DOI:
10.1039/C5GC01746G
(Paper)
Green Chem., 2016,
18, 176-185
Antimony recovery from the halophosphate fraction in lamp phosphor waste: a zero-waste approach
Received
29th July 2015
, Accepted 14th September 2015
First published on 14th September 2015
Abstract
Antimony is becoming an increasingly critical element as the supply-demand gap is expected to exceed 10% over the period 2015–2020. Antimony production is primarily concentrated in China (90%) and as the industrial demand for this metal surges, attention has to turn towards the recovery of antimony from (industrial) waste residues and end-of-life products in order to guarantee a sustainable supply of antimony. Although lamp phosphor waste is usually considered as a source of rare earths, it also contains significant amounts of antimony in the form of the white halophosphate phosphor (Ca,Sr)5(PO4)3(Cl,F):Sb3+,Mn2+ (HALO). HALO phosphor readily dissolves in dilute acidic conditions, making antimony far more accessible than in the main production route which is the energy intensive processing of stibnite ore (Sb2S3). HALO makes up 50 wt% of the lamp phosphor waste, but it has been systematically overlooked and treated as an undesired residue in the efforts to recover rare earths from lamp phosphor waste. In this paper, the feasibility of antimony recovery is discussed and an efficient process is proposed. The HALO phosphor is first dissolved in dilute HCl at room temperature, followed by a selective extraction of antimony with the ionic liquid Aliquat® 336. The remaining leachate is valorized as apatite which is a feed for the phosphate and fertilizer industry. A zero-waste valorization approach was followed, meaning that no residue or waste was accepted and that all the elements were converted into useful products. This paper thus emphasizes the potential of lamp phosphor waste as a secondary source of antimony and describes a sustainable process to recover it. The process can be integrated in lamp phosphor recycling schemes aimed at recovering rare earths.
Introduction
The word antimony comes from the Greek “anti monos”, meaning “never (found) alone” which explains why the metal was only isolated and correctly identified in the 16th century even though it had been known since ancient times as a cosmetic product.1 The abundance of antimony in the earth's crust is 0.2 ppm, making this element scarcer than the heavy rare-earth elements.2 Antimony (Sb) is mainly produced from stibnite (Sb2S3) which is found in quartz veins.1 Today, 90% of the global antimony supply is produced in China. This country also holds the largest reserves.1 Antimony is mostly used in the form of Sb2O3 as flame retardant due to its synergetic effect with halogenated flame retardants. Sb2O3 has become crucial to guarantee the safety of inherently flammable materials such as plastics, coatings and electronics and takes up the majority of the world's antimony production. Sb2O3 is also the main catalyst for the production of PET plastic and it is used as additive in glass and ceramics. Antimony metal is less important, although it is used in some types of lead alloys.1 Due to its economic importance and high supply risk, antimony was listed among the most critical elements in the critical raw material report from the European Commission (2014).3 The report forecasts that in the period 2015–2020 the supply-demand gap for antimony will be the most severe amongst all the considered strategic metals, exceeding 10% on an annual basis. Due to the strong concentration of production in China and the lack of new deposits, increasing attention should go to the recovery of antimony from secondary sources such as industrial waste residues and end-of-life consumer products. Recycling of critical metals and elemental sustainability is becoming increasingly important as resources become scarcer. In this context, lamp phosphor waste from collected end-of-life fluorescent lamps can be very interesting due to its relatively high antimony content (0.5–1 wt%) and its known rare-earth content, which adds value.4–7 Stibnite ore (Sb2S3) has an average grade of 2.7 wt%, but requires a lot of energy and chemicals to extract the antimony.1 The antimony in the lamp phosphor waste can be recovered in milder conditions (room temperature, dilute acid) and contains far less contaminants since lamp phosphors are synthesized using high purity products.8–12 Stibnite ore often contains impurities such as Pb, As and Fe.1
Lamp phosphors are fluorescent powders coated on the inside of the glass and are responsible for the emission of visible light in normal or compact fluorescent lamps (CFLs).8–12 Their luminescence is based on the adsorption of UV-radiation emitted by the mercury atoms in the gas filling of the lamps. Two important classes of lamp phosphors can be distinguished: the white halophosphate phosphors and the so-called “tri-band” phosphors which use a mixture of red, green and blue phosphors to generate white light. The white HALO phosphor was invented in 1942 and large amounts of this phosphor were produced as it found widespread use in fluorescent tubes.8–10,12 Small differences exist, but it's main structure is a fluoro–chloro apatite doped with manganese(II) and antimony(III).8–10 Its luminescence and shade of white can be adjusted by tuning the ratio of blue emitting antimony dopant and orange emitting manganese dopant or the fluorine-to-chlorine ratio.8–10,13 Some of the calcium can also be substituted by strontium to obtain narrower emission bands.8–10 The tri-band phosphors are more recent and have largely replaced halophosphate phosphors due to their better color rendering index, higher efficiency and excellent stability. Tri-band phosphors are composed of separate red (YOX), blue (BAM) and green (LAP/CAT) phosphors (Table 1), which together generate white light.10 The tri-band phosphors were crucial for the development of CFLs because the high wall temperature and higher UV flux of these lamps would quickly degrade the less resistant halophosphate phosphors. Their downside is that they rely on expensive rare-earth elements (REEs) for their luminescence, namely Eu(III), Tb(III) and Eu(II) for the red, green and blue phosphors, respectively. As a compromise between phosphor cost and performance, a double coating scheme can be applied.10 A base layer of inexpensive and more-easily-damaged halophosphate phosphor is coated with a resistant REE phosphor blend in order to protect it from the electron discharge and generate white light with high efficiency and good color rendering index. Despite the fact that since the 1990s the tri-band phosphors have largely replaced HALO phosphor, enormous amounts of HALO have been produced over the years and are either still in use or have been stockpiled or landfilled by recyclers.14,15 Today, processed lamp phosphor waste contains around 50 wt% of HALO, making it the largest component (Table 1).5
Table 1 Overview of the approximate lamp phosphor waste composition and average stoichiometry of the different commercial phosphors15
| Name |
Formula |
Fractiona (wt%) |
|
The remaining consists of SiO2 (fine glass particles) and Al2O3 (binder).
For convenience, a shorter formula for HALO is used in this paper: (Sr,Ca)5(PO4)3(Cl,F):Sb3+,Mn2+.
|
| HALOb |
Ca4.86Mn0.10Sb0.04Sr0.004(PO4)3Cl0.10F0.90 |
50 |
| YOX |
Y1.92Eu0.08O3 |
20 |
| BAM |
Ba0.86Eu0.14MgAl10O17 |
5 |
| LAP |
La0.60Ce0.27Tb0.13PO4 |
5 |
| CAT |
Ce0.63Tb0.37MgAl11O19 |
5 |
With annual sales of CFL's topping 2.5 × 109 units per year (2007), the volume of lamp phosphor waste is expected to grow rapidly as fluorescent lamps reach their end-of-life.16,17 Fluorescent lamps are considered as hazardous waste due to their mercury content, and have to be collected separately.14,15 While the glass and metal in the lamps is often recycled, the lamp phosphor powders are usually simply stockpiled or discarded despite their significant critical metal content (Table 1).14,15,18 However, the interest in this waste fraction has been growing since the rare-earth crisis of 2011.15,19 The REEs are present in the red (YOX), green (LAP/CAT) and blue (BAM) tri-band phosphors and many studies have been carried out to recover these valuable elements.14,15,18,20–27 Since 2012, the recycling of REEs from fluorescent lamps is also carried out on industrial scale by Solvay in France.5 However, the broadband antimony-containing phosphor (Sr,Ca)5(PO4)3(Cl,F):Sb3+,Mn2+ (HALO) does not contain rare earths and is therefore usually considered as a non-valuable residue even though it makes up around 50 wt% of the lamp phosphor waste (Table 1).14,15,18 As rare-earth prices have come down from their highs of 2011, more attention should go to this halophosphate phosphor as a potentially interesting secondary source of antimony. Antimony is crucial for many growing applications as described earlier but its recycling from end-of-life consumer waste has not been considered so far. The recovery of antimony should be integrated in REE recycling processes for fluorescent lamp phosphor waste in the larger attempt to valorize this valuable consumer waste product. The HALO content in these powders is too large to ignore or discard as a useless side product. In this paper, the feasibility of antimony recovery is discussed and a new process is presented to efficiently recover the antimony from the HALO phosphor using minimal amounts of chemicals. The resulting product is pure Sb2O3, which is the main commercial form of antimony and an apatite residue which is the primary feedstock for the phosphate and fertilizer industry. A zero-waste valorization strategy was followed, meaning that no waste was tolerated and all of the elements in HALO were valorized into useful products, not just antimony. The process was developed using commercial HALO phosphor in order to study the chemistry of this phosphor in detail. However, the implementation of the HALO valorization process in lamp phosphor recycling schemes is also discussed in this paper.
Experimental
Materials and chemicals
The lamp phosphors (Ca,Sr)5(PO4)3(Cl,F):Sb3+,Mn2+ (HALO), Y2O3:Eu3+ (YOX), LaPO4:Ce3+,Tb3+ (LAP) and BaMgAl10O17:Eu2+ (BAM) were purchased from Nichia (Japan) (>99%). The exact composition of the commercial HALO is shown in Table 2. CeMgAl11O19:Tb3+ (CAT), [A336][Cl] (Aliquat® 336) and SbCl3 (>99%) were obtained from Sigma-Aldrich (Diegem, Belgium). HCl (37%) and H2SO4 (96%) were purchased from Acros Organics (Geel, Belgium). NaOH (>99%) was purchased from VWR (Leuven, Belgium). The silicone solution in isopropanol was purchased from SERVA Electrophoresis GmbH (Germany) and the indium standard solution (1000 ppm) from Merck (Belgium). All chemicals were used as received without further purification.
Table 2 Composition of the HALO phosphor expressed in wt% and mol%
| Element |
Concentration (wt%) |
Concentration (mol%) |
| PO4 |
55.71 |
33.32 |
| Ca |
38.06 |
53.95 |
| F |
3.33 |
9.94 |
| Sb |
1.05 |
0.49 |
| Mn |
1.06 |
1.10 |
| Cl |
0.73 |
1.16 |
| Sr |
0.06 |
0.04 |
| Total |
100% |
100% |
Equipment and characterization
A TMS-200 thermoshaker (Nemus Life) was used to facilitate mixing of the samples and a centrifuge Heraeus Megafuge 1.0 was used to sediment the undissolved residue. The metal concentrations in the samples were determined by total reflection X-ray fluorescence spectroscopy (TXRF) on a Bruker S2 Picofox TXRF spectrometer equipped with a molybdenum source. For the sample preparation, plastic microtubes were filled with an amount of aqueous sample (100–200 mg) and 3 M HCl solution (200 μL) to avoid hydrolysis of Sb(III) ions. For the analysis of the ionic liquid (IL) phase, smaller samples were taken (50 mg) and diluted in EtOH (200 μL). Then, 50 μL of a 1000 ppm indium standard solution was added as an internal standard. Indium was chosen as a standard because this element has an X-ray energy very similar to both calcium and antimony, which reduces the effects caused by adsorption of secondary X-rays (matrix effects).28 The microtubes were vigorously shaken on a vibrating plate (IKA MS 3 basic). Finally, a 2 μL drop of this solution was put on a quartz plate, previously treated with a silicone/isopropanol solution (Serva®) to avoid spreading of the sample droplet on the quartz plate. The quartz plates were then dried for 30 min at 60 °C prior to analysis. Each sample was measured for 10 min.
Dissolution experiments
The solid material (e.g. HALO) was introduced in small glass vials (4 mL) together with the desired acidic solution (2 mL). A solid/liquid ratio of 25 mg g−1 was used in most experiments. The samples were then shaken (2000 rpm) at 25 °C for a certain amount of time (depending on the experiment). Finally, the vials were centrifuged for 5 min (5000 rpm) to sediment the residue and a sample was taken from the supernatant to determine the metal concentrations in solution (TXRF).
Solvent extraction experiments
A batch of leachate (20 mL) was prepared by dissolving HALO in 1 M HCl (25 mg g−1 solid/liquid ratio) and stirring the solution (500 rpm) at 25 °C until the dissolution was complete and a clear solution was obtained (5–10 min). The aqueous leachate was then divided into 2 g portions and contacted with the water saturated ionic liquid phase (1 g) using 4 mL vials with screw caps. The water-saturated IL was used to avoid volume changes when contacted with the leachate during solvent extraction experiments. The IL Aliquat® 336 was presaturated with water by contacting a large batch of pure IL with water, shaking it (1 h, 2000 rpm) and allowing it to phase separate. The extraction vials were then shaken for 30 min (2000 rpm) at 25 °C to guarantee a good mixing of the two phases. Finally, the vials were centrifuged for 10 min (5000 rpm) to accelerate phase separation and a sample was taken of the aqueous and IL phase to determine the metal concentrations (TXRF).
Scrubbing and stripping experiments
The loaded IL phase (1 g) was contacted with an aqueous solution (1 g) containing NaCl for scrubbing experiments or NaOH for stripping experiments. The vials were then shaken for 10 min (2000 rpm) at 25 °C. A sample of the IL phase was taken to determine the remaining metal concentration in the IL (TXRF).
Precipitation of the residue
The aqueous phase obtained after the extraction was used to conduct these precipitation experiments. NaOH was added to set the pH and the formed precipitate was settled by centrifugation. A sample of the supernatant was taken after 10 min and 24 h to determine the remaining metal concentrations (TXRF) in the effluent.
Results and discussion
Feasibility of antimony recycling from lamp phosphor waste
Before describing the recycling process, we chose to first discuss the feasibility of antimony recycling from lamp phosphor waste, as well as the general zero-waste valorization strategy. The first hurdle to antimony recycling is that antimony is only present in one phosphor (HALO). It is therefore important to avoid co-dissolution of the other components in the lamp phosphor waste as this would cause significant complications (Table 1). Luckily, it is well-known that the HALO phosphor is more easy to dissolve in acid than the other phosphors (HALO > YOX ≫ LAP/CAT/BAM).5,15,18,29 This was confirmed by our experiments in HCl solutions with separate commercial phosphors (Table 3).
Table 3 Time required for full dissolution of HALO in a 25 mg g−1 solution. The dissolutions were carried out at 25 °C while shaking (2000 rpm) at constant pH (HCl). The dissolution of the other phosphors in the same conditions (but in separate vials) is also shown
| pH |
Time for full HALO dissolution (min) |
YOX leaching (wt%) |
LAP/CAT/BAM leaching (wt%) |
|
Detection limit for rare-earths elements in TXRF < 10 ppb.
|
| 0 |
5 |
2.7 (5 min) |
<D.L.a (5 min) |
| 0.25 |
25 |
8.3 (25 min) |
<D.L.a (25 min) |
| 0.50 |
60 |
11.8 (60 min) |
<D.L.a (60 min) |
| 1.00 |
Partial hydrolysis Sb |
|
|
It is clear from these results that HALO is indeed more easily dissolved than the other phosphors. Amongst the other components, only YOX is partially dissolved. It is important to avoid the co-dissolution of YOX since it is a valuable phosphor and dissolving it together with HALO, which releases phosphoric acid, would cause the Y(III) and Eu(III) ions to reprecipitate as water-insoluble phosphates: YPO4 and EuPO4 (Ksp ≈ 10−25) (eqn (1) and (2)).30
| | | (Y,Eu)2O3 + 6HCl → 2(Y,Eu)Cl3 + 6H2O | (1) |
| | | (Y,Eu)3+ + H3PO4 → (Y,Eu)PO4(s) + 3H+ | (2) |
There are two ways to avoid partial co-dissolution of YOX and the consequent loss of yttrium and europium. The first is to minimize the dissolution of YOX by careful pH control (e.g. 5 min, pH 0), but this cannot fully exclude the dissolution of YOX (Table 3),5,29 The other approach is to selectively remove the YOX in a prior process step. This was not possible until recently because of the faster dissolution of HALO in acidic aqueous solutions. However, we recently developed a process to achieve this in the carboxyl-functionalized ionic liquid betainium bis(trifluoromethylsulfonyl)imide [Hbet][Tf2N], which can selectively dissolve metal oxides such as YOX.27 This selective dissolution of YOX allows the direct recycling of the most valuable component in the lamp phosphor waste and has the additional advantage of removing YOX from the lamp phosphor waste so that in a next step, HALO can be dissolved quickly and selectively in acid.27 The antimony recovery process described in this paper, can then be applied without further complications. This way, the valorization of HALO can fit in a general rare-earth recycling scheme for lamp phosphor waste, finally allowing the valorization of this major component of the lamp phosphor waste (50 wt%), instead of discarding it as a non-valuable waste residue.5,15,20
Waste valorization strategy
The aim of this work was to recover antimony from the HALO phosphor using a zero-waste approach.31 This is a key process requirement since it is not desirable to end up with more waste than the waste that is being treated in the first place. The key to HALO valorization lies in its resemblance to the industrially very important phosphate rock.31–37 The phosphor Ca5(PO4)3(Cl,F):Sb3+,Mn2+ (HALO) has a typical apatite structure, in which some of Ca(II) ions are substituted by Sb(III) and Mn(II) ions.8–10 Apatites have the general formula Ca5(PO4)3X (X = OH, F, Cl) and are the main component of phosphate rock which is the primary source of phosphoric acid and phosphate-based fertilizers.31–37 The importance of phosphate rock for the fertilizer industry is based on the H2SO4 (eqn (3)), H3PO4 (eqn (4)) or HNO3 (eqn (5)) route .37| | 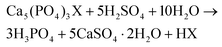 | (3) |
| | | Ca5(PO4)3X + 7H3PO4 → 5Ca(H2PO4)2 + HX | (4) |
| | | Ca5(PO4)3X + 10HNO3 → 5Ca(NO3)2 + 3H3PO4 + HX | (5) |
The sulfuric acid route yields phosphoric acid and phosphogypsum, while the phosphoric acid route directly forms a phosphate fertilizer with the formula Ca(H2PO4)2, called triple superphosphate (TSP) (Fig. 1).37 The nitric acid route is less common but can be used to avoid phosphogypsum formation. Ammonia and potash can be used to obtain NP and NPK fertilizers (Fig. 1).37 Phosphate rock digestion is also an industrial production route for hydrofluoric acid (HF). An important drawback of phosphate rock is that it usually contains traces of heavy metals (e.g. Cd, As, Pb) and radioactive elements such as U, Th and Ra.31,37,38 The fast depletion of phosphate rock reserves is also a major concern as it is the cornerstone of the entire phosphate and fertilizer industry.31–37
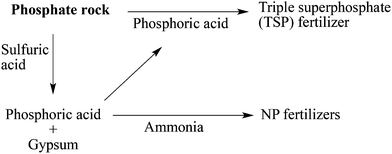 |
| | Fig. 1 Main industrial production routes for phosphate fertilizers. | |
Therefore we have developed a process which not only recovers the valuable and critical antimony from the HALO, but also amounts to a pure calcium phosphate product which can be integrated in the phosphate industry. The absence of radioactive elements in the HALO phosphor, make it an interesting source of phosphate and phosphoric acid. The production of new HALO phosphor has also faded out as this lamp phosphor has been replaced by the rare-earth containing tri-band phosphors. This means that the recycling of HALO present in lamps and waste stockpiles can be a net input to the phosphate cycle.31–37
Dissolution of HALO
Having discussed the general feasibility and strategy of the valorization process, we now describe in detail a potential recycling scheme. The first step is to dissolve the HALO and obtain a stable leachate. HCl and H2SO4 were considered since these are cheap and largely available acids. HNO3 is not a good choice as antimony(III) nitrate solutions are less stable and nitrate effluents have a large impact on the environment.39 When dissolving HALO in HCl, highly soluble Ca(II), Mn(II) and Sb(III) chlorides are formed (eqn (6)). The dissolution mechanism of apatites has been studied in detail, but at low pH values (pH < 0.5) it is sufficient to consider the full dissolution reactions as it occurs fast.40,41| | 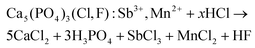 | (6) |
Raising the pH above 1 causes precipitation of insoluble Ca(II), Sb(III) and Mn(II) phosphates and also hydrolysis of Sb(III) ions.1,42–47 The room temperature hydrolysis of Sb(III)–HCl solutions was studied in detail by Hashimoto et al. and they showed that at low pH (1–4) Sb(III) ions are hydrolyzed to form SbOCl(s) and Sb4O5Cl2(s), and above pH 4.5 Sb2O3(s) is formed.47 Unfortunately, controlled hydrolysis of Sb(III) to isolate the antimony, is not an option in this case since the hydrolysis is incomplete at low pH and increasing the pH above pH 2 will cause parrallel precipitation of calcium and manganese phosphates. Therefore, the best strategy was to obtain a stable leachate, which could be further purified by solvent extraction. The experiments were carried out for leachates with a HALO content of 25 g L−1, but the solid/liquid ratio can be further increased since the solubility of these chloride salts is very high. If H2SO4 is used instead of HCl, calcium is precipitated as insoluble CaSO4(s)·2H2O (eqn (7)).
| | 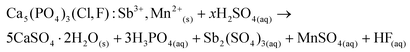 | (7) |
At 1 N acid, HALO is fully dissolved in both HCl and H2SO4, but calcium is precipitated with H2SO4 (Fig. 2). The removal of calcium with H2SO4 could be useful due to the large excess of calcium ions (110![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 Ca:Sb:Mn mol ratio).48,49 In aqueous sulfate solutions the remaining solubility of calcium is approximately 2000 ppm., but this can lowered by adding ethanol which decreases the solubility of CaSO4 (Fig. 3).50 In a 1:1 vol/vol EtOH–H2O solution, the soluble calcium could be reduced to 10 ppm in this HALO leachate.
:1:2 Ca:Sb:Mn mol ratio).48,49 In aqueous sulfate solutions the remaining solubility of calcium is approximately 2000 ppm., but this can lowered by adding ethanol which decreases the solubility of CaSO4 (Fig. 3).50 In a 1:1 vol/vol EtOH–H2O solution, the soluble calcium could be reduced to 10 ppm in this HALO leachate.
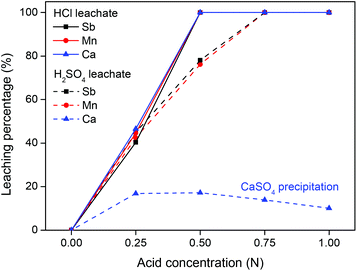 |
| | Fig. 2 Dissolution of HALO (25 mg g−1) in increasingly concentrated HCl and H2SO4 solutions. The samples were shaken (2000 rpm) for 1 h at 25 °C. | |
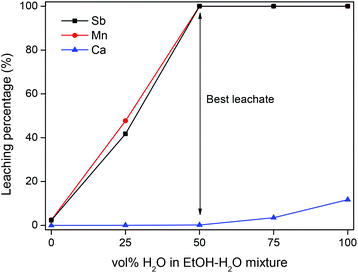 |
| | Fig. 3 Influence of the EtOH–H2O ratio on the dissolution of HALO with 1 N H2SO4. The samples were shaken (2000 rpm) for 1 h at 25 °C. | |
The absence of calcium in the leachate is advantageous, but the HCl dissolution route (1 M, 10 min) is still preferred, because in the sulfuric acid process large amounts of CaSO4·2H2O are created as a useless residue. Extraction of antimony with basic extractants from sulfate media is also less efficient than from chloride media.51,52 Antimony forms strong chloride complexes and can therefore be extracted as tetrachloroantimonate(III) complex SbCl4−.52,53 No waste residue is created with the HCl route, since neutralization of the HCl leachate with NaOH will precipitate a valuable calcium phosphate apatite product and a NaCl-containing effluent which can be discarded in sea water.
Separation of antimony
We previously mentioned that controlled hydrolysis of Sb(III) is not an option to isolate the antimony, because the hydrolysis is incomplete at low pH and increasing the pH above 2 will also cause precipitation of calcium and manganese phosphates. Therefore, antimony must be removed by other means such as solvent extraction or selective electrodeposition of antimony metal. The latter will not be discussed in this work as the demand for antimony metal is much lower than the demand for antimony(III) oxide.1 Electrodeposition of antimony metal in water can also cause formation of highly toxic stibine gas (SbH3) in certain conditions.54 Therefore, selective extraction of the Sb(III) ions is the best option. Sb(III) removal by solvent extraction has been studied previously, although the main focus has been to separate antimony from heavy and precious metal ions (Cd(II), Pb(II), Sn(II), Te(IV), Se(IV), Bi(IV), As(III), …) and from Cu(II) electroplating solutions.51,52,55–65 Most studies have focused on the use of neutral and acidic organophosphorus extractants or the use of secondary and tertiary amines as basic extractants (eqn (9) and (10)).51,52,55–65 These extractants are usually dissolved in organic solvents such as toluene, xylene or kerosene. Some studies have also proposed the use of ion exchange resins and sorbents but these are restricted to the removal of trace amounts of antimony from wastewater.66–68 While solvent extraction is certainly promising for the separation of Sb(III), the use of volatile and flammable organic solvents and highly concentrated acidic solutions often limits their applicability. Here, we propose the use of an ionic liquid (IL) as organic phase to selectively extract the Sb(III) ions from the leachate in mild conditions. Ionic liquids are ideal replacements for organic solvents in solvent extraction processes since they are reusable, non-volatile and non-flammable compounds. The commercial ionic liquid Aliquat® 336 was used here (undiluted). This ionic liquid consists mainly of trioctyl(methyl)ammonium chloride [N8881][Cl] but is in fact a mixture of C8 and C10 chains with C8 predominating. It is abbreviated here as [A336][Cl]. Aliquat® 336 has been used previously for solvent extraction studies of a wide variety of metal ions.53,69 It is a basic extractant, which means it extracts the Sb(III) ions as the anionic complex SbCl4− (eqn (8)).51–53| | | [A336]Cl(IL) + SbCl4−(aq) → [A336][SbCl4](IL) + Cl−(aq) | (8) |
This mechanism is similar to the one observed for secondary and tertiary amine extractants (eqn (9) and (10)).51–53
| | | R3N(org) + HCl(aq) → [R3NH][Cl](org) | (9) |
| | | [R3NH][Cl](org) + SbCl4−(aq) → [R3NH][SbCl4](org) + Cl−(aq) | (10) |
However, secondary and tertiary amines are generally less efficient than quaternary ammonium extractants such as the ionic liquid Aliquat® 336 and require higher chloride concentrations.53 The extraction of Sb(III), Ca(II) and Mn(II) by Aliquat® 336 was tested on a HALO leachate which was prepared by dissolving HALO (25 g L−1) in a diluted HCl solution (1 M). The composition of the leachate is shown in Table 4.
Table 4 Metal concentrations in the leachate (mg L−1) after dissolving HALO (25 g L−1) in a diluted HCl solution (1 M). The dissolution was carried out room temperature while stirring (500 rpm) for 10 min
| Ion |
Concentration (mg L−1) |
| Ca(II) |
9515 |
| Sr(II) |
15 |
| Sb(III) |
263 |
| Mn(II) |
265 |
The leachate was then contacted with water-saturated ionic liquid in a 2:1 leachate/IL phase ratio (wt:wt) and shaken 10 min (2000 rpm) at room temperature. The metal concentrations in the IL phase and the aqueous phase were measured by TXRF to determine the percentage extraction %E (eqn (11)).
| | 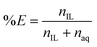 | (11) |
Here, nIL and naq are the number of moles in the IL phase and aqueous phase after extraction, respectively. First, the percentage extraction is plotted versus the chloride concentration by addition of NaCl to the aqueous phase (Fig. 4). The extraction of Sb(III) ions is very strong from the HCl leachate and 100% extraction efficiency is observed even without addition of NaCl. Mn(II) ions are only extracted at very high chloride concentrations as the complex MnCl4−.53 Ca(II) and Sr(II) ions do not form anionic chloro complexes in these conditions, and are therefore barely extracted in these conditions.
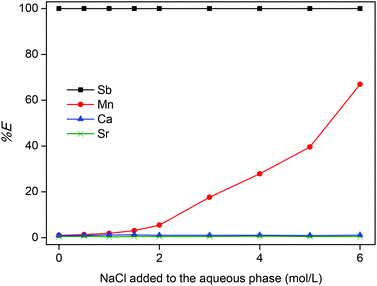 |
| | Fig. 4 Percentage extraction (%E) of Sb(III), Mn(II), Ca(II) and Sr(II) ions as function of the addition of NaCl to the HALO leachate (the aqueous phase). The HALO leachate was prepared by dissolving 25 g L−1 of HALO in 1 M HCl. A 2:1 phase ratio (wt:wt) was used for the aqueous phase and the IL phase [A336][Cl]. | |
The fact that the quaternary ammonium IL Aliquat® 336 allows direct extraction of Sb(III) from the HCl leachate without addition of extra NaCl or HCl, is a significant improvement compared to the use of secondary and tertiary amine extractants which require higher HCl concentrations to fully extract Sb(III).51–53 The use of an ionic liquid also eliminates the need for volatile and flammable organic solvents which reduces the environmental impact of the process. A pure ionic liquid phase is therefore preferred, but sometimes addition of an organic diluent such as toluene can be advantageous for example to diminish the viscosity in order to accelerate the extraction kinetics and facilitate phase separation. Diluting the IL with toluene did not significantly influence the extraction percentage of antimony (SbCl4−) (Fig. 5).
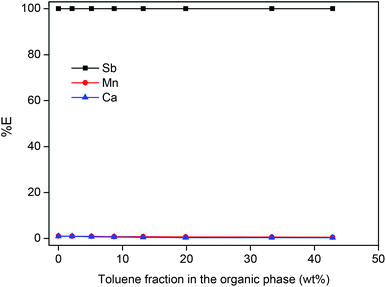 |
| | Fig. 5 Influence of toluene in the IL phase on the extraction of Sb(III), Mn(II) and Ca(II) ions. The extraction was done from a HALO leachate, prepared by dissolving 25 g L−1 of HALO in 1 M HCl. A 2:1 phase ratio (wt:wt) was used for the aqueous phase and the IL phase [A336][Cl]. | |
When the extraction is carried out directly from the leachate with a pure ionic liquid, the Ca(II) and Mn(II) extraction efficiency is 0.98% and 1.03%, respectively. This may seem low, but due to the large excess of Ca(II) in the leachate this is actually a large amount and it has to be removed in order to obtain a pure Sb2O3 end-product. Therefore, a scrubbing step is required, where the IL is contacted with an aqueous solution in order to remove the unwanted elements: Mn(II) and Ca(II) and the co-extracted HCl from the IL. Different scrubbing solutions were tested, but NaCl (1 M) is the best as it removes >99.9% of the Ca(II) and Mn(II) while keeping Sb(III) in the IL (Table 5).
Table 5 Scrubbing (removal) efficiency (%) for Sb(III), Mn(II) and Ca(II) from the ionic liquid using different scrubbing solutions
| Scrubbing solution |
Sb (%) |
Mn (%) |
Ca (%) |
| H2O |
4.5 |
>99.9 |
88.9 |
| HCl (1 M) |
1.1 |
>99.9 |
93.4 |
| NaCl (1 M) |
<0.1 |
>99.9 |
> 99.9 |
Next, the Sb(III) ions have to be removed (stripped) from the IL. This can be done by contacting the IL phase with an alkaline aqueous phase to transfer the Sb(III) ions to the water phase. Ammonia and NaOH solutions are frequently used bases for stripping of Sb(III) from amine extractants.52 Depending on the pH, the Sb(III) ions can then be hydrolyzed and precipitated as SbOCl(s) or Sb2O3(s). The stripping also simultaneously regenerates the IL (eqn (12) and (13)).46,47 NaOH is preferred here as it forms a (non-toxic) NaCl effluent.
| | 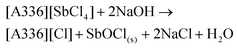 | (12) |
| | 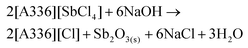 | (13) |
Fig. 6 shows the remaining antimony content in the IL after contacting the Sb(III)-loaded IL with a NaOH-containing aqueous phase. The stripping is sufficiently efficient (>99.99%) above pH 7. Higher pH values are not needed, since the further improvement in stripping efficiency is negligible.
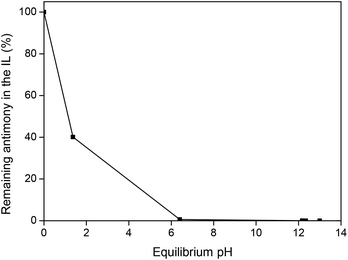 |
| | Fig. 6 Stripping of Sb(III) ions from the ionic liquid phase as function of the equilibrium pH, using a water phase with increasing amounts of NaOH. | |
The precipitation of Sb2O3 from aqueous chloride solutions at pH 4.5–12 is the main hydrometallurgical pathway to Sb2O3 and has been sufficiently demonstrated.1,42–47 It is widely used in the purification of antimony and the production of high-quality Sb2O3.1,42–47 The resulting Sb2O3 product, is the most important commercial form of antimony as it accounts for more than 90% of the antimony market. An important advantage of recovering antimony from lamp phosphor waste is that lamp phosphor waste contains much less contaminants compared to stibnite ore which is frequently polluted with elements such as lead, arsenic, iron and copper. The purity of the final Sb2O3 precipitate was >99.99 wt% with calcium and manganese levels below detection limit for TXRF (10 ppb and 1 ppb, respectively).
Zero-waste valorization of the residue
The HALO leachate (pH 0–0.5), from which the antimony has been initially extracted, is not discarded as waste but is neutralized with NaOH to obtain a clean effluent and a valuable residue. Fig. 7 shows the remaining concentration of calcium and manganese in the leachate as function of pH. At pH 7 (the desired effluent pH), the remaining manganese concentration is below detection limit of TXRF (1 ppb).70 Calcium removal is not complete at pH 7 (≈85% removal), because at this pH calcium phosphates are still partially soluble.71,72 However, since calcium is not toxic, it is more favorable to emit the manganese-free NaCl/CaCl2 effluent at pH 7 than to further increase the pH to remove the remaining calcium ions. The effluent is certainly free of fluoride at pH 7 due to large excess of Ca(II) ions and the low solubility of CaF2 (Ksp = 3.9 × 10−11).
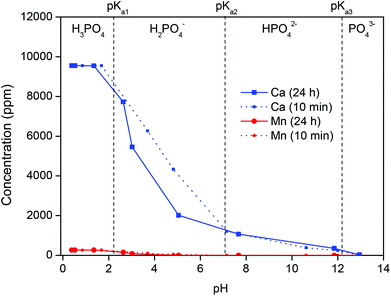 |
| | Fig. 7 The remaining metal concentration in solution is shown to evaluate the precipitation of the apatite residue as function of pH. The initial pH of the leachate after extraction was 0.37 and the pH was increased by using NaOH. | |
The composition of the effluent (excluding NaCl) and the maximum contaminant levels (MCL) allowed in drinking water in the USA, are shown in Table 6.73 Note that the quality requirements are stricter than the ones applicable to the disposal of wastewater. The effluent meets all regulatory limits and can therefore be discarded in the sea.73 Seawater contains 35 g L−1 (0.6 M) of NaCl on average.74
Table 6 Composition of the effluent (pH 7) as well as the maximum contaminant levels (MCL) permitted in drinking water (USA)73
| Element |
Effluent (mg L−1) |
MCL (mg L−1) |
| Ca |
1405 |
No regulatory limit |
| Sr |
15 |
No regulatory limit |
| Sb |
<0.005 |
0.006 |
| Mn |
<0.001 |
0.05 |
| F |
<0.02 |
4 |
| P |
<0.1 |
No regulatory limit |
The precipitate evolves over time as it ripens and this is accompanied by a decrease of the pH (Fig. 7). The main species to precipitate out of solution around pH 7 are CaHPO4 (eqn (14)) and a mixture of chloro-, fluoro- and hydroxoapatite: Ca5(PO4)3(OH,F,Cl) (eqn (15)).71,72
| | | CaCl2 + H3PO4 + 2NaOH → CaHPO4(s) + 2NaCl + 2H2O | (14) |
| | 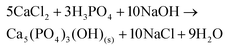 | (15) |
Manganese is co-precipitated and the stages of manganese precipitation can be visualized by the color change of the precipitate (Fig. 8). Below pH 2, no precipitation occurs and the solution is clear. Between pH 3 and 5 the Mn(II) ions are partially coprecipitated with the calcium phosphate, resulting in the typical pink color of Mn(II) salts. Above pH 7, manganese is fully removed as Mn(OH)2(s), which is cream colored and gradually turns brown upon standing (especially at high pH) due to the oxidation of Mn(II) and formation of Mn2O3.75 Note the strong color of the precipitate despite the small manganese content (<1 wt%).
 |
| | Fig. 8 Precipitation of the remaining elements present in the leachate after extraction of antimony. The pH was adjusted with NaOH. | |
The calcium phosphate precipitate can be used in the phosphate and fertilizer industry since it is very similar to phosphate rock which is also a mixture of calcium phosphates and apatites.31–37 Phosphate rock is the primary industrial feed for the production of phosphoric acid, phosphate fertilizers and hydrofluoric acid.37 The small amounts of manganese in the precipitate can be removed in a separate step, but it may be more interesting to keep it since manganese is an important plant micronutrient.76 The high purity of the recycled apatite, contrasts with the apatite in phosphate rock which contains traces of As, Pb and Cd.31,37,38 Phosphate rock is also known for its radioactivity caused by U, Th, Ra and their decay products.31,37,38 The precipitate produced in this process is thus a welcome source of phosphate. An overview of the entire process is shown in Fig. 9.
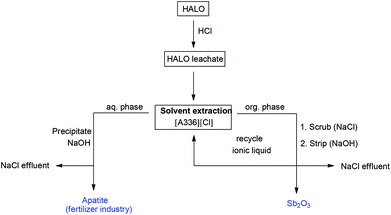 |
| | Fig. 9 Schematic overview of the proposed recycling process. After full dissolution in diluted HCl, the antimony is selective extracted from the leachate using the ionic liquid Aliquat® 336 and pure Sb2O3 is obtained after scrubbing and stripping. The remaining elements in the leachate are then reprecipitated as an apatite which can serve as feedstock in the fertilizer industry. The only waste of this process is an aqueous NaCl effluent which can be discarded in the sea. | |
Note that in this process only HCl and NaOH are consumed and no waste is created except the NaCl effluent. The ionic liquid [A336][Cl] is entirely reusable and non-volatile so no losses occur. This recycling process for the recovery of critical antimony from the HALO lamp phosphor is therefore a good example of a zero-waste valorization approach. It is in theory also possible to regenerate HCl and NaOH by electrolysis of the NaCl solution in a membrane cell (eqn (16)), followed by the recombination of H2 and Cl2 to form HCl (eqn (17))
| | | 2NaCl + H2O + E(V) → 2NaOH + H2 + Cl2 | (16) |
This would result in a completely closed-loop recycling process as the reagents can be regenerated from the effluent and only energy would be consumed. Although the electrolysis of NaCl and synthesis of HCl is carried out in this way industrially on very large scale, it is probably more interesting for medium-sized recycling plants to purchase HCl and NaOH and discard the clean NaCl solution in the sea.
Conclusion
Halophosphate is the largest component in lamp phosphor waste (50 wt%), but is usually discarded as a non-valuable residue in rare-earth recycling even though it contains industrially important antimony. The increasing supply risk for antimony combined with decreasing rare-earth prices should turn the attention of researchers to the valorization of this lamp phosphor waste component. Here, a process is proposed to efficiently recover the antimony (>99.99 wt% purity) and to valorize the remaining elements as a high-quality feedstock for the phosphate industry. This green process operates at room temperature and no waste is created except a NaCl effluent, which makes it a more sustainable source of antimony than the energy-intensive processing of stibnite ore. The process can be integrated in lamp phosphor recycling schemes aimed at recovering rare earths. The main advantage of this step-wise and selective approach to lamp phosphor recycling is that less chemicals and energy are consumed to obtain pure end-products. Selective valorization of the most valuable components is a better strategy than full dissolution followed by separation, when dealing with a highly complex feedstock such as lamp phosphor waste. It is our hope that this paper will highlight the potential of lamp phosphor waste as an overlooked source of antimony and stimulate the inclusion of antimony recovery in future lamp phosphor waste recycling schemes.
Acknowledgements
The authors wish to thank the KU Leuven (projects GOA/13/008 and IOF-KP RARE3) and the FWO Flanders (PhD fellowship to DD) for financial support.
Notes and references
- C. G. Anderson, Chem. Erde., 2012, 72, 3–8 CrossRef CAS.
-
G. B. Haxel, J. B. Hedrick and G. J. Orris, Rare Earth Elements Critical Resources for High Technology - Fact Sheet 087-02, U.S. Geological Survey, 2002 Search PubMed.
-
Report on Critical raw materials for the EU, European Commission, DG Enterprise & Industry, Brussels, 2014.
- G. Belardi, N. Ippolito, L. Piga and M. Serracino, Thermochim. Acta, 2014, 591, 22–30 CrossRef CAS.
-
J. J. Braconnier and A. Rollat, Solvay, European Patent, EP 2419377 A1, 2012 Search PubMed.
- T.-C. Chang, S.-F. Wang, S.-J. You and A. Cheng, J. Environ. Eng. Manage., 2007, 435–439 CAS.
- R. Dobrowolski and J. Mierzwa, Mater. Chem. Phys., 1993, 34, 270–273 CrossRef CAS.
-
S. T. Henderson, A. M. Marsden and L. Thorn Lighting, Lamps and lighting: a manual of lamps and lighting, Edward Arnold, London, 1972 Search PubMed.
-
K. Shinde, S. J. Dhoble, H. C. Swart and K. Park, in Phosphate Phosphors for Solid-State Lighting, Springer, Berlin Heidelberg, 2012, vol. 174, ch. 6, pp. 151–189 Search PubMed.
- A. M. Srivastava and T. J. Sommerer, Electrochem. Soc. Interface, 1998, 7, 28–31 CAS.
- C. R. Ronda, T. Jüstel and H. Nikol, J. Alloys Compd., 1998, 275–277, 669–676 CrossRef CAS.
-
S. Shionoya, W. M. Yen and H. Yamamoto, Phosphor Handbook, Taylor & Francis, Boca Raton, 2006 Search PubMed.
-
T. F. Soules, in Physics and Chemistry of Luminescent Materials, ed. C. R. Ronda, L. E. Shea and A. M. Srivastava, Electrochemical Society, Pennington, USA, 2000, vol. 40, pp. 56–68 Search PubMed.
- K. Binnemans and P. T. Jones, J. Rare Earths, 2014, 32, 195–200 CrossRef CAS.
- K. Binnemans, P. T. Jones, B. Blanpain, T. Van Gerven, Y. Yang, A. Walton and M. Buchert, J. Cleaner Prod., 2013, 51, 1–22 CrossRef CAS.
-
B. J. Baliga, The IGBT Device: Physics, Design and Applications of the Insulated Gate Bipolar Transistor, Elsevier Science, 2015 Search PubMed.
- Compact Fluorescent Lamps Could Nearly Halve Global Lighting Demand for Electricity, http://www.worldwatch.org/node/5918.
- Y. Wu, X. Yin, Q. Zhang, W. Wang and X. Mu, Resour., Conserv. Recycl., 2014, 88, 21–31 CrossRef.
- K. Binnemans, P. T. Jones, K. Acker, B. Blanpain, B. Mishra and D. Apelian, JOM, 2013, 65, 846–848 CrossRef.
- V. Innocenzi, I. De Michelis, B. Kopacek and F. Vegliò, Waste Manage., 2014, 34, 1237–1250 CrossRef CAS PubMed.
- H. Liu, S. Zhang, D. Pan, J. Tian, M. Yang, M. Wu and A. A. Volinsky, J. Hazard. Mater., 2014, 272, 96–101 CrossRef CAS PubMed.
- G. Mei, P. Rao, M. Mitsuaki and F. Toyohisa, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2009, 24, 418–423 CrossRef CAS.
- C. Tunsu, C. Ekberg and T. Retegan, Hydrometallurgy, 2014, 144–145, 91–98 CrossRef CAS.
- I. Urniezaite, G. Denafas and D. Jankunaite, Waste Manage. Res., 2010, 28, 609–614 CrossRef CAS PubMed.
- F. Yang, F. Kubota, Y. Baba, N. Kamiya and M. Goto, J. Hazard. Mater., 2013, 254–255, 79–88 CrossRef CAS PubMed.
- H. L. Yang, W. Wang, H. M. Cui, D. L. Zhang, Y. Liu and J. Chen, J. Chem. Technol. Biotechnol., 2012, 87, 198–205 CrossRef CAS.
- D. Dupont and K. Binnemans, Green Chem., 2015, 17, 856–868 RSC.
-
J. H. Hubbell and S. M. Seltzer, National Institute of Standards and Technology (NIST), NIST X-ray Attenuation Databases, http://physics.nist.gov/PhysRefData/XrayMassCoef/tab3.html, 2014.
-
R. Otto and A. Wojtalewicz, US patent2012/0027651 A1, 2012 Search PubMed.
- F. H. Firsching and S. N. Brune, J. Chem. Eng. Data, 1991, 36, 93–95 Search PubMed.
- K. Binnemans, P. T. Jones, B. Blanpain, T. Van Gerven and Y. Pontikes, J. Cleaner Prod., 2015, 99, 17–38 CrossRef CAS.
- C. J. Dawson and J. Hilton, Food Policy, 2011, 36(Supplement 1), S14–S22 CrossRef.
- R. H. E. M. Koppelaar and H. P. Weikard, Global Environ. Chang., 2013, 23, 1454–1466 CrossRef.
- L. Reijnders, Resour., Conserv. Recycl., 2014, 93, 32–49 CrossRef.
- R. W. Scholz, A. E. Ulrich, M. Eilittä and A. Roy, Sci. Total Environ., 2013, 461–462, 799–803 Search PubMed.
- P. Walan, S. Davidsson, S. Johansson and M. Höök, Resour., Conserv. Recycl., 2014, 93, 178–187 CrossRef.
-
T. P. Hignett, Fertilizer Manual, Springer, Netherlands, 2013 Search PubMed.
- M. Chen and T. E. Graedel, J. Cleaner Prod., 2015, 91, 337–346 CrossRef CAS.
-
N. C. Norman, Chemistry of Arsenic, Antimony and Bismuth, Springer, 1998 Search PubMed.
- S. V. Dorozhkin, Comments Inorg. Chem., 1999, 20, 285–299 CrossRef CAS.
- S. V. Dorozhkin, World J. Methodol., 2012, 2, 1–17 CrossRef PubMed.
- A. H. Abdullah, N. H. M. Noor, I. Ramli and M. Hashim, Mater. Chem. Phys., 2008, 111, 201–204 CrossRef CAS.
- X. Y. Chen, H. S. Huh and S. W. Lee, J. Solid State Chem., 2008, 181, 2127–2132 CrossRef CAS.
- M. Vuković, Z. Branković, D. Poleti, A. Rečnik and G. Branković, J. Sol–Gel Sci. Technol., 2014, 72, 527–533 CrossRef.
- M. Filella, N. Belzile and Y.-W. Chen, Earth-Sci. Rev., 2002, 59, 265–285 CrossRef CAS.
- J.-g. Yang and Y.-t. Wu, Hydrometallurgy, 2014, 143, 68–74 CrossRef CAS.
- H. Hashimoto, T. Nishimura and Y. Umetsu, Mater. Trans., 2003, 44, 1624–1629 CrossRef CAS.
- Y. Ling and G. P. Demopoulos, J. Chem. Eng. Data, 2004, 49, 1263–1268 CrossRef CAS.
- A. Al-Othman and G. P. Demopoulos, Hydrometallurgy, 2009, 96, 95–102 CrossRef CAS.
- V. Gomis, M. D. Saquete and J. García-Cano, Fluid Phase Equilib., 2013, 360, 248–252 CrossRef CAS.
- A. Alian and W. Sanad, Talanta, 1967, 14, 659–669 CrossRef CAS PubMed.
- B. M. Sargar, M. M. Rajmane and M. A. Anuse, J. Serb. Chem. Soc., 2004, 69, 283–298 CrossRef CAS.
- F. G. Seeley and D. J. Crouse, J. Chem. Eng. Data, 1966, 11, 424–429 CrossRef CAS.
- L. Tomlinson, J. Electrochem. Soc., 1964, 111, 592–596 CrossRef CAS.
-
P. Navarro, J. Simpson and F. J. Alguacil, Hydrometallurgy, 1999, 53, 121–131 Search PubMed.
- A. R. Byrne and D. Gorenc, Anal. Chim. Acta, 1972, 59, 81–89 CrossRef CAS.
- E. M. Donaldson and M. Wang, Talanta, 1986, 33, 35–44 CrossRef CAS PubMed.
- S. Facon, G. Cote and D. Bauer, Solvent Extr. Ion Exch., 1991, 9, 717–734 CrossRef CAS.
- A. P. Grimanis and I. Dore Hadzistelios, Anal. Chim. Acta, 1968, 41, 15–21 CrossRef CAS.
- J. N. Iyer and P. M. Dhadke, Indian J. Chem. Technol., 2003, 10, 665–669 CAS.
- N. V. Kirichenko, A. I. Nikolaev, V. G. Maiorov, A. V. Tyuremnov and E. G. Il'in, Russ. J. Inorg. Chem., 2013, 58, 474–480 CrossRef CAS.
- H. K. Lin, Hydrometallurgy, 2004, 73, 283–291 CrossRef CAS.
- S. G. Sarkar and P. M. Dhadke, Sep. Purif. Technol., 1999, 15, 131–138 CrossRef CAS.
- J. A. N. Szymanowski, Miner. Process. Extr. Metall. Rev., 1998, 18, 389–418 CrossRef CAS.
- R. G. Vibhute and S. M. Khopkar, Talanta, 1989, 36, 957–959 CrossRef CAS PubMed.
- N. V. Deorkar and L. L. Tavlarides, Hydrometallurgy, 1997, 46, 121–135 CrossRef CAS.
- P. Navarro and F. J. Alguacil, Hydrometallurgy, 2002, 66, 101–105 CrossRef CAS.
- L. Łukaszczyk and W. Żyrnicki, J. Pharm. Biomed. Anal., 2010, 52, 747–751 CrossRef PubMed.
- S. Wellens, B. Thijs and K. Binnemans, Green Chem., 2012, 14, 1657–1665 RSC.
- Bruker, Detection limits for the NIST 1640 reference standard, https://www.bruker.com/products/x-ray-diffraction-and-elemental-analysis/micro-xrf-and-txrf/s2-picofox/applications.html.
- J. F. De Rooij, J. C. Heughebaert and G. H. Nancollas, J. Colloid Interface Sci., 1984, 100, 350–358 CrossRef CAS.
-
J. W. Sausville, U.S. Patent, US3663473 A, 1972 Search PubMed.
-
(EPA), Environmental Protection Agency, National Primary Drinking Water Regulations, USA, 2009 Search PubMed.
- M. H. Sharqawy, J. H. Lienhard and S. M. Zubair, Desalin. Water Treat., 2010, 16, 354–380 CrossRef CAS.
-
G. Rayner-Canham, in Descriptive Inorganic Chemistry, Freeman, New York, 1996, ch. 8 Search PubMed.
- G. DalCorso, A. Manara, S. Piasentin and A. Furini, Metallomics, 2014, 6, 1770–1788 RSC.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence














