
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of sun-dried raisins (Vitis vinifera L.) on the in vitro composition of the gut microbiota†
Giuseppina
Mandalari
*ab,
Simona
Chessa
c,
Carlo
Bisignano
b,
Luisa
Chan
d and
Arianna
Carughi
e
aIFR-Extra, Institute of Food Research, Norwich Research Park, Colney Lane, NR4 7UA, Norwich, UK
bDepartment of Chemical, Biological, Pharmaceutical and Environmental Science, University of Messina, Salita Sperone 31, 98168 Messina, Italy. E-mail: gmandalari@unime.it; cbisignano@unime.it; Fax: +39 090 6766474; Tel: +39 090 6766593
cThe Model Gut, Institute of Food Research, Norwich Research Park, Colney Lane, NR4 7UA, Norwich, UK. E-mail: simona.chessa@ifr.ac.uk
dSecond Genome Inc., South San Francisco, CA, USA. E-mail: luisa@secondgenome.com
eSun-Maid Growers of California 13525 S. Bethel Ave, Kingsburg, CA 93631, USA. E-mail: arianna100@comcast.net
First published on 16th August 2016
Abstract
Modulation of the human gut microbiota has proven to have beneficial effects on host health. The aim of this work was to evaluate the effect of sun-dried raisins (SR) on the composition of the human gut microbiota. A full model of the gastrointestinal tract, which includes simulated mastication, a dynamic gastric model, a duodenal model and a colonic model of the human large intestine, was used. An increase in the numbers of bifidobacteria and lactobacilli was observed by plate-counting in response to the addition of either SR or FOS after 8 and 24 h fermentation. A significant decrease in Firmicutes and Bacteroidetes was observed in SR samples after 8 and 24 h fermentation. FOS resulted in the greatest production of short chain fatty acids. Sun-dried raisins demonstrated considerable potential to promote the colonization and proliferation of beneficial bacteria in the human large intestine and to stimulate the production of organic acids.
1. Introduction
The human gut microbiota is a complex and dynamic ecosystem comprised of thousands of different species of bacteria, greater than 90% of which belong to the phyla Bacteroidetes and Firmicutes.1,2 The composition of the intestinal microbiota is reportedly highly individual and strongly associated with mechanisms involved in maintaining host energy balance. Such processes include the fermentation of undigestible oligosaccharides, the regulation of lipid storage, the biosynthesis of vitamins, and the absorption of minerals.3,4 The findings of previous studies have suggested that a healthy gut microbiota could work to preclude the onset of certain chronic metabolic diseases, such as intestinal inflammation and metabolic syndrome.5,6 In contrast, the alteration of the gut microbial community structure and/or function, commonly known as dysbiosis, could lead to a range of pathologies, including inflammatory bowel disease.7 Intestinal inflammation and disturbed colonic fermentation in subjects suffering from irritable bowel syndrome have been found to be associated with (a) microbiotas limited in the abundance and diversity of Bacteroidetes and Firmicutes, and (b) reduced numbers of Clostridium coccoides.8 Commensal bacteria are known to inhibit the colonization of invading pathogens both passively, blocking host receptors, and actively, through production of metabolites such as short chain fatty acids (SCFA) and proteinaceous toxins.9 Prebiotics have proven beneficial in modulating the composition and metabolism of microbial populations inhabiting the human gut. Gibson & Roberfroid10 first described prebiotics as ‘non digestible food ingredients that beneficially affect the host by selectively stimulating the growth and/or activity of one or a limited number of bacteria in the colon, thus improving host health. The numbers of certain beneficial bacteria, such as bifidobacteria and lactobacilli have been shown to increase with the addition of prebiotics. The increased abundance of these advantageous strains helps prevent the colonization of gastrointestinal pathogens, regulate appetite, improve mineral absorption, and decrease serum lipid concentration.11While any food ingredient entering the large bowel has the potential to bear prebiotic benefits, the most common prebiotics are nondigestible fructo-oligosaccharides (FOS) and galacto-oligosaccharides (GOS).
Dried grapes (i.e., raisins; Vitis vinifera L.) are known to be rich in both single sugars (roughly equivalent amounts of fructose and glucose) and FOS (1–5%).12–14 In addition to significant amounts of tartaric acid, which in and of itself could influence the composition of the colonic microbiota,15 raisins also contain flavonols, such as quercetin and kaempferol, and phenolic acids, such as caftaric and coutaric acid.16 In 2010, Williamson and Carughi17 reported that raisins are richer in certain acids, such as protocatechuic and oxidized cinnamic acids, than their hydrated counterparts. The effects of dietary polyphenol-rich grape products on intestinal and gut microbial diversity have previously been evaluated in broiler chicks. The findings of these studies demonstrated that the bioconversion of polyphenols in the lower gut led to an increase in biodiversity.18 Furthermore, inclusion of dietary red wine polyphenols has been reported to have prebiotic effects in the distal gut, leading to a significant increase of beneficial bacteria after 4 weeks of daily consumption.19 We have previously reported the potential prebiotic effect of almond seeds and almond skins using an in vitro full model of gastrointestinal (GI) digestion. This model included both gastric and small intestinal environments, in addition to the colonic model of fermentation with representative human gut bacteria.20,21 The goal of the present study was to determine the effect(s) of sun-dried raisins on the composition of the human gut microbiota. A full dynamic gastric model (DGM), which provides a realistic and predictive simulation of the physical and chemical processing of the human stomach,22,23 was used in concert with a colon model simulating the human large intestine.
2. Materials and methods
2.1 Raisins
Sun-dried raisins (SR) were kindly provided by Sun-Maid Growers of California (CA, USA). The chemical composition of SR used in this study was as follows: carbohydrates (78%), water (15%), fiber (4%), and protein (3%).2.2 Chemicals and enzymes
Egg L-α-phosphatidylcholine (PC, lecithin grade 1, 99% purity) was obtained from Lipid Products (South Nutfield, Surrey, UK). Porcine gastric mucosa pepsin, bovine α-chymotrypsin, pancreatic α-amylase, porcine trypsin, porcine colipase, porcine pancreatic lipase and bile salts were obtained from Sigma (Poole, Dorset, UK). Lipase for the gastric phase of digestion was a gastric lipase analogue of fungal origin from Amano Enzyme Inc. (Nagoya, Japan). All other chemicals were of Analar quality.2.3 Simulated human digestion
In order to simulate the full GI tract, SR samples were processed in four sequential environments: mouth (oral processing), stomach (gastric digestion), small intestine (duodenal digestion) and large bowel (colonic fermentation).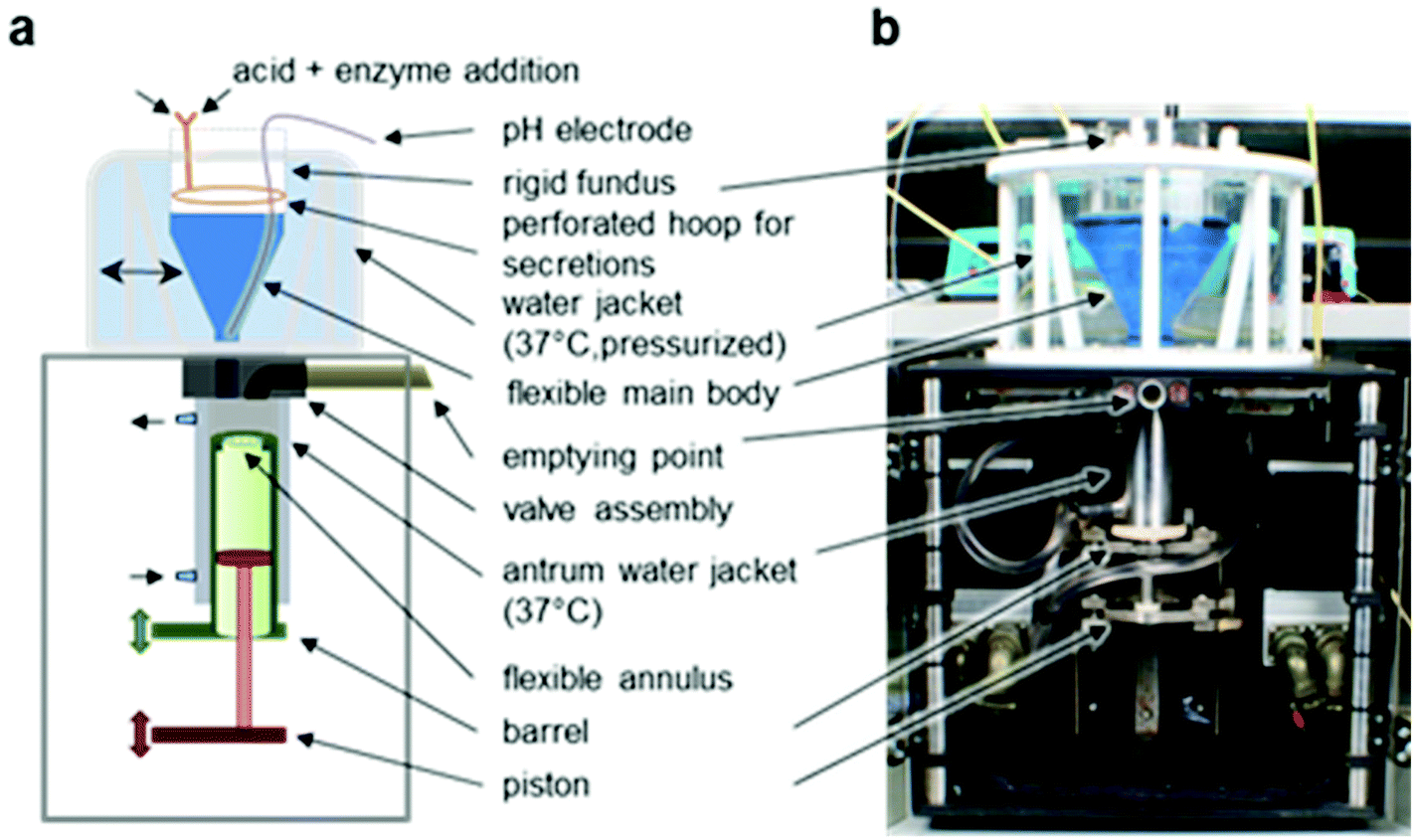 | ||
| Fig. 1 The Dynamic Gastric Model (DGM). (a) Schematic representation of the main components of the DGM. (b) Photographic image of the DGM.25 | ||
Every 2 h during duodenal incubation the sample was centrifuged (3700 rpm for 15 min, 5 °C), the supernatant was weighed and immediately frozen at −40 °C, and the insoluble residue was weighed and further incubated with fresh hepatic and pancreatic secretions. This was repeated four times over the duodenal incubation in an effort to eliminate residual monosaccharides from entering the large bowel. At the end of the incubation period the solid material was separated from the liquid fraction, weighed, and stored at −40 °C for further use in the colonic model. All liquid fractions obtained from the duodenal incubation were combined and freeze-dried for 7 days at room temperature. In order to precipitate the FOS and the non-digestible oligosaccharides that would end up in the large intestine in vivo, the freeze-dried extract was mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio with 90% ethanol and incubated overnight at room temperature. This liquid duodenal-ethanol sample was centrifuged the following morning and the precipitated solid material was added to the wet solid material.
:4 ratio with 90% ethanol and incubated overnight at room temperature. This liquid duodenal-ethanol sample was centrifuged the following morning and the precipitated solid material was added to the wet solid material.
2.4 Bacterial counts
Samples removed from the fermentation vessels were subjected to bacterial counts on selective agar plates for enumeration of different bacteria types, as follows: total anaerobes spp. were grown anaerobically at 37 °C on Wilkins–Chalgren anaerobe agar (Oxoid, UK) and counted after 3 days of incubation; bacteroides spp. were grown anaerobically at 37 °C on bacteroides bile esculin agar (Oxoid, UK) and counted after 5 days of incubation; clostridia spp. were grown anaerobically at 37 °C on reinforced clostridial agar (Oxoid, UK) and counted after 5 days of incubation; bifidobacteria spp. were grown anaerobically at 37 °C on Beerens’ agar (Oxoid, UK) and counted after 5 days of incubation; lactobacilli spp. were grown anaerobically at 37 °C on Rogosa agar (Oxoid, UK) and counted after 5 days of incubation.2.5 Microbiota profiling
Following colonic fermentation, DNA was isolated from samples using a MoBio PowerMag DNA Isolation Kit in accordance with the manufacturer's protocols (Carlsbad, CA), and DNA products were immediately stored at −20 °C. All DNA products were quantified using the Qubit® Quant-iT dsDNA Broad-Range Kit (Invitrogen, Life Technologies, Grand Island, NY) to ensure that they met minimum requirements in mass and purity.To enrich the sample in the V4 region of the 16S rRNA gene, DNA was amplified using fusion primers biased towards surrounding conserved regions and tailed with sequences amenable to Illumina (San Diego, CA) flow cell adapters and indexing barcodes. Each sample was polymerase chain reaction (PCR) amplified with two distinct V4 bar-coded fusion primers. The amplified PCR products from each sample were concentrated via a solid-phase reversible immobilization method of purifying amplicons, which were then quantified by capillary electrophoresis using an Agilent 2100 Bioanalyzer® (Santa Clara, CA). A pooled sample created by combining 35 distinct 16S rRNA gene V4-region amplified and barcoded samples was loaded into the Illumina MiSeq® reagent cartridge, and then onto the instrument along with the flow cell. After cluster formation on the MiSeq instrument, the amplicons were sequenced for 250 cycles with custom primers designed for paired-end sequencing.
2.6 Sequence processing
Using QIIME and custom scripts, sequences were quality filtered and de-multiplexed using exact matches to the supplied DNA barcodes.27 The resulting sequences were then queried against the Greengenes reference database28 of 16S rRNA gene sequences, and clustered at a threshold of 97% similarity by uclust (closed reference OTU picking). The longest sequence formed from each OTU was used as that particular OTU's representative sequence. The representative OTU sequences were then assigned taxonomic classification via Mothur's Bayesian classifier and trained against the Greengenes database clustered at 98%.2.7 OTU filters
Taxa were segregated and/or filtered based on their presence in at least one of the samples, or to their significantly increased abundance in one category compared to the alternate categories. ANOVA (analysis of variance) was employed to calculate p-values. Additionally, q-values were calculated using the Benjamini–Hochberg procedure to correct p-values and safeguard against false discovery rates. All statistical analyses were performed in the R programming environment.292.8 Summarization
The authors considered the abundance of each OTU and/or the incidence (the presence or absence) of each OTU. An ANOVA test coupled with Tukey's multiple comparisons of means was used to elucidate significant differences between genus richness and family abundance across multiple sample categories.2.9 Sampling normalization
The weighted UniFrac dissimilarity factors in differences in taxon abundance across samples but employs a pair-wise normalization by dividing the sum of differences by the sum of all abundances.30 The authors also considered the related UniFrac measure, which considers only the presence or absence of taxa.Two-dimensional ordinations and hierarchical clustering maps of the samples in the form of dendrograms were created to graphically summarize the inter-sample relationship. To create dendrograms, samples from the distance matrix were clustered hierarchically using the average neighbor (HC-AN) method. Principal coordinates analysis (PCoA) is a method of two-dimensional ordination plotting that is used to visualize complex relationships between samples. PCoA uses both the UniFrac and weighted UniFrac dissimilarity values to position the points relative to each other.
2.10 Whole microbiota significance testing
The Adonis PERMANOVA test was utilized to elucidate significant differences in discrete categorical or continuous variables associated with the whole microbiota (s).31 In this randomization/Monte Carlo permutation test, the samples are randomly reassigned to various sample categories, and the mean normalized cross-category differences from each permutation are compared to the true cross-category differences. The fraction of permutations with greater distinction among categories (larger cross-category differences) than that observed with the non-permuted data is reported as the p-value for the Adonis PERMANOVA test.2.11 Short chain fatty acid analysis
Periodically, one ml samples were removed from the batch culture fermenter and centrifuged at 15000g for 5 min. Twenty μl of the resulting supernatant was then injected into a high performance liquid chromatography (HPLC) system equipped with a refractive index detector, as previously reported.20 Quantification of organic acids was achieved by comparing real-time results with calibration curves for acetic, propionic, butyric, and lactic acids in concentrations ranging from 0.5 to 100 mM. Results are expressed in mmol L−1.32
3. Results
3.1 Plate counts
All samples were subjected to simulated passage through a full model of the GI tract. This model consisted of simulated oral processing, DGM digestion, duodenal digestion, and faecal batch culture fermentation. The average abundances of different types of bacteria enumerated upon being removed from fermentation vessels (10 μL) at times 0 h, 4 h, 8 h and 24 h are provided in Table 1. Compared to the control vessel, wherein no extra carbon source was added, the abundance of total anaerobes increased significantly with the addition of either FOS or SR after both 8 and 24 h of incubation. An increase in the numbers of bifidobacteria and lactobacilli was also observed in response to supplementation with either SR or FOS after 8 and 24 h of incubation. The bacteroides population significantly decreased after 4 h incubation in the presence of SR, whereas their numbers remained stable and similar to those at t = 0 after 8 and 24 h of incubation. There were no significant changes observed in the number of clostridia following incubation with FOS, whereas a slight increase relative to t = 0 was noticed following 4, 8, and 24 h incubation with SR. As expected, there were no observable shifts in the number of any of the bacterial groups examined in the control vessel, which was devoid of a carbon source.| FOS | SR | Control | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bacteria | 0 h | 4 h | 8 h | 24 h | 0 h | 4 h | 8 h | 24 h | 0 h | 4 h | 8 h | 24 h |
| Total Anaerobes | 1.97 × 108 | 3.06 × 108 | 1.91 × 109 | 2.16 × 109 | 1.40 × 108 | 6.88 × 108 | 2.15 × 109 | 1.30 × 109 | 1.04 × 108 | 1.02 × 108 | 2.40 × 108 | 1.66 × 108 |
| Bifidobacteria | 1.24 × 108 | 5.11 × 108 | 1.29 × 1010 | 3.16 × 109 | 1.10 × 108 | 4.07 × 108 | 6.40 × 109 | 4.61 × 109 | 1.13 × 108 | 2.35 × 107 | 2.23 × 107 | 2.43 × 107 |
| Lactobacilli | 6.89 × 107 | 7.35 × 108 | 4.24 × 109 | 1.86 × 109 | 5.30 × 107 | 3.09 × 108 | 3.13 × 108 | 1.51 × 108 | 6.09 × 107 | 5.68 × 107 | 5.37 × 107 | 2.24 × 107 |
| Bacteroides | 9.75 × 107 | 2.14 × 108 | 2.35 × 108 | 4.83 × 108 | 9.31 × 107 | 3.50 × 106 | 2.33 × 108 | 2.20 × 108 | 7.55 × 107 | 7.27 × 107 | 1.77 × 108 | 2.00 × 107 |
| Clostridia | 3.00 × 107 | 1.48 × 107 | 1.79 × 107 | 1.50 × 107 | 3.05 × 106 | 1.85 × 107 | 2.83 × 107 | 5.16 × 107 | 2.39 × 107 | 1.35 × 107 | 2.30 × 107 | 3.66 × 107 |
3.2 Taxonomic composition and changes in whole microbiota during fermentation
Between 91 and 210 bacterial genera were identified in the samples subjected to colonic fermentation, and no archaeal genera were detected. By using an ANOVA coupled with a Tukey Honest Significant Difference (HSD) test, a significant difference (p < 0.05; alpha = 0.05) in bacterial genus richness was observed during fermentation. A reduction in biodiversity by 13 species was observed in the SR-supplemented samples after 24 h fermentation, whereas an increase in diversity by 91 species was noticed in the FOS-supplemented samples. Members of the Firmicutes phylum dominated all samples (on average 49.4%). Over time (e.g., 0 h vs. 24 h), numerous samples gave rise to reversals in the presence/absence scoring of phylum-level OTU. This was the case for Actinobacteria, Bacteroidetes, Cyanobacteria, and Proteobacteria.A two dimensional PCoA plot graphically summarizing weighted UniFrac and unweighted UniFrac dissimilarity values is provided in Fig. 2. As the figure clearly depicts, there was a distinct separation between the microbiotas representing the SR- and FOS-augmented samples between 0 and 8 h, and 0 and 24 h, of fermentation. A gradual change in the microbiota composition was observed along the PCoA-1 axis (Fig. 3). Using the weighted UniFrac metric on the abundances of the 3535 taxa present in at least one fermentation sample, the Adonis PERMANOVA test yielded a p-value of 0.001, indicative of significant differences in the microbial community structure between the SR, FOS, and negative control samples. Moderate separation was observed between the microbiotas representing the negative control and SR samples along the PCoA-2 axis in the two dimensional PCoA plot of weighted UniFrac dissimilarity values. The accompanying Adonis PERMANOVA test yielded a p-value of 0.003 using the weighted UniFrac metric on the abundance of the 3535 taxa present in at least one sample, indicative of significant differences in the microbiota composition between these sample types. HC-AN analysis showed a distinct clustering of samples at the t = 0 fermentation time point, to the exclusion of all other time-points.
 | ||
| Fig. 2 Proportional abundance (%) of bacterial phyla over the course of a 24 h fermentation. Data represent the mean of three independent fermentations. | ||
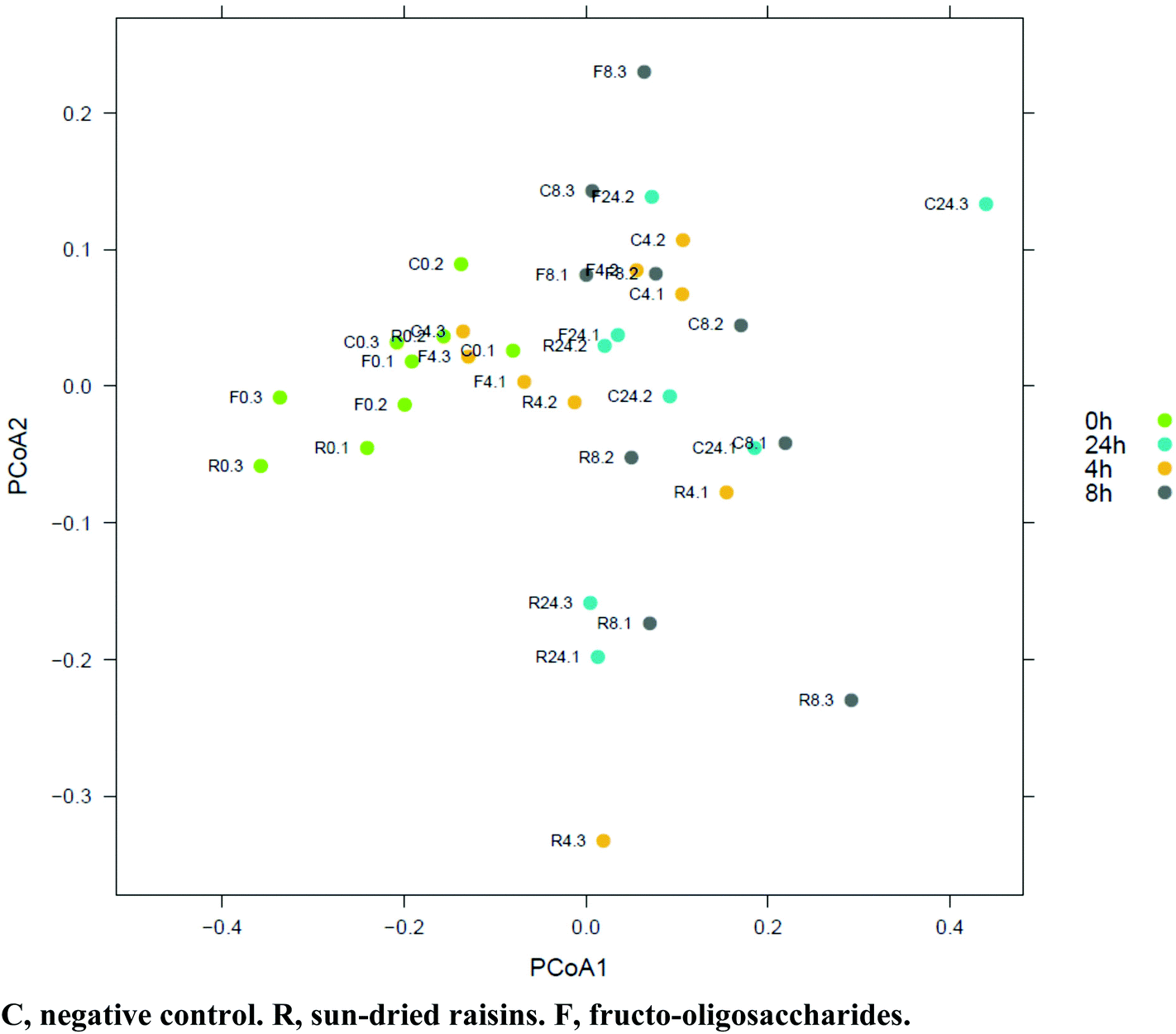 | ||
| Fig. 3 PCoA based on weighted UniFrac distance between samples (given abundances of 3535 taxa present in at least one sample). Axis 1, 36% of variation explained. Axis 2, 16% of variation explained. | ||
3.3 The effect of 4 h fermentation
A separation between the microbiotas representing samples fermented for 0 and 4 h was observed, with 104 OTU exhibiting significantly different abundance patterns between these time points. HC-AN analysis showed that samples collected from each time point (i.e., 0 h or 4 h) clustered together with the exclusion of samples collected at a different time. In particular, 60 of the 104 OTU exhibiting significant dissimilarity between the 0 h and 4 h time points belonged to the Firmicutes phylum. Comparative analysis showed that the 12 OTU profiles generating the lowest p-values in SR samples (0 versus 4 h fermentation) all belonged to one of three phyla: Firmicutes (7), Bacteroidetes (4), and Proteobacteria (1) (Fig. 4; Table S1† for identification). A significant increase in abundance was observed in nine of the differential OTU detected in the 0 h fermentation sample. In particular, the numbers of sequences belonging to Faecalibacterium prausnitzii and Bacteroides uniformis were significantly greater in all 0 h SR samples, whereas the number of sequences arising from Bacteroides fragilis was elevated significantly in all 4 h SR samples. Of the OTU observed with a shift in the abundance level, there was an observed tendency for those representing members of the Proteobacteria phylum to increase in abundance after 4 h of fermentation. In contrast, Bacteroidetes related OTU tended to decrease in abundance after 4 h of fermentation. Members of the Firmicutes phylum exhibited mixed responses across these time points.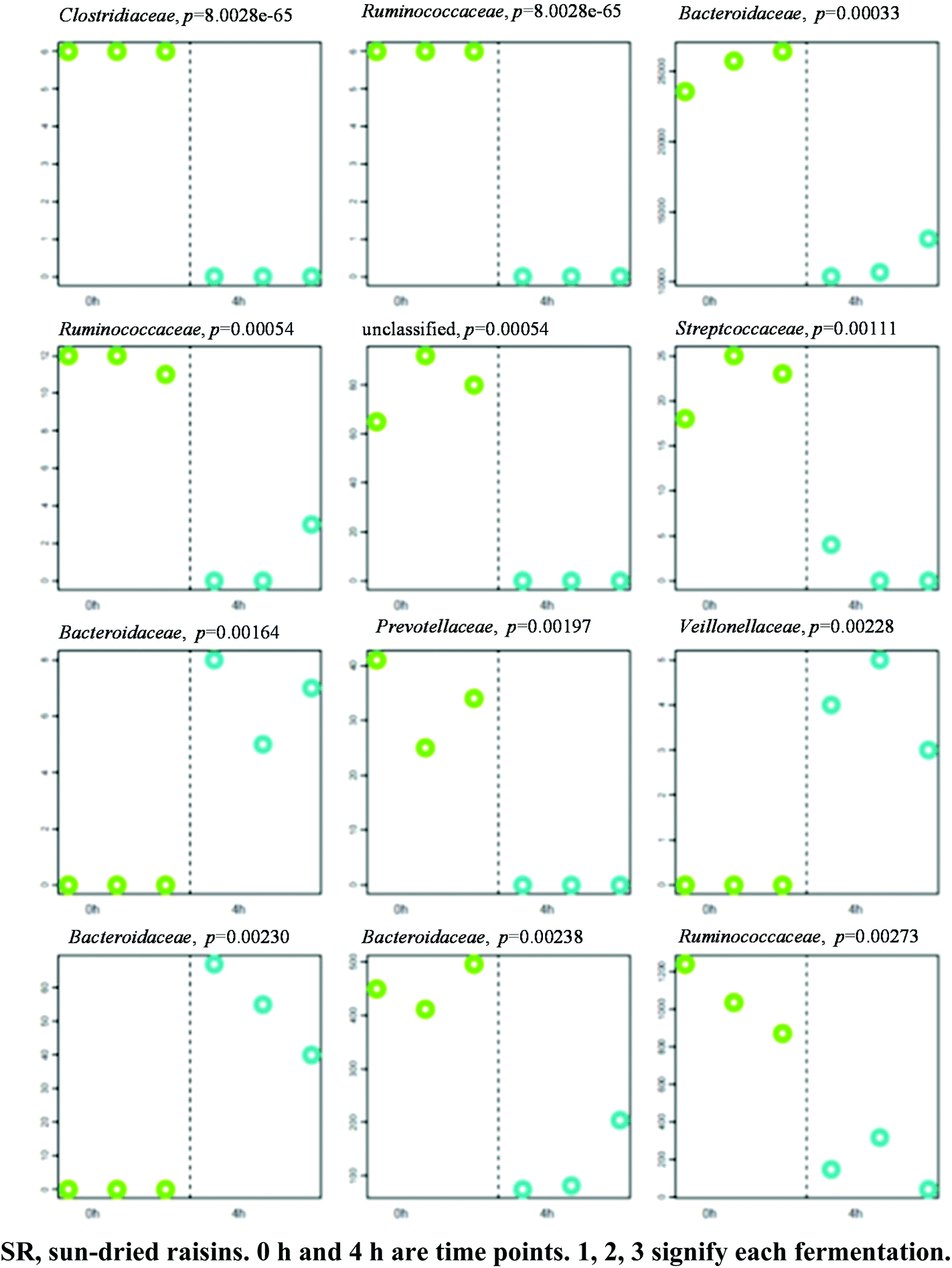 | ||
| Fig. 4 Profiles of the 12 OTUs (expressed as family's name) yielding the lowest p-values. P-Values shown atop each OTU plot have not been adjusted for multiple testing. The y-axis represents relative OTU abundance. Samples are grouped and shaded by category along the x-axis in the following order: SR 0.1, SR 0.2, SR 0.3, SR 4.1, SR 4.2, SR 4.3. | ||
3.4 The effect of 8 h fermentation
A separation between the microbiotas representing samples fermented for 0 and 8 h was observed, with 139 OTU exhibiting significantly different abundance patterns between these time points. Once again, HC-AN analysis showed that samples collected from each time point (i.e., 0 h or 8 h) clustered together to the exclusion of samples collected at the other time point. In particular, 92 of the 139 OTU exhibiting significant dissimilarity between the 0 h and 8 h time points belonged to the Firmicutes phylum. Comparative analysis showed that the 12 OTU profiles generating the lowest p-values in SR samples (0 versus 8 h fermentation) all belonged to one of two phyla: Firmicutes (11) or Bacteroidetes (1) (Fig. 5; Table S2† for identification). A significant increase in abundance was observed in ten of the selected OTU detected in the 0 h fermentation sample. In particular, the number of sequences belonging to Faecalibacterium prausnitzii and Bacteroides uniformis were significantly greater in all 0 h SR samples.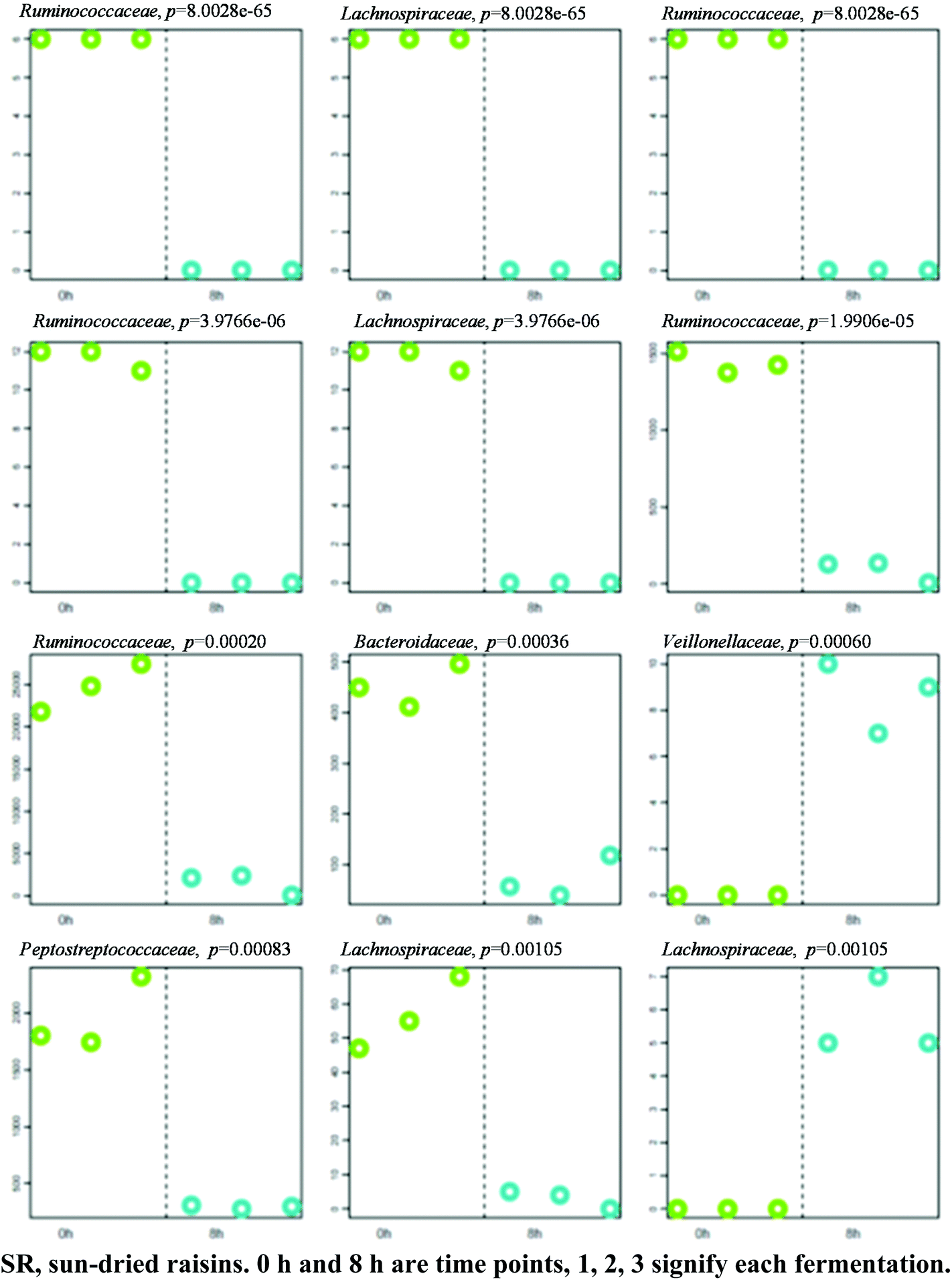 | ||
| Fig. 5 Profiles of 12 OTUs (expressed as family's name) generating the lowest p-values. P-Values shown atop each OTU plot have not been adjusted for multiple testing. The y-axis represents OTU abundance. Samples are grouped and shaded by category along the x-axis in the following order: SR 0.1, SR 0.2, SR 0.3, SR 8.1, SR 8.2, SR 8.3. | ||
Upon comparing the negative control samples with SR samples after 8 h of fermentation, 96 OTU exhibited significantly different abundance patterns between these diets. The vast majority (94 of 96) of OTU exhibiting a significant change in abundance belonged to the phylum Firmicutes, with members of the Ruminococcaceae family significantly more abundant in all of the negative control samples.
3.5 The effect of 24 h fermentation
A separation between the microbiotas representing samples fermented for 0 and 24 h was observed, with 164 OTU exhibiting significantly different abundance patterns between these time points. Here again, HC-AN analysis showed that samples collected from each time point (i.e., 0 h or 24 h) clustered together to the exclusion of samples collected at the other time point. In particular, 92 of the 139 OTU exhibiting significant dissimilarity between the 0 h and 8 h time points belonged to the Firmicutes phylum. Comparative analysis showed that the 12 OTU profiles generating the lowest p-values in SR samples (0 versus 24 h fermentation) all belonged to either the Firmicutes (11) or Bacteroidetes (1) phyla (Fig. 6; Table S3† for identification). A significant increase in abundance was observed in ten of the selected OTU detected in the 0 h fermentation sample. In particular, the number of sequences belonging to Faecalibacterium prausnitzii was significantly greater in all 0 h SR samples.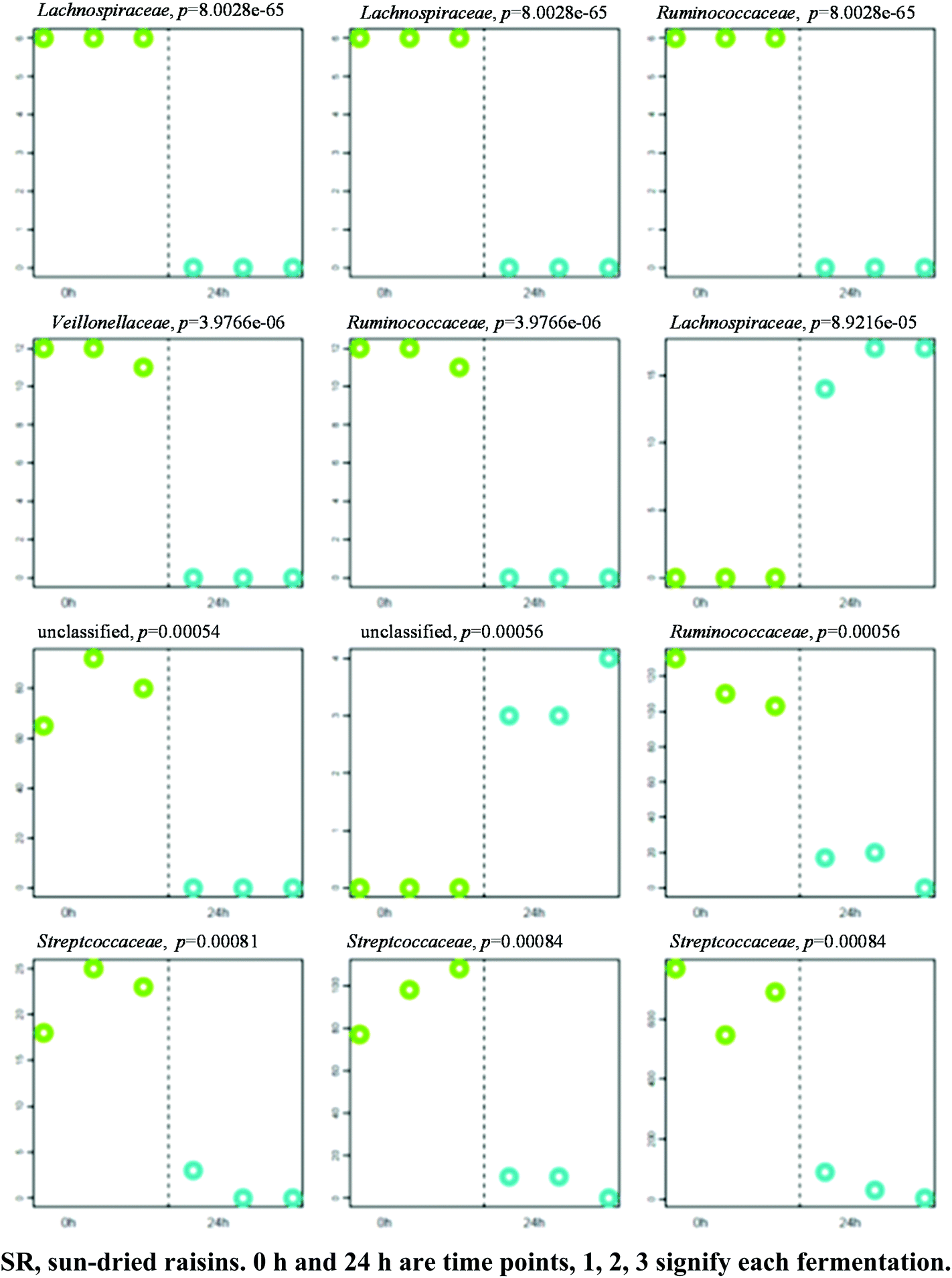 | ||
| Fig. 6 Profiles of 12 OTUs (expressed as family's name) generating the lowest p-values. P-Values shown atop each OTU plot have not been adjusted for multiple testing. The y-axis represents OTU abundance. Samples are grouped and shaded by category along the x-axis in the following order: SR 0.1, SR 0.2, SR 0.3, SR 24.1, SR 24.2, SR 24.3. | ||
When the negative control samples were compared with the SR samples after 24 h of fermentation, 38 OTU exhibited significantly different abundance patterns between these diets. Most (24 of 38) of the OTU exhibiting a significant change in abundance belonged to the phylum Firmicutes, the remainder belonging to the Bacteriodetes phylum. Blautia producta was significantly more abundant in all of the SR samples.
3.6 Changes in bifidobacteria and lactobacilli abundance during fermentation
Of the 4569 distinct OTU detected, 36 were species of Lactobacillus and 28 were species of Bifidobacterium. As was observed via plate count analysis, an increase in bifidobacteria abundance was detected at 4, 8, and 24 h of fermentation in both the SR- and FOS-augmented samples (Fig. 7). Significant increases in abundance of two strains of Bifidobacterium adolescentis, one strain of Bifidobacterium animalis, and one strain of Bifidobacterium bifidum were observed after fermentation with either SR or FOS. Fermentation in the presence of FOS also resulted in an increased abundance of two strains of Lactobacillus zeae.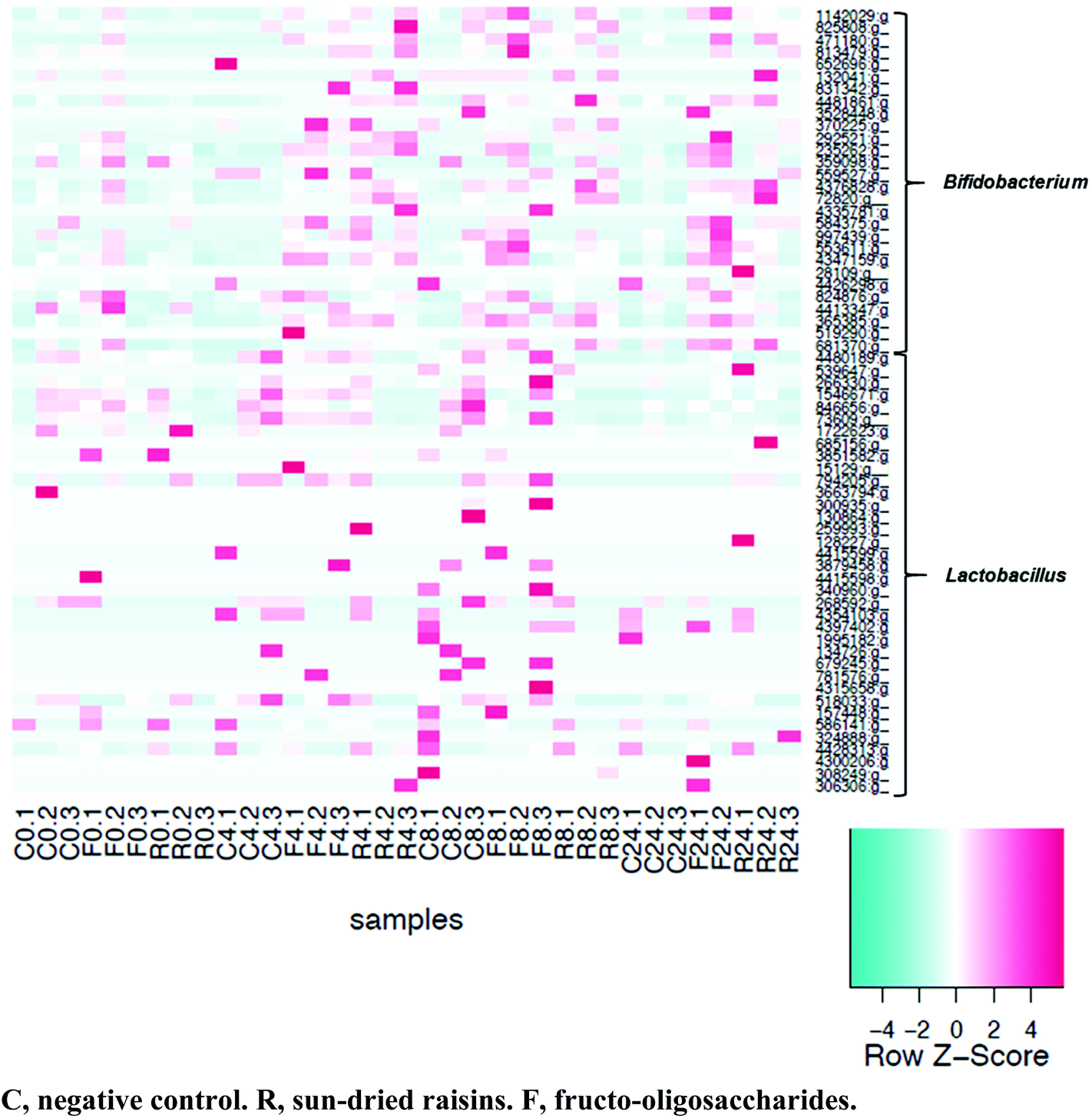 | ||
| Fig. 7 Heatmap of OTU classified as Lactobacillus or Bifidobacterium. Samples were arranged according to the amount of time fermented. Samples were not normalized for heatmap construction. | ||
3.7 Short chain fatty acid production during fermentation
The concentrations of lactic, acetic, propionic, and butyric acids produced at 0, 4, 8 and 24 h fermentation are provided in Table 2. Supplementation with FOS resulted in the greatest total short-chain fatty acid production at all time points tested, followed by SR, and finally the negative control. Fermentation with FOS resulted in the greatest yield of lactic and acetic acids. The concentrations of these acids increased sharply at 4 h and remained elevated through 24 h, which correlated directly with changes in the numbers of bifidobacteria and lactobacilli. The concentrations of lactic and acetic acids also increased after fermentation with SR. Propionic and butyric acid concentrations increased after 8 and 24 h of fermentation with SR, which was perhaps indicative of changes in the Eubacterium rectale population. In the absence of an additional carbon source (negative control), the concentrations of these organic acids did not change significantly.| Time (h) | Control | FOS | SR | |
|---|---|---|---|---|
| Total SCFA | 0 | 4.26 | 5.06 | 4.92 |
| 4 | 6.83 | 38.56 | 17.55 | |
| 8 | 12.63 | 70.93 | 35.88 | |
| 24 | 17.78 | 108.25 | 63.86 | |
| Lactic acid | 0 | 0.45 ± 0.02 | 0.45 ± 0.07 | 0.37 ± 0.01 |
| 4 | 0.48 ± 0.31 | 9.34 ± 0.41 | 3.68 ± 0.13 | |
| 8 | 0.56 ± 0.15 | 14.44 ± 1.26 | 7.56 ± 0.24 | |
| 24 | 0.59 ± 0.14 | 18.96 ± 1.59 | 11.58 ± 0.31 | |
| Acetic acid | 0 | 1.37 ± 0.12 | 1.69 ± 0.15 | 1.59 ± 0.12 |
| 4 | 3.46 ± 1.11 | 19.85 ± 1.29 | 7.26 ± 1.26 | |
| 8 | 6.64 ± 1.29 | 39.96 ± 2.24 | 19.24 ± 2.14 | |
| 24 | 8.96 ± 1.25 | 58.96 ± 2.51 | 36.48 ± 0.24 | |
| Propionic acid | 0 | 0.85 ± 0.17 | 0.96 ± 0.12 | 1.17 ± 0.08 |
| 4 | 1.24 ± 0.14 | 3.48 ± 0.11 | 2.36 ± 0.04 | |
| 8 | 2.89 ± 0.28 | 6.89 ± 0.56 | 3.45 ± 0.27 | |
| 24 | 4.69 ± 0.54 | 14.85 ± 1.38 | 6.84 ± 1.24 | |
| Butyric acid | 0 | 1.59 ± 0.21 | 1.96 ± 0.18 | 1.79 ± 0.06 |
| 4 | 1.65 ± 0.21 | 5.89 ± 0.63 | 4.25 ± 1.01 | |
| 8 | 2.54 ± 0.14 | 9.64 ± 0.42 | 5.63 ± 1.25 | |
| 24 | 3.54 ± 0.19 | 15.48 ± 2.36 | 8.96 ± 1.36 |
4. Discussion
The results of this investigation support the notion that sun-dried raisins could exert a beneficial, prebiotic effect on the human gut via modulation of its microbiota, and as such, increased the production of organic acids. To be truly meaningful and valuable to the scientific community, any evaluation of novel functional food ingredients should consider the availability of compounds able to be (a) hydrolysed by human enzymes and (b) absorbed in the small intestine. In the present study, the authors utilized a full model of the human GI tract, the one combining simulated gastric and duodenal digestion followed by colonic fermentation. The carbohydrate-digestive capacity of the human gut microbiota has been established and a number of glycoside hydrolases encoded by the human genome identified.33 However, in the present study, the arsenal microbiota was not used in combination with human digestive enzymes.SR contain small amounts of FOS, a well-known and commercially available prebiotic. Many claims have been put forth regarding the health benefits of inulin-type fructans, which are present in a wide range of food plants. The basis for most of these claims center on gastrointestinal functionality, including improved gut function, increased stool frequency, faecal bulking, and regularity.34,35 Raisins contain 2.0–3.5 g per 100 g of tartaric acid (TA). Studies have demonstrated that the inclusion of TA in the diet has a positive impact on colonic health. One such investigation compared the effect of a low-fiber, grape-free diet to one including either 120 g of sun-dried raisins or 5 g cream of tartar (roughly equivalent to the amount of TA in the raisins) on intestinal function in healthy adults. The authors found that both diets effectively minimized the intestinal transit time.15 Unlike other fruit acids (e.g., malic, citric), TA bypasses the small intestine and is fermented by colonic bacteria to short-chain fatty acids. As was discussed above, these acids play a significant role in maintaining colonic well-being.
Phenolic compounds have been shown to be metabolized primarily by microbiota residing in the gut, and as such these compounds significantly affect intestinal health.36 A recent study reported that polyphenols contained in red wine altered the intestinal Bacteroidetes/Firmicutes balance and significantly increased the concentration of Firmicutes and Bacteroidetes found in stool samples.19 Polyphenol consumption resulted in no discernable effect on Lactobacillus spp. abundance. In the present study, the authors demonstrate that fermentation with SR and FOS results in a slightly divergent shift in microbiota composition over time. Although both SR and FOS caused an alteration of the structure of the microbiota composition compared to the negative control, it seemed that the effects of SR and FOS were diversified. Plotted graphically in PCoA format, dissimilarity indices seem to show SR and FOS samples slowly clustering apart from the negative control samples. Incubation with both SR and FOS resulted in a decrease in Bacteroidetes, whereas Actinobacteria and Proteobacteria increased. Bacteroidetes also decreased in the negative control vessel, as well as Actinobacteria. The numbers of the predominant phylum Firmicutes decreased after supplementation with SR and in the negative control vessel, whereas they increased with FOS after 8 and 24 h supplementation. There was a tendency for FOS to promote the growth of Bifidobacteria and Lactobacilli, resulting in increased concentrations of acetic and lactic acids over the fermentation period. Supplementation with SR tended to promote the proliferation of Bifidobacterium spp., while the numbers of Lactobacillus spp. remained unchanged. This observation suggests that the polyphenols present in SR, rather than FOS, were responsible for the modulation of the gut microbiota.
Previous studies have reported that catechin intake did not affect the growth of Lactobacillus spp. in vitro, and Procyanidin-rich extracts did not stimulate the growth of Lactobacilli in healthy adults.37,38 In the present study, the abundance of Firmicutes did not change significantly following fermentation with either FOS or SR. However, a marked decrease in Bacteroidetes and an increase in Proteobacteria and Actinobacteria was observed after 24 h fermentation with either supplement.
The phylum Proteobacteria encompasses many Gram-negative pathogens whose functionality seems to increase under dysbiosis conditions, and the detrimental effect of sulphate-reducing bacteria on ulcerative colitis has been reported.39,40 However, an increase in the numbers of Desulfovibrionales associated with a high-fat diet, has not yet been linked to any serious health issues.41 In the present study, all three substrates determined an increase in Proteobacteria numbers, although the proportional abundance (%) in FOS was much lower compared with SR and control samples (Fig. 2).
The increase in the abundance of Actinobateria could explain the elevated levels of short chain fatty acids. Of these, propionate is thought to lower serum cholesterol levels and stimulate satiety.42 Although appropriate levels of propionate have been shown to be beneficial by improving insulin sensitivity, lowering liver and plasma fatty acids and reducing food intake, an excessive amount of this organic acid was associated with adverse effects such as propionic acidemia and neurobehavioural disorders.43
An increased abundance of butyrate-producing strains related to the Blautia coccoides–Eubacterium rectale group was observed following fermentation with SR (compared to the negative control samples). Previously, the presence of butyrate in the human colon has been hypothesized to play a role in the prevention of colon cancer and ulcerative colitis.44,45 Certain members of the Lachnospiraceae and Ruminococcaceae families have been identified as active members of the gut environment. These are by far the most abundant Firmicute families, accounting for ca. 50% and 30% of phylotypes, respectively.46 Certain species within the Lachnospiraceae, including Eubacterium rectale, E. ventriosum, Coprococcus sp., and Roseburia sp., have been shown to be associated with butyrate production. The depleted abundance of certain Ruminococcaceae, such as Faecalibacterium prausnitzii, has been correlated to many cases of Chron's disease.47 In the present study, the numbers of Faecalibacterium prausnitzii decreased significantly after 4, 8, and 24 h fermentation with SR. Similarly, the Ruminococcaceae family, as a whole, was more abundant in the control samples than those supplemented with SR. Fermentation with SR also induced an increase in the number of Roseburia spp. This genus has recently received considerable attention as it is seemingly capable of modulating the activity of gut microbiota towards improved host health.
5. Conclusions
We have demonstrated that sun-dried raisins exhibit immense potential in their capacity to (a) promote the colonization and proliferation of beneficial bacteria in the human GI tract, and (b) stimulate the production of advantageous organic acids. More in-depth studies on the digestibility of raisins and the long-term effects associated with raisins’ supplementation need to be performed in vivo with human test subjects.Competing interest
AC is scientific advisor for Sun-Maid Growers of California; G. Mandalari, S. Chessa, C. Bisignano and L. Chan, no conflicts of interest.Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.Acknowledgements
The work was supported by Sun-Maid Growers of California (USA) and the University of Messina Research & Mobility 2015 Project (project code RES_AND_MOB_2015_DE_LUCA). The sun-dried raisins used in this study were generously provided by the Sun-Maid Growers of California (CA, USA). The authors are grateful to the Second Genome Solutions team for data analysis and discussion.References
- R. E. Ley, D. A. Peterson and J. I. Gordon, Cell, 2006, 124, 837–848 CrossRef CAS PubMed.
- J. R. Marchesi, Environ. Microbiol., 2011, 13, 3088–3102 CrossRef PubMed.
- L. V. Hooper, T. Midtvedt and J. I. Gordon, Annu. Rev. Nutr., 2002, 22, 283–307 CrossRef CAS PubMed.
- P. J. Turnbaugh, R. E. Ley, M. Hamady, C. M. Fraser-Liggett, R. Knight and J. I. Gordon, Nature, 2007, 449, 804–810 CrossRef CAS PubMed.
- A. Costabile, S. Santarelli, S. P. Claus, J. Sanderson, B. N. Hudspith, J. Brostoff, J. L. Ward, A. Lovegrove, P. R. Shewry, H. E. Jones, A. M. Whitley and G. R. Gibson, PLoS One, 2014, 9, e111225 Search PubMed.
- P. J. Turnbaugh, F. Backhed, L. Fulton and J. I. Gordon, Cell Host Microbe, 2008, 3, 213–223 CAS.
- L. F. Butto’ and D. Haller, Int. J. Med. Microbiol., 2016 Search PubMed , pii: S1438-4221(16)30015-17.
- K. Ponnusamy, J. N. Choi, J. Kim, S. Y. Lee and L. H. Lee, J. Med. Microbiol., 2011, 60, 817–827 CrossRef CAS PubMed.
- I. Sekirov and B. B. Finlay, J. Physiol., 2009, 587, 4159–4167 CrossRef CAS PubMed.
- G. R. Gibson and M. B. Roberfroid, J. Nutr., 1995, 125, 1401–1412 CAS.
- M. Nyman, Br. J. Nutr., 2002, 87, S163–S168 CrossRef CAS PubMed.
- A. Carughi, FASEB J., 2009, 23, 716–719 Search PubMed.
- M. E. Camire and M. P. Dougherty, J. Agric. Food Chem., 2003, 51, 834–837 CrossRef CAS PubMed.
- J. G. Muir, S. J. Shepherd, O. Rosella, R. Rose, J. S. Barrett and P. R. Gibson, J. Agric. Food Chem., 2007, 55, 6619–6627 CrossRef CAS PubMed.
- G. A. Spiller, J. A. Story, E. J. Furumoto, J. C. Chezem and M. Spiller, Br. J. Nutr., 2003, 90, 803–807 CrossRef CAS PubMed.
- F. Karadeniz, R. W. Durst and R. E. Wrolstad, J. Agric. Food Chem., 2000, 48, 5343–5350 CrossRef CAS PubMed.
- G. Williamson and A. Carughi, Nutr. Res., 2010, 30, 511–519 CrossRef CAS PubMed.
- A. Viveros, S. Chamorro, M. Pizarro, I. Arija, C. Centeno and A. Brenes, Poult. Sci., 2011, 90, 566–578 CrossRef CAS PubMed.
- M. I. Queipo-Ortuno, M. Boto-Ordonez, M. Murri, J. M. Gomez-Zumaquero, M. C. Postigo, R. Estruch, F. Cardona Diaz and F. J. Tinahones, Am. J. Clin. Nutr., 2012, 95, 1323–1334 CrossRef CAS PubMed.
- G. Mandalari, C. Nueno-Palop, G. Bisignano, M. S. J. Wickham and A. Narbad, Appl. Environ. Microbiol., 2008, 74, 4264–4270 CrossRef CAS PubMed.
- G. Mandalari, R. M. Faulks, C. Bisignano, K. W. Waldron, A. Narbad and M. S. J. Wickham, FEMS Microbiol. Lett., 2010, 304, 116–122 CrossRef CAS PubMed.
- I. Pitino, C. L. Randazzo, G. Mandalari, R. B. Lo Curto, R. M. Faulks, Y. LeMarc, C. Bisignano, C. Caggia and M. S. J. Wickham, Food Microbiol., 2010, 27, 1121–1127 CrossRef PubMed.
- M. Vardakou, A. Mercuri, S. A. Barker, D. Q. Craig, R. M. Faulks and M. S. J. Wickham, AAPS PharmSciTech, 2011, 12, 620–626 CrossRef PubMed.
- G. Mandalari, C. Bisignano, A. Filocamo, S. Chessa, M. Saro’, G. Torre, R. M. Faulks and P. Dugo, Nutr., 2013, 29, 338–344 CrossRef CAS PubMed.
- E. C. Thuenemann, G. Mandalari, G. T. Rich and R. M. Faulks, The Impact of Food Bioactives on Gut Health: in vitro and ex vivo Models, Springer, 2015, Open 47–59 Search PubMed.
- G. Mandalari, R. M. Faulks, G. T. Rich, V. Lo Turco, D. R. Picout, R. B. Lo Curto, G. Bisignano, P. Dugo, G. Dugo, K. W. Waldron, P. R. Ellis and M. S. J. Wickham, J. Agric. Food. Chem., 2008, 56, 3409–3416 CrossRef CAS PubMed.
- J. G. Caporaso, J. Kuczynski, J. Stombaugh, K. Bittinger, F. D. Bushman, E. K. Costello, N. Fierer, A. G. Pena, J. K. Goodrich, J. I. Gordon, G. A. Huttley, S. T. Kelley, D. Knights, J. E. Koenig, R. E. Ley, C. A. Lozupone, D. McDonald, B. D. Muegge, M. Pirrung, J. Reeder, J. R. Sevinsky, P. J. Turnbaugh, W. A. Walters, J. Widmann, T. Yatsunenko, J. Zaneveld and R. Knight, Nat. Methods, 2010, 7, 335–336 CrossRef CAS PubMed.
- D. McDonald, M. N. Price, J. Goodrich, E. P. Nawrocki, T. Z. DeSantis, A. Probst, G. L. Anderson, R. Knight and P. Hugenholtz, ISME J., 2012, 6, 610–618 CrossRef CAS PubMed.
- R. Development Core Team, Vienna, Austria, 2008, ISBN 3-900051-07-0, http://www.R-project.org.
- C. Lozupone, M. Hamady and R. Knight, BMC Bioinf., 2006, 7, 371 CrossRef PubMed.
- M. J. Anderson, Aust. J. Ecol., 2001, 26, 32–46 Search PubMed.
- C. E. Rycroft, M. R. Jones, G. R. Gibson and R. A. Rastall, J. Appl. Microbiol., 2001, 91, 878–887 CrossRef CAS PubMed.
- A. El Kaoutari, F. Armougom, J. I. Gordon, D. Raoult and B. Hernissat, Nat. Rev. Microbiol., 2013, 11, 497–504 CrossRef CAS PubMed.
- J. Van Loo, P. Coussement, L. De Leenheer, H. Hoebregs and G. Smits, Crit. Rev. Food Sci. Nutr., 1995, 35, 525–552 CrossRef CAS PubMed.
- E. Gomez, K. M. Tuohy, G. R. Gibson, A. Klinder and A. Costabile, J. Appl. Microbiol., 2010, 108, 2014–2021 Search PubMed.
- M. V. Selma, J. C. Espin and F. A. Tomas-Barberan, J. Agric. Food Chem., 2009, 57, 6485–6501 CrossRef CAS PubMed.
- X. Tzounis, J. Vulevic, G. G. Kuhnle, T. George, J. Leonczak, G. R. Gibson, C. Kwik-Uribe and J. P. Spencer, Br. J. Nutr., 2008, 99, 782–792 CrossRef CAS PubMed.
- J. Yamakoshi, S. Tokutake, M. Kikuchi, Y. Kubota, M. Konishi and T. Mitsuoka, Microb. Ecol. Health Dis., 2001, 13, 25–31 CrossRef CAS.
- S. Maccaferri, A. Klinder, S. Cacciatore, R. Chitarrari, H. Honda, C. Luchinat, I. Bertini, P. Carnevali, G. R. Gibson, P. Brigidi and A. Costabile, Mol. Nutr. Food Res., 2012, 56, 1342–1352 CAS.
- K. M. Tuohy, D. J. Hinton, S. J. Davies, M. J. Crabbe, G. R. Gibson and J. M. Ames, Mol. Nutr. Food Res., 2006, 50, 847–857 CAS.
- M. A. Hildebrandt, C. Hoffmann, S. A. Sherril-Mix, S. A. Keilbaugh, M. Hamady, Y. Y. Chen, R. Knight, R. S. Ahima, F. Bushman and G. D. Wu, Gastroenterology, 2009, 137, 1716–1724 CrossRef CAS PubMed.
- E. Hosseini, C. Grootaert, W. Verstraete and T. Van De Wiele, Nutr. Rev., 2011, 69, 245–258 CrossRef PubMed.
- S. H. Al-Lahham, M. P. Peppelenbosch, H. Roelofsen, R. J. Vonk and K. Venema, Biochim. Biophys. Acta, 2010, 1801, 1175–1183 CrossRef CAS PubMed.
- G. A. Weaver, J. A. Krause, T. L. Miller and M. J. Wolin, Gut, 1988, 29, 1539–1543 CrossRef CAS PubMed.
- E. J. Simpson, M. A. Chapman, J. Dawson, D. Berry, I. A. Macdonald and A. Cole, Gut, 2000, 46, 73–77 CrossRef CAS PubMed.
- A. Biddle, L. Stewart, J. Blanchard and S. Leschine, Diversity, 2013, 5, 627–640 CrossRef.
- T. Fujimoto, H. Imaeda, K. Takahashi, E. Kasumi, S. Bamba, Y. Fujiyama and A. Andoh, J. Gastroenterol. Hepatol., 2013, 28, 613–619 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6fo01137c |
| This journal is © The Royal Society of Chemistry 2016 |