
Ex situ hydrodeoxygenation in biomass pyrolysis using molybdenum oxide and low pressure hydrogen†
Michael W.
Nolte
a,
Jing
Zhang
a and
Brent H.
Shanks
*ab
aDepartment of Chemical and Biological Engineering, Iowa State University, Ames, IA 50011, USA. E-mail: bshanks@iastate.edu; Fax: +1 515 294 2689; Tel: +1 515 294 1895
bCenter for Biorenewable Chemicals (CBiRC), Iowa State University, Ames, IA 50011, USA
First published on 2nd September 2015
Abstract
Biomass pyrolysis vapors were hydrodeoxygenated using MoO3 and low pressure H2 (1.8 bar Ptotal) in a tandem microreactor. High yields of mostly linear alkanes (C1–C6) and aromatics were achieved from the pyrolysis of cellulose, lignin, and corn stover feedstocks.
The pyrolysis of biomass in order to produce a liquid intermediate for the production of fuels and chemicals is attracting attention as a potential partial replacement for petroleum. However, the unique properties of biomass pyrolysis oil (or bio-oil) makes the processing of it in a traditional petroleum refinery a significant challenge. Notably, the acidic products will corrode steel vessels and piping, the water and alkali and alkaline earth metals (AAEMs) in bio-oil will be detrimental to typical silica and alumina-supported metal catalysts, and the high oxygen content will require significantly more hydroprocessing for removal, as well as other undesirable characteristics.1 Therefore, bio-oil requires at least some degree of preprocessing before introduction into a petroleum refinery.
One attractive method of upgrading bio-oil is through hydrodeoxygenation (HDO), which can reduce the oxygen content as well as acid concentration. Ideally in HDO, the oxygen in bio-oil is removed as water, while the carbon is retained.2 Efficient HDO reactions minimize hydrogen consumption in the process and are performed under lower hydrogen pressures thereby reducing capital and operation costs.2 Several catalyst systems have been investigated for HDO reactions with bio-oil model compounds as well as hydroprocessing of liquid bio-oil. Past efforts of hydroprocessing bio-oil have been thoroughly reviewed in the literature.3
In more recent work, zeolites have been found to generate aromatics in cellulose catalytic fast pyrolysis.4,5 However, hydrocarbon yields were low (up to ∼30 C%) and coke yields could be very high (as high as 60 C%).4,5 With a lignin feed, the coke yields were as high as 80 C%.4 Precious and non-precious metal catalysts have been shown to be active in key hydrogenation and hydrogenolysis reactions. Various supported Pt catalysts converted m-cresol to toluene at 60–80% selectivity at 0.5 atm H2 in N2 with minor products being methylcyclohexanol, methylcyclohexane, and phenol.6 The authors found the reaction to be sensitive to the partial pressure of H2, where an increase in H2 partial pressure led to an increase in m-cresol conversion and yield of the saturated ring products.6 Elliot et al. were able to recover high yields of naphthenes, aromatics, and alkanes using a hydrotreating step followed by a hydrocracking step, although the process was operated at a pressure of 13.8 MPa.7 Sitthisa and Resasco tested Cu, Pd, and Ni supported on SiO2 for the HDO of furfural.8 Copper was most active in the hydrogenation to furfuryl alcohol, but not HDO to methylfuran, while Pd and Ni gave the highest selectivity toward the decarbonylation product furan.8 Sulfided Mo and CoMo catalysts showed activity in the HDO of guaiacol at a hydrogen pressure of 4 MPa.9 Molybdenum sulfide gave similar selectivities to the products cyclohexane, methylcyclopentane, and benzene, while CoMoS showed >80% selectivity to benzene with much lower yields of saturated ring products.9 High surface area phosphided Ni and Mo catalysts yielded a mixture of HDO products from 4-methylphenol.10 A Ni2P catalyst was found to be more active than a MoP catalyst with the activity increasing with higher H2 pressure due to the minimization of coke formation.10
Using a high pressure hydropyrolysis reactor, Venkatakrishnan et al. were able to achieve a high carbon balance from the catalytic hydrodeoxygenation of cellulose and poplar wood feedstocks.11 The hydrodeoxygenation catalyst was Pt–Mo supported on multiwalled carbon nanotubes (MWCNTs) and was situated ex situ of the pyrolysis reactor to allow for independent pressure and temperature control.11 Relatively high hydrocarbon yields were obtained from HDO of cellulose (72.6 C%) and HDO of poplar wood (53.8 C%). From cellulose, the major hydrocarbon products were linear alkanes, including n-hexane (17.6 C%), which was postulated to have formed from the HDO of levoglucosan. The liquid fuel range (C4+) yields were 55% and 32% from cellulose and poplar, respectively. To achieve the high hydrocarbon yields, the HDO was performed at a total pressure of 2.7 MPa.11 To our knowledge, hydrodeoxygenation of biomass fast pyrolysis vapors at near atmospheric hydrogen pressure has not been reported. Ideally, the HDO would be performed at low pressure and result in products with a high selectivity to unsaturated hydrocarbons so as to minimize H2 consumption.
Prasomsri et al. used MoO3 and low pressure H2 for HDO of bio-oil model compounds.12 Remarkably, the catalyst was able to directly deoxygenate the model compounds, even removing the ring oxygen in furan compounds, without fully saturating the products.12 Under the reaction conditions used, the postulated reaction pathway was via a reverse Mars-van Krevelen mechanism, whereby H2 interacts with surface oxygen forming water and an active vacant site on the catalyst. The oxygen in the reactant then fills the vacancy, leading to oxygen transfer to the catalyst and the formation of the unsaturated product. Therefore, the catalyst requires a constant stream of H2 for continuous reduction and vacant site formation. However, it is possible to over reduce the Mo, as was found by the authors, where the catalyst gradually deactivated due to the formation of inactive Mo+4. Also, the rate of catalyst deactivation was found to increase as the reaction temperature increased. In a subsequent study looking into the surface modification of the MoO3, the authors postulated that the formation of oxycarbohydrides on the surface stabilized the active Mo+5 form, thereby slowing the reduction to Mo+4 and prolonging the activity of the catalyst.13 Molybdenum carbide catalysts have also been found to be effective in the HDO reactions.14 Using anisole as a reactant, Lee et al. obtained benzene selectivities over 90% at a range of reaction temperatures and conversions.14 However, Mo2C did yield cyclohexane at selectivities of 2–7%, while MoO3 was not found to catalyze the formation of any cyclic hydrogenation products from anisole.12,14
These studies suggest that molybdenum-based catalysts have unique properties that make them suitable for use as HDO catalysts. Notably, the catalytic material is cost effective, active at moderate temperatures (200–400 °C) and low H2 pressure while generally being more hydrogen efficient by minimizing hydrogenation reactions. However, to our knowledge these catalysts have only been tested with relatively simple model compounds that contain only one or two oxygen atoms. In this work, molybdenum oxide (MoO3, Sigma Aldrich, >99.5%) was employed for ex situ HDO of cellulose, lignin, and corn stover pyrolysis vapors. First, the optimal catalytic conditions were found for HDO of cellulose pyrolysis vapors and then these conditions were applied in HDO of lignin and whole biomass (corn stover) pyrolysis vapors.
A single-shot tandem micropyrolyzer (Rx-3050tr, Frontier Labs, Japan) was used for pyrolysis and ex situ HDO. A description and a schematic of the tandem micropyrolyzer has been shown previously15 and a more detailed description of the procedures used in this work can be found in the ESI.† Briefly, the tandem micropyrolyzer consisted of two reactors in series with the first reactor being used to pyrolyze the biomass and the second reactor containing a fixed catalyst bed. In all experiments, 250–300 μg of feedstock was pyrolyzed at 500 °C and the catalyst bed temperature was varied between 300–400 °C. Helium was used as the carrier gas in the pyrolyzer with H2 being added at a secondary gas inlet between the two reactors. The pyrolysis product distribution from cellulose has previously been reported using helium only as the carrier gas.16 The addition of hydrogen did not significantly change the distribution or yields of the pyrolytic products. The data in this study is presented on a carbon yield basis (C%). The char yields were measured to be 10.1 C% for cellulose, 55.5 C% for lignin, and 43.0 C% for corn stover. Since the carbon in the char remained in the first reactor and could not be converted by the catalyst, only the carbon from the volatile pyrolysis products could be converted to hydrocarbons and was indicated by the maximum theoretical hydrocarbon yield. To see the extent of HDO, the experiments with catalyst are presented with the control case of fast pyrolysis without any subsequent catalytic conversion.
The effect of catalyst loading on the degree of HDO was investigated for catalyst loadings of 200, 300, and 400 mg of MoO3. It was found that a loading of 200 mg was able to fully catalyze HDO in cellulose pyrolysis. Therefore, a 200 mg catalyst loading was used for the subsequent experiments. It has been reported previously in the literature that ∼320 °C was an optimal reaction temperature for MoO3 catalysts due to a tradeoff between the rate of deoxygenation and catalyst deactivation.12 In this work, the catalytic reaction temperature was set at 300, 350, or 400 °C. The different catalyst temperatures were compared based on passing four biomass injections through the catalyst bed and observing the products resulting from each injection. That is, the biomass injections were performed by pyrolyzing a biomass sample with the vapors reacting on the same catalyst bed without any type of regeneration/calcination between injections. The time between each injection was about one hour and was determined by the GC separation program completing and cooling back down to the initial oven temperature for the next injection. The catalyst bed temperature was held constant at the specified reaction temperature and the gas flow through the catalyst bed was maintained at 60 mL min−1 of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v He:H2 for the entirety of the experiment, including during and between sample injections.
:1 v/v He:H2 for the entirety of the experiment, including during and between sample injections.
At 300 °C, there was no apparent hydrocarbon formation from the cellulose vapor feed (Fig. S1†). The main products included dehydration products of levoglucosan, such as levoglucosenone, as well as the standard low molecular weight compounds produced during cellulose pyrolysis. Conversely, complete HDO to hydrocarbons occurred at 350 °C and 400 °C after the second injection. At 400 °C, the products from the first injection of cellulose included the same low molecular weight oxygenates and dehydration products as seen at 300 °C (Fig. 1). However, the total oxygenate yield was much less than the control case of cellulose pyrolysis without catalyst. In the first injection, there was also some CO, CO2, and a small amount of hydrocarbons (∼7 C%) formed. From the presence of the dehydration products, it appeared the fresh MoO3 predominately acted as an acid catalyst to catalyze the dehydration of levoglucosan to levoglucosenone and other dehydration products. Ammonia temperature programmed desorption (NH3-TPD) was performed to determine the quantity and strength of the acid groups on the fresh and reduced molybdenum oxide catalysts (Fig. S2†). The fresh MoO3 was found to contain some acidity. By reducing the catalyst at 350 °C under flow of 1:10 v/v H2:Ar at 130 mL min−1 for one hour, the acidity greatly diminished as compared with the fresh catalyst. In the literature, it was postulated that HDO with MoO3 occurs through a reverse Mars-van Krevelen mechanism and not through acid–base chemistry.12 The reverse Mars-van Krevelen mechanism would be consistent with the initial induction period needed to form an adequate number of vacant sites for reaction, and therefore, lead to total HDO of the feed for the 2nd injection.
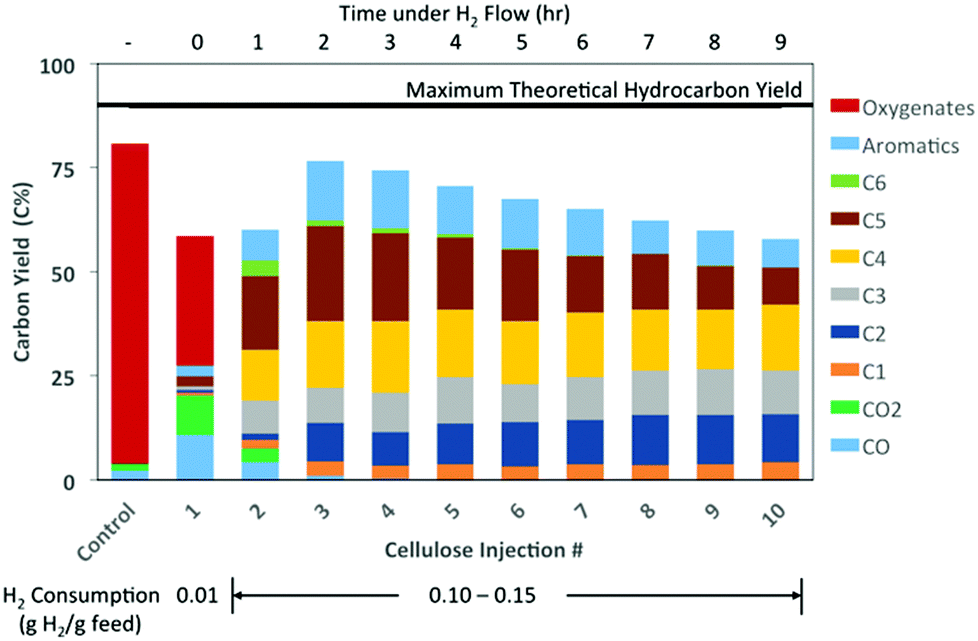 | ||
| Fig. 1 Product distribution from HDO of cellulose pyrolysis vapors at 400 °C. Reaction conditions: 500 °C pyrolysis, 60 mL min−1 1:1 v/v He:H2 flow (1.8 bar Ptotal), no catalyst prereduction. | ||
To validate this hypothesis, further HDO tests were performed at a catalyst bed temperature of 400 °C, but in this case the MoO3 was reduced for 1 h before the first cellulose injection (Fig. 2A). At the first cellulose injection, apart from CO and CO2, no other oxygenated products were observed. Instead, the same alkanes and aromatics were obtained at relatively similar yields as was observed previously in Fig. 1. Only a small amount of olefins (<3 C%) was observed for the first injection. After the first injection, the olefin yields along with the CO and CO2 yields decreased to nearly zero, which was coupled with notable increases in the yields of ethane and butanes, and smaller increases in n-hexane and aromatic yields. For injections 2–4, the yields of each of the hydrocarbons remained relatively constant. It has been shown in the literature that the MoO3 catalyst deactivates due to the formation of inactive MoO2.12,13 At 400 °C, Prasomsri et al. found MoO3 to rapidly deactivate within the first 3 hours on stream in m-cresol HDO.13 In this study, the catalyst was found to retain a similar activity level through 4 cycles under H2 flow (Fig. 2). For additional cycles the formation of some hydrocarbon products (i.e., C5+) decreased with each subsequent injection (Fig. 1). However, for other products, such as for C4 and lighter, their yields remained relatively constant through 10 cycles.
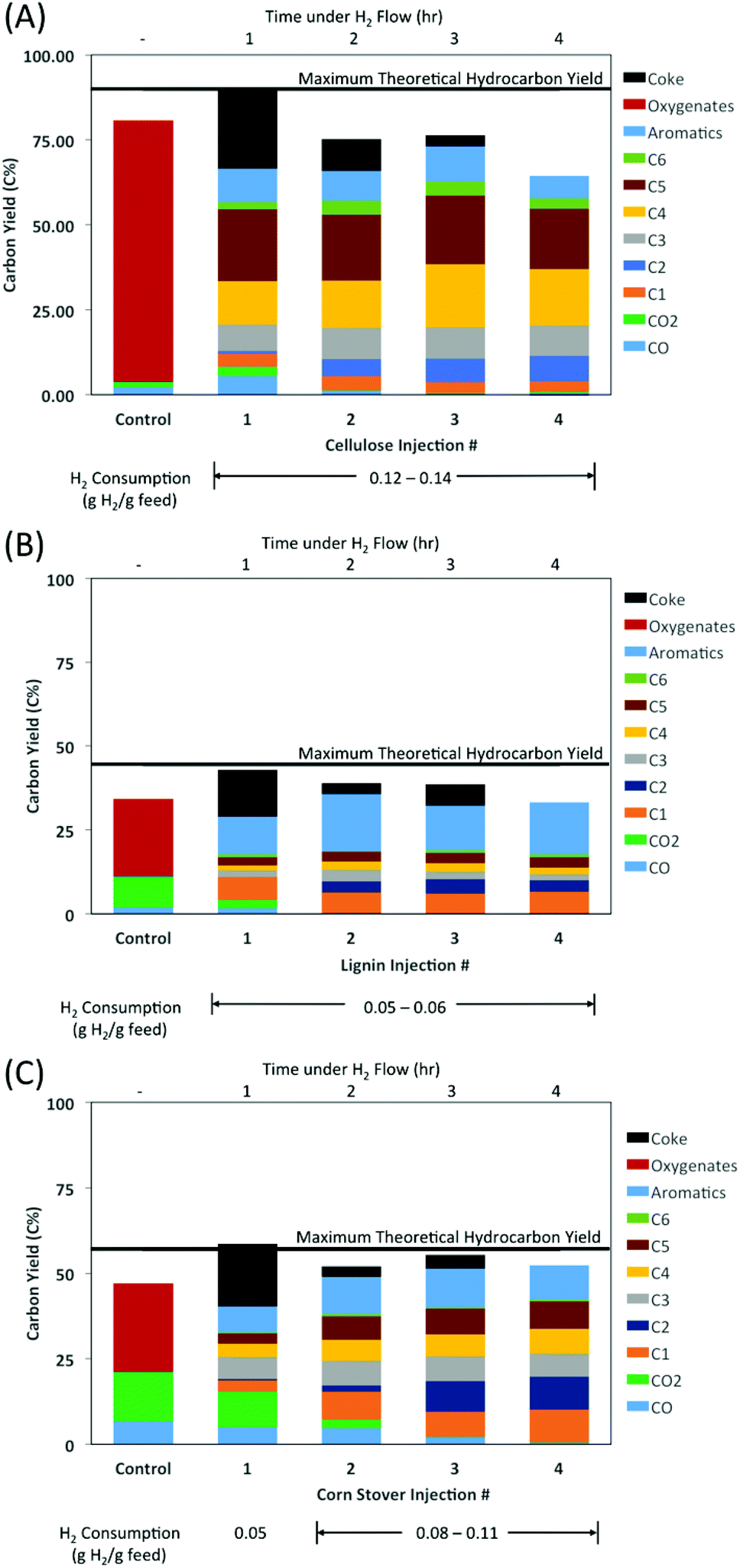 | ||
| Fig. 2 Product distribution from the HDO of pyrolysis vapors from (A) cellulose, (B) lignin, and (C) corn stover. Reaction conditions: 500 °C pyrolysis, 400 °C catalyst bed, 60 mL min−1 1:1 v/v He:H2 flow (1.8 bar Ptotal), 1 h catalyst prereduction. | ||
The HDO of lignin (Fig. 2B) and corn stover (Fig. 2C) was performed using the same reaction conditions as the HDO of cellulose. That is, a reaction temperature of 400 °C was used with the MoO3 being prereduced for one hour before the first injection. As was the case with cellulose, none of the oxygenates (except for CO and CO2) seen in the control case were observed in the first feed injection or any subsequent injection.
In each feedstock, CO and CO2 were present in the first injection, along with methane, but the yield of ethane was relatively low. After the first injection, CO and CO2 yields decreased to nearly zero, while the methane and ethane yields greatly increased.
For all feedstocks, the formation of linear alkanes appeared to be much more favored than the formation of branched alkanes. From cellulose, the yields of n-butane were ∼10–14 times greater than for i-butane, and n-pentane yields were ∼2–5 times greater than i-pentane yields. For lignin, the yields of n-butane and n-pentane were 2–4 times greater than i-butane and i-pentane, respectively. For cornstover, the ratio between n-butane and i-butane was about 5–7 while the ratio between n-pentane and i-pentane was about 2–3, which was directionally similar to the additive results from the individual cellulose and lignin experiments. For C6 compounds, only the linear n-hexane was identified, and not any branched isomers. In a high pressure deoxygenation study using a Pt–Mo/MWCNT catalyst, the yields of n-alkanes were even greater than i-alkanes, where n-butane and n-pentane yields were 26 and 14 times higher than their respective i-isomers from a cellulose feed.11 Venkatakrishnan et al. also observed cyclic alkane products, i.e., cyclopentane and cyclohexane derivatives, in addition to the n- and i-isomers.11 However, in this study there were no detectable cyclic alkane products.
Among the aromatic products, benzene and toluene were obtained in similar yields and accounted for the bulk of the aromatics yield from cellulose, representing 70–80% of the total aromatic yield. In lignin and cornstover HDO, benzene and toluene were also the predominant aromatics (50–60% of the total aromatics), however, ethyl benzene (20–25%), and to a lesser extent the xylenes (combined 8–15%), comprised a noticeable fraction of the total aromatic yield.
The yield of larger hydrocarbon products (C4+) was 44–53 C% from cellulose. The respective yields from lignin and corn stover were slightly lower at 16–23 C% and 15–26 C%. These yields, obtained at a low hydrogen pressure, are comparable to the high pressure hydrodeoxygenation study of Venkatakrishnan et al., where the C4+ yield was 55 C% from the cellulose feed and 32.1 C% from the poplar feed.11
The measured coke yields were relatively significant upon the first injection for all of the feedstocks, where the yield was ∼14–24 C%. Subsequent injections resulted in much less additional coke formation. Catalyst coking did not appear to inhibit catalyst performance, as hydrocarbon yields were higher for the 2nd injection than for the 1st, and remained relatively high for the 3rd and 4th injections.
The majority of the products appeared to evolve from the catalyst bed fairly rapidly, however a small portion of the compounds was slower to react/desorb. The low, background stream of products resulted in a raised baseline and noticeable peak tailing in the MS and FID chromatograms, especially for the lower boiling point compounds. In cellulose HDO, the baseline returned to normal ∼15–20 min post-injection, but for lignin HDO, the baseline normalized much faster, ∼5–10 min post-injection. For corn stover, the baseline normalized after about 10–15 min. Prasomsri et al. previously showed that HDO of furanic compounds, which are major products from cellulose pyrolysis, occurred at a lower rate than for the non-furanic compounds.12 Although, the measured specific HDO rate of the lignin model compound, anisole, was only slightly higher than the furanic compounds.12 Therefore, the prolonged elevated baseline from a cellulose injection as compared to a lignin injection, may be due to slower reaction rates for the furanic and other cellulose pyrolytic products (e.g., anhydrosugars) and also the fact that cellulose contains a greater amount of oxygen than lignin (∼50 wt% O content of cellulose pyrolysis vapors vs. ∼33 wt% for lignin).
The decrease in coke yield for the later injections may also be due to the compounds slowly evolving off the catalyst bed that were too difficult to quantify. The coke yield was calculated by measuring the total amount of CO2 generated during oxidative treatment following an injection and then subtracting the coke yields from previous injections to find the coke yield for the injection of interest. If carbon on the catalyst surface had desorbed at some point, then coke yields for the later injection would be underestimated. Some coke may also have been oxidized from the catalyst between pyrolysis experiments. After the completion of one run, the reactor was opened to retrieve the sample cup and load in the sample cup for the next run. In the process, a small amount of air may have entered the reactor and been swept through the catalyst bed. Under the current experimental conditions, it was too difficult to measure if any CO2 had come off the catalyst bed during this action. The unaccounted fraction is hypothesized to mainly include the slowly evolving products that were too difficult to accurately quantify and the coke lost between experiments.
The hydrogen consumption was calculated from the product distributions for each injection of the feed. Details for the hydrogen consumption calculations are given in the ESI.† Generally, about 0.05–0.15 g H2 was consumed per g feedstock. The H2 consumption was comparable to the 0.05 g H2 per g feed consumption reported by Venkatakrishnan et al. for cellulose and poplar HDO.11 The higher H2 consumption reported in this study may be related to the higher yields of shorter chain hydrocarbons, which will consume a slightly higher amount of hydrogen to form. Although, in this study only aromatics were detected and not cyclohexane or any other saturated cyclic compounds as was observed by Venkatakrishnan et al.11
Conclusions
The ex situ hydrodeoxygenation of cellulose, lignin, and corn stover pyrolysis vapors was performed using low pressure H2 and a MoO3 catalyst. The MoO3 catalyst was found to be very effective at producing hydrocarbons at higher yields than have been previously reported for ex situ HDO of bio-oil when a catalyst bed temperature of 400 °C was used prior to condensation. An initial induction period was needed to reduce the catalyst to a more active form in order to fully deoxygenate the pyrolysis vapors from the first feed injection. The products consisted of mainly linear alkanes (C1 to C6) and aromatics with the total hydrocarbon yield being ∼75–90 C% from the volatile pyrolysis products (excluding char) for the three feedstocks. Even though high catalyst loadings were used in this work and further optimization is required, this catalytic system exhibits potential for hydrodeoxygenation in biomass pyrolysis.Acknowledgements
Funding for this work was from the Mike and Jean Steffenson professorship.Notes and references
- M. S. Talmadge, R. M. Baldwin, M. J. Biddy, R. L. McCormick, G. T. Beckham, G. A. Ferguson, S. Czernik, K. A. Magrini-Bair, T. D. Foust, P. D. Metelski, C. Hetrick and M. R. Nimlos, Green Chem., 2014, 16, 407–453 RSC.
- D. A. Ruddy, J. A. Schaidle, J. R. Ferrell III, J. Wang, L. Moens and J. E. Hensley, Green Chem., 2014, 16, 454–490 RSC.
- D. C. Elliott, Energy Fuels, 2007, 21, 1792–1815 CrossRef CAS.
- K. Wang, K. H. Kim and R. C. Brown, Green Chem., 2014, 16, 727–735 RSC.
- T. R. Carlson, G. A. Tompsett, W. C. Conner and G. W. Huber, Top. Catal., 2009, 52, 241–252 CrossRef CAS.
- A. J. Foster, P. T. M. Do and R. F. Lobo, Top. Catal., 2012, 55, 118–128 CrossRef CAS.
- D. C. Elliot, T. R. Hart, G. G. Neuenschwander, L. J. Rotness and A. H. Zacher, Environ. Prog. & Sustainable Energy, 2009, 28, 441–449 Search PubMed.
- S. Sitthisa and D. E. Resasco, Catal. Lett., 2011, 141, 784–791 CrossRef CAS.
- V. N. Bui, D. Laurenti, P. Afanasiev and C. Geantet, Appl. Catal., B, 2011, 101, 239–245 CrossRef CAS.
- V. M. L. Whiffen and K. J. Smith, Top. Catal., 2012, 55, 981–990 CrossRef CAS.
- V. K. Venkatakrishnan, W. N. Delgass, F. H. Ribeiro and R. Agrawal, Green Chem., 2015, 17, 178–183 RSC.
- T. Prasomsri, T. Nimmanwudipong and Y. Roman-Leshkov, Energy Environ. Sci., 2013, 6, 1732–1738 CAS.
- T. Prasomsri, M. Shetty, K. Murugappan and Y. Roman-Leshkov, Energy Environ. Sci., 2014, 7, 2660–2669 CAS.
- W.-S. Lee, Z. Wang, R. J. Wu and A. Bhan, J. Catal., 2014, 319, 44–53 CrossRef CAS.
- K. Wang, J. Zhang, B. H. Shanks and R. C. Brown, Green Chem., 2015, 17, 557–564 RSC.
- J. Zhang, M. W. Nolte and B. H. Shanks, ACS Sustainable Chem. Eng., 2014, 2, 2820–2830 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods and materials, HDO of cellulose at 300 °C, NH3-TPD of MoO3, and detailed product distributions. See DOI: 10.1039/c5gc01614b |
| This journal is © The Royal Society of Chemistry 2016 |