
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCross-dehydrogenative coupling reaction using copper oxide impregnated on magnetite in deep eutectic solvents†
Xavier
Marset
,
Juana M.
Pérez
and
Diego J.
Ramón
*
Instituto de Síntesis Orgánica (ISO), and Departamento de Química Orgánica, Facultad de Ciencias, Universidad de Alicante, Apdo. 99, E-03080-Alicante, Spain. E-mail: djramon@ua.es
First published on 9th September 2015
Abstract
The synthesis of different tetrahydroisoquinolines using choline chloride![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethylene glycol as a deep eutectic solvent (DES) and copper(II) oxide impregnated on magnetite as a catalyst has been accomplished successfully. The copper catalyst amount is the lowest loading ever reported. The presence of DES showed to be essential since the reaction in the absence of this medium did not proceed. A direct proportional relationship was found between the conductivity of DES medium and the yield obtained. The DES and the catalyst could be reused up to ten times without any detrimental effect on the yield of the reaction, with the aerobic conditions making the protocol highly sustainable, where the only waste is water.
:ethylene glycol as a deep eutectic solvent (DES) and copper(II) oxide impregnated on magnetite as a catalyst has been accomplished successfully. The copper catalyst amount is the lowest loading ever reported. The presence of DES showed to be essential since the reaction in the absence of this medium did not proceed. A direct proportional relationship was found between the conductivity of DES medium and the yield obtained. The DES and the catalyst could be reused up to ten times without any detrimental effect on the yield of the reaction, with the aerobic conditions making the protocol highly sustainable, where the only waste is water.
Introduction
Tetrahydroisoquinolines are widely present in Nature.1 The synthesis of these compounds has been paid much attention in industrial and academic research due to their biological and pharmaceutical applications, such as anticancer2 and anticonvulsant agents,3 enzyme inhibitors,4 ligand receptors5 and therapeutic agents.6The C–C bond formation via C–H activation7 is one of the most challenging reactions in organic synthesis. Various strategies for transition-metal-catalysed C–H bond activation have been of significant interest, as they are environmentally friendly processes, and no functionalization step is needed. The C(sp3)–H bond activation at the α-position of nitrogen has been broadly used in different transformations. The key step, in this transformation, is the generation of an iminium intermediate assisted by the lone pair of the nitrogen atom, via a single-electron transfer (SET) mechanism.8
For this purpose an important number of different methods have been developed, with metal-catalysed protocols being well established. Different catalysts derived from vanadium,9 iron,10 copper,11 ruthenium,12 rhodium,13 palladium,14 antimonium,15 iridium16 or gold,17 among others have been recently introduced. In all cases, the protocols needed a highly reactive oxidizing agent such as peroxides or high oxygen pressure. Moreover, the lack of recyclability, and the high catalyst loading (5–20 mol%) made these protocols unsustainable for large chemical production. The metal-free version using organic radical promoters, recently published, has similar drawbacks.18
Within the framework of green chemistry, solvents occupy a strategic place. In order to be qualified as a green medium, the components of this solvent have to meet different criteria such as availability, non-toxicity, biodegradability, recyclability, inflammability, renewability and low price, among others. DES (Deep Eutectic Solvent) is an environmentally benign alternative to hazardous (organic) solvents and, in many cases, might replace them. DESs are liquid systems formed from a eutectic mixture of a solid Lewis or Brønsted acids and bases which can contain a variety of anionic and/or cationic species.19 These two components are capable of self-association, often through a strong bond interaction, to form an eutectic mixture with a melting or phase transition point lower than that of each individual component.20 The properties of a solvent, such as conductivity, viscosity, vapour pressure and thermal stability can be fine-tuned by appropriately choosing the mixture components, with the large-scale preparation being feasible. Besides these interesting advantages, the application of DES in organic synthesis is in its infancy,21 with the related metal-catalysed process being nearly unknown.22 Only very recently, the superparamagnetic CuFeO2 has been used as a catalyst for the multicomponent synthesis of imidazo[1,2-a]pyridines in DMU-citric acid medium.23
Here, we introduced a recyclable copper-impregnated magnetite catalyst for the C–H activation in choline chloride:ethylene glycol as medium using bio-renewable air as the only oxidizing agent.
Results and discussion
To start with this study, 2-(4-fluorophenyl)-1,2,3,4-tetrahydroisoquinoline (1a) and phenylacetylene (2a) using copper impregnated on magnetite as a catalyst was selected as the model reaction for the optimization of the conditions (Table 1).|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | DES (molar ratio) | T (°C) |
3ab (%) |
4ab (%) |
| a Reaction carried out using compounds 1a (0.5 mmol) and 2a (1 mmol) in 1 mL of DES. b Conversion determined by 1H-NMR. c Reaction carried out using compounds 1a (0.5 mmol) and 2a (0.5 mmol) in 1 mL of DES. d Reaction carried out using 1a (0.5 mmol) and 2a (2.5 mmol) in 1 mL of DES. e 99% of conversion after 7 days of reaction. f Reaction carried out under an argon atmosphere. g Reaction carried out using visible LED light irradiation. h Reaction carried out under microwave irradiation for 10 h at 80 W. i Reaction carried out under ultrasound bath for 8 h. | |||||
| 1 | CuO–Fe3O4 (1.82) | ChCl:urea (1:2) |
50 | 55 | 29 |
| 2 | CuO–Fe3O4 (1.82) | AcChCl:urea (1:2) |
50 | 50 | 13 |
| 3 | CuO–Fe3O4 (1.82) | ChCl:glycerol (1:2) |
50 | 59 | 11 |
| 4 | CuO–Fe3O4 (1.82) | ChCl:ethylene glycol (1:2) |
50 | 83 | 2 |
| 5 | CuO–Fe3O4 (1.82) | Ph3P+MeBr–:glycerol (1:2) |
50 | 60 | 11 |
| 6 | CuO–Fe3O4 (1.82) | ChCl:resorcinol (1:1) |
50 | 10 | 21 |
| 7 | CuO–Fe3O4 (0.91) | ChCl:ethylene glycol (1:2) |
50 | 83 | 3 |
| 8 | CuO–Fe3O4 (0.37) | ChCl:ethylene glycol (1:2) |
50 | 56 | 21 |
| 9 | CuO–Fe3O4 (3.64) | ChCl:ethylene glycol (1:2) |
50 | 95 | 3 |
| 10c | CuO–Fe3O4 (1.82) | ChCl:ethylene glycol (1:2) |
50 | 75 | 2 |
| 11d | CuO–Fe3O4 (1.82) | ChCl:ethylene glycol (1:2) |
50 | 92 | 2 |
| 12 | CuO–Fe3O4 (3.64) | ChCl:ethylene glycol (1:2) |
RT | 57e | 0 |
| 13 | CuO–Fe3O4 (3.64) | ChCl:ethylene glycol (1:2) |
100 | 38 | 0 |
| 14f | CuO–Fe3O4 (3.64) | ChCl:ethylene glycol (1:2) |
50 | 42 | 0 |
| 15g | CuO–Fe3O4 (3.64) | ChCl:ethylene glycol (1:2) |
50 | 49 | 9 |
| 16h | CuO–Fe3O4 (3.64) | ChCl:ethylene glycol (1:2) |
50 | 32 | 6 |
| 17i | CuO–Fe3O4 (3.64) | ChCl:ethylene glycol (1:2) |
53 | 17 | 0 |
| 18 | CuO–Fe3O4 (3.64) | Ethylene glycol | 50 | 40 | 9 |

Initially, the reaction was performed using different DESs (entries 1–6), obtaining the best result with the mixture choline chloride (ChCl):ethylene glycol (1:2) (entry 4), with the only by-product observed being the corresponding lactam 4a. Then, the amount of catalyst was evaluated obtaining similar results when the loading was decreased (entry 7). However, a further decrease of catalyst loading down to 0.37 mol% (entry 8) led to lower yield. Increasing the amount of copper to 3.64 mol% (entry 9) the yield could be improved. It should be pointed out that even this high amount of copper catalyst is one of the lowest metal catalyst loadings reported so far in the literature. The addition of only one equivalent of alkyne led to the decrease of the reaction yield (entry 10), and the addition of an excess of alkyne did not improve it (entry 11). The study of the temperature of reaction was carried out obtaining, after seven days of reaction at room temperature, a full conversion of the starting material (entry 12). Increasing the temperature up to 100 °C decreased the yield (entry 13). The reaction was carried out under an argon atmosphere (entry 14) obtaining a very low yield, highlighting the capital role of oxygen in the air as the final oxidizing agent. To finish with the optimization of the reaction conditions, the reaction was tested using LED irradiation (photoredox conditions), microwave irradiation and an ultrasound bath (entries 15–17), but the yield did not improve. Finally, the reaction was repeated in ethylene glycol obtaining a modest result (entry 18).
To prove the essential role of DES [ChCl:(CH2OH)2], other VOC solvents were tested as reaction medium (Fig. 1). In all cases a mixture of products 3a and 4a was obtained in a ratio close to 1:1, highlighting the role of DES for minimizing the lactam formation. It should be pointed out that when the reaction was performed in 1,4-dioxane as a solvent the main product was 4a.
 | ||
| Fig. 1 Obtained yield in different solvents. | ||
We also found an interesting correlation between the DES conductivity and the yield of the desired product (Fig. 2), in such a way that a higher conductivity affords a better yield. Since an iminium intermediate is generated in the reaction media, a better conductivity means an easier movement of ions that could explain the increase in the yield. Nevertheless, the correlation between obtained yields and conductivities of VOC solvents and of water did not fit with the aforementioned plot. It should be pointed out that the reaction using ChCl:1,2-propanediol:water (1:1:1, conductivity 12.09 mS cm−1) or ChCl:glycerol:water (1:2:1, conductivity 13.78 mS cm−1) gave the product 3a in 46 and 53% yield respectively. Although these two mixtures have higher conductivity than the previous DES used, the presence of water changed the direct proportion between yield and conductivity probably due to the highly nucleophilic character of water.
 | ||
| Fig. 2 Relationship between solvent conductivity and yield. | ||
Once the optimal conditions were determined, the reaction was repeated with a variety of catalysts prepared by the simple impregnation protocol24 (Table 2). The reaction without a catalyst gave a poor yield (entry 2). Then, the activity of the support was evaluated using magnetite as the unique catalyst. Nanoparticles or microparticles of magnetite (entries 3 and 4) were used with the results showing the inactivity of the support, reaching a low conversion of product and highest amount of compound 4a, as the only by-product detected by GC-MS. Once the activity of magnetite was tested, different metal oxides impregnated on magnetite (entries 5–17) were evaluated as catalysts, observing that none of them gave better results than the copper catalyst (entry 1). After that, different copper salts were tested (entries 18–20), obtaining moderate to good results, but poorer than the one obtained by the heterogeneous copper oxide impregnated on magnetite.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) |
3ab (%) |
Entry | Catalyst (mol%) |
3ab (%) |
| a Reaction carried out using compounds 1a (0.5 mmol) and 2a (1 mmol) in 1 mL of DES. b Yield determined by 1H-NMR, yield of oxidised compound 4a in brackets. | |||||
| 1 | CuO–Fe3O4 (3.64) | 95 (3) | 12 | IrO2–Fe3O4 (0.26) | 33 (47) |
| 2 | — | 6 (20) | 13 | PtO/PtO2–Fe3O4 (1.08) | 33 (7) |
| 3 | Nano-Fe3O4 (259.15) | 0 (34) | 14 | Au2O3–Fe3O4 (0.28) | 13 (4) |
| 4 | Micro-Fe3O4 (259.15) | 15 (48) | 15 | PdO/Cu–Fe3O4 (3.05/1.79) | 35 (28) |
| 5 | CoO–Fe3O4 (2.83) | 4 (31) | 16 | NiO/Cu–Fe3O4 (1.82/1.76) | 78 (2) |
| 6 | NiO–Fe3O4 (2.06) | 30 (30) | 17 | WO3–Fe3O4 (1.13) | 37 (8) |
| 7 | Ru2O3–Fe3O4 (2.64) | 8 (28) | 18 | CuCl2 (8.5) | 88 (5) |
| 8 | Rh2O3–Fe3O4 (0.84) | 0 (45) | 18 | CuO (4.04) | 46 (10) |
| 9 | PdO–Fe3O4 (2.43) | 46 (7) | 20 | Cu(OAc)2 (3.64) | 80 (6) |
| 10 | Ag2O–Fe3O4 (2.5) | 13 (0) | 21 | CuO (3.64) + Fe3O4 (255.26) | 51 (9) |
| 11 | OsO2–Fe3O4 (1.03) | 5 (21) | |||

After that, the addition of a mixture of CuO and Fe3O4, was evaluated (entry 21), obtaining a decrease in the conversion compared to the impregnated catalyst, which seems to be related to a synergic effect between the metal oxide and support in the catalyst.
In order to establish the reusability of the catalyst and DES, the reaction with nitromethane (see Table 5, entry 1) was repeated under standard conditions (Fig. 3). When the reaction was completed, the mixture was extracted with cyclopentyl methyl ether, recently reported as a potential green alternative solvent.25 All organic compounds were removed and the mixture of DES and catalyst, lower phase in the decantation, was reused under the same reaction conditions. This catalytic mixture could be recycled up to 10 times without any decrease in the yield. When just the catalyst was recovered, by magnetic decantation, and fresh solvent was used, the obtained yield showed an important decrease, pointing out the sharp decrease of the catalyst after 4 reaction cycles. In fact, the ICP-MS analysis of the crude reaction solution showed the leaching of a small amount of copper (14.2 ppm; 3.6% of the initial amount) and iron (0.30 ppm; 0.001% of initial amount), the values were completely different from the reported solubility of these metal oxides (3.68 ppm for CuO and 10.85 ppm for Fe3O4).26 The higher solubility in DES of copper species seems to show that the heterogeneous catalyst is only a reservoir of highly active copper clusters. To try to understand more this effect, the standard reaction was performed as usual (Table 2, entry 1) and, after 36 h, only the catalyst was removed by magnetic decantation, with the yield of 3a being estimated in 40% by GC-MS. The mixture was heated again for 36 h, and after the usual work up the yield of compound 3a increased up to 65%, with the oxidised by-product 4a reaching 25%. At the end of the first cycle, the catalyst was removed, by magnetic decantation, as well as the organics by cyclopentyl methyl ether extraction (yield of compound 3a 93%). Then, the used DES medium was employed alone in other cycles (without catalyst) and the final product 3a was obtained in 52% (29% for by-product 4a). These two experiments showed that there was a partial leaching of active species, capable of performing the oxidative step. However, and due to the great amount of by-product, these leached species seemed to be less effective to catalyze the final nucleophilic addition.
 | ||
| Fig. 3 Recycling of the CuO–Fe3O4 catalyst and CuO–Fe3O4 + DES. | ||
In order to study the effect of the reaction conditions on the copper heterogeneous catalyst, the nanosize distribution of the copper nanoparticles was measured after one reaction cycle. A uniform size distribution was found, 60% of nanoparticles have an average size between 2–4 nm. In the fresh catalyst 63% of nanoparticles have an average size between 2–6 nm, showing a small overall decrease in the particle size with the reaction cycles which is in concordance with a partial solubilization–readsorption of copper species (Fig. 4).
 | ||
| Fig. 4 Particle size distribution of fresh and recycled catalyst. | ||
The XPS and Auger Electron Spectroscopy (AES, see ESI†) studies of the catalyst showed the transformation of the initial Cu(0) onto the corresponding copper(I) oxide and Cu(OH)2 in the recycled catalyst (Fig. 5) with CuO being the main species in both cases. These changes in particle size as well as the initial oxidation state did not seem to affect the activity of the catalyst, since it could be reused ten times without losing its activity.
 | ||
| Fig. 5 XPS of fresh and recycled copper catalyst. | ||
Once the best conditions were established, the scope of the reaction was evaluated. First of all, different pro-electrophiles were tested by modifying the nitrogen substituent at the tetrahydroisoquinoline ring (Table 3). The reaction was carried out with different N-substituted substrates. When the substituent was an aryl group, bearing both, electron-withdrawing or electron-donating groups (entries 1 and 3), the results were excellent. In the case of phenyl derivatives, the yield was moderate. On the other hand, when the reaction was carried out with the free amine or with a strong electron-withdrawing group such as tosyl, the reaction did not take place at all (entries 4 and 5), recovering the starting material unchanged.

Having studied the scope of pro-electrophiles, we tested a variety of alkynes as pro-nucleophiles (Table 4). Once again, the reaction took place obtaining moderate to good yields when the alkyne had an electron-rich (entry 1) or electron-poor (entries 2–5) aryl substituents. Not only aryl substituents were tested, but also olefinic and aliphatic ones (entries 6–9) and the reaction still worked smoothly. It has to be pointed out that, in the case of using a dialkyne, the reaction was selective in such a way that only one of the two alkynes reacted (entry 8).
|
|
|||
|---|---|---|---|
| Entry | R′ | Product | Yieldb (%) |
| a Reaction carried out using compounds 1a (0.5 mmol) and 2a (1 mmol) in 1 mL of DES. b Isolated yield after distillation. c THPO stands for 2-(tetrahydro-2H-pyran-2-yl)oxy. | |||
| 1 | 4-MeOC6H4 | 3f | 57 |
| 2 | 4-BrC6H4 | 3g | 61 |
| 3 | 4-CF3C6H4 | 3h | 68 |
| 4 | 3-ClC6H4 | 3i | 58 |
| 5 | 2-BrC6H4 | 3j | 64 |
| 6 | 1-C6H9 | 3k | 37 |
| 7 | C6H11 | 3l | 83 |
| 8 | HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CC5H10 CC5H10 |
3m | 71 |
| 9 | THPOCH2c |
3n | 65 |

After the study of alkynes, as pro-nucleophiles, was completed, we decided to check other types of reagents (Table 5), such as nitroalkanes (entry 1), heterocycles (entry 2), phosphonates (entry 3), silyl enol ethers (entry 4), ketones (entries 5 and 6) and fluoroborates (entry 7), proving that this methodology can be applied to a wide range of substrates with very different properties and obtaining similar results. It should be noted that both, silyl enol ether and ketone (entries 4 and 5) afford the same product 3r but with different diastereomeric ratios. Only the starting material alongside a small amount of by-product 4a was detected from the crude of the reaction, when a moderate or low yield of product was obtained.
|
|
|||
|---|---|---|---|
| Entry | Nu-H | Product | Yieldb (%) |
|
a Reaction carried out using compounds 1a (0.5 mmol) and 4 (1 mmol) in 1 mL of DES.
b Isolated yield after distillation.
c Yield after 7 days at RT.
d Mixture of isomers syn:anti (1:1.25).
e Mixture of isomers syn:anti (1.4:1).
|
|||
| 1 | MeNO2 | 3o | 95/15c |
| 2 |

|
3p | 84 |
| 3 |

|
3q | 45 |
| 4 |

|
3r | 50d |
| 5 |

|
3r | 38e |
| 6 |

|
3s | 24 |
| 7 |

|
3t | 51 |
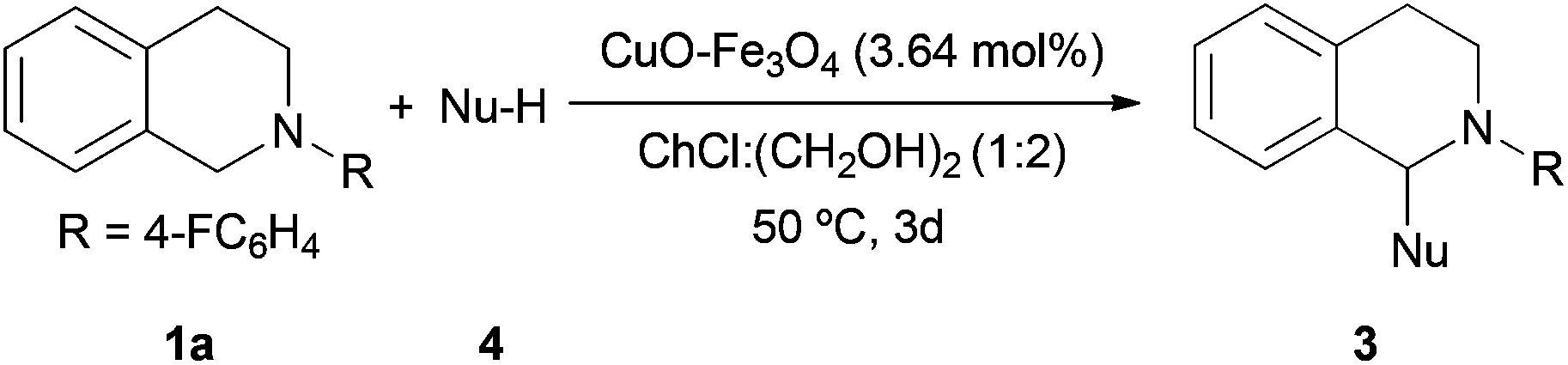
Experimental
General
XPS analyses were carried out on a VG-MicrotechMutilab. XRD analyses were carried out on a Bruker D-8 Avance diffractometer with a Göebel mirror, a high temperature chamber (up to 900 °C), an X-ray generator Kristalloflex K 760-80F (3 kW, 20–60 kV and 5–80 mA).The TEM images were obtained on a JEOL, model JEM-2010 equipped with an X-ray detector Oxford INCA Energy TEM 100 for microanalysis (EDS). XRF analyses were obtained on a Philips Magix PRO (PW2400) X-ray spectrometer equipped with a rhodium X-ray tube and a beryllium window. BET isotherms were carried out on an Autosorb-6 (Quantachrome), using N2. The melting points were obtained by using a Reichert Thermovar apparatus. The NMR spectra were recorded on a Bruker AC-300 (300 MHz for 1H and 75 MHz for 13C) using CDCl3 as a solvent and TMS as an internal standard for 1H and 13C; chemical shifts are given in δ (parts per million) and coupling constants (J) in hertz. FT-IR spectra were recorded on a JASCO 4100LE (Pike Miracle ATR) spectrophotometer. Mass spectra (EI) were recorded at 70 eV on a Shimadzu QP-5000 spectrometer, giving fragment ions in m/z with relative intensities (%) in parentheses. The chromatographic analyses (GLC) were carried out on a Hewlett Packard HP-5890 instrument equipped with a flame ionization detector and 12 m HP-1 capillary column (0.2 mm diam, 0.33 mm film thickness, OV-1 stationary phase), using nitrogen (2 mL min−1) as a carrier gas, Tinjector = 275 °C, Tdetector = 300 °C, Tcolumn = 60 °C (3 min) and 60–270 °C (15 °C min−1), P = 40 kPa. Thin layer chromatography (TLC) was carried out on Schleicher & Schuell F1400/LS 254 plates coated with a 0.2 mm layer of silica gel; detection was carried out by UV254 light. Column chromatography was performed using silica gel 60 of 40–63 mesh. All reagents were commercially available (Acros, Aldrich, Fluorochem) and were used as received. The ICP-MS analyses were carried out on a Thermo Elemental VGPQ-ExCell spectrometer. The elemental analysis was performed on an Elemental Microanalyzer Thermo Finnigan Flash 1112 Series.
Synthetic procedures
:1) as an eluent.
On the other hand, in order to recycle only the catalyst, we added water to the reaction mixture, dissolving the DES and decanting the solution with the aid of a magnet, the catalyst remained in the reaction vessel. Then, fresh DES, nitromethane and compound 1a were added to the vessel, carrying out the new reaction under standard conditions.
Conclusions
In conclusion, we have demonstrated that the appropriate mixture of DES and copper(II) oxide impregnated on magnetite is a good catalytic system to perform the cross-dehydrogenative coupling reaction between tetrahydroisoquinolines with a broad range of pro-nucleophiles in a highly selective way. The little amount of copper catalyst used is the lowest copper catalyst loading ever published. A direct proportional relationship between yield and conductivity was found in the absence of water. The high recyclability of the mixture (solvent + catalyst), as well as the use of cyclopentyl methyl ether for the workup drives this protocol towards Green Chemistry. Moreover, the protocol uses only the oxygen present in air, showing the high activity of the catalytic system and providing the first example of a biorenewable approach, with water being the only stoichiometric waste. All these facts made the sustainability of the whole process extremely high, compared with any other alternative.Acknowledgements
This work was supported by the Spanish Ministerio de Economía y Competitividad (MICINN; CTQ2011-24151) and the University of Alicante. J. M. P. thanks the MICINN (FPI program) for her fellowship.Notes and references
- The Chemistry and Biology of Isoquinoline Alkaloids, ed. J. D. Phillipson and M. F. Roberts, Springer-Verlag, Berlin, 1985 Search PubMed.
- (a) Y. Li, H. B. Zhang, W. L. Huang, X. Zhen and Y. M. Li, Chin. Chem. Lett., 2008, 19, 169–171 CrossRef CAS; (b) M. P. Chelopo, S. A. Pawar, M. K. Sokhela, T. Govender, H. G. Kruger and G. E. M. Maguire, Eur. J. Med. Chem., 2013, 66, 407–414 CrossRef CAS PubMed; (c) T. Ramanivas, B. Sushma, V. L. Nayak, K. C. Shekar and A. K. Srivastava, Eur. J. Med. Chem., 2015, 92, 608–618 CrossRef CAS PubMed.
- R. Gitto, R. Caruso, B. Pagano, L. D. Luca, R. Citraro, E. Russo, G. D. Sarro and A. Chimirri, J. Med. Chem., 2006, 49, 5618–5622 CrossRef CAS PubMed.
- G. L. Grunewald, V. H. Dahanukar, T. M. Caldwell and K. R. Criscione, J. Med. Chem., 1997, 40, 3997–4005 CrossRef CAS PubMed.
- (a) A. J. Bojarski, M. J. Mokrosz, S. C. Minol, A. Koziol, A. Wesolowska, E. Tatarczynska, A. Klodzinska and E. Chojnacka-Wójcik, Bioorg. Med. Chem., 2002, 10, 87–95 CrossRef CAS PubMed; (b) J. Renaud, S. F. Bischoff, T. Buhl, P. Floersheim, B. Fournier, C. Halleux, J. Kallen, H. Keller, J.-M. Schaeppi and W. Stark, J. Med. Chem., 2003, 46, 2945–2957 CrossRef CAS PubMed; (c) M. E. Ashford, V. H. Nguyen, I. Greguric, T. Q. Pham, P. A. Keller and A. Katsifis, Org. Biomol. Chem., 2014, 12, 783–794 RSC.
- J. H. Kang, BMB Rep., 2011, 114–119 Search PubMed.
- (a) H. Nakamura, Synlett, 2015, 1649–1664 CrossRef CAS; (b) B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308–1318 CrossRef CAS PubMed.
- C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed.
- K. M. Jones, P. Karier and M. Klussmann, ChemCatChem, 2012, 4, 51–54 CrossRef CAS.
- (a) T. Zeng, G. Song, A. Moores and C.-J. Li, Synlett, 2010, 2002–2008 CAS; (b) W. Han, P. Mayer and A. R. Ofial, Adv. Synth. Catal., 2010, 352, 1667–1676 CrossRef CAS; (c) P. Liu, C.-Y. Zhou, S. Xiang and C.-M. Che, Chem. Commun., 2010, 46, 2739–2741 RSC; (d) M. O. Ratnikov, X. Xu and M. P. Doyle, J. Am. Chem. Soc., 2013, 135, 9475–9479 CrossRef CAS PubMed.
- (a) Z. Li and C.-J. Li, J. Am. Chem. Soc., 2004, 126, 11810–11811 CrossRef CAS PubMed; (b) Z. Li and C.-J. Li, Org. Lett., 2004, 6, 4997–4999 Search PubMed; (c) Z. Li, D. S. Bohle and C.-J. Li, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8928–8933 CrossRef CAS PubMed; (d) Z. Li, P. D. MacLeod and C.-J. Li, Tetrahedron: Asymmetry, 2006, 17, 590–597 CrossRef CAS; (e) O. Baslé and C.-J. Li, Green Chem., 2007, 9, 1047–1050 Search PubMed; (f) D. Sureshkumar, A. Sud and M. Klussmann, Synlett, 2009, 1558–1561 CAS; (g) W. Su, J. Yu, Z. Li and Z. Jiang, J. Org. Chem., 2011, 76, 9144–9150 CrossRef CAS PubMed; (h) E. Boess, C. Schmitz and M. Klussmann, J. Am. Chem. Soc., 2012, 134, 5317–5325 CrossRef CAS PubMed; (i) R. Hudson, S. Ishikawa, C.-J. Li and A. Moores, Synlett, 2013, 1637–1642 CAS; (j) J. Yu, Z. Li, K. Jiang, M. Liu and W. Su, Tetrahedron Lett., 2013, 54, 2006–2009 CrossRef CAS; (k) F.-F. Wang, C.-P. Luo, G. Deng and L. Yang, Green Chem., 2014, 16, 2428–2431 RSC; (l) G. H. Dang, D. T. Nguyen, D. T. Le, T. Truong and N. T. S. Phan, J. Mol. Catal. A: Chem., 2014, 395, 300–306 CrossRef CAS; (m) X. Liu, J. Zhang, S. Ma, Y. Ma and R. Wang, Chem. Commun., 2014, 50, 15714–15717 RSC; (n) S. Sun, C. Li, P. E. Floreacing, H. Lou and L. Liu, Org. Lett., 2015, 17, 1684–1687 CrossRef CAS PubMed; (o) I. Perepichka, S. Kundu, Z. Hearne and C.-J. Li, Org. Biomol. Chem., 2015, 13, 447–451 RSC.
- (a) Q.-Y. Meng, Q. Liu, J.-J. Zhong, H.-H. Zhang, Z.-J. Li, B. Chen, C.-H. Tung and L. Z. Wu, Org. Lett., 2012, 14, 5992–5995 CrossRef CAS PubMed; (b) M. Rueping, R. M. Koenigs, K. Poscharny, D. C. Fabry, D. Leonori and C. Vila, Chem. – Eur. J., 2012, 18, 5170–5174 CrossRef CAS PubMed; (c) D. B. Freeman, L. Furst, A. G. Condie and C. R. J. Stephenson, Org. Lett., 2012, 14, 94–97 CrossRef CAS PubMed; (d) P. Kohls, D. Jadhav, G. Pandey and O. Reiser, Org. Lett., 2012, 14, 672–675 CrossRef CAS PubMed; (e) J. W. Trucker, Y. Zhang, T. F. Jamison and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2012, 51, 4144–4147 CrossRef PubMed; (f) L. R. Espelt, E. M. Wiensch and T. P. Yoon, J. Org. Chem., 2013, 78, 4107–4114 CrossRef PubMed; (g) Q.-Y. Meng, J.-J. Zhong, Q. Liu, X.-W. Gao, H.-H. Zhang, T. Lei, A.-J. Li, K. Feng, B. Chen, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2013, 135, 19052–19055 CrossRef CAS PubMed; (h) G. Bergonzini, C. S. Schindler, C.-J. Wallentin, E. N. Jacobsen and C. R. J. Stephenson, Chem. Sci., 2014, 5, 112–116 RSC.
- R. Pollice, N. Dastbaravardeh, N. Marquise, M. D. Mihovilovic and M. Schnürch, ACS Catal., 2015, 5, 587–595 CrossRef CAS PubMed.
- J. Xuan, T.-T. Zeng, Z.-J. Feng, Q.-H. Deng, J.-R. Chen, L.-Q. Lu, W.-J. Xiao and H. Alper, Angew. Chem., Int. Ed., 2015, 54, 1625–1628 CrossRef CAS PubMed.
- (a) A. Tanoue, W.-J. Yoo and S. Kobayashi, Adv. Synth. Catal., 2013, 355, 269–273 CAS; (b) C. Huo, C. Wang, M. Wu, X. Jia, X. Wang, Y. Yuan and H. Xie, Org. Biomol. Chem., 2014, 12, 3123–3128 RSC.
- (a) A. G. Condie, J. C. Gozález-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464–1465 CrossRef CAS PubMed; (b) Z.-J. Feng, J. Xuan, X.-D. Xia, W. Ding, W. Guo, J.-R. Chen, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Org. Biomol. Chem., 2014, 12, 2037–2040 RSC.
- T. Amaya, T. Ito and T. Hirao, Heterocycles, 2012, 86, 927–932 CrossRef CAS.
- (a) A. S.-K. Tsang, P. Jensen, J. M. Hook, A. S. K. Hashmi and M. H. Todd, Pure Appl. Chem., 2011, 83, 655–665 CrossRef CAS; (b) W. Fu, W. Guo, G. Zou and C. Xu, J. Fluorine Chem., 2012, 140, 88–94 CrossRef CAS; (c) J. Dhineshkumar, M. Lamani, K. Alagiri and K. R. Prabhu, Org. Lett., 2013, 15, 1092–1095 CrossRef CAS PubMed; (d) G. Zhang, Y. Ma, S. Wang, W. Kong and R. Wang, Chem. Sci., 2013, 4, 2645–2651 RSC; (e) A. Tanoue, W.-J. Yoo and S. Kobayashi, Org. Lett., 2014, 16, 2346–2349 CrossRef CAS PubMed; (f) L. Huang and J. Zhao, RSC Adv., 2013, 3, 23377–23388 RSC; (g) H.-M. Huang, Y.-J. Li, Q. Ye, W.-B. Yu, L. Han, J.-H. Jia and J.-R. Gao, J. Org. Chem., 2014, 79, 1084–1092 CrossRef CAS PubMed; (h) J. F. Franz, W. B. Kraus and K. Zeitler, Chem. Commun., 2015, 51, 8280–8283 RSC.
- E. L. Smith, A. P. Abbot and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- (a) C. Ruβ and B. König, Green Chem., 2012, 14, 2969–2982 RSC; (b) Q. Zhang, K. D. O. Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC; (c) Y. Dai, J. van Spronsen, G.-J. Witkamp, R. Verpoorte and Y. H. Choi, Anal. Chim. Acta, 2013, 766, 61–68 CrossRef CAS PubMed; (d) M. Francisco, A. van den Bruinhorst and M. C. Kroon, Angew. Chem., Int. Ed., 2013, 52, 3074–3085 CrossRef CAS PubMed; (e) B. Tang and K. H. Row, Monatsh. Chem., 2013, 144, 1427–1454 CrossRef CAS; (f) a. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef CAS.
- P. Liu, J.-W. Hao, L.-P. Mo and Z.-H. Zhang, RSC Adv., 2015, 5, 48675–48705 RSC.
- (a) C. Vidal, F. J. Suárez and J. García-Álvarez, Catal. Commun., 2014, 44, 76–79 CrossRef CAS; (b) M. J. Rodríguez-Álvarez, C. Vidal, J. Díez and J. García-Álvarez, Chem. Commun., 2014, 50, 12927–12929 RSC; (c) C. Vidal, L. Merz and J. García-Álvarez, Green Chem., 2015, 17, 3870–3878 RSC; (d) J. García-Alvarez, in Green Technologies for the Environment, eds. R. Luqie and O. Obre, ACS Books, New York, 2015, pp. 37–53 Search PubMed.
- J. Lu, X.-T. Li, E.-Q. Ma, L.-P. Mo and Z.-H. Zhang, ChemCatChem, 2014, 6, 2854–2859 CrossRef CAS.
- D. J. Ramón, Johnson Matthey Technol. Rev., 2015, 59, 120–122 CrossRef.
- (a) K. Watanabe, N. Yamagiwa and Y. Torisawa, Org. Process Res. Dev., 2007, 11, 251 CrossRef CAS; (b) A. Kadam, M. Nguyen, M. Kopach, P. Richardson, F. Gallou and Z.-K. Wan, Green Chem., 2013, 15, 1880 RSC.
- A. P. Abbot, G. Capper, D. L. Davies, K. J. McKenzie and S. U. Obi, J. Chem. Eng. Data, 2006, 51, 1280–1282 CrossRef.
- I. Perepichka, S. Kundu, Z. Hearne and C. J. Li, Org. Biomol. Chem., 2015, 13, 447–451 CAS.
- S. O'Sullivan, E. Doni, T. Tuttle and J. A. Murphy, Angew. Chem., Int. Ed., 2014, 53, 474–478 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images, characterization data, and 1H-NMR and 13C-NMR data. See DOI: 10.1039/c5gc01745a |
| This journal is © The Royal Society of Chemistry 2016 |