2014, 16, 727–735 RSC.
- J. Jae, G. A. Tompsett, Y. C. Lin, T. R. Carlson, J. Shen, T. Zhang, B. Yang, C. E. Wyman, W. C. Conner and G. W. Huber, Energy Environ. Sci., 2010, 3, 358–365 CAS.
- V. M. Roberts, V. Stein, T. Reiner, A. Lemonidou, X. Li and J. A. Lercher, Chem. – Eur. J., 2011, 17, 5939–5948 CrossRef CAS PubMed.
- R. Beauchet, F. Monteil-Rivera and J. M. Lavoie, Bioresour. Technol., 2012, 121, 328–334 CrossRef CAS PubMed.
- W. Xu, S. J. Miller, P. K. Agrawal and C. W. Jones, ChemSusChem, 2012, 5, 667–675 CrossRef CAS PubMed.
- J. Zakzeski, A. L. Jongerius, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemSusChem, 2012, 5, 1602–1609 CrossRef CAS PubMed.
- A. W. Scaroni, M. R. Khan, S. Eser and L. R. Radovic, in Coal Pyrolysis, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag, Weinheim, 2012, pp. 407–420 Search PubMed.
- T. Kaneko, F. Derbyshire, E. Makino, D. Gray, M. Tamura and K. Li, in Coal Liquefaction, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag, Weinheim, 2012, pp. 17–30 Search PubMed.
- S. Vasireddy, B. Morreale, A. Cugini, C. Song and J. J. Spivey, Energy Environ. Sci., 2011, 4, 311–345 Search PubMed.
- H. Fiege, H. W. Voges, T. Hamamoto, S. Umemura, T. Iwata, H. Miki, Y. Fujita, H. J. Buysch, D. Garbe and W. Paulus, in Phenol Derivatives, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag, Weinheim, 2012, pp. 525–526 Search PubMed.
- W. Xu, S. J. Miller, P. K. Agrawal and C. W. Jones, Catal. Lett., 2014, 144, 434–438 CrossRef CAS.
- C. Zhao, D. M. Camaioni and J. A. Lercher, J. Catal., 2012, 288, 92–103 CrossRef CAS.
- A. Vjunov, M. Y. Hu, J. Feng, D. M. Camaioni, D. Mei, J. Z. Hu, C. Zhao and J. A. Lercher, Angew. Chem., Int. Ed., 2014, 53, 479–482 Search PubMed.
- P. A. Zapata, J. Faria, M. Pilar Ruiz, R. E. Jentoft and D. E. Resasco, J. Am. Chem. Soc., 2012, 134, 8570–8578 CrossRef CAS PubMed.
- G. Alfke, W. W. Irion and O. S. Neuwirth, in Oil Refining, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag, Weinheim, 2012, pp. 211–223 Search PubMed.
- P. A. Jacobs, M. Dusselier and B. F. Sels, Angew. Chem., Int. Ed., 2014, 53, 8621–8626 CrossRef CAS PubMed; M. Dusselier, P. Van Wouwe, A. Dewaele, P. A. Jacobs and B. F. Sels, Science, 2015, 349, 78–80 CrossRef PubMed.
- E. Klemm, J. Wang and G. Emig, Microporous Mesoporous Mater., 1998, 26, 11–21 CrossRef CAS.
- P. B. Venuto and E. L. Wu, J. Catal., 1969, 15, 205–208 CrossRef CAS.
- J. M. Serra, E. Guillon and A. Corma, J. Catal., 2004, 227, 459–469 CrossRef CAS.
- W. Vermeiren and J. P. Gilson, Top. Catal., 2009, 52, 1131–1161 CrossRef CAS.
- C. Hu, J. Li, W. Jia, M. Liu, Z. Hao and Z. Zhu, Chin. J. Chem., 2015, 33, 247–252 CrossRef CAS.
- H. G. Karge, J. Ladebeck, Z. Sarbakt and K. Hatada, Zeolites, 1982, 2, 94–102 CrossRef CAS.
- N. Y. Chen, W. E. Garwood and F. G. Dwyer, in Shape Selective Catalysis in Industrial Applications, Marcel Dekker, United States, 2nd edn, 1996, pp. 101–105 Search PubMed.
- T. Onodera, T. Sakai, Y. Yamasaki, K. Sumitani and M. Sueoka, US, 4320242, 1982 Search PubMed.
- K. Kase, T. Idai, K. Ishii and E. Takahashi, US, 4847435, 1989 Search PubMed.
- L. T. Mika, E. Cséfalvay and I. T. Horváth, Top. Catal., 2015, 247, 33–46 CrossRef CAS.
- S. K. Jana, T. Okamoto, T. Kugita and S. Namba, Appl. Catal., A, 2005, 288, 80–85 CrossRef CAS.
- C. B. Phillips and R. Datta, Ind. Eng. Chem. Res., 1997, 36, 4466–4475 CrossRef CAS.
- V. Bolis, J. C. Vedrine, J. P. Van de Berg, J. P. Wolthuizen and E. G. Derouane, J. Chem. Soc., Faraday Trans., 1980, 76, 1606–1616 RSC.
- R. F. Parton, J. M. Jacobs, D. R. Huybrechts and P. A. Jacobs, in Zeolites as Catalysts, Sorbents, and Detergents Builders, ed. H. G. Karge and J. Weitkamp, Elsevier, Amsterdam, 1989, pp. 163–192 Search PubMed.
- M. Guisnet, H. S. Cerqueira and F. R. Ribiero, in Deactivation and Regeneration of Zeolite Catalysts, ed. M. Guisnet and F. Ramao Ribiero, Imperial College Press, London, 2011, pp. 139–145 Search PubMed.
- C. Baerlocher and L. B. McCusker, Database of Zeolite Structures, http://www.iza-structure.org/databases/, accessed March 2015 Search PubMed.
- D. Verboekend and J. Pérez-Ramírez, ChemSusChem, 2014, 7, 753–764 CrossRef CAS PubMed.
- E. Derouane, J. Catal., 1986, 100, 541–544 CrossRef CAS.
- C. B. Khouw and M. E. Davis, in Selectivity in Catalysis, ed. M. E. Davis and S. L. Suib, American Chemical Society, Washington, DC, the United States, 1993, pp. 206–211 Search PubMed.
- N. Y. Chen and W. E. Garwood, J. Catal., 1978, 52, 453–458 CrossRef CAS.
- B. Smit and T. L. M. Maesen, Chem. Rev., 2008, 108, 4125–4184 CrossRef CAS PubMed.
- M. Osman, L. Atanda, M. M. Hossain and S. Al-Khattaf, Chem. Eng. J., 2013, 222, 498–511 CrossRef CAS.
- M. Weber, M. Weber and M. Kleine-Boymann, in Phenol, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag, Weinheim, 2012, pp. 505–508 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Thermo-gravimetric analysis, catalytic tests, X-ray diffraction patterns, 27Al MAS NMR spectra, and N2 adsorption isotherms. See DOI: 10.1039/c5gc01868d |
| This journal is © The Royal Society of Chemistry 2016 |