
Naturally nano: synthesis of versatile bio-inpired monodisperse microspheres from Bacillus spores and their applications†
Zhiming
Zeng
a,
Yin
Zhong
a,
Huicui
Yang
a,
Ruihua
Fei
a,
Rui
Zhou
b,
Rafael
Luque
c and
Yonggang
Hu
*a
aState Key Laboratory of Agricultural Microbiology, College of Life Science and Technology, Huazhong Agricultural University, Wuhan 430070, China. E-mail: Yongganghu@mail.hzau.edu.cn
bState Key Laboratory of Agricultural Microbiology, College of Veterinary Medicine, Huazhong Agricultural University, Wuhan 430070, China
cDepartamento de Quimica Organica, Universidad de Cordoba, Campus de Rabanales, Edificio Marie Curie (C-3), Ctra Nnal IV-A, Km 396, E14014, Cordoba, Spain. E-mail: q62alsor@uco.es
First published on 20th August 2015
Abstract
A novel class of bionanocomposites based on monodisperse microparticles containing metal nanoparticles including Au, Pd, Ag and Pt were synthesized and characterized using a simple and efficient approach. The versatile nanocomposites exhibited unique possibilities as new biosensor for highly selective and sensitive immunoassays as well as a remarkable enhancement in catalytic activity in the reduction of 4-nitrophenol selected as model reaction which illustrated their potential in various different applications.
Introduction
Uniform sized colloidal nanoparticles (NPs) have attracted a great deal of attention for both fundamental research and technological applications. The large surface-to-volume ratio and the high collision frequency of NPs, associated to their greater mobility, accounts for a high surface energy. These properties cause serious stability problems including a tendency to aggregate (to minimize such high surface energies), changes in shape as well as reduction and eventual loss of their initial activity.1 An alternative method to stabilize NPs entails the design of nanomaterials by loading NPs onto the surfaces of supporting materials. The symbiotic effects of the nanocomposites can provide new insights that overcome the main limitations of unsupported nanoparticles as well as yield novel properties and potential applications. These nanocarriers can be modified with affinity groups, such as –NH2,2 –SH,3 –COOH,4 and –CN,5 using various activation methods. However, the direct fabrication of suitable nanocarriers with a homogenous distribution of functionalities on their surfaces still remains a significant challenge.6 The development of novel nanocarriers to form new nanocomposites with high performance and low costs is nevertheless of great importance and relevance towards advances in the design of functional innovative nanomaterials with specific applications.Functional monodisperse microparticles experienced increasing interest in recent years due to their relevant applications in micro-electromechanical systems,7 chemical release systems,8 optical materials,9 biological applications,10,11 environmental analysis12 as well as their use as catalyst supports.13 Various types of microspheres have been prepared as nanocarriers to load various NPs to obtain new nanocomposites such as optoelectronic elements and catalysts, with high stability, recyclability, and long-term stability.11,14–17
Two general chemical approaches have been generally proposed for the preparation of functional monodisperse microparticles. The first approach involves pre-polymer modification as compared to the second based on polymerization of monomers with desired functional groups.18 Both methodologies essentially have inherent drawbacks such as the usage of hazardous chemicals and the need for multiple steps of preparation. The stressing environmental concerns push us to develop an eco-friendly, simple and cost-effectively method for the preparation of large quantities of highly uniform, functional monodisperse microparticles.19,20
The use of living single cells such as fungi, yeast, bacteria and spores was considered an environment friendly, simple and nontoxic alternative for the preparation of novel functional microspheres.21–26 Several nanoparticles including SiO2, TiO2, Ga2O3, Au, halloysite nanotubes and NdFeB have been assembled onto biological cells to form nanocomposites. In particular, the design of bacterial endospore-based nanocomposites has recently attracted a significant interest due to the controllable protective properties of endospore shells.23–25 These cell-based nanocomposites exhibited attractive application prospects in scientifically and technologically important areas including cell therapy, cell-based sensors, electronic devices, and biocatalysis as well as fundamental studies in single cell-based biology. However, to the best of our knowledge, this field is still in its infancy and no reports can be found the preparation of spore-based microspheres loaded with metal nanoparticles and their applications. In this regard, the concept of using spores of B. subtilis and B. amyloliquefaciens as biology derived materials for the preparation of functional microspheres can be highly advantageous.
Herein, we propose a novel biological strategy that makes use of bacterium spores from Bacillus subtilis and Bacillus amyloliquefaciens to prepare highly monodispersed microspheres as prospective metal nanoparticle (NPs)-supports for different applications (Scheme 1). Both Bacillus species, not regarded as pathogens, are classified as a novel food ingredient and employed as probiotics for both human and animal consumption.27,28
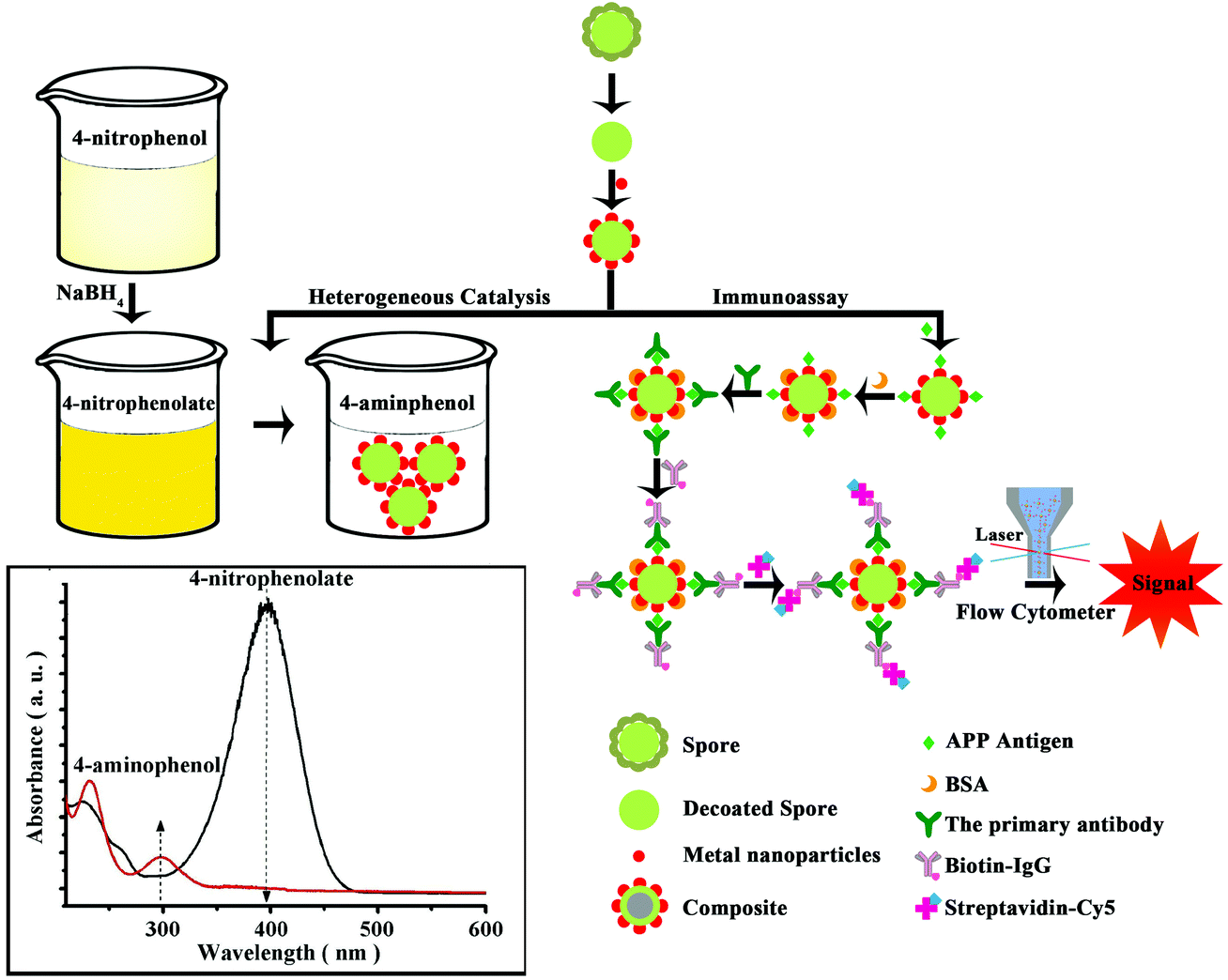 | ||
| Scheme 1 Spore-based hybrid microspheres loaded with highly dispersed noble metal nanoparticles and their applications in heterogeneous catalysis and immunoassay. | ||
Experimental
Materials
Chloroauric acid, sodium borohydride, hydrochloric acid, potassium dihydrogen phosphate, disodium hydrogen phosphate, sodium hydroxide, sodium chloride, disodium salt of EDTA, ethanol, anhydrous calcium chloride, glacial acetic acid, concentrated hydrochloric acid, sodium dodecyl sulfate, Tryptone, Yeast extract and agar were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China) and dithiothreitol from Regal Biotechnology Co., Ltd (Anhui, China). All materials were of analytical grade and used as received without any further purification. Ultrapure water was purified by a Millipore Simplicity system and had an electrical resistance of 18 MΩ cm.Preparation of spore-based highly monodisperse microspheres
Bacillus subtilis strain and Bacillus amyloliquefaciens strains were kindly provided by State Key Laboratory of Agricultural Microbiology, Huazhong Agricultural University. To prepare the spores, B. subtilis and B. amyloliquefaciens (CCTCC AB 2013062) strains stored at −20 °C were grown for 12 h in beef extract peptone liquid medium at 37 °C under stirring (160 rpm). The activated strains were spread on beef-peptone agar plates and incubated at 37 °C for 7 days to promote extensive sporulation and autolysis of vegetative cells. Lawns were then scraped and washed from plates into deionized water. Vegetative cell debris were largely removed by five washes (50 mL of deionized water) with resuspension and centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 min. After that, the collected spores were resuspended in deionized water and stored at 4 °C for further use.
000 rpm for 5 min. After that, the collected spores were resuspended in deionized water and stored at 4 °C for further use.
The stored spores were diluted with deionized water and treated under ultrasounds for 20 min, followed by centrifugation at 8000 rpm (10 min). The supernatant was removed. The remained spores were subsequently incubated in decoating buffer (containing 0.1 mol L−1 NaCl, 0.1 mol L−1 NaOH, 1% sodium dodecyl sulfate and 0.1 mol L−1 dithiothreitol) with magnetic force stirring for 1 h at 70 °C. The protein fragments of the outer layer were lysed using a lysing buffer. The as-prepared spores were then inactivated using a high-pressure (0.1 MPa) steam sterilizer for 30 min at 121 °C. After centrifugation, the supernatant was discarded and the treated spores (TSs) were repeatedly washed with deionized water (six times) and then stored at 4 °C until needed. The number of TSs was determined by direct counting with a Burker chamber under an optical microscope. The total volume of solution had a concentration of about 1.08 × 109 particles per mL.
Preparation of noble metal NPs/nanocubes
Au NPs were synthesized by reduction of the HAuCl4 solution with sodium citrate according to the procedure described in Frens's method.29 Briefly, 100 mL of 0.01% (w/v) of the HAuCl4 solution was heated under refluxing conditions for 15 min, and then 1% (w/v) sodium citrate solution was added. Heating continued for 15 additional minutes after the colored solution remained unchanged. Ag NPs were prepared as follows: a solution of 5 mmol L−1 AgNO3 (100 mL) was added to 300 mL of vigorously stirred ice-cold 2 mmol L−1 NaBH4. A solution of 1% polyvinylalcohol (50 mL) was added during the reduction. The mixture was then refluxed for ca. 1 h to decompose any NaBH4 excess. Comparably, Pd NPs were synthesized following the addition of 2.5 g polyvinylpyrrolidone to 150 mL of ethanol in a flask until complete dissolution, followed by the addition of 50 mg Palladium acetate. The mixture was reacted in an oil bath at 110 °C for 2 h.Pt NPs were prepared from a solution of 1 mmol L−1 K2PtCl6 (3 mL) in 21 mL double distilled water, followed by the addition of KBr (0.0357 g) under stirring (for 1 h) and Polyvinylpyrrolidone (0.033 g). The reaction was conducted in an oil bath at 130 °C for 3 h.
Pd nanocubes were synthesized in an oil bath. K2PdCl4 (0.01 mol L−1, 1.00 mL), deionized water (17.50 mL) and CTAB (0.10 mol L−1, 2.50 mL) were mixed in a flask at 80 °C for 15 minutes until the solution temperature was equilibrated. Ascorbic acid (0.10 mol L−1, 0.16 mL) was subsequently added into the flask. The resultant mixture solution stirred for 2 h and then cooled down at room temperature. NPs/nanocubes solutions were stored at 4 °C prior to their use.
Preparation of NPs/nanocubes nanocomposites
The solution of NPs/nanocubes was mixed with an as-prepared aqueous dispersion of microspheres under ultrasonication in a water bath for a few minutes. The final nanocomposites were separated out after six centrifugation/wash/redispersion cycles to remove any residual free NPs/nanocubes and eventually redispersed in deionized water and stored at 4 °C for further uses.Characterization
A transmission electron microscope (TEM; JEM-100CXII, JEOL Ltd, Tokyo, Japan), a field emitting scanning electron microscope (SEM; X-650, Hitachi, Ltd, Tokyo, Japan), a field-emission high resolution transmission electron microscope (JEM-2100F, JEOL Ltd, Japan) and a high-resolution transmission electron microscope (JEM-2100 (HR), JEOL Ltd, Tokyo, Japan) were utilized to characterize the morphology and structure of the obtained nanomaterials.The thermal stability of TSs was analyzed using a thermogravimetric-differential scanning calorimetry (TG-DSC; PerkinElmer Instruments (Shanghai) Co., Ltd, Shanghai, China).
Crystal structure identification was carried out using an X-ray diffractometer (XRD; D/MAX-RB, Rigaku, Tokyo, Japan) with Cu Kα radiation (λ = 1.54056 Å) in 2θ ranging from 10 to 85°. All samples for XRD measurements were supported on glass substrates.
Fourier transform infrared spectra (FTIR) in the range of 4500–250 cm−1 were recorded using a FTIR spectrometer (330FTIR, Thermo Fisher Scientific, Waltham, MA, USA) in KBr pressed disks.
Surface enhanced Raman scattering spectroscopy (SERS) measurements were performed using a laser Raman spectrometer (IN VIA, Renishaw, Gloucestershire, UK) with an incident 514 nm laser light perpendicular to the substrate.
UV-visible (UV-vis) absorption spectra were recorded on a UV–vis spectrophotometer (Beckman Coulter, Brea, CA, USA) in aqueous media. A zetasizer (Nano ZS90, Malvern Instruments Ltd, Malvern, UK) was used to determine the size distribution of NPs/nanocubes and the zeta potentials of spores.
Acid–base titrations were conducted using an automatic potentiometric titrator (Metrohm titrator 836, Metrohm, Herisau, Switzerland). Potentiometric titrations of concentrated (3.2 mg dry weight per mL) spores suspension were carried out under N2 atmosphere at room temperature. The suspension was titrated using 0.50 mol L−1 NaOH and 1.02 mol L−1 HCl solutions. A known amount of HCl was added at the beginning of the experiment to lower the pH to approximately 2.2. The spore suspension was equilibrated for 40 min and titrated to pH 12 with NaOH. Therefore, the total adsorption site (2.2 ≤ pKa ≤ 12.0) concentrations on adsorbent surfaces were calculated according to the changes in pH values of the suspension and the amount of added OH−. The total site concentrations of functional groups on spores’ surfaces were calculated according to the titration curves using a non-electrostatic approach. The data optimization program FITEQL was used to model titration data in order to obtain site concentrations and acidity constants for proton-active functional groups present on the spore surface.
The heat of Au NPs adsorption on the surface of spores was measured in an isothermal microcalorimeter NANO ITC2G (ITC; TA Instruments, New Castle, DE, USA). The calorimeter was positioned at a temperature controlled environment (25 ± 0.1 °C), and calibrated by a standard electric pulse. The spore suspension (1.0 mL, 0.86 mg dry mass per mL) was filled into a 1 mL stainless steel titration ampoule and was stirred at 120 rpm by a twisted stirrer paddle. The ampoule was lowered into the calorimeter and allowed to equilibrate for at least 120 min. After a highly stable heat flow (i.e. the signal deviation was less than 200 nW in 30 min) was obtained, the titration started by injections of the Au NPs solution into the ampoule. A total volume of 250 μL Au NPs solution was titrated at a rate of 1 μL s−1 and the time interval between each injection was 20 min. The output signal collected as heat power and versus time was continuously acquired by a NanoAnalyze software. Control experiments (i.e. the dilution heat) were performed by titrating Au NPS solution into ultrapure water.
Adsorption experiments were conducted as follows: 30 μL spores suspensions containing 0.27 mg spores and a series of Au NPs stock solutions were mixed in a 2 mL centrifuge tube. The final Au NPs concentrations in the suspension ranged from 7.2 to 43.2 mg L−1. The mixture was stirred at 25 °C (140 rpm) for 12 h. After centrifugation at 8000 rpm (3 min), the concentration of Au NPs in the supernatant was analyzed using a Thermo Scientific Multiskan Go instrument.
Catalytic reactions for 4-nitrophenol reduction with Au microspheres
An aqueous solution of sodium borohydride (NaBH4, 0.5 mol L−1, 3.0 mL) was added to a solution of 4-nitrophenol (0.1 mmol L−1, 3.0 mL). A certain amount of Au NPs, determined by inductively coupled plasma mass spectrometry (ICP-AES; PerkinElmer, Waltham, MA, USA), was then added to this solution. Immediately after the addition of Au NPs, UV-vis spectra of the mixture were recorded with a UV-vis spectrometer. Kinetic studies were performed by measuring the change in intensity of the absorbance at 400 nm with time. The spectra were recorded every minute in the 250–550 nm range.For reusability experiments, the reaction solution composed of NaBH4 (0.5 mol L−1, 3.0 mL), 4-nitrophenol (0.1 mmol L−1, 3.0 mL) and Au-microspheres (3.1 μg) was centrifuged upon reaction completion (typically 8 minutes). Au-microspheres were then collected and redispersed in a fresh mixture of the reactants for another catalytic run. All of the experiments were carried out at room temperature without stirring.
Immunoassay experiments
A BioTek Synergy HT Multi microplate reader (BioTek Instruments, Inc., Winooski, VT, USA) was used to measure the immobilization of recombinant ApxIVA protein on spores. A VORTEX-5 Vortex Shaker (Kylin-Bell Lab Instruments Co., Ltd, Haimen, China) was utilized to disperse spores. The incubation procedures were carried out using a heating platform (Jing Hong Laboratory Instrument Co., Ltd, Shanghai, China). Samples were analyzed with a FACS Calibur flow cytometer (Becton, Dickinson and Company, Franklin L., NJ, USA) using an excitation wavelength of 635 nm.Immobilization of recombinant ApxIVA proteins onto Au microspheres
Recombinant ApxIVA proteins were immobilized onto Au microspheres in a 2 mL Eppendorf tube. First, 200 μL of 0.1 mg mL−1 recombinant ApxIVA protein diluted in coating solution was covalently attached to the surface of Au microspheres. The Eppendorf tube was stirred (160 rpm) at 37 °C for 3 h. After removing the supernatant, Au microspheres were washed three times with 1 mL PBST to remove any residual ApxIVA protein. To discourage non-specific binding, Au microspheres were blocked with 1 mL blocking buffer (BSA dissolved in PBS at pH 7.4) and then the tube was incubated (160 rpm) at 37 °C, after which the modified Au microspheres were washed three times with 1 mL PBST.Procedures for the immunoassay
Firstly, a series of dilutions of pig serum (200 μL) were prepared into 2 mL Eppendorf tubes containing 200 μL Au microspheres-ApxIVA and stirred (160 rpm) at 37 °C for 30 min. The tubes were then centrifuged at 8000 rpm for 3 min. The supernatant was removed and the microspheres were washed three times with 1 mL PBST. Secondly, 200 μL biotin-IgG was added to each Eppendorf tube. These tubes were incubated under stirring (160 rpm) at 37 °C for 30 min followed by washing (3 times) of Au microspheres. Thirdly, 200 μL Cy™5-conjugated streptavidin was transferred to each tube and incubated with shaking (160 rpm) at 37 °C for 30 min in the absence of light. The washing steps were also repeated three times and the fluorescent encoded Au microspheres were resuspended with 1 mL PBS. Following the above mentioned immune reaction, the fluorescence signal was measured using a FACS Calibur flow cytometer (at least 10000 Au microspheres were analyzed).
Results and discussion
TEM and SEM images of B. subtilis 168 spores are shown in Fig. 1A1 and A3, respectively. These images revealed the presence of a highly cross-linked layer of vegetative cell-type peptidoglycan,30 covered by a soft and flexible outer layer composed of proteins.31 In order to prevent metal NP aggregation on the surface of the spores, the outer layer from the core surface was firstly sonicated and subsequently peeled off. The protein fragments of the outer layer were then lysed using a lysing buffer.32 Images shown in Fig. 1A2 and A4 confirmed that clear, smoothly shaped and essentially solid oval spheres (ca. 0.6 ± 0.1 μm × 1.0 ± 0.2 μm) with a narrow size distribution could be obtained for TSs upon surface removal. | ||
| Fig. 1 Nanoparticles, nanocubes, native spores, TSs, hybrid microspheres were characterized using TEM, SEM, HAABF-STEM, HAADF-STEM, EDS, and HRTEM. A1 and A2 were the TEM images of native spores and TSs, respectively. A3, A4 and A5 were the SEM images of native spores, TSs and Au microspheres, respectively. TEM images of Au, Ag, Pt, Pd, and Pd nanocubes correspond to B1–F1. B2–F2, B3–F3, B4–F4, B5–F5. B6–F6 are images of TEM, HAABF–STEM, HAADF–STEM, EDS and HRTEM of hybrid microspheres, respectively. Hybrid microspheres loading Au, Ag, Pt, Pd, and Pd nanocubes were respectively corresponding to B, C, D, E, and F. Insets in images B1–F1 correspond to the size distribution of NPs/nanocubes. | ||
FTIR spectroscopy provided additional insights into the surface characteristics of TSs, pointing to the presence of carboxylic acids (RCOOH; 3410, 1727, 1652, and 1383 cm−1), peptide bonds (–CO–NH–; 1652, and 1568 cm−1) and amino groups (–NH2, and –NH; 3100, and 3300 cm−1) among others (Fig. 2A). These findings indicate the presence of a range of anchoring sites extensively utilized to stabilize various metals (e.g. Au, Pd, etc.) and biomolecules.2,4
 | ||
| Fig. 2 (A) FTIR spectra of the treated spores (black line) and the Au microspheres (red line). (B) Potentiometric titration curves for TSs and native spores. (C) TG-DSC curves of the treated spores. (D) XRD patterns of treated spores (black line) and Au microspheres (red line). (E) UV–vis spectra of colloidal gold nanoparticles (black line) and Au microspheres (red line). (F) SERS spectra of the treated spores (black line) and the Au microspheres (red line). | ||
Acidity constants (pKa) and site concentrations of functional groups on spore walls (Fig. 2B) further confirmed the presence of carboxylic, amino and hydroxyl groups on the spore surfaces, with typical deprotonation constants of (4 < pKa < 6, acids) and (9 < pKa < 11, amino or hydroxyl),33,34 respectively. Interestingly, the concentraction of carboxylic groups could be improved to 1.74 × 10−3 mol g−1 (pKa of 4.68; pKa of 5.99) as compared to a 1.65 × 10−3 mol g−1 observed for native spores (pKa of 5.94). Comparatively, the concentration of amino/hydroxyl groups on TSs (pKa of 9.00) were found to decrease to 3.41 × 10−3 mol g−1 (from 3.94 × 10−3 mol g−1 for native spores, pKa of 10.35). With a remarkably superior TSs zeta potential (in water) of −48.2 mV as compared to untreated native spores (−39.09 mV), these results pointed out a significantly improved dispersity and stability of TSs in aqueous media most probably related to the presence of carboxylic functional groups on their surface. In any case, TSs were additionally found to be stable and highly dispersible in a number of organic solvents (i.e. ethanol, methanol, acetone, formaldehyde, acetonitrile, formic acid and dimethylformamide) with the possibility to be redispersed after solvent removal (Fig. S2, ESI†).
The thermal stability of TSs was subsequently analysed by means of TG-DSC (Fig. 2C). Two main mass loss contributions could be observed in the TG curve of the microspheres upon heating. The first mass loss (before 100 °C) accounted for a ca. 10.0% weight loss and can be assigned to the removal of physisorbed water molecules. An endothermic peak (85 °C) could be also visualized in the DSC curves. The second weight loss (ca. 5.0%), accompanied by an endothermic peak at 247.9 °C in the DSC curves, corresponded to the removal of binded water molecules as well as to a potential partial removal of volatile organics. The unusual two-step weight loss of water can be related to the presence of multiple concentric shells in the spores, which can absorb water.35–38 However, the degradation of the material starts at ca. 248 °C, indicating a remarkable thermal stability of TSs which can be particularly useful for catalytic-related applications.
Noble metal NPs including Au, Ag, Pt, Pd, and Pd nanocubes were subsequently incorporated into TSs. As example, Fig. 1B1, C1, D1, E1 and F1 clearly illustrate the formation of NPs with small sizes and a narrow NP size distribution. The average sizes of Au, Ag, Pt and Pd were found to be ca. 20.5 nm, 3.2 nm, 2.9 nm, 2.0 nm, respectively. Pd nanocubes incorporated into TSs possessed an average size of 32.4 nm. These nanocomposites were characterized in detail using HRTEM and derived HAABF-STEM, HAADF-STEM and EDS (Fig. 1). In most cases, all supported NPs were nicely and homogeneously dispersed on the surface of TSs, preserving the uniform morphology and shape of the parent TSs.
The incorporated NPs structures and interactions on the TSs surface, taking Au microspheres as an example, was further investigated using XRD, UV-vis absorption spectroscopy, FTIR, SERS and ITC. Wide-angle XRD pattern of AuNPs loaded onto TSs exhibited four peaks which could be indexed as the (111), (200), (220) and (311) reflections of the face-centered-cubic structure of crystalline Au0 (Fig. 2D). Among the peaks observed, the intensity of the (111) peak was the highest, indicating a predominant (111) crystal facet in Au-TSs. The absence of large and intense peaks at higher angles further suggested the absence of large Au NPs on Au-microspheres. These results were further confirmed by UV–vis and FTIR experiments which showed almost identical spectra for Au microsphere materials as compared to Au NPs in diluted aqueous solution and TSs, respectively (Fig. 2A and E). Only slight band shifts were observed for FT-IR spectra, although the characteristic peaks at 3100 and 3300 cm−1 for TSs (amino and hydroxyl groups) were missing in the FTIR spectra of Au microspheres.
Furthermore, SERS characterization of the bionanocomposites (Fig. 2F) pointed out bands at 953 and 1388 cm−1 (C–COO− and symmetric COO− stretching vibrations, respectively) enhanced after Au incorporation, in good agreement with previous reports.39 CH2 wagging (1289 cm−1) and bending (1323 cm−1) bands were also enhanced in Au-microsphere materials as additional evidence of the metal–carboxylate interactions. Similarly, the enhanced broad and strong band at 1560 cm−1 (–NH3+ bending) and corresponding decrease of NH2 stretching vibration bands (2927 and 3172 cm−1) are consistent with metal-amino group interactions.
Thermodynamic investigations of the interaction between Au NPs and TSs using ITC supported an exothermic adsorption of Au onto TSs under the investigated conditions, with ΔHads measured values ranging from −884 to −78 kJ mol−1 Au (see ESI† for full details). As presented in Table 1, the maximum number of layers was 3.82 (hence confirming multilayer Au adsorption), and the maximum adsorption at monolayer of Au NPs onto TSs was 39 mg g−1.
| v m (mg g−1) | n | c (L mg−1) | R | |
|---|---|---|---|---|
| TSs: treated spores; vm: maximum adsorption at monolayer; n: adsorbent sets a limit to the maximum number of layers that can be adsorbed; c: BET constant expressive of the energy of interaction with surface; R: correlation coefficient. | ||||
| TSs | 39.0 | 3.82 | 4.34 × 10−4 | 0.9845 |
These results and investigations confirmed the already proved involvement of carboxylic and amino groups in the incorporation and stabilization through physic-chemical interactions of metal NPs onto TSs surfaces. Particularly, with major repulsive electrostatic forces (surfaces of both Au NPs and TSs are negatively charged) and weak – if not negligible – van der Waals interactions,40 the affinity between Au NPs and TSs (i.e. hydrophobicity/hydrophilicity) may account as main physisorption phenomena in the synthesized bionanocomposites.
After full characterization, the synthesized bionanocomposites were subsequently employed in two remarkably different applications to illustrate the versatility of the proposed system: an immunoassay of antibodies against Actinobacillus pleuropneumoniae (only for Au-microspheres) as well as a heterogeneously catalyzed model reaction (e.g. hydrogenation of 4-nitrophenol).
Microsphere-based assays are among the most promising techniques for clinic diagnostics and research purposes. They are suitable for complex samples including food and body fluids due to a superior removal of background, being also suitable to analyse low volume samples and trace amounts of analytes. The antibody immunoassay of a protein of APP Repeats-in-Toxin IV (ApxIVA) was selected as a model system to demonstrate the use of the spore-based microspheres as a new platform for biosensor developments. APP is the etiological agent of a highly contagious disease, porcine pleuropneumonia caused by Actinobacillus pleuropneumoniae, which can cause severe economic losses in the swine industry worldwide.41 ApxIVA is highly specific to APP species. ApxIVA protein antibodies can be used not only to detect all serotypes of APP, but also to differentiate between naturally infected and inactivated-vaccine immunized pigs.
An Au microsphere (0.6 ± 0.1 μm × 1.2 ± 0.2 μm) from B. amyloliquefaciens spores was used as a model platform for immunoassay (Fig. 3, see also ESI†). Under optimum conditions, the calibration plot obtained for standard positive serum was approximately linear within the dilution range 1:160–1:10240 (Fig. 3A). The limit of detection (LOD) for the assay was 1:20480, remarkably improved as compared to that of ApxIVA–ELISA (1:320) (S/N = 3).
 | ||
| Fig. 3 Utilisation of Au microspheres in immunoassay experiments. (A) Calibration curve of the base-10 logarithm of fluorescence response versus the base-10 logarithm of the dilution factor of diluted APP positive serum. (B) Specificity study for APP immunoassay. Sera are separately infected with: APP, Porcine circovirus type 2 (PCV2), Japanese encephalitis virus (JEV), Pseudorabies virus (PRV), Escherichia coli (E. coli), Streptococcus suis serotype 2 (S. s 2), Haemophilus parasuis (H. ps). Experimental conditions: spores: 3.705 × 109 CFU mL−1; BSA: 20 mg mL−1; Blocking time: 1.5 h; Biotin-IgG: 1.0 μg mL−1; SA-Cy5: 0.5 μg mL−1. | ||
To evaluate diagnostic specificity, sera infected with agents responsible of various porcine diseases were detected by this proposed method. These diseases included APP, Porcine circovirus type 2 (PCV2), Japanese encephalitis virus (JEV), Pseudorabies virus (PRV), Escherichia coli (E. coli), Streptococcus suis serotype 2 (S. s 2), Haemophilus parasuis (H. ps). As shown in Fig. 3B, sera infected with any of these diseases presented weak fluorescence signals, similar to that obtained for the standard negative serum of APP. These signals were insignificant with respect to the fluorescence signal yielded by the 1:5120-diluted standard positive serum of APP, indicating a highly specificity for APP. The ability of the immunosensor to analyze clinical samples was also tested on porcine serum (Table S1, ESI†). The immunosensor yielded an efficiency of 88.9%, sensitivity of 92.6% and specificity of 85.2% as compared to ApxIVA–ELISA. These results clearly indicate that the proposed method has a high sensitivity and specificity to detect ApxIVA-targeting antibodies of APP in clinical diagnostic tests.
Additionally, the Au-catalyzed reduction of 4-nitrophenol (4-NP) in the presence of NaBH4 at room temperature was selected as a model system to illustrate the potential of as-prepared TSs as recoverable catalyst-supports and alternative to reported literature systems. This proposed reaction was exclusively selected to demonstrate the versatility and possibilities of the synthesized bionanocomposites in essentially different applications (immunoassay and catalysis) and to allow a ready comparison with literature reported catalysts. No changes in absorption could be observed even after 20 h in the absence of catalyst or when TSs was utilized in the reaction, indicating that 4-NP was not converted in the absence of the nanocomposite catalyst (even using a large excess of NaBH4). Comparatively, the addition of small amounts of Au microspheres (containing 3.1 μg Au) significantly decreased the absorption band at 400 nm, indicating the reduction of 4-NP and the formation of 4-aminophenol (4-AP, band at 295 nm), respectively. UV-vis spectra are shown as a function of the reaction time for a typical reduction process in Fig. 4A. These spectra show an isosbestic point (313 nm), which suggests that the catalytic reduction of 4-NP exclusively generates 4-AP without any by-products. A large excess of NaBH4 is used in the reaction, expected to be of pseudo-first order. Calculated reaction rate constant (k) was 0.396 min−1 for the Au nanocomposites loaded with 3.1 μg Au. These results demonstrate that the bio-inspired Au microspheres exhibit a high catalytic activity, remarkably superior to most literature reported Au as well as Ag and Pd-based catalysts at room temperature (Table 2)42–50 The obtained values were only comparable to certain reported Au nanomaterials (Au@MIL-100 (Fe) and 2%Au@2%Ag/MOF) while exclusively inferior to those reported for Pd/C systems (Table 2).
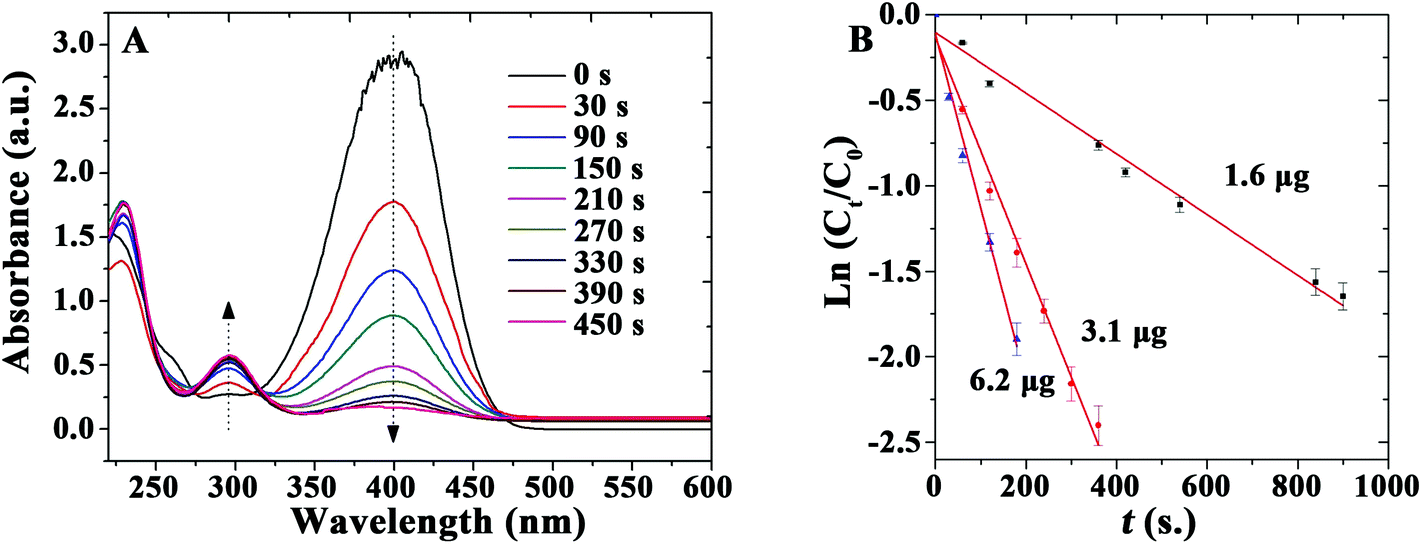 | ||
| Fig. 4 (A) UV-vis spectra of the reduction of 4-nitrophenol in aqueous solution. (B) Reaction rate constant k determined using different amounts of catalyst: ▲ = 6.2 μg; ● = 3.1 μg; ■ = 1.6 μg. | ||
| Material | Reaction conditions | Reaction rate constant (k, min−1) | Ref. |
|---|---|---|---|
| Au-microspheres | 3 mL NaBH4 0.5 M, 3 mL 4-NP 0.1 M, 3.1 μg Au | 39.6 × 10−2 | This work |
| 2%Au@2%Ag/MOF (I22) | 162.3 mg NaBH4 in 3.3 mL H2O, 15 mL of 4-NP 0.18 M, 8 mL H2O, 3.46 mg catalyst | 29 × 10−2 | 42 |
| Au/Tween/GO | 1 mL NaBH4 0.16 M, 1 mL 4-NP 0.007 M, 2.16 mM Au | 25.37 × 10−2 | 44 |
| Au/Tween | 1 mL NaBH4 0.16 M, 1 mL 4-NP 0.007 M, 2.16 mM Au | 9.05 × 10−2 | 44 |
| Au@MIL-100 (Fe) | 162.3 mg NaBH4 in 11.3 mL H2O, 15 mL 4-NP 0.18 mM, 3.4 mg catalyst | 33 × 10−2 | 45 |
| Au NPs/GO | 1 mL NaBH4 0.16 M, 1 mL 4-NP 0.007 M, 18 mg L−1 Au | 18.8 × 10−2 | 47 |
| Au nanoplates/GO | NaBH4(excess), 4-NP 5 × 10−5 M, 2.5 mg L−1 Au | 4.67 × 10−2 | 47 |
| Ag/rGO | NaBH4(excess), 4-NP 5 × 10−5 M, 2.5 mg L−1 Ag | 27.1 × 10−2 | 48 |
| Ag@SiO2 | 1 × 10−2 M NaBH4, 5 × 10−5 M 4-NP, Ag | 37 × 10−2 | 43 |
| Ag/Poly(CAN-VA) | 1 mL NaBH4 3 × 10−5 M, 0.1 mL 4-NP 3 × 10−6 M, 3 mL H2O, 0.01 g catalyst | 3.9 × 10−2 | 46 |
| Pd/Al-SBA-15 | 5 mL NaBH4 (6 × 10−4 mol−1), 45 mL 4-NP (0.18 mmol−1) 0.07 g catalyst | 6.1 × 10−2 | 49 |
| Pd/C | 4-NP 1.67 × 10−3 M, 0.0024 mM Pd | 52.8 × 10−2 | 50 |
In addition, the effect of the amount of Au contained in the materials on the catalyzed reduction was subsequently studied (Fig. 4B). The rate constant k was calculated to be 0.108, 0.396, and 0.606 min−1 for the reaction using the same catalyst containing 1.6, 3.1, and 6.2 μg of Au, respectively. These results indicate that an increase in the quantity of catalyst can further improve reaction rates under the investigated reaction conditions.
Turnover frequencies (TOF) were also worked out to estimate the catalytic performance of the nanocomposites with different loadings and at different temperatures (Table 3). Pd-microspheres exhibited the highest catalytic activity (TOF 132.5 min−1) as compared to Au, Ag, and Pt bionanocomposites. Differences in TOF values are believed to result from the synergistic interaction between NPs and TSs as previously reported.44,51 Most importantly, TOF values were also found to be significantly superior to those of most relevant literature reports for the reduction of 4-NP,51–57 both for Pd-containing materials (i.e. Pd/C, 99 min−1; Pd/Al-SBA-15, 82 min−1)49,50 as well as Au-containing nanomaterials and only comparable to those of surface-protected SiO2-based supports.58
| Catalysts | TOF/min−1 (25 °C) | TOF /min−1 (90 °C) | Wt% |
|---|---|---|---|
| Au microspheres | 1.69 | 12.66 | 0.25 |
| 3.28 | 16.41 | 0.04 | |
| 5.99 | 23.94 | 0.01 | |
| Pt microspheres | 3.74 | 7.47 | 0.84 |
| 5.42 | 9.75 | 0.32 | |
| 9.08 | 21.68 | 0.01 | |
| Pd nanocubes microspheres | 20.23 | 62.81 | 0.11 |
| 33.64 | 85.14 | 0.02 | |
| 50.73 | 100.21 | 0.01 | |
| Pd microspheres | 11.67 | 35.00 | 0.97 |
| 20.14 | 47.00 | 0.24 | |
| 32.08 | 132.50 | 0.03 | |
| Ag microspheres | 1.02 | 6.07 | 0.29 |
| 2.54 | 7.08 | 0.11 | |
| 4.68 | 14.05 | 0.005 |
The excelling catalytic performances of the bionanocomposites may be attributed to a high dispersion of small-sized NPs stabilized on the TSs support (which also prevented aggregation after successive reactions). According to previous studies on natural biopolymers as catalysis support (e.g. chitosan),59 the interaction between Au and the nitrogen and oxygen functionalities present in TSs may also positively contribute to the excelling catalytic activities observed in TSs with respect to other supports.51–57,59,60
The recyclability of catalysts is also important for their practical applications. After several reuses, the catalytic activity of Au microspheres was essentially unchanged after nine reaction cycles (Fig. 5), with only a nominal decrease in conversion from 95 to 85% (probably due to minimum loss of nanocomposite during the repeated reuse/recycle processes). The catalyst recovered after a ninth reaction cycle was also characterised to evaluate the stability of the bio-nanocomposites under the investigated conditions as compared to fresh catalysts (Fig. 6). The morphology and optical absorption as well as shapes and sizes of Au NPs on TSs remained virtually unchanged after several cycles of catalytic reaction as clearly visualized in Fig. 6. ICP-AES analysis further demonstrated no obvious leaching of NPs in the reaction medium (up to the detection limit, 0.5 ppm) which confirmed their excelling stabilities and reusabilities under the investigated conditions.
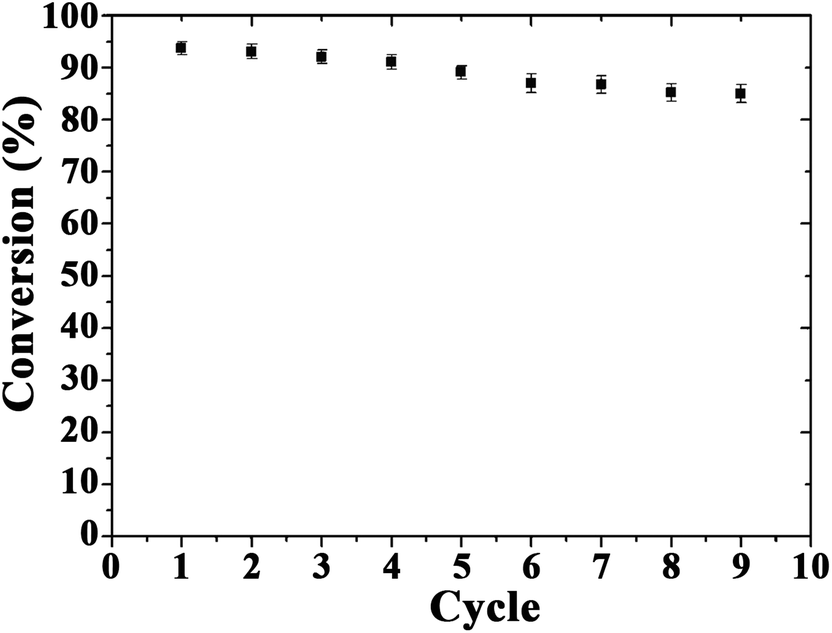 | ||
| Fig. 5 Reuses of the Au-bionanocomposite catalyst in the reduction of 4-nitrophenol. Reaction conditions: NaBH4 (0.5 mol L−1), 3.0 mL; 4-nitrophenol (0.1 mmol L−1), 3.0 mL; Au, 3.1 μg. | ||
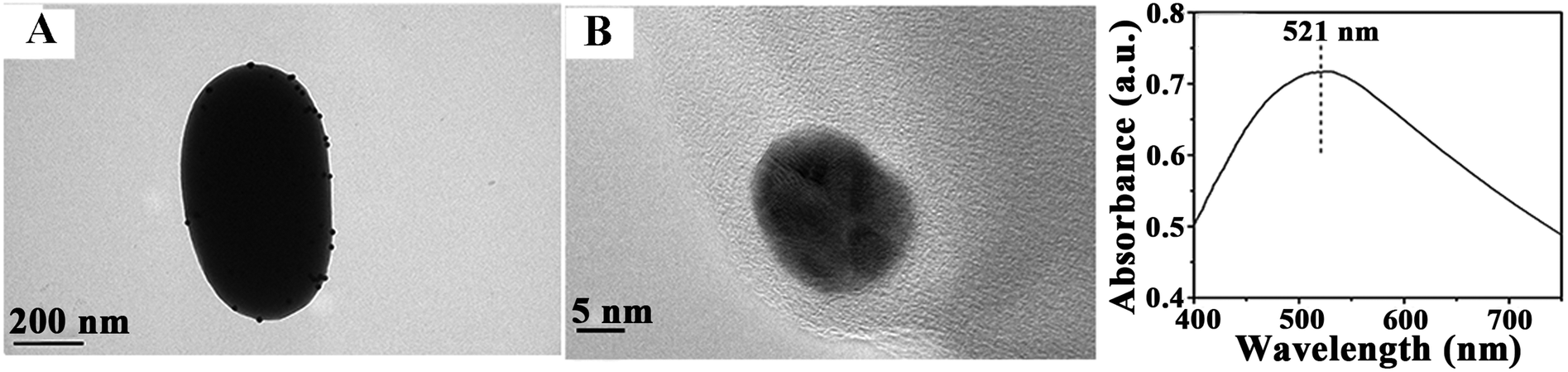 | ||
| Fig. 6 Characterisation of the reused Au-bionanocomposite after 9 reaction runs. (A) TEM and (B) HRTEM as well as UV–vis spectra clearly showing the typical characteristics and properties observed for a freshly synthesized Au-microsphere. | ||
Conclusions
In summary, this contribution introduces an innovative biological approach using Bacillus spores for the preparation of highly monodisperse microparticles featuring different functionalities (carboxylic, amino and hydroxyl groups). Compared to conventional methods, advantages of this approach include simplicity, low-cost, potential for high-throughput and environment friendliness. TSs were demonstrated as a highly suitable support with an excellent mechanical strength (due to the presence of a protective shell) to stabilize metal NPs, thereby producing highly versatile, stable and recyclable bio-nanocomposites for various applications. These included heterogenous catalysis in a simple model hydrogenation of 4-nitrophenol at room temperature as well as antibody immunoassays, demonstrating the remarkable potential of these novel class of bio-derived systems as advanced functional nanomaterials. Importantly, the spore-based microspheres could be massively produced using cell cultures via fermentation at industrial scale in a potentially cost-effective and environmentally friendly manner paving the way to a more extended utilization of the proposed systems in various applications.Acknowledgements
This work was supported by the National Basic Research Program of China (973 Program grant No. 2012CB518802 for RZ), the National Natural Science Foundation of China Grants (No. 31421064), and Special Fund for Agro-scientific Research in the Public Interest (201303034-8). Zhiming Zeng, Ying Zhong, and Huicui Yang contributed equally to this work. We also thank Prof. Yanlong Gu for discussions as well as the reviewers of this manuscript for their valuable inputs and suggested insightful revisions to improve the quality of our work.References
- (a) J. Watt, S. Cheong, M. F. Toney, B. Ingham, J. Cookson, P. T. Bishop and R. D. Tilley, ACS Nano, 2010, 4, 396402 CrossRef PubMed; (b) R. J. White, R. Luque, V. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481–494 RSC.
- K. Esumi, R. Isono and T. Yoshimura, Langmuir, 2004, 20, 237–243 CrossRef CAS PubMed.
- W. Liu, X. Yang and L. Xie, J. Colloid Interface Sci., 2007, 313, 494–502 CrossRef CAS PubMed.
- W. Liu, X. Yang and W. Huang, J. Colloid Interface Sci., 2006, 304, 160–165 CrossRef CAS PubMed.
- F. Dong, W. Guo, S.-K. Park and C.-S. Ha, Chem. Commun., 2012, 48, 1108–1110 RSC.
- K. K. R. Datta, B. V. S. Reddy, K. Ariga and A. Vinu, Angew. Chem., Int. Ed., 2010, 49, 5961–5965 CrossRef CAS PubMed.
- C. J. Martinez, B. Hockey, C. B. Montgomery and S. Semancik, Langmuir, 2005, 21, 7937–7944 CrossRef CAS PubMed.
- J.-W. Kim, A. Fernández-Nieves, N. Dan, A. S. Utada, M. Marquez and D. A. Weitz, Nano Lett., 2007, 7, 2876–2880 CrossRef CAS PubMed.
- P. M. Ajayan, L. S. Schadler, C. Giannaris and A. Rubio, Adv. Mater., 2000, 12, 750–753 CrossRef CAS.
- S.-H. Kim, J. W. Shim and S.-M. Yang, Angew. Chem., Int. Ed., 2011, 123, 1203–1206 CrossRef.
- A. Sukhanova and I. Nabiev, Crit. Rev. Oncol. Hemat., 2008, 68, 39–59 CrossRef PubMed.
- A. Álvarez-Diaz, A. Salinas-Castillo, M. Camprubí-Robles, J. M. Costa-Fernández, R. Pereiro, R. Mallavia and A. Sanz-Medel, Anal. Chem., 2011, 83, 2712–2718 CrossRef PubMed.
- J. Kim, S. Park, J. E. Lee, S. M. Jin, J. H. Lee, I. S. Lee, I. Yang, J.-S. Kim, S. K. Kim, M.-H. Cho and T. Hyeon, Angew. Chem., Int. Ed., 2006, 118, 7918–7922 CrossRef.
- R. Wilson, A. R. Cossins and D. G. Spiller, Angew. Chem., Int. Ed., 2006, 45, 6104–6117 CrossRef CAS PubMed.
- D. Jańczewski, N. Tomczak, M.-Y. Han and G. J. Vancso, Eur. Polym. J., 2009, 45, 1912–1917 CrossRef.
- A. J. Amali, B. Sharma and R. K. Rana, Chem. – Eur. J., 2014, 20, 12239–12244 CrossRef CAS PubMed.
- J. M. Köhler, A. März, J. Popp, A. Knauer, I. Kraus, J. Faerber and C. Serra, Anal. Chem., 2013, 85, 313–318 CrossRef PubMed.
- B. Karagoz, D. Gunes and N. Bicak, Macromol. Chem. Phys., 2010, 211, 1999–2007 CrossRef CAS.
- S. Liu, R. Deng, W. Li and J. Zhu, Adv. Funct. Mater., 2012, 22, 1692–1697 CrossRef CAS.
- A. R. Abate, M. Kutsovsky, S. Seiffert, M. Windbergs, L. F. V. Pinto, A. Rotem, A. S. Utada and D. A. Weitz, Adv. Mater., 2011, 23, 1757–1760 CrossRef CAS PubMed.
- V. Berry and R. F. Saraf, Angew. Chem., Int. Ed., 2005, 117, 6826–6831 CrossRef.
- A. I. Zamaleeva, I. R. Sharipova, R. V. Shamagsumova, A. N. Ivanov, G. A. Evtugyn, D. G. Ishmuchametova and R. F. Fakhrullin, Anal. Methods, 2011, 3, 509–513 RSC.
- S. H. Yang, D. Hong, J. Lee, E. H. Ko and I. S. Choi, Small, 2013, 9, 178–186 CrossRef CAS PubMed.
- D. Hong, M. Park, S. H. Yang, J. Lee, Y.-G. Kim and I. S. Choi, Trends Biotechnol., 2013, 31, 442–447 CrossRef CAS PubMed.
- J. H. Park, S. H. Yang, J. Lee, E. H. Ko, D. Hong and I. S. Choi, Adv. Mater., 2014, 26, 2001–2010 CrossRef CAS PubMed.
- S. A. Konnova, I. R. Sharipova, T. A. Demina, Y. N. Osin, D. R. Yarullina, O. N. Ilinskaya, Y. M. Lvov and R. F. Fakhrullin, Chem. Commun., 2013, 49, 4208–4210 RSC.
- P. Mazza, Boll. Chim. Farm., 1994, 133, 3–18 CAS.
- J.-M. Huang, R. M. La Ragione, W. A. Cooley, S. Todryk and S. M. Cutting, Vaccine, 2008, 26, 6043–6052 CrossRef CAS PubMed.
- G. Frens, Nat. Phys. Sci., 1973, 241, 20–22 CrossRef CAS.
- A. Moir, J. Appl. Microbiol., 2006, 101, 526–530 CrossRef CAS PubMed.
- A. O. Henriques, J. Moran and P. Charles, Annu. Rev. Microbiol., 2007, 61, 555–588 CrossRef CAS PubMed.
- A. Monroe and P. Setlow, J. Bacteriol., 2006, 188, 7609–7616 CrossRef CAS PubMed.
- N. Yee, L. G. Benning, V. R. Phoenix and F. G. Ferris, Environ. Sci. Technol., 2004, 38, 775–782 CrossRef CAS PubMed.
- J. B. Fein, C. J. Daughney, N. Yee and T. A. Davis, Geochim. Cosmochim. Acta, 1997, 61, 3319–3328 CrossRef CAS.
- A. J. Westphal, P. B. Price, T. J. Leighton and K. E. Wheeler, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3461–3466 CrossRef CAS PubMed.
- E. Hockly, V. M. Richon, B. Woodman, D. L. Smith, X. B. Zhou, E. Rosa, K. Sathasivam, S. Ghazi-Noori, A. Mahal, P. A. S. Lowden, J. S. Steffan, J. L. Marsh, L. M. Thompson, C. M. Lewis, P. A. Marks and G. P. Bates, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 2041–2046 CrossRef CAS PubMed.
- M. Plomp, T. J. Leighton, K. E. Wheeler and A. J. Malkin, Biophys. J., 2005, 88, 603–608 CrossRef CAS PubMed.
- O. Sahin, E. H. Yong, A. Driks and L. Mahadevan, J. R. Soc., Interface, 2012, 9, 3156–3160 CrossRef PubMed.
- C. Jing and Y. Fang, Chem. Phys., 2007, 332, 27–32 CrossRef CAS.
- C. J. van Oss, Colloids Surf., B, 1995, 5, 91–110 CrossRef CAS.
- J. T. Bossé, H. Janson, B. J. Sheehan, A. J. Beddek, A. N. Rycroft, J. Simon Kroll and P. R. Langford, Microbes Infect., 2002, 4, 225–235 CrossRef.
- H.-L. Jiang, T. Akita, T. Ishida, M. Haruta and Q. Xu, J. Am. Chem. Soc., 2011, 133, 1304–1306 CrossRef CAS PubMed.
- B. P. Bastakoti, S. Guragain, S.-i. Yusa and K. Nakashima, RSC Adv., 2012, 2, 5938–5940 RSC.
- W. Lu, R. Ning, X. Qin, Y. Zhang, G. Chang, S. Liu, Y. Luo and X. Sun, J. Hazard. Mater., 2011, 197, 320–326 CrossRef CAS PubMed.
- F. Ke, J. Zhu, L.-G. Qiu and X. Jiang, Chem. Commun., 2013, 49, 1267–1269 RSC.
- H. Xiao and Y. Xia, Polym. Eng. Sci., 2010, 50, 1767–1772 CAS.
- Y. Zhang, S. Liu, W. Lu, L. Wang, J. Tian and X. Sun, Catal. Sci. Technol., 2011, 1, 1142–1144 CAS.
- K. C. Hsu and D. H. Chen, Nanoscale Res. Lett., 2014, 9, 484–493 CrossRef PubMed.
- M. Al-Naji, A. Roibu, M. Goepel, W.-D. Einicke, R. Luque and R. Glaser, Catal. Sci. Technol., 2015, 5, 2085–2091 CAS.
- Y. Fang and E. Wang, Nanoscale, 2013, 5, 1843–1848 Search PubMed.
- S. Carrettin, Y. Hao, V. Aguilar-Guerrero, B. C. Gates, S. Trasobares, J. J. Calvino and A. Corma, Chem. – Eur. J., 2007, 13, 7771–7779 CrossRef CAS PubMed.
- J. Morère, M. J. Tenorio, M. J. Torralvo, C. Pando, J. A. R. Renuncio and A. Cabañas, J. Supercrit. Fluids, 2011, 56, 213–222 CrossRef.
- H. He and C. Gao, Molecules, 2010, 15, 4679 CrossRef CAS PubMed.
- S. Harish, J. Mathiyarasu, K. L. N. Phani and V. Yegnaraman, Catal. Lett., 2009, 128, 197–202 CrossRef CAS.
- Y. Mei, Y. Lu, F. Polzer, M. Ballauff and M. Drechsler, Chem. Mater., 2007, 19, 1062–1069 CrossRef CAS.
- Z. Jin, F. Wang, F. Wang, J. Wang, J. C. Yu and J. Wang, Adv. Funct. Mater., 2013, 23, 2137–2144 CrossRef CAS.
- W. Hu, B. Liu, Q. Wang, Y. Liu, Y. Liu, P. Jing, S. Yu, L. Liu and J. Zhang, Chem. Commun., 2013, 49, 7596–7598 RSC.
- Q. Zhang, I. Lee, J. Ge, F. Zaera and Y. Yin, Adv. Funct. Mater., 2010, 20, 2201–2214 CrossRef CAS.
- A. Corma, P. Concepción, I. Domínguez, V. Forné and M. J. Sabater, J. Catal., 2007, 251, 39–47 CrossRef CAS.
- J. Ge, T. Huynh, Y. Hu and Y. Yin, Nano Lett., 2008, 8, 931–934 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5gc00630a |
| This journal is © The Royal Society of Chemistry 2016 |