
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient visible-light-driven CO2 reduction to CO using a Ru(II)–Re(I) supramolecular photocatalyst in an aqueous solution†
Akinobu
Nakada
a,
Kazuhide
Koike
bc,
Kazuhiko
Maeda
a and
Osamu
Ishitani
*ac
aDepartment of Chemistry, Graduate School of Science and Engineering, Tokyo Institute of Technology, 2-12-1-NE-1 O-okayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: ishitani@chem.titech.ac.jp; Fax: +81 3 5734 2284; Tel: +81 3 5734 2240
bNational Institute of Advanced Industrial Science and Technology, 16-1 Onogawa, Tsukuba 305-8569, Japan
cCREST, Japan Science and Technology Agency (JST), 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 28th August 2015
Abstract
In an aqueous solution, [Ru(dmb)2–(BL)–Re(CO)3Cl]2+ (BL = bridging ligand) most efficiently photocatalyzed the reduction of CO2 to CO under visible-light irradiation using 2-(1,3-dimethyl-2,3-dihydro-1H-benzimidazol-2-yl)benzoic acid (BI(CO2H)H) as a water-soluble sacrificial reductant (ΦCO = 13%, TON = 130). Since BI(CO2H)H could efficiently produce one-electron-reduced species of [Ru(diimine)3]2+-type complexes under visible-light irradiation even in an aqueous solution, that is one of the main reasons why the photocatalytic system induced the highly efficient CO2 reduction. This result strongly indicates that BI(CO2H)H should be a useful reductant for evaluating the real abilities of various photocatalytic systems in water as well.
The photocatalytic reduction of CO2 using water as a reductant and sunlight as an energy source is a promising technology for solving the serious problems of global warming, as well as energy and carbon-resource shortages. Although various photocatalytic systems involving transition-metal complexes as a photosensitizer and/or a catalyst have been reported1,2 besides the semiconductor photocatalyst,3 most of the systems using metal complexes have been tested only in organic solvents with a sacrificial reductant. For future practical implementation of photocatalytic reduction technology, photocatalytic reactions must proceed using water as the reductant in aqueous solution. As the first step toward this objective, efficient photocatalysts for CO2 reduction that can function in an aqueous solution should be developed, even if they require a sacrificial reductant. Several photocatalytic systems based on a metal-complex catalyst with a [Ru(bpy)3]2+-type photosensitizer (bpy = 2,2′-bipyridine) in aqueous solutions have been tested for CO2 reduction4–12 and for hydrogen evolution from water13–19 in the presence of ascorbate ion (asc−) as a sacrificial reductant. Unfortunately, most of these systems exhibited very low efficiency, durability, and selectivity for CO2 reduction.
Ru(II)–Re(I) supramolecular photocatalysts constructed with both a Ru photosensitizer and Re catalyst units can efficiently and selectively reduce CO2 to CO in a dimethylformamide (DMF) and triethanolamine (TEOA) mixed solution; they also exhibit high durability.20–26 In particular, the use of 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH) as a sacrificial reductant achieved an extraordinarily high quantum yield of CO formation (ΦCO = 45%) by suppression of the back electron transfer from one-electron-reduced species (OERS) of the photocatalyst to the one-electron-oxidized species (OEOS) of the reductant because of the fast deprotonation of the OEOS.25
We recently reported selective photocatalytic CO2 reduction to formic acid in an aqueous solution using a Ru(II)–Re(I) supramolecular photocatalyst and asc− as a reductant.4 In this system, however, the efficiency of the CO2 reduction was much lower than that using 1-benzyl-1,4-dihydronicotinamide (BNAH) as a reductant in the DMF–TEOA mixed solution. Although asc− is, to the best of our knowledge, the only reported reductant that can be used in an aqueous solution for photocatalytic CO2 reduction using a [Ru(bpy)3]2+-type photosensitizer, back electron transfer from the reduced photosensitizer to the OEOS of asc− is efficient because of the stability of the OEOS, and the final product of the oxidized ascorbate (dehydroascorbic acid) accepts the electron from the reduced photosensitizer and/or reaction intermediates.4,16,18,19 Moreover, we observed that asc− accelerated a photochemical ligand-substitution reaction of the Ru(II) photosensitizer, which caused deactivation of the photocatalytic system.4 These properties of asc− as an inhibitor should make it difficult to evaluate the “real” photocatalytic activities of the systems constructed with such metal complexes in an aqueous solution.
Herein, we report 2-(1,3-dimethyl-2,3-dihydro-1H-benzimidazol-2-yl)benzoic acid (BI(CO2H)H, Chart 1) as a suitable water-soluble reductant for the photocatalytic CO2 reduction; this reductant efficiently quenched the excited state of the Ru(II) photosensitizer unit, giving the OERS of the photosensitizer with a high yield. When this reductant was used, a Ru(II)–Re(I) supramolecular photocatalyst (RuRe, Chart 1) functioned as an efficient (Φ = 13%) and durable (TON = 130) photocatalyst for CO2 reduction, selectively giving CO even in an aqueous solution.
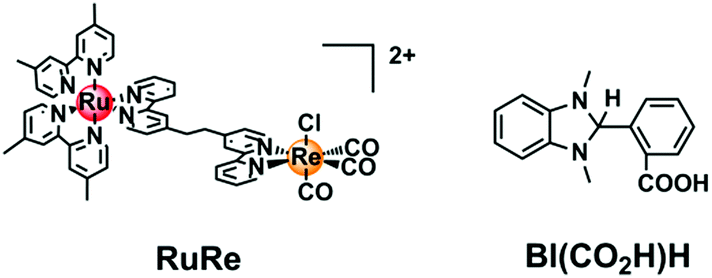 | ||
| Chart 1 The Ru(II)–Re(I) supramolecular photocatalyst RuRe and the reductant BI(CO2H)H used in this study. | ||
As the first step in investigating the photocatalytic reaction, we evaluated the solubility of BI(CO2H)H in aqueous solutions: to dissolve BI(CO2H)H in an aqueous solution, more than an equal amount of NaOH should be added to the solution. This indicates that BI(CO2H)H dissolves as the carboxylate ion, BI(CO2−)H. The pH at the equivalence point was 8.9 for 10 mM BI(CO2H)H. Because bubbling of the solution containing BI(CO2H)H (10 mM) with CO2 induced precipitation of BI(CO2H)H even in the presence of 0.1 M NaOH, the concentration of CO2 in the solution should be controlled for the photocatalytic reaction. Suitable conditions were achieved using the following procedure: a CO2-saturated NaOH (0.1 M) aqueous solution was mixed with the same amount of an aqueous solution containing BI(CO2H)H (20 mM) and NaOH (0.1 M), which was bubbled with Ar, giving a solution at pH = 9.8 in which all of the added BI(CO2H)H was completely dissolved.
In a typical run of the photocatalytic reaction, an aqueous solution containing RuRe (0.05 mM), BI(CO2H)H (10 mM), NaOH (0.1 M), and CO2 was irradiated at λex > 500 nm using a high-pressure Hg lamp combined with a K2CrO4 (30% w/w, d = 1 cm) filter. The Ru photosensitizer unit of RuRe was selectively excited because BI(CO2−)H and the Re catalyst unit could not absorb the λ > 500 nm light (Fig. S1, ESI†). The irradiation-time dependences of CO, formate, and H2 production are shown in Fig. 1a. CO was the main product, and the turnover number of CO formation (TONCO) based on the amount of photocatalyst used after 6 h of irradiation reached 130. The quantum yield of the photocatalytic CO formation was 13% under the optimized conditions using 480 nm monochromatic light (see ESI†). To the best of our knowledge, this value is 5.6 times greater than that of the best reported for photocatalytic CO2 reduction in an aqueous solution under visible-light irradiation.11 H2 was also produced as a by-product during irradiation with long induction periods of up to 3 h. Fig. 1b shows the UV–vis absorption spectra of the reaction solution after irradiation, where the peak at approximately 460 nm is attributed to the MLCT absorption band of the Ru(II) unit. This result indicates that the Ru photosensitizer unit decomposed during the induction period. [RuII(bpy)2(X)(Y)]n+-type complexes have been reported to function as catalysts for the photocatalytic formation of H2 in solutions containing water.27 The decomposition product(s) of the Ru unit can therefore be reasonably assumed to catalyze H2 evolution after 3 h of irradiation.
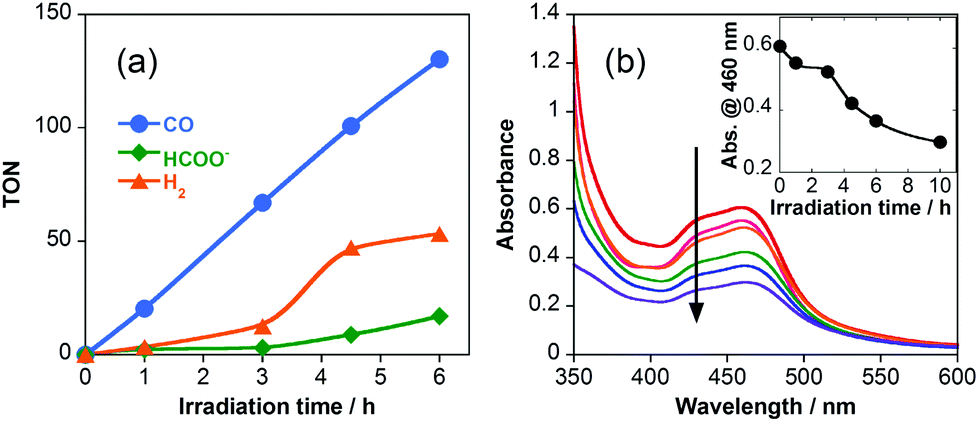 | ||
| Fig. 1 (a) Time-dependent production of CO (blue), HCOO− (green), and H2 (orange) in the photocatalytic reaction using RuRe. A 4 mL of NaOH (0.1 M) aqueous solution containing RuRe (0.05 mM) and BI(CO2−)H (10 mM) was irradiated at λex > 500 nm under a CO2 atmosphere. (b) UV–vis absorption changes during the photocatalytic reaction. The inset shows time-dependent absorbance changes at 460 nm. | ||
Table 1 summarizes the results of the photocatalytic reaction and its control experiments. As previously described, the irradiation to RuRe in the presence of BI(CO2−)H under a CO2 atmosphere photocatalytically produced CO as the main product (entry 1, Table 1). On the other hand, in the control experiments without irradiation, RuRe, BI(CO2−)H, or CO2, i.e., under an Ar atmosphere, did not give any CO2 reduction products (entries 2–5). Notably, much less CO was produced with larger amounts of H2 and formate when only a mononuclear model complex of the Ru photosensitizer unit, [Ru(dmb)2(mmb)]2+ (Ru, dmb = 4,4′-dimethyl-2,2′-bipyridine; mmb = 4-methyl-2,2′-bipyridine), was used instead of RuRe (entry 6). The formation of H2 and formate is attributable to products obtained from the photocatalytic reaction system consisting of Ru as a redox photosensitizer and the decomposition product(s) of the Ru as the catalyst. A mononuclear model complex of the Re catalyst unit, Re{4,4′-(CH2PO3H2)2bpy}(CO)3Cl (Re), did not drive CO2 reduction (entry 7) because Re cannot absorb the irradiated light. A mixed system of two mononuclear model complexes Ru and Re did not work well (entry 8). This strongly suggests that the strategy of the supramolecular photocatalysts, i.e., connecting a photosensitizer and a catalyst with appropriate chemical bonding, is useful for constructing various efficient photocatalytic systems not only in organic solutions20,24 but also in aqueous solution.4
| Entry | Complexb | BI(CO2−)Hc | hν | CO2 | Product/μmol | ||
|---|---|---|---|---|---|---|---|
| CO | HCOO− | H2 | |||||
| a Four milliliters of the reaction solutions were irradiated for 3 h. b The complex concentration was 0.05 mM. c The BI(CO2−)H concentration was 10 mM. d λ ex > 500 nm. e [Ru(dmb)2(mmb)]2+ (dmb = 4,4′-dimethyl-2,2′-bipyridine, mmb = 4-methyl-2,2′-bipyridine). f Re{4,4′-(CH2PO3H2)2bpy}(CO)3Cl. ○ = with, × = without. | |||||||
| 1 | RuRe | ○ | ○ | ○ | 13.5 | 0.6 | 2.5 |
| 2 | RuRe | ○ | × | ○ | N.D. | N.D. | <0.1 |
| 3 | RuRe | ○ | ○ | × | N.D. | N.D. | 0.5 |
| 4 | RuRe | × | ○ | ○ | N.D. | N.D. | N.D. |
| 5 | × | ○ | ○ | ○ | N.D. | N.D. | <0.1 |
| 6 | Ru | ○ | ○ | ○ | 1.0 | 4.5 | 19.9 |
| 7 | Re | ○ | ○ | ○ | N.D. | N.D. | <0.1 |
| 8 | Ru + Ref | ○ | ○ | ○ | 1.5 | 0.4 | 1.0 |
To clarify the carbon sources of the produced CO and formate, we conducted 13CO2 labeling experiments. GC-mass spectra (Fig. S2, ESI†) show carbon monoxide and formic acid and/or formate produced by the photocatalytic reactions using RuRe under a 13CO2 (99%, 609 mmHg) atmosphere and under an ordinary CO2 atmosphere. These results clearly indicate that 93% of CO was obtained by CO2 reduction. On the other hand, almost no formate was produced from CO2. The formate might be produced by the partial decomposition of BI(CO2−)H; the oxidation products of BI(CO2−)H are described in greater detail below. The carbonyl ligands of Re(bpy)(CO)3Cl have been reported to be gradually substituted by 13CO during the photocatalytic reduction of 13CO2,28 and the amount of 12CO produced in the photocatalytic reduction was only 3.7 times the molar equivalents of RuRe added. Therefore, a similar ligand exchange between the carbonyl ligands and the produced CO on the Re center should proceed in the photocatalytic reduction of 13CO2 in the presence of RuRe; this exchange should be the main carbon source of the produced 12CO. Another carbon source of 12CO might be the contaminant, i.e., 12CO2 in the used 13CO2 gas (the 13C content was 99%).
We evaluated the reducing power of BI(CO2−)H in the aqueous solution using cyclic voltammetry, where irreversible oxidation waves of BI(CO2−)H and asc− were observed (Fig. S3, ESI†). The peak potential of BI(CO2−)H was negatively shifted by 340 mV compared to that of asc−; therefore, BI(CO2−)H has a much stronger reducing power compared with that of asc−, which is one of the properties that makes BI(CO2−)H a suitable sacrificial reductant for the photocatalytic CO2 reaction in aqueous solution. Actually, BI(CO2−)H served as an efficient quencher of emission from the 3MLCT excited state of the Ru photosensitizer unit of RuRe in an aqueous solution (eqn (1) and Fig. S4, ESI†). The emission quenching rate constant (kq) was determined as 4.6 × 109 M−1 s−1 from the slope of the linear Stern–Volmer plots (Fig. S4, inset, ESI†), eqn (2), and the emission lifetime of RuRe (τem = 366 ns, Fig. S5, ESI†). Notably, the kq value was similar to the diffusion-controlled rate constant in water (7.4 × 109 M−1 s−1 at 25 °C), and the kq with ascorbate instead of BI(CO2−)H was 2.4 × 107 M−1 s−1.
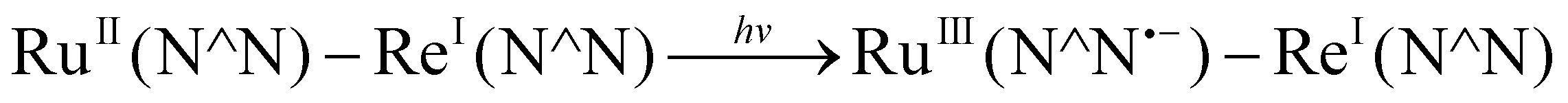 | (1a) |
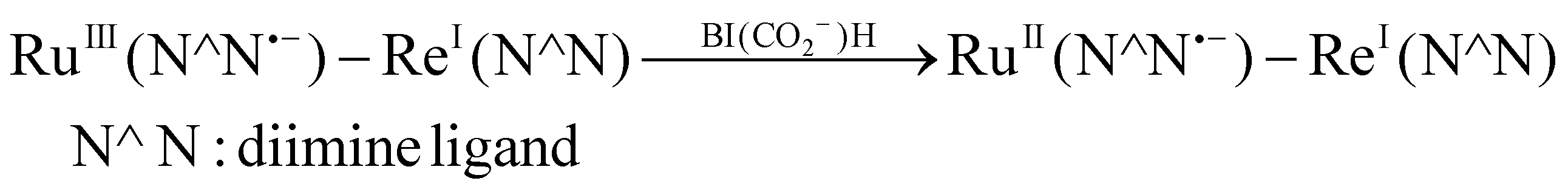 | (1b) |
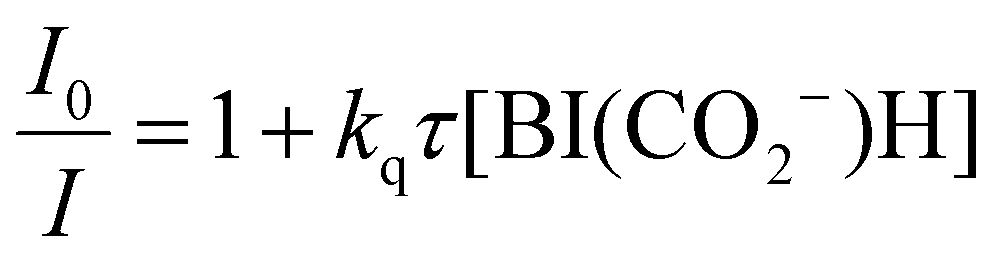 | (2) |
The first reduction potentials (E1/2) of Ru and Re(dmb)(CO)3Cl measured in MeCN were −1.73 V and −1.71 V vs. Ag/AgNO3, respectively (Fig. S6, ESI†).29 Therefore, the intramolecular electron transfer from the OERS of the Ru unit to the Re unit (eqn (3)) should be thermodynamically favourable. Taking into account this fact and the results of the control experiments described previously, we can conclude that the CO2 reduction proceeded on the Re unit:
| RuII(N^N˙−) − ReI(N^N) → RuII(N^N) − ReI(N^N˙−) | (3) |
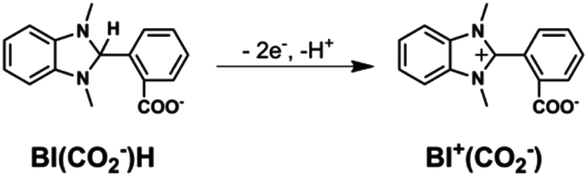
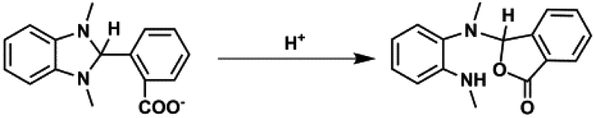
1H NMR spectra of the reaction solution (Fig. S7, ESI†) show that BI(CO2−)H was converted into a two-electron-oxidized compound BI+(CO2−) in 79% yield after the photocatalytic reaction for 22.5 h (eqn (4)). Other smaller signals possibly associated with the formation of the hydrolysis product of BI(CO2−)H (eqn (5)) were also observed. Another possible product is the fragment(s) of BI(CO2−)H generated by the elimination of formate detected after the photocatalytic reaction:
 | (4) |
 | (5) |
On the basis of the quantitative analysis with the 1H NMR spectra, the amount of BI+(CO2−) produced was very similar to the combined amounts of CO and H2 produced during the photocatalytic reaction (Fig. 2). This similarity clearly indicates that BI(CO2−)H acted as a two-electron donor for the photocatalytic formation of CO and H2 because both require two-electron reduction. Given the results of both the 13CO2 labeling experiments and the 1H NMR analysis, we conclude that the material balance of the photocatalytic CO formation is as shown in eqn (6):
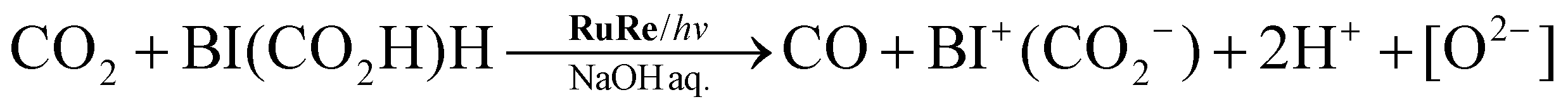 | (6) |
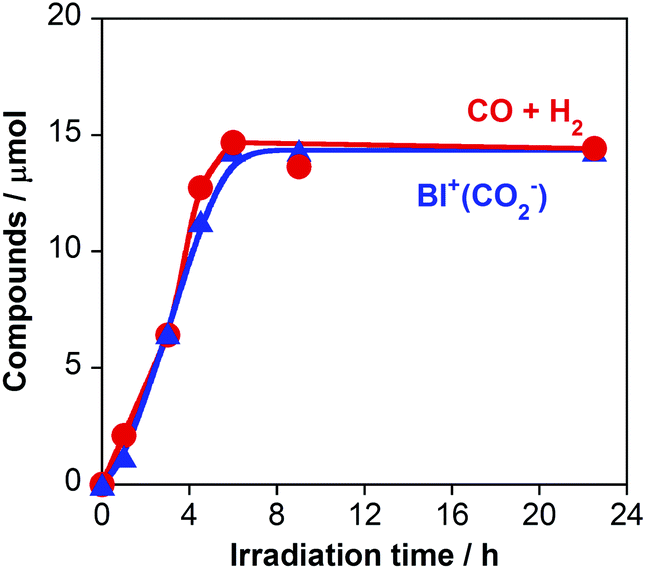 | ||
| Fig. 2 Amounts of CO + H2 (red) and BI+(CO2−) (blue) produced during the photocatalytic reaction: a 2 mL of D2O solution containing RuRe (0.05 mM), BI(CO2−)H (10 mM), and NaOD (0.1 M) was irradiated at λex > 500 nm under a CO2 atmosphere. | ||
As previously described, the photocatalysis of RuRe (ΦCO = 13%, TON = 130) when BI(CO2−)H was used as the reductant was substantially improved compared to the reported performance of a Ru(II)–Re(I) supramolecular system with asc− (ΦHCOOH = 0.2%, TONHCOOH = 25). The reasons for the low photocatalytic activities in the case of asc− were described previously; one of them is the efficient back electron transfer from the reduced Ru(II) photosensitizer unit to the oxidized asc−. To clarify the improvement of the photochemical reduction process of the Ru photosensitizer unit by BI(CO2−)H, we monitored the UV–vis absorption spectral changes of an aqueous solution containing the mononuclear model complex Ru and BI(CO2−)H during irradiation under an Ar atmosphere. A new absorption peak at λmax = 510 nm, which is attributed to the OERS of Ru, was observed during the irradiation (Fig. S8, ESI†). Notably, no such accumulation of the OERS was observed in the case where asc− (200 mM) was used instead of BI(CO2−)H (10 mM).4 Because the quenching efficiencies of emission from the excited Ru photosensitizer unit in the experiments were similar in both cases (94% by 10 mM of BI(CO2−)H and 87%4 by 200 mM of asc−), the efficiency of the back electron transfer from the OERS of Ru to the oxidized BI(CO2−)H should be much lower compared to that in the case where asc− was used. This is one of the main reasons why BI(CO2−)H remarkably improved the quantum yield for CO2 reduction compared to that achieved with asc−.
Another significant difference between the cases where BI(CO2−)H and asc− were used is the main product of CO2 reduction: CO in the case of BI(CO2−)H and formate in the case of asc−. Kaneko and co-workers reported that, in the electrocatalytic CO2 reduction with Re(bpy)(CO)3Br as a catalyst in an aqueous solution, formic acid was the main product during the electrolysis at −1.3 V vs. SCE, whereas CO became the main product at more negative applied potentials.30 This result might indicate that the electron-supply rate to the Re catalyst affects distribution of the reduction products, i.e., formate might become a main product under a slow electron-supply condition. If this is true, we can understand the difference of the main product between the photocatalytic systems in which BI(CO2−)H and asc− are used as the reductants. The formation speed of the OERS of the Ru(II) photosensitizer unit was much lower in the case where asc− was used than in the BI(CO2−)H system, which should cause slow electron supply to the Re catalytic unit.
Conclusions
A Ru(II)–Re(I) binuclear complex exhibited high photocatalytic activity with 13% quantum yield for CO2 reduction to CO even in aqueous solution. The new sacrificial reductant BI(CO2−)H enabled the efficient production of the reduced photosensitizer unit, which allowed us to observe the real photocatalytic activities of the Ru(II)–Re(I) supramolecular photocatalyst in water. We believe that the water-suitable Ru(II)–Re(I) supramolecular photocatalyst can be used in a Z-scheme hybrid system31,32 with a semiconductor photocatalyst for CO2 reduction, where water is used as an electron donor.Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Artificial photosynthesis (AnApple)” (no. 24107005) from the Japan Society for the Promotion of Science (JSPS).References
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 Search PubMed.
- G. Sahara and O. Ishitani, Inorg. Chem., 2015, 54, 5096–5104 CrossRef CAS PubMed.
- J. Mao, K. Li and T. Peng, Catal. Sci. Technol., 2013, 3, 2481–2498 CAS.
- A. Nakada, K. Koike, T. Nakashima, T. Morimoto and O. Ishitani, Inorg. Chem., 2015, 54, 1800–1807 CrossRef CAS PubMed.
- A. H. A. Tinnemans, T. P. M. Koster, D. H. M. W. Thewissen and A. Mackor, Recl. Trav. Chim. Pays-Bas, 1984, 103, 288–295 CrossRef CAS.
- J. L. Grant, K. Goswami, L. O. Spreer, J. W. Otvos and M. Calvin, J. Chem. Soc., Dalton Trans., 1987, 2105–2109 RSC.
- C. A. Craig, L. O. Spreer, J. W. Otvos and M. Calvin, J. Phys. Chem., 1990, 94, 7957–7960 CrossRef CAS.
- E. Kimura, X. Bu, M. Shionoya, S. Wada and S. Maruyama, Inorg. Chem., 1992, 31, 4542–4546 CrossRef CAS.
- E. Kimura, S. Wada, M. Shionoya and Y. Okazaki, Inorg. Chem., 1994, 33, 770–778 CrossRef CAS.
- K. Mochizuki, S. Manaka, I. Takeda and T. Kondo, Inorg. Chem., 1996, 35, 5132–5136 CrossRef CAS.
- D. J. Boston, C. Xu, D. W. Armstrong and F. M. MacDonnell, J. Am. Chem. Soc., 2013, 135, 16252–16255 CrossRef CAS PubMed.
- N. Ikuta, S. Y. Takizawa and S. Murata, Photochem. Photobiol. Sci., 2014, 13, 691–702 CAS.
- G. M. Brown, B. S. Brunschwig, C. Creutz, J. F. Endicott and N. Sutin, J. Am. Chem. Soc., 1979, 101, 1298–1300 CrossRef CAS.
- S. Fukuzumi, T. Kobayashi and T. Suenobu, Angew. Chem., Int. Ed., 2011, 50, 728–731 CrossRef CAS PubMed.
- Y. Sano, A. Onoda and T. Hayashi, Chem. Commun., 2011, 47, 8229–8231 RSC.
- M. Guttentag, A. Rodenberg, R. Kopelent, B. Probst, C. Buchwalder, M. Brandstätter, P. Hamm and R. Alberto, Eur. J. Inorg. Chem., 2012, 2012, 59–64 CrossRef CAS.
- B. Shan, T. Baine, X. A. N. Ma, X. Zhao and R. H. Schmehl, Inorg. Chem., 2013, 52, 4853–4859 CrossRef CAS PubMed.
- M. Guttentag, A. Rodenberg, C. Bachmann, A. Senn, P. Hamm and R. Alberto, Dalton Trans., 2013, 42, 334–337 RSC.
- C. Bachmann, B. Probst, M. Guttentag and R. Alberto, Chem. Commun., 2014, 50, 6737–6739 RSC.
- B. Gholamkhass, H. Mametsuka, K. Koike, T. Tanabe, M. Furue and O. Ishitani, Inorg. Chem., 2005, 44, 2326–2336 CrossRef CAS PubMed.
- S. Sato, K. Koike, H. Inoue and O. Ishitani, Photochem. Photobiol. Sci., 2007, 6, 454–461 CAS.
- K. Koike, S. Naito, S. Sato, Y. Tamaki and O. Ishitani, J. Photochem. Photobiol., A, 2009, 207, 109–114 CrossRef CAS.
- H. Takeda and O. Ishitani, Coord. Chem. Rev., 2010, 254, 346–354 CrossRef CAS.
- Y. Tamaki, K. Watanabe, K. Koike, H. Inoue, T. Morimoto and O. Ishitani, Faraday Discuss, 2012, 155, 115–127 RSC.
- Y. Tamaki, K. Koike, T. Morimoto and O. Ishitani, J. Catal., 2013, 304, 22–28 CrossRef CAS.
- E. Kato, H. Takeda, K. Koike, K. Ohkubo and O. Ishitani, Chem. Sci., 2015, 6, 3003–3012 RSC.
- J.-M. Lehn and R. Ziessel, J. Organomet. Chem., 1990, 382, 157–173 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990–2012 CrossRef CAS.
- The reduction potentials of the complexes could not be measured in an aqueous solution because of competing hydrogen evolution on the working electrode.
- T. Yoshida, K. Tsutsumida, S. Teratani, K. Yasufuku and M. Kaneko, J. Chem. Soc., Chem. Commun., 1993, 631–633 RSC.
- K. Sekizawa, K. Maeda, K. Domen, K. Koike and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 4596–4599 CrossRef CAS PubMed.
- F. Yoshitomi, K. Sekizawa, K. Maeda and O. Ishitani, ACS Appl. Mater. Interfaces, 2015, 7, 13092–13097 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, photophysical data, results of 13CO2 labeling experiments, 1H NMR observation of the reductant, and UV-vis absorption changes. See DOI: 10.1039/c5gc01720c |
| This journal is © The Royal Society of Chemistry 2016 |