
Luteolin suppresses the JAK/STAT pathway in a cellular model of intestinal inflammation
Carla
Nunes
 *,
Leonor
Almeida
,
Rui M.
Barbosa
and
João
Laranjinha
*,
Leonor
Almeida
,
Rui M.
Barbosa
and
João
Laranjinha
Center for Neurosciences and Cell Biology and Faculty of Pharmacy, University of Coimbra, 3000-548 Coimbra, Portugal. E-mail: carlapumky@gmail.com; Fax: +351 239 488 503
First published on 14th December 2016
Abstract
Current treatment strategies for inflammatory bowel diseases (IBDs) are associated with a lower efficacy and with several side effects that strongly affect the quality of life of IBD patients. Consequently, the development of new therapies, combining efficacy and safety is an important goal in the field of intestinal inflammation. In this context, evidence supports that polyphenols can be promising candidates due to their ability to modulate intracellular inflammatory signalling cascades. Luteolin, a naturally occurring flavonoid, exhibits anti-inflammatory properties in several models of inflammation. However, its action against intestinal inflammation has been poorly explored. Therefore, there is a lack of scientific knowledge about the potential impact of luteolin in the intestinal inflammation, particularly regarding the underlying molecular mechanisms by which luteolin can exert its anti-inflammatory action. We assessed the potential anti-inflammatory effect of luteolin in a cellular model of intestinal inflammation using cytokine-stimulated HT-29 colon epithelial cells, and the underlying key molecular mechanisms were identified. Luteolin significantly inhibited interleukine-8 (IL-8) production, cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) expression and nitric oxide (˙NO) overproduction induced by cytokines, indicating that luteolin negatively modulates key inflammatory signalling cascades underlying intestinal inflammation. Mechanistically, the inhibition of the JAK/STAT pathway was identified as a critical mechanism by which luteolin exerts its intestinal anti-inflammatory action. This study uncovers novel molecular mechanisms by which luteolin may act against intestinal inflammation, which might support the use of luteolin as a future therapeutic strategy in IBD.
1 Introduction
Inflammatory bowel diseases (IBDs), whose major forms are Crohn's disease and ulcerative colitis, are chronic inflammatory disorders of the gastrointestinal tract characterized by periods of remission and of relapses, which affect millions of individuals in developed countries worldwide.1Although its etiology is not yet completely understood, it is believed that a complex interplay among genetic, immunological, microbial and environmental factors leads to an uncontrolled and persistent inflammation that results in the disruption of the intestinal epithelial barrier and excessive tissue injury observed in IBD.2 In this context, an abnormal and enhanced activation of both, the nuclear factor κB (NF-κB) pathway3 and also the kinase/signal transducer and activator of the transcription (JAK/STAT) pathway,4 has been observed in IBD patients, suggesting an important role of these inflammatory cascades in the pathogenesis of IBD.
Nowadays, the current treatment strategies for IBD are not curative, its main purpose being to induce and then maintain the remission of symptoms, trying to improve the quality of life of IBD patients. Moreover, these conventional therapies are associated with problems of lower efficacy and/or serious side effects in a large number of patients, limiting their use.5,6 Consequently, the development of new therapies, combining efficacy and safety is an important goal in the IBD research. In this context, it seems that compounds able to inhibit the overactivation of the NF-κB pathway and/or JAK/STAT pathway, the two major inflammatory pathways to date involved in intestinal inflammation, can be useful in the management of IBD.
During the last two decades, dietary polyphenols have been a focus of intensive research in the context of prevention and treatment of several oxidative stress and inflammatory-related diseases.7 Initially, these health promoting effects of polyphenols were solely assigned to their well-known in vitro antioxidant properties. More recently, mainly because of their poor bioavailability, alternative pathways have been unravelled and increasing evidence strongly supports that the health benefits of polyphenols are essentially mediated by their ability to modulate key signalling cascades and gene transcription.8–13
Luteolin (3′,4′,5,7-tetrahydroxyflavone) (Fig. 1) is a flavone that exists in high concentrations, mainly in the glycosylated forms, in celery, green pepper, spinach, chamomile tea, carrots, broccoli, etc. The glycosylated forms of luteolin are hydrolyzed into luteolin in the intestinal tract.14 A healthy diet seems to contain between 2 mg–125 mg of luteolin per day.15,16
 | ||
| Fig. 1 Chemical structure of luteolin (3′,4′,5,7-tetrahydroxyflavone). | ||
Although several studies show that luteolin is a strong antioxidant,17,18 anticancer19,20 and anti-inflammatory agent,21–24 its anti-inflammatory effect specifically against intestinal inflammation has been poorly explored.25–27 Hence, little is known about the potential impact of luteolin in the intestinal inflammation and about the underlying molecular mechanism by which luteolin can exert its anti-inflammatory action.
In this way and also considering that the gastrointestinal tract is the compartment where the concentration of luteolin might achieve its higher concentration in the body, we propose to identify key molecular mechanisms underlying the potential anti-inflammatory properties of luteolin against intestinal inflammation, by using a cellular model of intestinal inflammation, consisting of intestinal epithelial cells (IECs) stimulated with a cocktail of cytokines relevant in the intestinal inflammation (tumor necrosis factor α, TNF-α; interferon-γ, IFN-γ; interleukin-1, IL-1).
2 Materials and methods
2.1 Materials
Luteolin was obtained from Sigma-Aldrich (St Louis, MO, USA). Its purity was ≥98%, as determined by TLC. Luteolin solution was prepared in DMSO every day and used freshly. For the experiments, the final concentration in DMSO did not exceed 0.1%. Controls with 0.1% DMSO were performed. DMSO, at this concentration, did not affect HT-29 cells.Cell culture reagents, namely fetal bovine serum (FBS) and Dulbecco's Modified Eagle Medium (DMEM) with Glutamax were purchased from Biochrom GmbH (Berlin, Germany). Trypsin and cytokines (TNF-α, IL-1, IFN-γ) were obtained from Gibco-Invitrogen (Grand Island, NY, USA). The human interleukin-8 (IL-8) ELISA kit was obtained from R&D Systems, Inc (Minneapolis, MN, USA). The antibody anti-Lamin B1 and the alkaline phosphatase-conjugated secondary antibodies were purchased from Abcam (Cambridge, CB, UK). Rabbit polyclonal antibody to anti-inducible nitric oxide synthase (iNOS), goat polyclonal antibody to anti-phospho-STAT1 (Tyr 701), and goat polyclonal antibody to anti-phospho-JAK-1 were purchased from Santa Cruz (Santa Cruz, CA, USA). Rabbit polyclonal antibody to IκB and rabbit monoclonal antibody to phospho-p38 MAPK (Thr180/Tyr182) were obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA). Mouse monoclonal antibody to cyclooxygenase-2 (COX-2) was obtained from Millipore (Billerica, USA) and mouse monoclonal antibody to actin was purchased from Sigma-Aldrich (St Louis, MO, USA). All other chemicals were obtained from Sigma-Aldrich (St Louis, MO, USA) with the highest purity available.
2.2 Cell culture
The HT-29 cell line was purchased from Sigma-Aldrich (St Louis, MO, USA). The HT-29 cells are a well characterised epithelial cell line derived from a primary colon tumour, which exhibit the characteristics of the normal intestinal epithelium.28 Cells were grown in DMEM with FBS (10% v/v) and no antibiotic supplements onto 75 cm2 flasks at 37 °C, under a humidified atmosphere of 5% CO2. Twenty-four hours before each experiment, cells were starved in serum-free medium.In all assays, the HT-29 cells were pre-treated for 30 min with two concentrations of luteolin (50 μM and 100 μM) before exposure to a mixture of cytokines (20 ng ml−1 TNF-α; 10 ng ml−1 IL-1; 50 ng ml−1 INF-γ). The inflammatory stimulus was maintained for different time intervals, depending on the assay.
2.3 Interleukin-8 (IL-8) analysis
IL-8 protein secretion was measured in the supernatant of cell cultures by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions. Measurements were performed in duplicate.2.4 Measurement of intracellular content of IkB, iNOS, COX-2, phospho-JAK-1, phopho-STAT-1 and phospho-p38 MAPK by western blot
Whole and nuclear cellular protein extracts from several experiments were prepared as previously described12 and then analyzed by western blotting.The intracellular content of IkB, iNOS, COX-2, phospho-JAK-1 and phospho-p38 MAPK were assessed by western blotting in whole cell lysates. Phospho-STAT-1 (Tyr701) levels were analysed in nuclear extracts and also in whole cell lysates.
The cellular protein concentration was quantified by using the Bio-Rad protein assay reagent, according to the manufacturer's (Bio-Rad, USA) instructions.
Reduced and denatured proteins were separated by SDS/PAGE electrophoresis on a 10%–12% (v/v) acrylamide gel and transferred onto PVDF membranes using the Trans-Blot Turbo Transfer System of Bio-Rad (Bio-Rad, USA).
To avoid non-specific binding, the membranes were blocked for 1 h at room temperature with 5% (w/v) non-fat dried milk in TBS buffer supplemented with Tween 20 [25 mM Tris-HCl pH 7.6, 150 mM NaCl, 0.1% (v/v) Tween 20] and then incubated with antibodies against IkB, iNOS, COX-2, phospho-JAK-1, phopho-STAT-1 and phospho-p38 MAPK, overnight at 4 °C with constant low shaking.
Then, membranes were washed three times with TBS-T and incubated with a phosphatase alkaline-labelled secondary antibody for 2 h at room temperature. Immunoreactive complexes were detected by fluorescence after blot exposure to the ECF substrate in a Typhoon 9000 scanner (Amersham Biosciences) and analyzed with the ImageQuant TM software from Amersham Biosciences. Actin and lamin B1 were used as internal standards to monitor protein loading per lane.
2.5 Measurement of nitric oxide production
Considering that in aqueous oxygenated solutions ˙NO is oxidized to nitrite, a highly sensitive and selective approach was used to quantify the ˙NO-derived nitrite accumulated in the cell culture medium as previously described.11 Nitrite in the cell culture supernatants suffers reductive cleavage by an iodide/tri-iodide containing reaction mixture followed by the quantification of the released ˙NO by gas-phase chemiluminescence.29 The reaction mixture, consisting of 45 mmol L−1 potassium iodide (KI) and 10 mmol L−1 iodine (I2) in glacial acetic acid, was kept at 56 °C in a septum-sealed reaction vessel, continuously bubbled with nitrogen. The outlet of the gas stream was passed through a scrubbing bottle containing sodium hydroxide (1 mol L−1; 0 °C) in order to trap traces of acid and iodine before transfer into the analyzer (CLD 88 Eco Medics, Switzerland), where ˙NO undergoes a chemiluminescence reaction with ozone (O3). The ˙NO signal output was sampled at 2 Hz. Peak integration was performed by using EDAQ Power Chrom software.2.6 Statistical analysis
All data were expressed as mean ± SEM of at least three independent assays, each one in duplicate or triplicate. Significant levels between groups were assessed by one-way analysis of variance (ANOVA). A value of p < 0.05 was considered statistically significant. Calculations were performed with GraphPad Prism software.3 Results
3.1 Luteolin partially suppressed cytokine-induced IL-8 overproduction
IL-8, a chemotactic cytokine for T lymphocytes and neutrophils, is highly expressed in the colon of patients with active IBD, playing an important role in the pathogenesis of this disease.30,31 Therefore, the effect of luteolin (50 μM and 100 μM) on the IL-8 secretion was investigated in HT-29 cells stimulated for 16 h with the cytokine mixture (20 ng ml−1 TNF-α; 10 ng ml−1 IL-1; 50 ng ml−1 INF-γ). It should be noted that the concentrations of luteolin used had no effect on the viability of HT-39 cells (data not shown).IL-8 production was deeply increased by cytokines (92.5 ± 19.2 ng mL−1) but, as shown in Fig. 2, luteolin was able to significantly inhibit the overproduction of IL-8 in a dose-dependent manner (50 μM luteolin – 45.2 ± 3.5 ng mL−1 and 100 μM luteolin – 30.1 ± 3.7 ng mL−1).
 | ||
| Fig. 2 Luteolin inhibits the IL-8 production induced by cytokines in HT-29 cells. Cells were treated with two different concentrations of luteolin (50 μM and 100 μM) for 30 min and then stimulated with cytokines (20 ng ml−1 TNF-α; 10 ng ml−1 IL-1; 50 ng ml−1 INF-γ). After 16 hours, IL-8 protein secretion in the culture supernatant was quantified by using an ELISA kit. Data represent the mean ± SEM from at least three independent experiments. Statistical significance: ***p < 0.001 as compared to control cells; &p < 0.05 as compared to cells stimulated by cytokines. | ||
3.2 Luteolin inhibited COX-2 induction by cytokines
COX-2, an enzyme inducible by a pro-inflammatory stimulus, is up-regulated in active IBD, playing a key role in the pathogenesis of this disease.32–34 Therefore, the effect of luteolin on the COX-2 expression induced by cytokines was evaluated by western blotting. The COX-2 protein was not detectable in the non-stimulated cells (Fig. 3). However, stimulation of HT-29 cells with cytokines for 16 hours induced a significant up-regulation of COX-2 levels (Fig. 3). Pre-treatment with luteolin, in a concentration-dependent manner, inhibited this COX-2 up-expression induced by cytokines (Fig. 3). In fact, considering the cells treated only with cytokines as 100%, we can observe that pre-treatment with 50 μM and 100 μM of luteolin decreased the COX-2 expression to 30.4% and 9.2%, respectively. | ||
| Fig. 3 Luteolin inhibits COX-2 expression induced by cytokines in HT-29 cells. Cells were treated as described in Fig. 2. COX-2 expression was evaluated after 16 hours in whole protein extracts by western blot as described in “Materials and methods”. In the bar graph, the relative expression of COX-2 normalized to the actin level represents the mean ± SEM from at least three independent experiments and are expressed as a percentage of cells incubated with cytokines (100%). Statistical significance: ***p < 0.001 as compared to control cells; &&&p < 0.001 as compared to cells stimulated with cytokines. | ||
3.3 Luteolin inhibited the iNOS protein expression induced by cytokines, leading to a significant reduction of ˙NO overproduction
The overproduction of ˙NO via iNOS up-regulation seems to play a critical role in the pathogenesis of inflammatory diseases, including IBD.35,36 Therefore, we firstly evaluated the effect of luteolin on ˙NO production by HT-29 cells stimulated with the cytokines. Cells were incubated with luteolin for 30 min before being stimulated with the cytokine mixture for another 24 h. As shown in Fig. 4, a significant increase in the ˙NO level was observed after treatment of the cells with the mixture of cytokines (control 1.8 ± 0.08 nmol per 106 cells; cytokines 3 ± 0.04 nmol per 106 cells). Pre-treatment with luteolin inhibited cytokine-stimulated ˙NO production, with the ˙NO levels similar to that obtained in control cells (50 μM luteolin – 1.8 ± 0.1 nmols per 106 cells; 100 μM luteolin – 1.6 ± 0.2 nmols per 106 cells) (Fig. 4). | ||
| Fig. 4 Luteolin significantly reduces the ·NO overproduction induced by cytokines in HT-29 cells. Cells were treated as described in Fig. 2. After 24 hours of incubation, the supernatants were collected and the ˙NO production was measured by chemiluminescence as described in “Materials and methods”. The bar graph represents the mean ± SEM from at least three independent experiments. The statistical significance: ***p < 0.001 as compared to control cells and &&&p < 0.001 as compared to cells stimulated with cytokines. | ||
To determine whether the inhibitory effect of luteolin on ˙NO production was due to a decrease of the iNOS levels induced by cytokines, the expression of iNOS, under various experimental conditions was also evaluated by western blotting.
The iNOS protein was not detectable in the unstimulated cells. However, stimulation with cytokines for 16 hours induced an up-regulation of iNOS levels. Pre-treatment with luteolin showed a significant concentration-dependent inhibition of iNOS protein expression induced by cytokines (Fig. 5). In fact, considering the cells treated only with the cytokines as 100%, 50 μM and 100 μM of luteolin decreased the iNOS expression to 18.2 and 0.65%, respectively (Fig. 5).
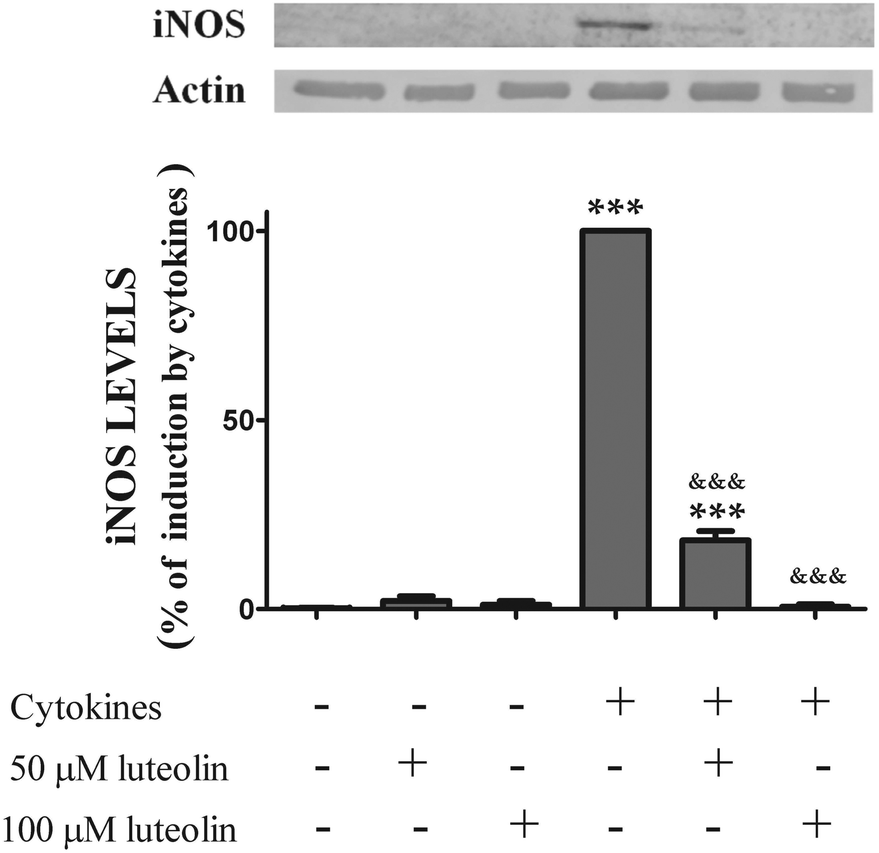 | ||
| Fig. 5 Luteolin inhibits iNOS expression induced by cytokines in HT-29 cells. Cells were treated as described in Fig. 2. iNOS expression was evaluated after 16 hours in whole protein extracts by western blot as described in “Materials and methods”. In the bar graph, the relative expression of iNOS normalized to the actin level represents the mean ± SEM from at least three independent experiments and are expressed as a percentage of cells incubated with cytokines (100%). Statistical significance: ***p < 0.001 as compared to control cells; &&&p < 0.001 as compared to cells stimulated with cytokines. | ||
3.4 Luteolin did not inhibit cytokine-induced IκB degradation
Next, we investigated the molecular mechanisms by which luteolin inhibited the overproduction of pro-inflammatory mediators (IL-8 and ˙NO) and the expression of inflammatory enzymes (iNOS and COX-2) as shown above.NF-κB is one of the key orchestrators of pro-inflammatory gene expression37 and it is also implicated in the pathogenesis of inflammatory diseases as IBD.3 The first approach was to examine the potential effect of luteolin in the NF-κB pathway activation.
Under normal physiological conditions, NF-κB is sequestrated in the cytoplasm in an inactive form as NF-κB-IκB. The activation of the NF-κB pathway involves the IκB phosphorylation, which results in its degradation followed by translocation of the NF-κB to the nucleus, where it induces the expression of the pro-inflammatory mediators mentioned above.38,39 Thus, IκB degradation, a crucial step for the NF-κB activation, was evaluated by western blotting. Luteolin alone did not change the expression pattern of IκB in the absence of cytokines (Fig. 6). As expected, cytokines induced a rapid degradation (30 min) of the IκB protein (28.8 ± 3.9% of IκB content) (Fig. 6). However, as shown in Fig. 6, luteolin did not affect the IκB degradation induced by cytokines (50 μM luteolin – 24.1 ± 7.4% of IκB content and 100 μM luteolin – 23.9 ± 6.2% of IκB content).
 | ||
| Fig. 6 Luteolin did not inhibit the IκB degradation induced by cytokines in HT-29 cells. Cells were treated with luteolin (50 μM and 100 μM) for 30 min and then stimulated with the cocktail of cytokines for 30 minutes. IkB degradation was evaluated in cytoplasmic extracts by western blotting, as described in “Materials and methods”. In the bar graph, the relative expression of IκB normalized to the actin level was expressed as mean ± SEM from at least five independent experiments. Statistical significance: ***p < 0.001 as compared to control cells. | ||
3.5 Luteolin decreased the levels of cytokine-induced phosphorylated STAT1
Besides NF-κB, another key inflammatory signalling pathway is the JAK/STAT cascade. Moreover, INF-γ, one of the cytokines used in this study as inflammatory stimulus, is an important activator of the JAK/STAT pathway.40,41 Following INF-γ stimulation, STAT-1 is phosphorylated (Tyr701) by tyrosine kinases of the JAK family, namely by JAK-1. This phosphorylation induces STAT-1 dimerization and subsequent nuclear translocation, where STAT1 binds to DNA, inducing the transcription of several genes that encoding pro-inflammatory cytokines as IL-8,42 inflammatory enzymes such as iNOS and COX-2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 43–45 and also mediators of cell death.46
43–45 and also mediators of cell death.46
Therefore, the effect of luteolin on the nuclear levels of phosphorylated (activated) STAT-1 was examined. First, the activation time course of STAT-1 induced by cytokines in HT-29 cells was evaluated. Phospho-STAT-1 (Tyr701) was not detectable in the nucleus of non-stimulated cells (Fig. 7). The phosphorylation and nuclear translocation of STAT-1 induced by cytokines were significant at 30 minutes and remained elevated for at least 6 h in HT-29 cells (data not shown). Consequently, cells were incubated with luteolin and then challenged with cytokines for 30 min and 6 h.
 | ||
| Fig. 7 Luteolin decreases the levels of phosphorylated-STAT1 (Tyr701) in the nucleus of cytokine-stimulated HT-29 cells. Cells were treated as described in Fig. 2. After 30 min (A) and after 6 h (B), nuclear protein extracts were obtained and analysed by western blot using an anti-phospho-STAT-1 (tyr701) antibody. In the bar graph, the relative expression of phosphorylated STAT-1 normalized to the lamin B1 level represents the mean ± SEM from at least three independent experiments and is expressed as a percentage of cells incubated with cytokines (100%). Statistical significance: ***p < 0.001 as compared to control cells and &&&p < 0.001 as compared to cells stimulated with cytokines. | ||
Pre-treatment with luteolin significantly decreased the nuclear levels of this activated transcription factor in the cells stimulated with cytokines for 30 minutes (50 μM luteolin – 84.5 ± 6.5% and 100 μM luteolin – 54.8 ± 7.5% of cells incubated with cytokines) (Fig. 7A). This protective effect exerted by luteolin was stronger at 6 h (50 μM luteolin – 70.9 ± 6.7% and 100 μM luteolin – 31.7 ± 5.9% of cells incubated with cytokines) (Fig. 7B). With the aim to know whether this effect exerted by luteolin was due to suppression of the STAT-1 phosphorylation induced by cytokines or to an inhibition of the nuclear translocation of STAT-1, we also examined the levels of phospho-STAT-1 (Tyr701) in the whole cell lysates. As shown in Fig. 8, luteolin effectively prevented the phosphorylation of STAT-1 at Tyr701 induced by cytokines (50 μM luteolin – 70.6 ± 4.6% and 100 μM luteolin – 31.4 ± 3.5% of cells incubated with cytokines).
 | ||
| Fig. 8 Luteolin decreases the total levels of phosphorylated-STAT1 (Tyr701) in cytokine-stimulated HT-29 cells. Cells were treated as described in Fig. 2. After 6 h of incubation, whole protein extracts were obtained and analysed by western blot using an anti-phospho-STAT-1 (tyr701) antibody. In the bar graph, the relative expression of phospho-STAT-1 normalized to the actin level represents the mean ± SEM from at least three independent experiments and is expressed as a percentage of cells incubated with cytokines (100%). Statistical significance: ***p < 0.001 as compared to control cells and &&&p < 0.001 as compared to cells stimulated with cytokines. | ||
3.6 Luteolin suppressed the cytokine-induced JAK1 phosphorylation
Considering that STAT-1 phosphorylation at tyrosine 701 depends on the activation of JAKs, next the effect of luteolin on the cellular levels of JAK-1 phosphorylated induced by cytokines was evaluated.Luteolin, in a concentration-dependent manner, suppressed the JAK-1 phosphorylation induced by cytokines after 30 minutes (Fig. 9), which suggests that luteolin is able to inhibit the JAK/STAT cascade at least from the step of JAK-1 phosphorylation. We recognize that the quality of western blot is not perfect, which may be related with the polyclonal antibody to phospho-JAK-1. However, the bands are visible and allowed reliable quantification.
 | ||
| Fig. 9 Luteolin decreases the total levels of phosphorylated-JAK-1 in cytokine-stimulated HT-29 cells. Cells were incubated as described in Fig. 1. After 30 min of incubation, whole protein extracts were obtained and analysed by western blot using an anti-phospho-JAK-1 antibody. In the bar graph, the relative expression of phospho-JAK-1 normalized to the actin level represents the mean ± SEM from three independent experiments and is expressed as a percentage of cells incubated with cytokines (100%). Statistical significance: &p < 0.05 and &&&p < 0.001 as compared to cells stimulated with cytokines. | ||
3.7 Luteolin did not inhibit cytokine-induced p38 MAPK phosphorylation
The p38 MAPK seems to play a key role in the regulation of the JAK/STAT pathway.47 The cocktail of cytokines induced p38 MAPK phosphorylation (activation) at 30 min of incubation (Fig. 10). However, as shown in Fig. 10, cytokine-induced p38 MAPK phosphorylation was not affected by the treatment with luteolin (50 μM luteolin – 93.2 ± 5.2% and 100 μM luteolin – 100.5 ± 1.3% of cells incubated with cytokines). | ||
| Fig. 10 Luteolin has no effect on the levels of p-p38 MAPK induced by cytokines in HT-29 cells. Cells were incubated as described in Fig. 1. After 30 min of incubation, whole protein extracts were obtained and analysed by western blot using an anti-phospho-p38 MAPK antibody. In the bar graph, the relative expression of phospho-p38 MAPK normalized to the actin level represents the mean ± SEM from three independent experiments and is expressed as a percentage of cells incubated with cytokines (100%). Statistical significance: ***p < 0.001 as compared to control cells. | ||
4 Discussion
IBD is a multifactorial chronic inflammatory disease of the gastrointestinal tract that is characterized by an increased local production of cytokines, namely TNF-α, IFN-γ and IL-1, which induce an inflammatory cascade, leading to IECs’ damage and, influencing their viability and normal function. Therefore, in this study, we used HT-29 colon epithelial cells stimulated with a cytokine mixture (TNF-α, IFN-γ and IL-1) as a cellular model of intestinal inflammation, with the aim to evaluate the potential intestinal anti-inflammatory effect of luteolin and to identify the underlying key molecular mechanisms.Although over the last few years, several studies showed that luteolin can be an important anti-inflammatory agent,21–24 its anti-inflammatory action specifically against intestinal inflammation and IBD has been poorly explored25–27 and, in fact, the gastrointestinal system is likely the site in the body in which luteolin may achieve its higher concentration and in an unmodified polyphenolic structure. Moreover, and most importantly, the molecular mechanisms by which luteolin can exert its anti-inflammatory action are not completely understood, and the results described in the literature are frequently contradictory and open to criticism.
In this way, we firstly show that luteolin is effective in inhibiting cytokine-induced key inflammatory mediators, namely IL-8, COX-2, iNOS and ˙NO, supporting a significant anti-inflammatory effect of luteolin against intestinal inflammation.
As IL-8 is a key chemokine, the inhibition of the IL-8 overproduction by luteolin suggests that luteolin can interfere with the recruitment of neutrophils and T-cells, fighting the exacerbation and progression of intestinal inflammation. This anti-inflammatory effect exerted by luteolin is consistent with studies in the literature using several models of inflammation.25,48,49 Likewise, the inhibition of COX-2 and iNOS expression by luteolin reported in the present study is in agreement with the results shown by other authors at different inflammatory models.50–53 However, the key question is the identification of the signalling pathway modulated by luteolin that prevents the expression of such inflammatory modulators.
The COX enzymes are the rate-limiting enzymes in the conversion of arachidonic acid into prostaglandins, and it is well known that's involved in IBD.32–34 So, the inhibition of cytokine-induced COX-2 expression by luteolin indicates that luteolin can positively modulate the arachidonic acid pathway under inflammatory conditions.
˙NO can exert beneficial or harmful effects as a function of its concentration and the redox environment in which it is produced. ˙NO is produced from L-arginine by nitric oxide synthase (NOS) family.54,55 Neuronal NOS (nNOS) and endothelial NOS (eNOS) are constitutively expressed and produce small amounts of ˙NO for short periods of time, being involved in a plethora of physiological processes. The expression of iNOS is inducible by cytokines producing high amounts of ˙NO for a longer period of time and therefore it has been involved in pathophysiologic processes.56 Particularly in the case of IECs, ˙NO overproduction via iNOS up-regulation plays an important role in the chronic inflammatory process associated with IBD.35,36,57 Here, we proved the capacity of luteolin to inhibit the iNOS protein expression induced by cytokines, leading to a significant reduction in the ˙NO overproduction. In this regard, it is noteworthy that luteolin seems to not affect the activity of constitutive NOS, because luteolin did not alter the basal levels of ˙NO in the HT-29 cells. This is relevant because ˙NO, at low concentration, is an important modulator of key signalling pathways and it is necessary for maintaining the physiological functions of the gut, including gastrointestinal motility, secretion and absorption.
COX-2 overexpression and ˙NO overproduction by iNOS are not only involved in the inflammatory processes but are also closely associated with colorectal cancer frequently developed by IBD patients.34,58,59 Therefore, the results presented here strongly suggest that luteolin can efficiently fight the intestinal inflammation, inducing and maintaining the period of remission of IBD, and also decreases the risk of development of colorectal cancer in these patients.
Next to these observational studies, our main goal was to provide new insights into cellular signalling mechanisms underlying the capacity of luteolin to protect the intestinal epithelial cells against the pro-inflammatory stimulus.
The NF-κB signalling pathway would be a likely candidate for the anti-inflammatory action of luteolin because it is one of the key regulators of the expression of important genes involved in inflammation, regulating the expression of cytokines, including IL-8, and pro-inflammatory enzymes such as COX-2 and iNOS, among others.37,60 Also, it is well known that NF-κB is strongly activated in the inflamed gut of IBD patients.3,61–64 However, under our experimental conditions, luteolin could not prevent the cytokine-induced degradation of the IκB, a crucial step for the classical activation of the NF-kB signalling cascade. Our results are, hence, apparently in contradiction to those reported by other authors that showed the suppression of the IκB degradation by luteolin in several models of inflammation.25,26,65–67 However these apparently contradictory results can be probably be explained by the differences in the cell type and/or in the pro-inflammatory stimulus. In this regard, it is important to specifically refer to the work developed by Kim and collaborators25 that reported the suppression of the IκB degradation and the consequent inhibition of the NF-κB pathway activation by luteolin in TNF-α-stimulated HT-29 cells, the same cell line that we used in the present study. In this specific case, the different pro-inflammatory stimulus used by these authors (TNF-α) and by us (TNF-α, IFN-γ and IL-1) probably can explain the difference between the results obtained.
Nonetheless, we cannot completely exclude a possible inhibition of the NF-kB pathway by luteolin at the transcriptional activity level in our inflammatory mode. In fact, in rat-1 fibroblasts, luteolin neither inhibited the NF-kB nuclear translocation nor the DNA binding activity of NF-kB p65, but it was able to inhibit the NF-kB transcriptional activity by preventing the interaction between the p65 subunit of NF-kB and the transcriptional co-activator CBP.68
The JAK/STAT pathway is another crucial signalling cascade in the IBD context.4 In fact, the JAK/STAT pathway has been associated not only with the inflammatory processes in IBD but also with colorectal cancer development and progression that frequently occurs in these patients.69,70 In this context, Malilas and colleagues reported recently that cancer upregulated gene 2 (CUG2), an oncogene, promotes enhanced migration of colon cancer cells and also resistance to doxorubicin, an anticancer drug, through STAT1 activation.69
Tofacitinib, an oral JAK inhibitor, currently used in the treatment of rheumatoid arthritis,71 also showed promising results for the treatment of IBD, especially ulcerative colitis,72,73 suggesting that the inhibition of this pathway can be a new and innovative therapeutic approach for IBD.
Despite the importance of this inflammatory pathway in the intestine, the potential inhibition of the JAK/STAT cascade by luteolin was essentially explored in microglial cells, where luteolin attenuated the inflammatory response of brain microglial cells by suppressing the STAT activation.51,74 Luteolin also inhibited the JAK/STAT pathway in human cholangiocarcinoma cells.75 In this way, to the best of our knowledge, no study showed yet in a clear way, the effect of luteolin on the JAK/STAT pathway in the context of intestinal inflammation. So, here, we demonstrated for the first time, in a cellular model of intestinal inflammation, the ability of luteolin to reduce the levels of STAT-1 phosphorylated at Tyr701 induced by cytokines in the nucleus of HT-29 cells. This effect seems to be related, at least partially, with the suppression of cytokine-induced JAK-1 phosphorylation by luteolin.
As a caution note, although not explored in this study, we cannot exclude a potential activation induced by luteolin of negative regulators of the JAK/STAT pathway such as protein inhibitors of activated STATs (PIASs), protein tyrosine phosphatases (PTPs) and suppressors of cytokine signalling (SOCS) proteins.
Moreover, there is evidence that p38 MAPK can mediate the phosphorylation of STAT1 at serine 727, which is not only a crucial modification to the maximization of its transcriptional activity47 but that also enhances the interaction between STAT1 and PIAS1, promoting small ubiquitin-related modifiers-1 (SUMO-1) conjugation to STAT1 and the consequent inhibition of STAT1 activity.76 This fact suggests that the phosphorylation of Ser727 by p38 MAPK can trigger a negative feedback loop through PIAS1 recruitment and SUMOylation of STAT1. In this context, considering that luteolin did not inhibit the phosphorylation of p38 MAPK induced by cytokines, we can speculate that the anti-inflammatory effect of luteolin may benefit from this negative feedback mechanism mediated by p38 MAPK.
5 Conclusions
In conclusion, our results clearly show that luteolin is effective in inhibiting cytokine-induced key inflammatory mediators such as IL-8, COX-2, iNOS and ˙NO, which indicates a significant anti-inflammatory effect of this flavone against intestinal inflammation. We also show that this intestinal anti-inflammatory action is, at least partially, related to the suppression of the JAK/STAT inflammatory pathway. Considering the abundance reached by luteolin in the intestine, this study contributes to enlightening important and unexplored mechanisms by which luteolin may fight the intestinal inflammation, allowing the development of luteolin as a therapeutic strategy to limit the inflammatory processes and also to prevent colorectal cancer development in IBD patients.Abbreviations
| CUG2 | Cancer upregulated gene 2 |
| COX-2 | Cyclooxygenase-2 |
| DMEM | Dulbecco's Modified Eagle Medium |
| FBS | Foetal bovine serum |
| IBD | Inflammatory bowel disease |
| IECs | Intestinal epithelial cells |
| IL | Interleukin |
| INF-γ | Interferon-γ |
| iNOS | Inducible nitric oxide synthase |
| JAK | Janus kinase |
| NF-κB | Nuclear factor κB |
| PIAS | Protein inhibitors of activated STATs |
| PTPs | Protein tyrosine phosphatases |
| SOCS | Suppressor of cytokine signalling |
| STAT | Signal transducer and activator of transcription |
| SUMO-1 | Small ubiquitin-related modifiers-1 |
| TNF-α | Tumor necrosis factor-α |
| Tyr | Tyrosine |
Conflict of interest
The authors have no conflicts of interest to declare.Acknowledgements
This work is funded by FEDER funds through the Operational Programme Competitiveness Factors – COMPETE and national funds by FCT – Foundation for Science and Technology under the project PTDC/BBB-BQB/3217/2012 and strategic project UID/NEU/04539/2013. Carla Nunes acknowledges the FCT fellowship, SFRH/BPD/46149/2008.Notes and references
- T. A. Malik, Surg. Clin. North Am., 2015, 95, 1105–1122 CrossRef PubMed , v.
- H. S. de Souza and C. Fiocchi, Nat. Rev. Gastroenterol. Hepatol., 2016, 13, 13–27 CrossRef CAS PubMed.
- I. Atreya, R. Atreya and M. F. Neurath, J. Intern. Med., 2008, 263, 591–596 CrossRef CAS PubMed.
- M. Coskun, M. Salem, J. Pedersen and O. H. Nielsen, Pharmacol. Res., 2013, 76, 1–8 CrossRef CAS PubMed.
- R. A. Speight and J. C. Mansfield, Clin. Med., 2013, 13, 378–382 CrossRef PubMed.
- A. B. Pithadia and S. Jain, Pharmacol. Rep., 2011, 63, 629–642 CrossRef CAS PubMed.
- J. Gonzalez-Gallego, S. Sanchez-Campos and M. J. Tunon, Nutr. Hosp., 2007, 22, 287–293 CAS.
- C. G. Fraga, M. Galleano, S. V. Verstraeten and P. I. Oteiza, Mol. Aspects Med., 2010, 31, 435–445 CrossRef CAS PubMed.
- I. Rahman, S. K. Biswas and P. A. Kirkham, Biochem. Pharmacol., 2006, 72, 1439–1452 CrossRef CAS PubMed.
- B. Romier, Y. J. Schneider, Y. Larondelle and A. During, Nutr. Rev., 2009, 67, 363–378 CrossRef PubMed.
- C. Nunes, E. Ferreira, V. Freitas, L. Almeida, R. M. Barbosa and J. Laranjinha, Food Funct., 2013, 4, 373–383 CAS.
- C. Nunes, N. Teixeira, D. Serra, V. Freitas, L. Almeida and J. Laranjinha, Toxicol. Res., 2016, 5, 53 RSC.
- S. Upadhyay and M. Dixit, Oxid. Med. Cell. Longevity, 2015, 2015, 504253 Search PubMed.
- J. Y. Ying, S. J. Gu and T. W. Yao, Yao Xue Xue Bao, 2008, 43, 335–342 CAS.
- C. Manach, A. Scalbert, C. Morand, C. Remesy and L. Jimenez, Am. J. Clin. Nutr., 2004, 79, 727–747 CAS.
- J. A. Ross and C. M. Kasum, Annu. Rev. Nutr., 2002, 22, 19–34 CrossRef CAS PubMed.
- S. Gal, D. Lichtenberg, A. Bor and I. Pinchuk, Chem. Phys. Lipids, 2007, 150, 186–203 CrossRef CAS PubMed.
- G. B. Sun, X. Sun, M. Wang, J. X. Ye, J. Y. Si, H. B. Xu, X. B. Meng, M. Qin, J. Sun, H. W. Wang and X. B. Sun, Toxicol. Appl. Pharmacol., 2012, 265, 229–240 CrossRef CAS PubMed.
- J. Lu, G. Li, K. He, W. Jiang, C. Xu, Z. Li, H. Wang, W. Wang, X. Teng and L. Teng, J. Transl. Med., 2015, 13, 42 CrossRef PubMed.
- M. J. Tuorkey, Eur. J. Cancer Prev., 2016, 25, 65–76 CrossRef CAS PubMed.
- M. Hytti, N. Piippo, E. Korhonen, P. Honkakoski, K. Kaarniranta and A. Kauppinen, Sci. Rep., 2015, 5, 17645 CrossRef CAS PubMed.
- Z. Jia, P. Nallasamy, D. Liu, H. Shah, J. Z. Li, R. Chitrakar, H. Si, J. McCormick, H. Zhu, W. Zhen and Y. Li, J. Nutr. Biochem., 2015, 26, 293–302 CrossRef CAS PubMed.
- T. Y. Lin, C. W. Lu and S. J. Wang, Neurotoxicology, 2016, 55, 48–57 CrossRef CAS PubMed.
- J. Sung and J. Lee, J. Med. Food, 2015, 18, 557–564 CrossRef CAS PubMed.
- J. A. Kim, D. K. Kim, O. H. Kang, Y. A. Choi, H. J. Park, S. C. Choi, T. H. Kim, K. J. Yun, Y. H. Nah and Y. M. Lee, Int. Immunopharmacol., 2005, 5, 209–217 CrossRef CAS PubMed.
- J. S. Kim and C. Jobin, Immunology, 2005, 115, 375–387 CrossRef CAS PubMed.
- Y. Nishitani, K. Yamamoto, M. Yoshida, T. Azuma, K. Kanazawa, T. Hashimoto and M. Mizuno, BioFactors, 2013, 39, 522–533 CrossRef CAS PubMed.
- I. Chantret, A. Barbat, E. Dussaulx, M. G. Brattain and A. Zweibaum, Cancer Res., 1988, 48, 1936–1942 CAS.
- M. Feelisch, T. Rassaf, S. Mnaimneh, N. Singh, N. S. Bryan, D. Jourd'Heuil and M. Kelm, FASEB J., 2002, 16, 1775–1785 CrossRef CAS PubMed.
- L. Mazzucchelli, C. Hauser, K. Zgraggen, H. Wagner, M. Hess, J. A. Laissue and C. Mueller, Am. J. Pathol., 1994, 144, 997–1007 CAS.
- K. Mitsuyama, A. Toyonaga, E. Sasaki, K. Watanabe, H. Tateishi, T. Nishiyama, T. Saiki, H. Ikeda, O. Tsuruta and K. Tanikawa, Clin. Exp. Immunol., 1994, 96, 432–436 CrossRef CAS PubMed.
- M. Romero, R. Artigiani, H. Costa, C. T. Oshima, S. Miszputen and M. Franco, Arq. Gastroenterol., 2008, 45, 295–300 CrossRef PubMed.
- I. I. Singer, D. W. Kawka, S. Schloemann, T. Tessner, T. Riehl and W. F. Stenson, Gastroenterology, 1998, 115, 297–306 CrossRef CAS.
- D. Wang and R. N. Dubois, Oncogene, 2010, 29, 781–788 CrossRef CAS PubMed.
- N. Avdagic, A. Zaciragic, N. Babic, M. Hukic, M. Seremet, O. Lepara and E. Nakas-Icindic, Bosnian J. Basic Med. Sci., 2013, 13, 5–9 CAS.
- S. S. Dhillon, L. A. Mastropaolo, R. Murchie, C. Griffiths, C. Thoni, A. Elkadri, W. Xu, A. Mack, T. Walters, C. Guo, D. Mack, H. Huynh, S. Baksh, M. S. Silverberg, J. H. Brumell, S. B. Snapper and A. M. Muise, Clin. Transl. Gastroenterol., 2014, 5, e46 CrossRef CAS PubMed.
- H. L. Pahl, Oncogene, 1999, 18, 6853–6866 CrossRef CAS PubMed.
- J. M. Muller, H. W. Ziegler-Heitbrock and P. A. Baeuerle, Immunobiology, 1993, 187, 233–256 CrossRef CAS PubMed.
- S. Wang, Z. Liu, L. Wang and X. Zhang, Cell. Mol. Immunol., 2009, 6, 327–334 CrossRef CAS PubMed.
- C. Schindler and C. Plumlee, Semin. Cell Dev. Biol., 2008, 19, 311–318 CrossRef CAS PubMed.
- I. Rauch, M. Muller and T. Decker, JAKSTAT, 2013, 2, e23820 Search PubMed.
- N. M. Gharavi, J. A. Alva, K. P. Mouillesseaux, C. Lai, M. Yeh, W. Yeung, J. Johnson, W. L. Szeto, L. Hong, M. Fishbein, L. Wei, L. M. Pfeffer and J. A. Berliner, J. Biol. Chem., 2007, 282, 31460–31468 CrossRef CAS PubMed.
- E. Tedeschi, M. Menegazzi, Y. Yao, H. Suzuki, U. Forstermann and H. Kleinert, Mol. Pharmacol., 2004, 65, 111–120 CrossRef CAS PubMed.
- X. Pan, X. Cao, N. Li, Y. Xu, Q. Wu, J. Bai, Z. Yin, L. Luo and L. Lan, Inflammation Res., 2014, 63, 597–608 CrossRef CAS PubMed.
- M. Stempelj, M. Kedinger, L. Augenlicht and L. Klampfer, J. Biol. Chem., 2007, 282, 9797–9804 CrossRef CAS PubMed.
- H. S. Kim and M. S. Lee, Cell. Signalling, 2007, 19, 454–465 CrossRef CAS PubMed.
- K. C. Goh, S. J. Haque and B. R. Williams, EMBO J., 1999, 18, 5601–5608 CrossRef CAS PubMed.
- A. Miniati, Z. Weng, B. Zhang, A. Therianou, M. Vasiadi, E. Nicolaidou, A. J. Stratigos, C. Antoniou and T. C. Theoharides, Clin. Exp. Dermatol., 2014, 39, 54–57 CrossRef CAS PubMed.
- Z. Weng, A. B. Patel, M. Vasiadi, A. Therianou and T. C. Theoharides, PLoS One, 2014, 9, e90739 Search PubMed.
- O. H. Kang, J. G. Choi, J. H. Lee and D. Y. Kwon, Molecules, 2010, 15, 385–398 CrossRef CAS PubMed.
- T. K. Kao, Y. C. Ou, S. Y. Lin, H. C. Pan, P. J. Song, S. L. Raung, C. Y. Lai, S. L. Liao, H. C. Lu and C. J. Chen, J. Nutr. Biochem., 2011, 22, 612–624 CrossRef CAS PubMed.
- A. K. Pandurangan, S. A. Kumar, P. Dharmalingam and S. Ganapasam, Pharmacogn. Mag., 2014, 10, S306–S310 CrossRef PubMed.
- C. M. Park and Y. S. Song, Nutr. Res. Pract., 2013, 7, 423–429 CrossRef CAS PubMed.
- C. Nathan and Q. W. Xie, Cell, 1994, 78, 915–918 CrossRef CAS PubMed.
- W. K. Alderton, C. E. Cooper and R. G. Knowles, Biochem. J., 2001, 357, 593–615 CrossRef CAS PubMed.
- S. Moncada, R. M. Palmer and E. A. Higgs, Pharmacol. Rev., 1991, 43, 109–142 CAS.
- R. K. Cross and K. T. Wilson, Inflammatory Bowel Dis., 2003, 9, 179–189 CrossRef.
- J. R. Kanwar, R. K. Kanwar, H. Burrow and S. Baratchi, Curr. Med. Chem., 2009, 16, 2373–2394 CrossRef CAS PubMed.
- H. M. Roelofs, R. H. Te Morsche, B. W. van Heumen, F. M. Nagengast and W. H. Peters, BMC Gastroenterol., 2014, 14, 1 Search PubMed.
- L. L. Yates and D. C. Gorecki, Acta Biochim. Pol., 2006, 53, 651–662 CAS.
- R. D. Ellis, J. R. Goodlad, G. A. Limb, J. J. Powell, R. P. Thompson and N. A. Punchard, Inflammation Res., 1998, 47, 440–445 CrossRef CAS PubMed.
- M. F. Neurath, I. Fuss, G. Schurmann, S. Pettersson, K. Arnold, H. Muller-Lobeck, W. Strober, C. Herfarth and K. H. Buschenfelde, Ann. N. Y. Acad. Sci., 1998, 859, 149–159 CrossRef CAS PubMed.
- G. Rogler, K. Brand, D. Vogl, S. Page, R. Hofmeister, T. Andus, R. Knuechel, P. A. Baeuerle, J. Scholmerich and V. Gross, Gastroenterology, 1998, 115, 357–369 CrossRef CAS.
- S. Schreiber, S. Nikolaus and J. Hampe, Gut, 1998, 42, 477–484 CrossRef CAS PubMed.
- J. Chen, J. Zhao, L. Chen, N. Dong, Z. Ying, Z. Cai, D. Ji, Y. Zhang, L. Dong, Y. Li, L. Jiang, M. J. Holtzman and C. Chen, J. Transl. Med., 2015, 13, 293 CrossRef PubMed.
- A. Xagorari, A. Papapetropoulos, A. Mauromatis, M. Economou, T. Fotsis and C. Roussos, J. Pharmacol. Exp. Ther., 2001, 296, 181–187 CAS.
- J. S. Kim, H. J. Lee, M. H. Lee, J. Kim, C. Jin and J. H. Ryu, Planta Med., 2006, 72, 65–68 CrossRef CAS PubMed.
- S. H. Kim, K. J. Shin, D. Kim, Y. H. Kim, M. S. Han, T. G. Lee, E. Kim, S. H. Ryu and P. G. Suh, Biochem. Pharmacol., 2003, 66, 955–963 CrossRef CAS PubMed.
- W. Malilas, S. S. Koh, S. Kim, R. Srisuttee, I. R. Cho, J. Moon, H. S. Yoo, S. Oh, R. N. Johnston and Y. H. Chung, Int. J. Oncol., 2013, 43, 1111–1116 CAS.
- M. L. Slattery, A. Lundgreen, S. A. Kadlubar, K. L. Bondurant and R. K. Wolff, Mol. Carcinog., 2013, 52, 155–166 CrossRef PubMed.
- D. Vyas, K. M. O'Dell, J. L. Bandy and E. G. Boyce, Ann. Pharmacother., 2013, 47, 1524–1531 CrossRef CAS PubMed.
- J. Panes, C. Su, A. G. Bushmakin, J. C. Cappelleri, C. Mamolo and P. Healey, BMC Gastroenterol., 2015, 15, 14 CrossRef CAS PubMed.
- W. J. Sandborn, S. Ghosh, J. Panes, I. Vranic, C. Su, S. Rousell and W. Niezychowski, N. Engl. J. Med., 2012, 367, 616–624 CrossRef CAS PubMed.
- K. Rezai-Zadeh, J. Ehrhart, Y. Bai, P. R. Sanberg, P. Bickford, J. Tan and R. D. Shytle, J. Neuroinflammation, 2008, 5, 41 CrossRef PubMed.
- P. Aneknan, V. Kukongviriyapan, A. Prawan, S. Kongpetch, B. Sripa and L. Senggunprai, Asian Pac. J. Cancer Prev., 2014, 15, 5071–5076 CrossRef PubMed.
- S. Vanhatupa, D. Ungureanu, M. Paakkunainen and O. Silvennoinen, Biochem. J., 2008, 409, 179–185 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |