DOI:
10.1039/C5FO00807G
(Paper)
Food Funct., 2016,
7, 355-363
Tenuigenin exhibits anti-inflammatory activity via inhibiting MAPK and NF-κB and inducing Nrf2/HO-1 signaling in macrophages
Received
2nd July 2015
, Accepted 9th October 2015
First published on 26th October 2015
Abstract
Tenuigenin (TNG), isolated from the root of the Chinese herb Polygala tenuifolia, possesses various biological and pharmacological activities, including anti-oxidation and anti-inflammation activities. In this study, we aimed to further investigate whether its anti-inflammatory activity is associated with the inhibition of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) expression in lipopolysaccharide (LPS)-induced RAW 264.7 cells. Our results showed that TNG treatment dramatically reduced prostaglandin E2 (PGE2) and NO production, decreased iNOS and COX-2 gene expression, inhibited JNK1/2, ERK1/2, p38 and NF-κB (p65) phosphorylation, and blocked IκBα phosphorylation and degradation. Further studies revealed that TNG dramatically up-regulated heme oxygenase (HO)-1 and nuclear factor erythroid 2-related factor 2 (Nrf2) expression, which was related to the induction of Nrf2 nuclear translocation and decreased Keap1 protein expression. Additionally, treatment with JNK1/2, ERK1/2 or p38 inhibitors had no effect on the TNG-induced HO-1 protein expression. Furthermore, the LPS-induced iNOS and COX-2 expression levels were inhibited by TNG, which was partially reversed by the HO-1-siRNA and HO-1 inhibitors. Together, these results showed that TNG's anti-inflammatory activity is related to the inhibition of iNOS and COX-2 expression via down-regulation of the MAPK and NF-κB, and up-regulation of the Nrf2/HO-1 signaling pathways.
Introduction
Inflammation is a crucial factor of the innate immune responses that attenuate tissue damage. However, the longer the inflammation persists, the severer the levels of tissue damage.1,2 The sources of inflammation are widespread, including lipopolysaccharide (LPS), which is known to induce a disturbance in immune and inflammatory responses. Numerous studies have shown that LPS, a pathogenic component of endotoxin in the outer membrane of Gram-negative bacteria, is capable of activating multiple signaling pathways.3 Nuclear factor-kappa B (NF-κB), which is an important pro-inflammatory transcription factor, consists of p50/p65 and IκBα. Under physical conditions, heterodimers of NF-κB components, mainly p50/p65, are located in the cytoplasm in an inactive form through their linkage to an inhibitor of the κB (IκB) protein.4 However, LPS induces IκB phosphorylation and degradation to release NF-κB dimers into the nucleus to express specific target genes, such as pro-inflammatory cytokines, inducible nitric oxide synthase (iNOS), and the cyclooxygenase-2 (COX-2) protein.5 Moreover, the three mitogen-activated protein kinase (MAPK) signaling pathways, which consist of c-Jun NH2-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), and p38, also lead to the overexpression of inflammatory mediators, including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), NO and prostaglandin (PG) E2. COX-2 and iNOS are responsible for the production of PGE2 and NO, respectively.6,7 Hence, inhibition of the production of TNF-α, IL-6, IL-1β, iNOS and COX-2 is responsible for therapeutic actions against various inflammatory diseases.8 Furthermore, the levels of iNOS and COX-2 are crucial indicators for the assessment of the inflammatory extent in macrophages.9
Increasing evidence shows that heme oxygenase (HO)-1, an anti-oxidant enzyme that plays an essential role in heme degradation, is an important mechanism for the cellular pathophysiological conditions of oxidative stress and inflammation.10,11 In addition, HO-1 is essential for decreasing the levels of proinflammatory COX-2 and iNOS, which contribute to reducing COX-2-derived PGE2 and iNOS-derived NO production.12 HO-1 is mediated by nuclear factor-erythroid 2 related factor 2 (Nrf2). Nrf2 plays a critical role in improving cellular oxidative stress and inflammatory damage by regulating antioxidant responses.13,14 Under normal conditions, Nrf2 is located in the cytoplasm with its repressor protein, Kelch-like ECH-associated protein 1 (Keap1). Under stress conditions, Nrf2 dissociates from Keap1, translocates into the nucleus and binds to the antioxidant response element (ARE), which results in induction of phase II detoxifying enzymes and cytoprotective genes, such as HO-1.15
Tenuigenin (TNG) is a major active component isolated from the root of the Chinese herb Polygala tenuifolia, which is called ‘Yuan Zhi’ in accordance with the Chinese Materia Medica, and has been used to improve insomnia, neurosis, and dementia.16 Moreover, several reports have shown that TNG possesses a variety of biological and pharmacological activities, including anti-oxidation and anti-inflammation activities.17,18 Furthermore, our previous experimental data suggested that TNG ameliorated LPS-stimulated acute lung injury (ALI) by the inhibition of inflammatory responses, which suppressed the NF-κB and MAPK signaling pathways. Additionally, TNG significantly inhibited the production of TNF-α, IL-6 and IL-1β in LPS-induced RAW 264.7 cells.19 To further investigate the anti-inflammatory mechanism of TNG, we sought to demonstrate that its anti-inflammatory activity is related to the inhibition of iNOS and COX-2 expression via down-regulation of the MAPK and NF-κB and up-regulation of the Nrf2/HO-1 signaling pathways.
Materials and methods
Reagents
Tenuigenin (TNG), purity >98%, was purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Jilin, China). LPS (Escherichia coli 055:B5) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Antibodies against iNOS, COX-2, p-(JNK1/2), JNK1/2, p-(ERK1/2), ERK1/2, p-p38, p38, p-p65, Keap1, Nrf2, HO-1, Lamin B and β-actin were obtained from Cell Signaling (Boston, MA, USA). The PGE2 enzyme-linked immunosorbent assay (ELISA) kit was obtained from Biolegend (San Diego, CA, USA). Tin protoporphyrin IX (SnPP IX, HO-1 inhibitor) was purchased from Calbiochem (La Jolla, CA, USA). The control siRNA, HO-1-siRNA, and Nrf2-siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Penicillin and streptomycin, foetal bovine serum (FBS) and Dulbecco's modified Eagle's medium (DMEM) for cell culture use were purchased from Invitrogen-Gibco (Grand Island, NY). Faststart Universal SYBR Green Master was purchased from Roche (Basel, Switzerland). The Prime-Script RT-PCR kit was obtained from Takara (Dalian, China). Dimethyl sulfoxide (DMSO), SP600125, SB203580, and U0126 (specific inhibitors of the JNK1/2, p38, and ERK1/2, respectively) were purchased from Sigma-Aldrich (St. Louis, MO). All other chemicals, unless specifically stated elsewhere, were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
The RAW 264.7 mouse macrophage cell line was obtained from the China Cell Line Bank (Beijing, China) and was cultured in DMEM medium added with 10% fetal bovine serum (FBS), 100 U ml−1 of penicillin and 100 U ml−1 of streptomycin, and 3 mM glutamine at 37 °C under a humidified atmosphere containing 5% CO2.
Nitrite assay and measurement of PGE2 levels
Cells were grown in 96-well plates (1 × 104 cells per well), subjected to LPS for 18 h, and were then treated with or without TNG for 6 h. The supernatant was mixed with an equal volume of Griess reagent (Sigma) for 20 min, and nitrite was measured at 525 nm using a microplate reader. The levels of PGE2 were measured by ELISA in accordance with the manufacturer's instructions. Briefly, cells were seeded into 24-well plates (2 × 105 cells per well), subjected to LPS for 18 h, and were then treated with or without TNG for 6 h to assay PGE2 production. The supernatant was collected and analyzed using a specific ELISA kit.
Total RNA extraction and qPCR
Total RNA was extracted from cells using the Trizol reagent in accordance with the manufacturer's instructions. After the concentration of RNA was determined by using a spectrophotometer, 1 μg of RNA was transformed into cDNA using the Prime-Script RT-PCR kit (Takara). β-Actin acted as a control for the amount of total mRNA. The following PCR primer sequences (forward and reverse, respectively) were used: iNOS: 5′-ACA TCG ACC CGT CCA CAG TAT-3′ and 5′-CAG AGG GGT AGG CTT GTC TC-3′, COX2: 5′-ACA CAC TCT ATC ACT GGC ACC-3′ and 5′-TTC AGG GCG AAG CGT TTGC-3′; β-actin: 5′-TCT GTG TGG ATT GTG GCT CTA-3′ and 5′-CTG CTT GCT GAT CCA CAT CTG-3′. PCR reactions were carried out using the SYBR green working solution and quantitatively measured with the Applied Biosystems 7300 real-time PCR system and software (Applied Biosystems, Carlsbad, CA, USA). The following thermal cycler parameters were used: 95 °C for 10 min, followed by 40 cycles of 95 °C for 10 s, 60 °C for 30 s. Gene expression changes were calculated by the comparative Ct method and the values were analyzed by normalizing with β-actin mRNA expression.
Isolation of nuclear and cytosolic fractions
The cytoplasmic and the nuclear extracts were prepared using an NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (Pierce Biotechnology, Rockford, IL, USA), following the manufacturer's instructions. All steps were performed on ice or at 4 °C.
Nrf2-siRNA transfection
For Nrf2-siRNA or HO-1-siRNA transfection, RAW 264.7 cells were grown in 6-well plates (2 × 105 cells per well) until the confluence of cells reached approximately 50%. The cells were then subjected to transient transfection with Nrf2-negative control siRNA, Nrf2-siRNA or HO-1-siRNA using the siRNA transfection reagent Lipofectamine™ 2000, following the manufacturer's protocol. After 18 h, the transfected cells were exposed to TNG for 6 h, followed by lysis buffer for western blot analysis.
Western blot analysis
The cells were lysed in RIPA with protease and phosphatase inhibitors for 30 min. The protein concentrations were measured using a BCA protein assay kit (Beyotime, China), and 40 μg of proteins were electrophoretically transferred onto a PVDF membrane following separation on a 10% SDS-polyacrylamide gel. The membrane was blocked with a blocking solution (5% (w/v) nonfat dry milk) for 2 h, followed by an overnight incubation at 4 °C with a specific primary antibody. The following day, the membrane was incubated for an additional 1 h with an HRP-conjugated secondary antibody (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5000 dilution) at room temperature after thoroughly washing three times with TBST. Bands were detected by ECL (Amersham Pharmacia Biotech, Piscataway, NJ) and band intensities were quantified using ImageJ gel analysis software. All experiments were performed in triplicate for each experimental condition.
:5000 dilution) at room temperature after thoroughly washing three times with TBST. Bands were detected by ECL (Amersham Pharmacia Biotech, Piscataway, NJ) and band intensities were quantified using ImageJ gel analysis software. All experiments were performed in triplicate for each experimental condition.
Statistical analysis
All data in this study were presented as means ± SEM for three independent experiments. The results were analyzed using two-tailed Student's t-test. Statistical significance was defined as p < 0.05 or p < 0.01.
Results
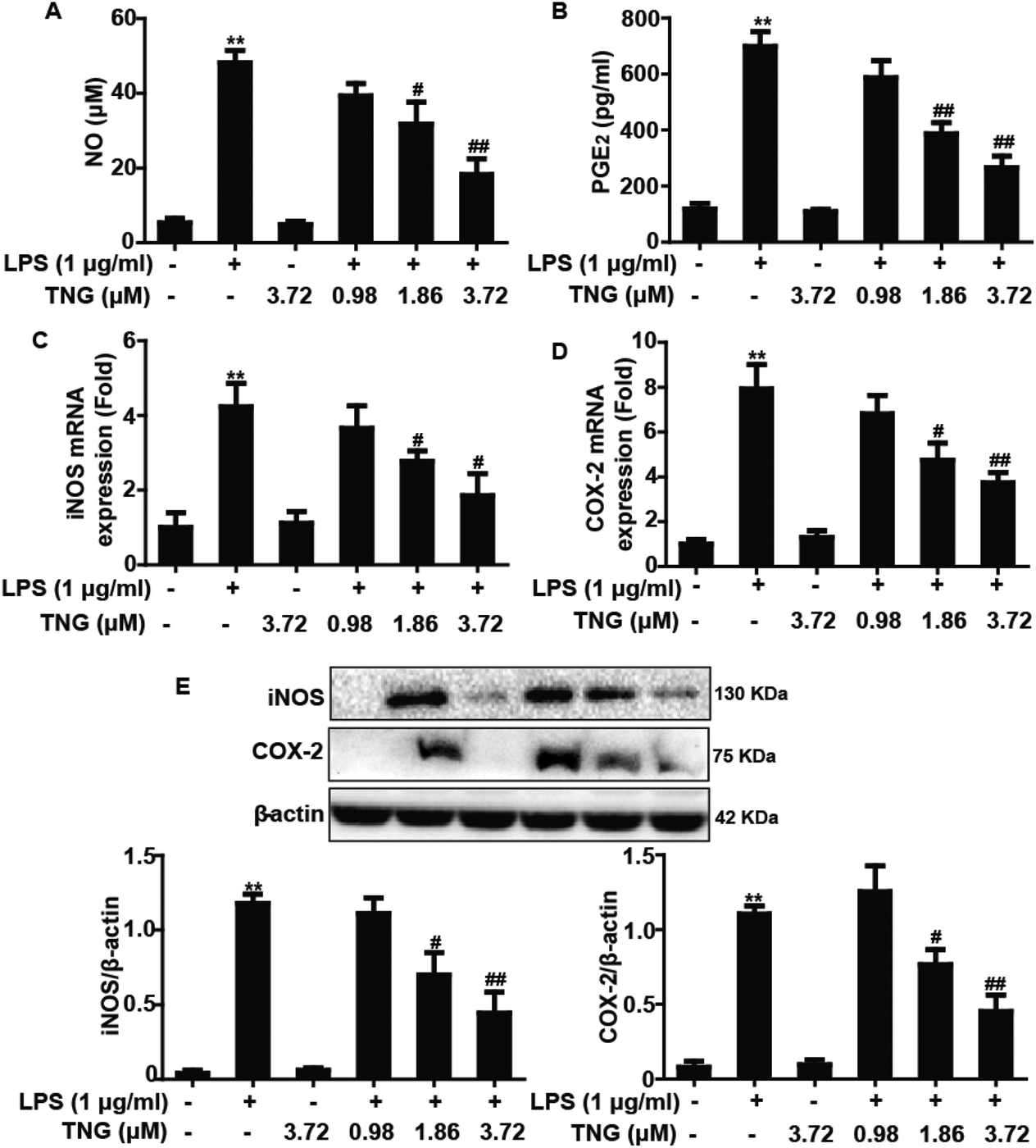
Effects of TNG treatment on LPS-induced NO and PGE2 production and iNOS and COX-2 expression in RAW 264.7 cells
To investigate whether the inhibition of NO and PGE2 production by TNG is due to a decrease in iNOS and COX-2 expression levels, which are essential for inflammation as they produce iNOS-derived NO and COX-2-derived PGE2, we determined the NO and PGE2 production and the iNOS and COX-2 expression after exposure to LPS-induced RAW 264.7 cells with TNG (0.98, 1.86 or 3.72 μM, which were determined in our previous study).19 Treatment of RAW 264.7 cells with LPS alone resulted in a significant increase in NO (812.5%) and PGE2 (647.7%) production, induced an obvious expression of iNOS and COX-2 mRNA (420.2% and 553.4%, respectively) and protein (1831.4% and 1036.7%, respectively), which were suppressed by 1.86 and 3.72 μM of TNG treatments in RAW 264.7 cells compared with the LPS-treated group (Fig. 1).
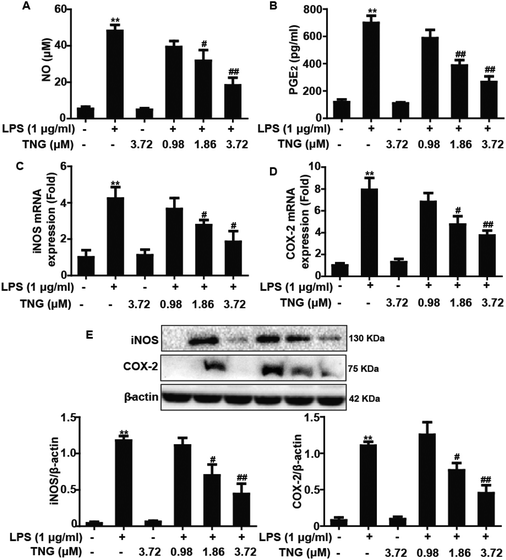 |
| | Fig. 1 Effects of TNG treatment on LPS-induced NO and PGE2 production and iNOS and COX-2 expression in RAW 264.7 cells. RAW 264.7 cells were exposed to LPS for 18 h, and were then treated with or without TNG for 6 h. (A) The effect of TNG on NO production was analyzed by the Griess reaction; (B) PGE2 production was measured by ELISA. Total RNA was extracted from RAW 264.7 cells and gene expression was quantified using real-time PCR. (C, D) Effects of TNG on LPS-induced iNOS and COX-2 mRNA expression. (E) The effects of TNG on LPS-induced iNOS and COX-2 protein expression were analyzed by western blot. All data were presented as means ± SEM of three independent experiments. **p < 0.01 vs. the control group; #p < 0.05 and ##p < 0.01 vs. the LPS group. | |
Effects of TNG treatment on LPS-induced NF-κB activation in RAW 264.7 cells
NF-κB is a vital transcription factor that is essential for COX-2 and iNOS gene expression. Therefore, we investigated whether TNG pretreatment effectively blocked IκBα phosphorylation and degradation as well as suppressed the phosphorylation of NF-κB (p65), which were induced by LPS. Compared to the LPS-treated group, 1.86 and 3.72 μM of TNG pretreatments decreased IκBα phosphorylation (43.5% and 62.4%, respectively) and degradation (30.8% and 53.8%, respectively) and inhibited the phosphorylation of NF-κB (p65) (32.5% and 76.5%, respectively), whereas the 0.98 μM TNG pretreatment did not exhibit these effects (Fig. 2).
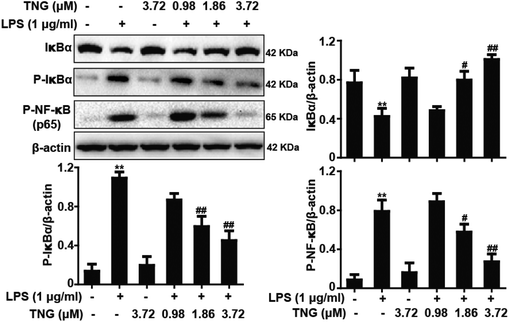 |
| | Fig. 2 Effects of TNG treatment on LPS-induced NF-κB activation in RAW 264.7 cells. RAW 264.7 cells were pretreated with TNG (0.98, 1.86 and 3.72 μM) for 6 h and then were exposed to LPS for 2 h. The protein levels were analyzed by western blot. Quantifications of IκBα, P-IκBα, and P-NF-κB (p65) expression were normalized to that of β-actin. Similar data were obtained in three independent experiments, and one of the three representative experiments is shown. **p < 0.01 vs. the control group; #p < 0.05 and ##p < 0.01 vs. the LPS group. | |
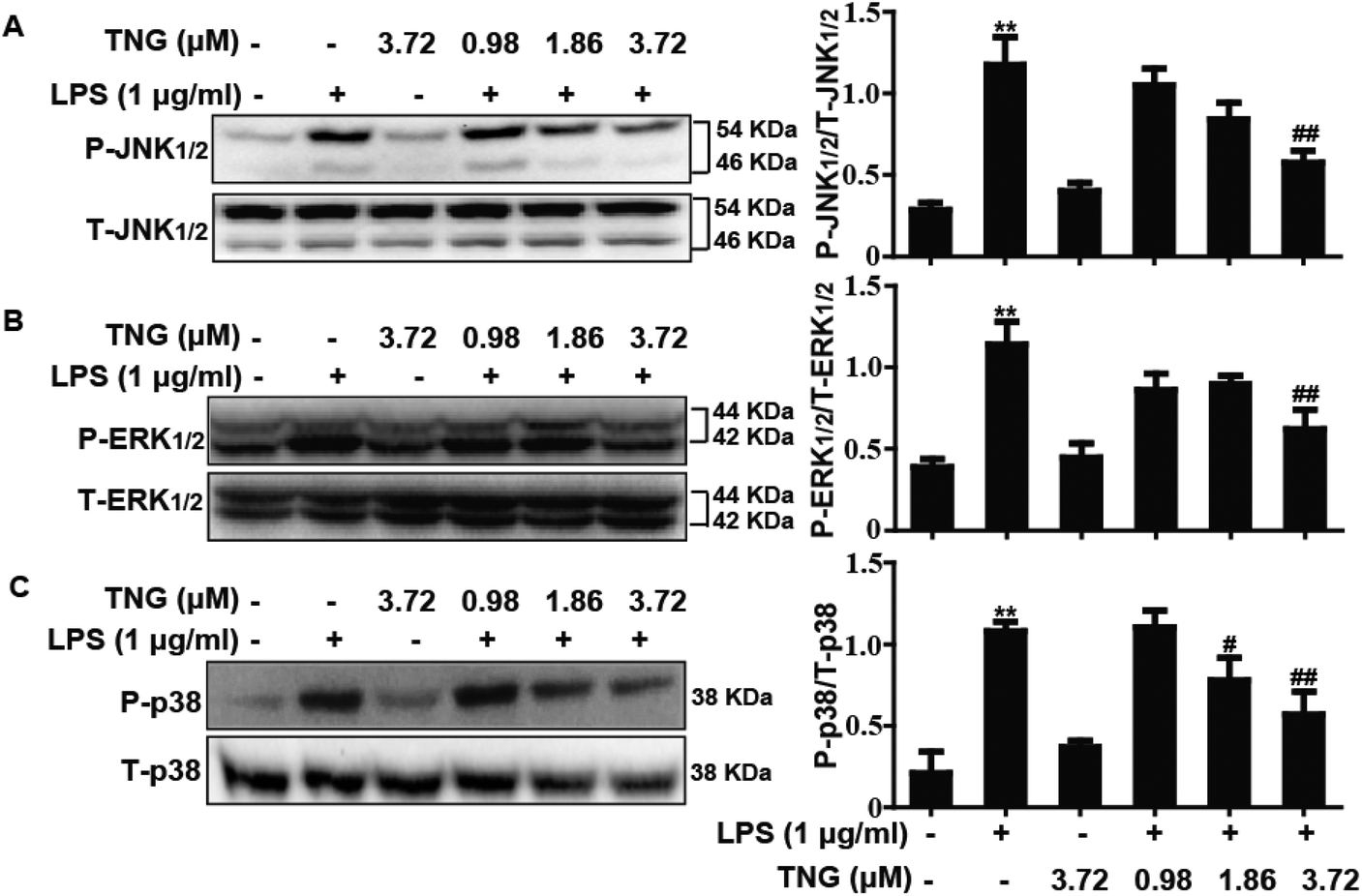
Effects of TNG treatment on LPS-induced MAPK activation in RAW 264.7 cells
MAPK pathways play an important role in the inflammatory process and are associated with the release of pro-inflammatory mediators, such as iNOS and COX-2. Thus, we investigated whether TNG affected the MAPK pathways in LPS-induced RAW 264.7 cells. As shown in Fig. 3, compared with the control group, the phosphorylation of JNK1/2 (340.4%), ERK1/2 (281.1%) and p38 (460.5%) was increased after exposure to LPS, whereas the 3.72 μM TNG pretreatment effectively decreased their expression to 172.3%, 140.3% and 250.0%, respectively. However, the 0.98 μM TNG pretreatment did not exhibit these effects (data not shown), and the 1.86 μM TNG pretreatment only decreased the phosphorylation of p38 to 312.1%.
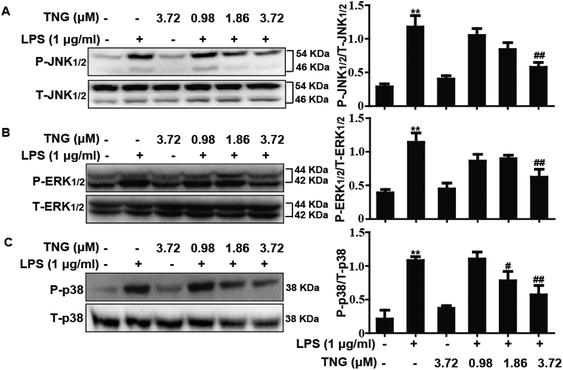 |
| | Fig. 3 Effects of TNG treatment on LPS-induced MAPK activation in RAW 264.7 cells. Cells were pre-treated with TNG (0.98, 1.86 and 3.72 μM) for 6 h and were then exposed to LPS for 1 h. The protein levels were analyzed by western blot. Quantification of the relative expression of (A) P-JNK1/2/T-JNK1/2, (B) P-ERK1/2/T-ERK1/2 and (C) P-p38/T-p38. **p < 0.01 vs. the control group; #p < 0.05 and ##p < 0.01 vs. the LPS group. | |
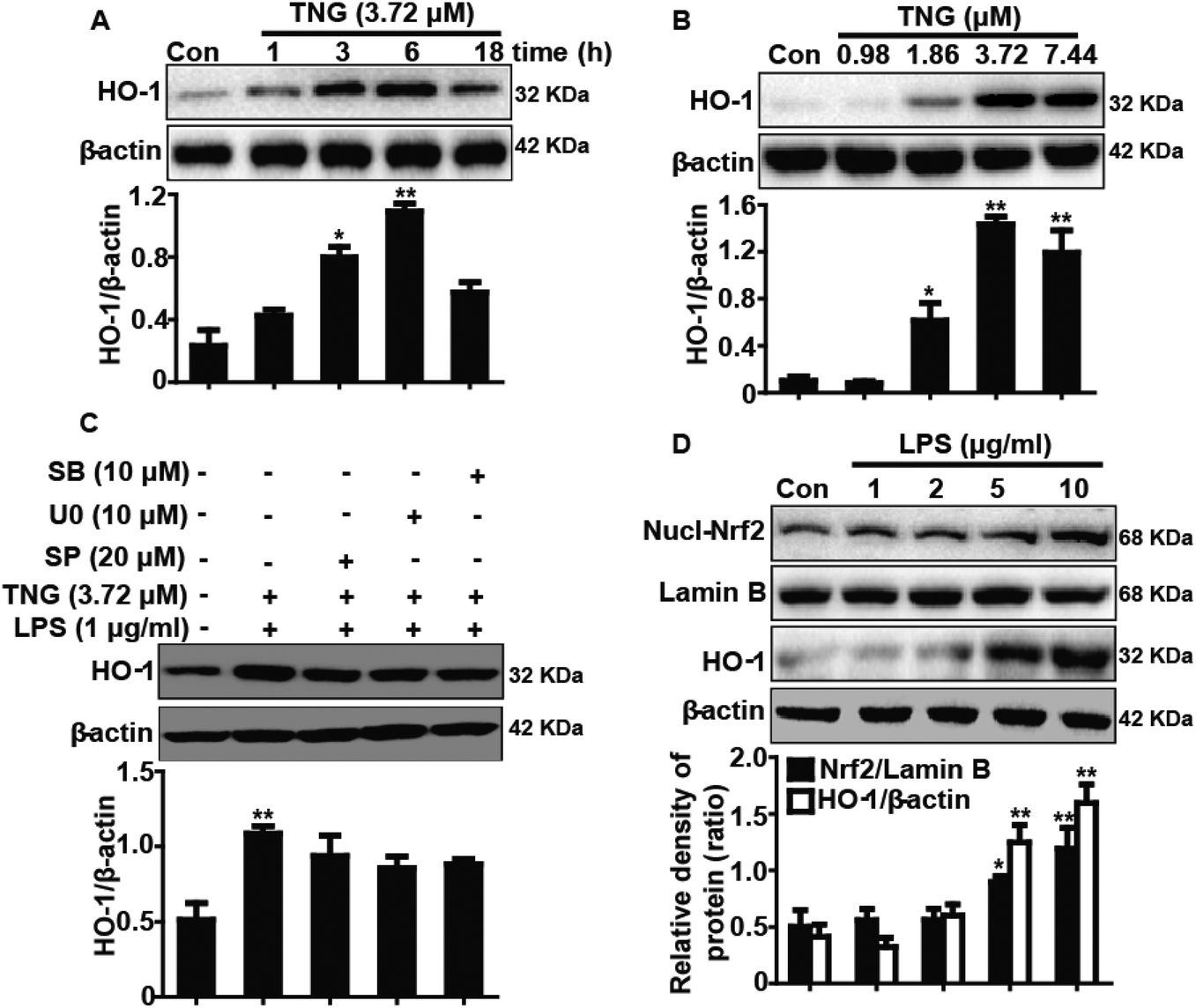
Effects of TNG treatment on HO-1 protein expression in RAW 264.7 cells
Because HO-1 is essential for inhibiting inflammatory responses, we examined whether the TNG-induced HO-1 expression increased the resistance of RAW 264.7 cells to inflammatory damage. To determine the optimal exposure period for enhancing HO-1 protein expression, cells were treated with TNG (3.72 μM) for different periods (Fig. 4A). In addition, to investigate the most effective concentration for increasing HO-1 protein expression, cells were treated with TNG (0.98, 1.86, 3.72 and 7.44 μM) for 6 h (Fig. 4B). Treatment with TNG (3.72 μM) for 6 h exhibited the highest induction of HO-1. Moreover, because the addition of LPS can activate the MAPK pathway and its activation can induce Nrf2/HO-1 up-regulation, to eliminate the probable effect of LPS-activated MAPKs on HO-1 expression by TNG, MAPK inhibitors and LPS were added to cells to investigate their effects. Exposure of cells to U0126 (ERK1/2 inhibitor, 10 μM), SB203580 (p38 inhibitor, 10 μM), or SP600125 (JNK1/2 inhibitor, 20 μM) minimally affected HO-1 expression (Fig. 4C), which eliminated the probable role of MAPKs in HO-1 induction by TNG. Furthermore, because the Nrf2/HO-1 signal pathway could be activated by LPS, we first treated RAW 264.7 cells with 1, 2, 5, or 10 μg ml−1 LPS and found that 1 μg ml−1 of LPS has almost no effect on Nrf2 transcription and HO-1 protein expression (Fig. 4D). This result indicated that cells simultaneously subjected to TNG and LPS (1 μg ml−1) induced Nrf2/HO-1 activation, which only exhibited the role of induction by TNG.
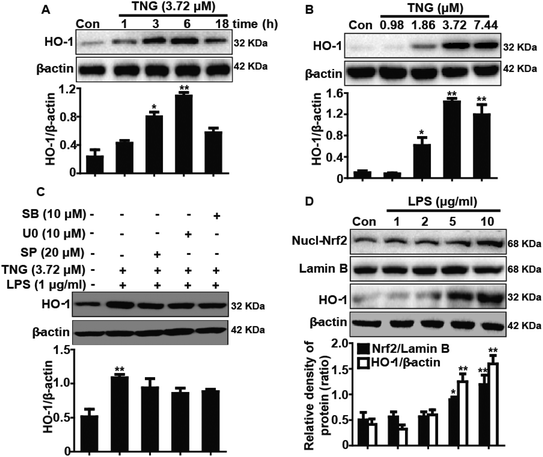 |
| | Fig. 4 Effects of TNG treatment on HO-1 protein expression in RAW 264.7 cells. (A, B) Cells were treated with TNG (3.72 μM) for the indicated time periods, or subjected to various concentrations of TNG (0.98, 1.86, 3.72 and 7.44 μM) for 6 h. HO-1 protein levels were measured by western blot. (C) Cells were pretreated with or without SP600125 (20 μM), U0126 (10 μM), and SB203580 (10 μM) for 18 h, and were then subjected to TNG for 6 h, followed by exposure to LPS for an additional 1 h. HO-1 protein levels were measured by western blot. (D) Cells were subjected to LPS (1, 2, 5 and 10 μg ml−1) for 24 h, and then the total expressed HO-1 protein and the nuclear-expressed Nrf2 protein were analyzed by western blot. β-Actin and Lamin B acted as internal controls, respectively. All data were presented as means ± SEM of three independent experiments. *p < 0.05 and **p < 0.01 vs. the control group. | |
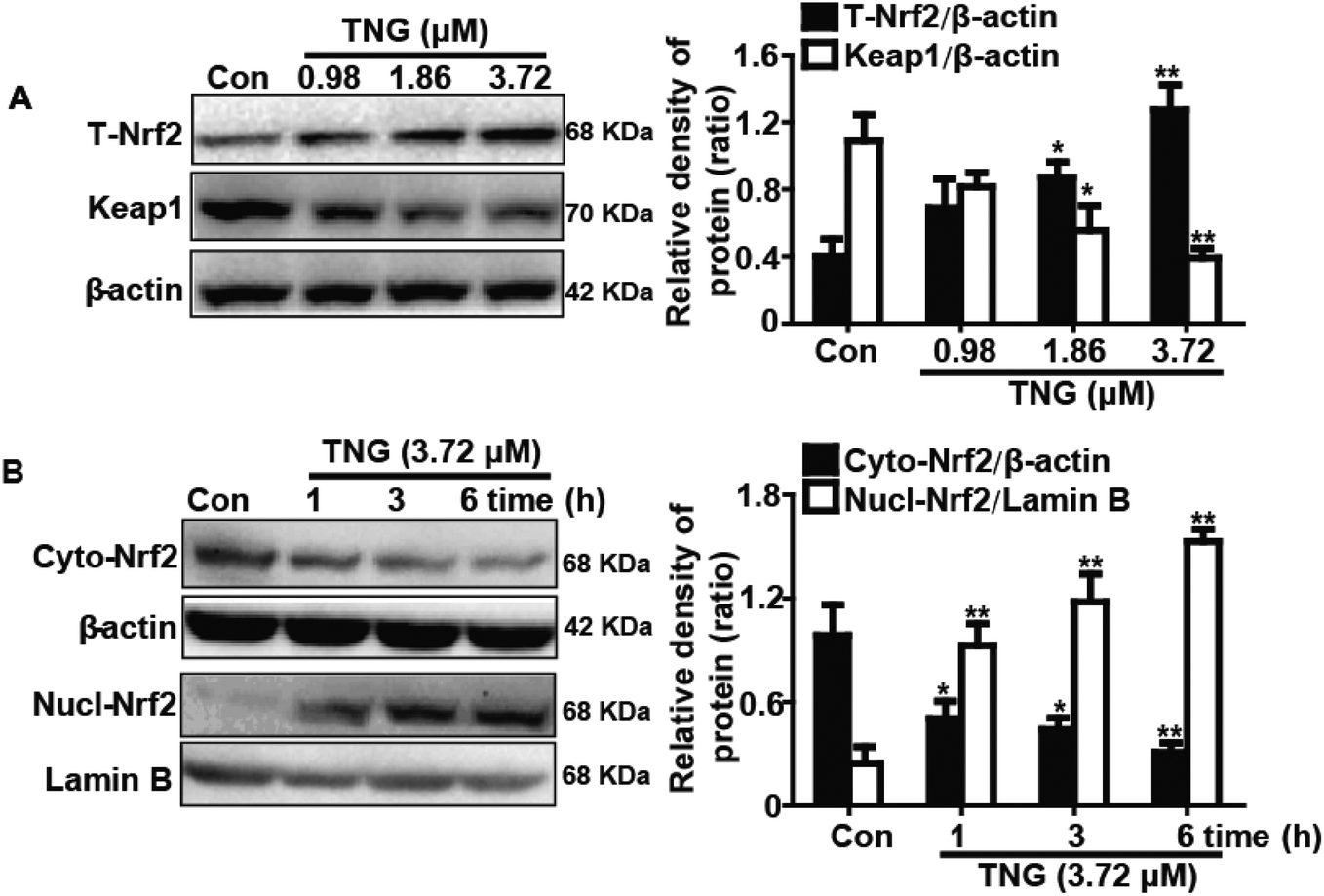
Effects of TNG on Nrf2 protein expression and Keap1 degradation in RAW 264.7 cells
Nrf2 regulates antioxidant responses by transcriptionally activating HO-1 gene expression. In addition, Keap1 negatively regulates Nrf2 by inhibiting Nrf2 activation. Consequently, we examined whether TNG could induce Nrf2 activation and Keap1 degradation in association with HO-1 up-regulation. RAW 264.7 cells were treated with TNG (0.98, 1.86 or 3.72 μM) for 6 h, and then total protein was extracted from the cells for western blot analysis. TNG (1.86 and 3.72 μM) significantly increased the total Nrf2 (201.4% and 310.0%, respectively) and Keap1 degradation (53.4% and 60.6%, respectively) when compared with untreated control cells (Fig. 5A). Furthermore, we determined that 3.72 μM TNG resulted in a significant decrease (50.1%, 55.6% and 66.7%, respectively) in the cytoplasmic levels and a concomitant increase (246.6%, 354.8% and 507.4%, respectively) in the nuclear levels of Nrf2 in a time-dependent manner (Fig. 5B).
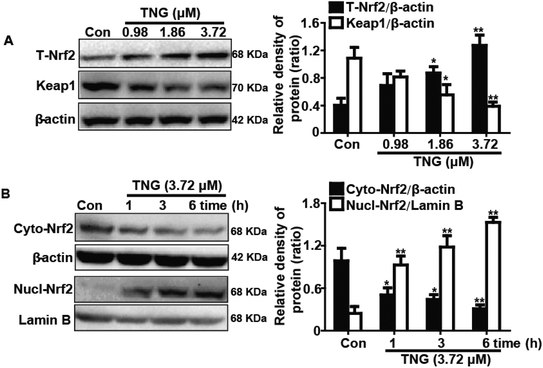 |
| | Fig. 5 Effects of TNG on Nrf2 protein expression and Keap1 degradation in RAW 264.7 cells. (A) Cells were subjected to various concentrations of TNG (0.98, 1.86 and 3.72 μM) for 6 h, and the total expressed Nrf2 and Keap1 protein were analyzed by western blot. (B) Cells were treated with TNG (3.72 μM) for the indicated time periods, and the nuclear and cytoplasmic levels of Nrf2 were examined by western blot analysis. β-Actin and Lamin B acted as internal controls, respectively. All data were presented as means ± SEM of three independent experiments. *p < 0.05 and **p < 0.01 vs. the control group. | |
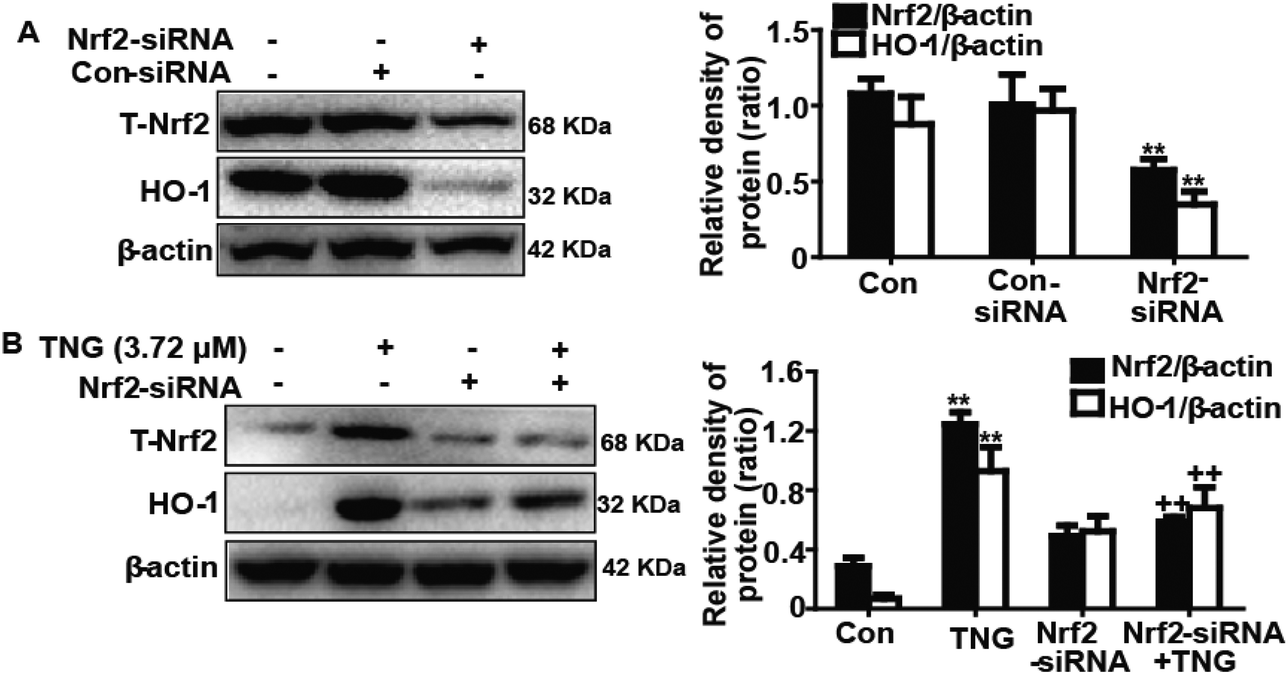
Effects of Nrf2-siRNA transfection on TNG-induced HO-1 protein expression in RAW 264.7 cells
To further investigate the role of Nrf2 in TNG-induced HO-1 protein expression, we used siRNA to knockdown Nrf2 in the RAW 264.7 cells. The cells were subjected to a transient transfection with control or Nrf2 siRNA. Western blot analysis indicated that Nrf2 siRNA reduced the total Nrf2 and HO-1 protein expression compared with the negative control (Fig. 6A). Moreover, we investigated whether Nrf2 siRNA transfection inhibited TNG-induced HO-1 protein expression. Our experiments found that the TNG-enhanced HO-1 protein expression was markedly reduced to 24.2% in Nrf2 siRNA-transfected cells when compared with nonspecific control-siRNA (Fig. 6B). The results of this assay provide further evidence that the transcriptional activator Nrf2 can modulate HO-1 gene expression.
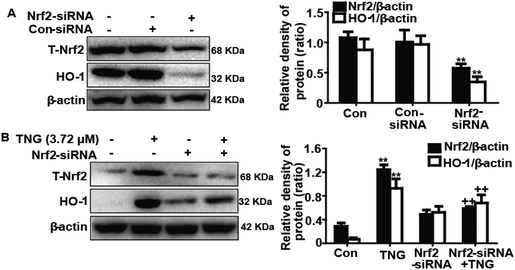 |
| | Fig. 6 Effects of Nrf2-siRNA transfection on TNG-induced HO-1 protein expression in RAW 264.7 cells. (A) Cells were subjected to a transient transfection with Nrf2-negative control siRNA or Nrf2-siRNA. (B) Nrf2 mediates TNG-enhanced HO-1 protein expression. Nrf2-siRNA or Nrf2-negative control siRNA were transfected into cells for 18 h followed by treatment with TNG (3.72 μM) for 6 h, and proteins were analyzed by western blot. All data were presented as means ± SEM of three independent experiments. **p < 0.01 vs. the control group; ++p < 0.01 vs. the TNG group. | |
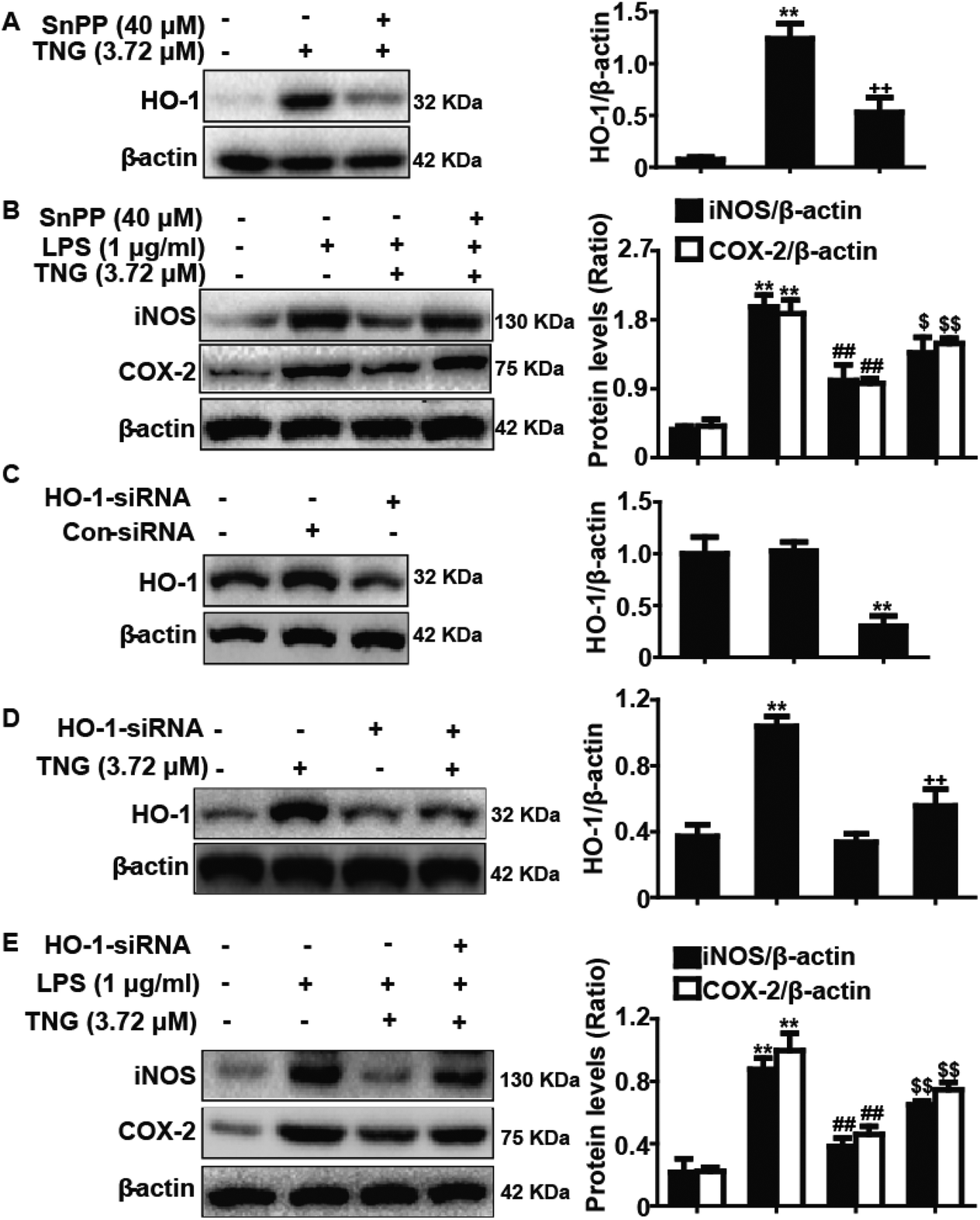
Effects of SnPP and HO-1-siRNA on TNG-inhibited iNOS and COX-2 protein expression in RAW 264.7 cells
Increasing evidence suggests that HO-1 plays a crucial role in inhibiting inflammatory responses. Accordingly, we determined whether the anti-inflammatory activity of TNG was related to increased HO-1 expression using HO-1-siRNA and SnPP. SnPP, tin protoporphyrin IX, which is a competitive inhibitor of HO-1 activity, and its mechanism of action have been previously reported.20 In our studies, RAW 264.7 cells were exposed to HO-1-siRNA or SnPP (40 μM) for 18 h and were then treated with TNG (3.72 μM) for 6 h, and HO-1 protein expression was analyzed by western blot. The results showed that the TNG-induced HO-1 protein expression was reduced by HO-1-siRNA and SnPP treatment (Fig. 7A, C and D). In addition, cells were pretreated with or without HO-1-siRNA or SnPP for 1 h, and were then subjected to LPS (1 μg ml−1) for 18 h, followed by exposure to TNG (3.72 μM) for an additional 6 h. The inhibitory effects of TNG on LPS-induced iNOS and COX-2 protein expression were partially reversed by HO-1-siRNA and SnPP (Fig. 7B and E).
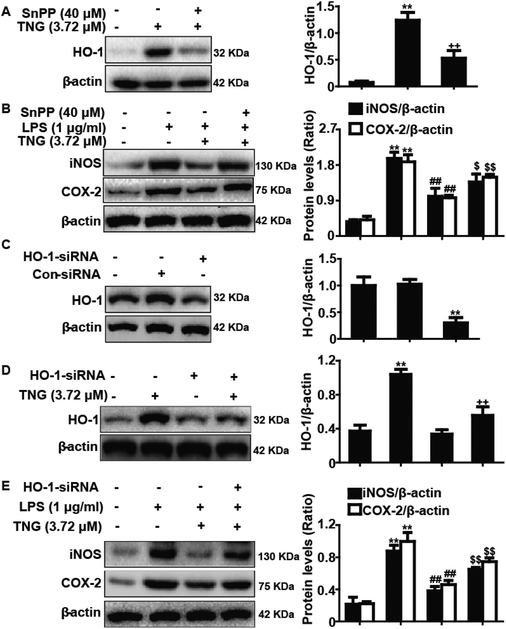 |
| | Fig. 7 Effects of SnPP and HO-1-siRNA on TNG-inhibited iNOS and COX-2 protein expression in RAW 264.7 cells. (A) Cells were exposed to SnPP (40 μM) for 18 h and were then treated with TNG (3.72 μM) for 6 h, and the HO-1 protein expression was analyzed by western blot. (B) Cells were exposed to SnPP (40 μM) for 1 h and were then subjected to LPS (1 μg ml−1) for 18 h, followed by treatment with TNG (3.72 μM) for 6 h. The iNOS and COX-2 protein expression were analyzed by western blot. (C) Control siRNA or HO-1-siRNA was transfected into cells for 24 h and was collected. HO-1 protein expression was analyzed by western blot. (D) Cells were subjected to a transient transfection with control siRNA or HO-1-siRNA for 18 h, followed by treatment with TNG (3.72 μM) for 6 h. HO-1 protein expression was analyzed by western blot. (E) Control siRNA or HO-1-siRNA was transfected into cells for 1 h, and the cells were then subjected to LPS (1 μg ml−1) for 18 h, followed by treatment with TNG (3.72 μM) for 6 h. The iNOS and COX-2 protein expression was analyzed by western blot. All data were presented as means ± SEM of three independent experiments. **p < 0.01 vs. the control group; ##p < 0.01 vs. the LPS group; ++p < 0.01 vs. the TNG group; $p < 0.05 and $$p < 0.01 vs. the LPS + TNG group. | |
Discussion
Inflammation, a complex biological process, is closely involved in the release of inflammatory mediators, including NO and PGE2, which are synthesized by iNOS and COX-2, respectively.7 Reports have suggested that iNOS and COX-2 cause inflammation by producing iNOS-derived NO and COX-2-derived PGE2, which result in a variety of pathological diseases that induce tissue damage.21 In addition, macrophages, which are an important type of immune cells, produce massive cytokines and inflammatory mediators when infected with pathogens, such as LPS.22 Therefore, the inhibition of iNOS and COX-2 expression in the macrophages may be an essential therapeutic target for developing anti-inflammatory agents. In our studies, we demonstrated that treatment with TNG significantly decreased COX-2 and iNOS mRNA and protein expression and reduced NO and PGE2 production in LPS-induced RAW cells.
NF-κB, an essential regulatory transcription factor, is crucial for modulating iNOS and COX-2 expression.23 Activation of NF-κB results in IκB phosphorylation and subsequent degradation by kinase (IKK), which are capable of accelerating NF-κB dimers to enter the nucleus and promote the expression of specific target genes, such as iNOS and COX-2.24 Hence, in the current study, the amounts of phospho-IκBα and phospho-p65 were measured by western blot, which showed that TNG markedly blocked the phosphorylation and subsequent degradation of IκBα and suppressed NF-κB (p65) phosphorylation. Moreover, abundant reports have suggested that MAPKs, a group of signaling molecules that include p38, ERK1/2 and JNK1/2, play a pivotal role in pro-inflammatory responses.25,26 JNK1/2, ERK1/2 and p38 phosphorylation are essential for the regulation of iNOS and COX-2 expression.27 Our results suggested that TNG significantly decreased JNK1/2, ERK1/2 and p38 phosphorylation in LPS-induced RAW 264.7 cells.
Recent reports showed that HO-1 plays an important role in the regulation of the inflammatory responses by inhibiting the production of pro-inflammatory cytokines and the expression of iNOS and COX-2.28 Our results demonstrated that different concentrations and exposure periods of TNG treatment dramatically enhanced HO-1 induction in RAW 264.7 cells. Additionally, several signaling molecules, including MAPKs (JNK1/2, ERK1/2 and p38), are involved in regulating HO-1 gene expression.29 Because the addition of LPS can activate MAPK pathways, which can induce Nrf2/HO-1 up-regulation, we investigated the probable effect of LPS-activated MAPKs on HO-1 expression by TNG. In this study, treatment with MAPK inhibitors and LPS had no significant effect on the level of HO-1 expression, which eliminated the probable effect of MAPKs on HO-1 expression by TNG. Furthermore, recent reports indicated that a higher dose of LPS exposure also activates HO-1 and Nrf2 expression.14 Consequently, Nrf2 and HO-1 expression induced by LPS was observed in our experiment. The results showed that LPS did not lead to an increase in the expression of Nrf2 and HO-1, which indicated that Nrf2 and HO-1 activation only were induced by TNG.
HO-1 expression is regulated by Nrf2, which is bound to Keap1, a repressor of Nrf2 in the cytoplasm.30 Moreover, Keap1 inactivation is suggested to be a mechanism for the dissociation of the Nrf2-Keap1 complex, which leads to nuclear translocation of Nrf2 and induction of HO-1 expression.31 Our results indicated that TNG enhanced the nuclear translocation of Nrf2 and reduced Keap1 protein expression, thereby stimulating the transcriptional activity of Nrf2. We also confirmed the role of Nrf2 as a key regulator in TNG-induced HO-1 expression using Nrf2-siRNA knock down to silence the Nrf2 expression. Together, these results suggested that TNG-induced HO-1 and Nrf2 expression is not associated with MAPK pathway activation but is associated with Keap1 degradation. Furthermore, recent reports suggested that the Nrf2/HO-1 signaling pathway plays a critical role in attenuating inflammation-associated pathogenesis by inhibiting iNOS and COX-2 expression.32 Because TNG treatment also induced Nrf2/HO-1 activation, we hypothesized that TNG inhibited iNOS and COX-2 expression, which is associated with the TNG-mediated Nrf2/HO-1 signaling pathway. Our observations suggested that TNG had an inhibitory effect on iNOS and COX-2 expression, which was partially reversed by SnPP (40 μM) and HO-1-siRNA.
Conclusion
In conclusion, TNG treatment effectively inhibited LPS-induced MAPK and NF-κB activation and up-regulated Nrf2/HO-1 expression, which may be closely associated with the inhibition of LPS-induced COX-2 and iNOS expression, thereby preventing the inflammatory response and providing a potential novel strategy for the treatment of inflammatory diseases.
Declaration of interest
The authors report no conflict of interest. The authors alone are responsible for the content of this manuscript.
Acknowledgements
This work was supported by a grant from the Natural Science Foundation of Jilin (no. 20150520050JH).
Notes and references
- C. N. Serhan, N. Chiang and T. E. Van Dyke, Nat. Rev. Immunol., 2008, 8, 349–361 CrossRef CAS PubMed.
- S. Reuter, S. C. Gupta, M. M. Chaturvedi and B. B. Aggarwal, Free Radicals Biol. Med., 2010, 49, 1603–1616 CrossRef CAS PubMed.
- P. R. Mayeux, J. Toxicol. Environ. Health, 1997, 51, 415–435 CrossRef CAS PubMed.
- T. Collins and M. I. Cybulsky, J. Clin. Invest., 2001, 107, 255–264 CrossRef CAS PubMed.
- T. D. Gilmore, Oncogene, 2006, 25, 6680–6684 CrossRef CAS PubMed.
- S. Becker, S. Mundandhara, R. B. Devlin and M. Madden, Toxicol. Appl. Pharmacol., 2005, 207, 269–275 CrossRef PubMed.
- J. B. Kim, A. R. Han, E. Y. Park, J. Y. Kim, W. Cho, J. Lee, E. K. Seo and K. T. Lee, Biol. Pharm. Bull., 2007, 30, 2345–2351 CAS.
- H. G. Choi, D. S. Lee, B. Li, Y. H. Choi, S. H. Lee and Y. C. Kim, Int. Immunopharmacol., 2012, 13, 271–279 CrossRef CAS PubMed.
- C. S. Williams, M. Mann and R. N. DuBois, Oncogene, 1999, 18, 7908–7916 CrossRef CAS PubMed.
- L. E. Otterbein, F. H. Bach, J. Alam, M. Soares, H. Tao Lu, M. Wysk, R. J. Davis, R. A. Flavell and A. M. Choi, Nat. Med., 2000, 6, 422–428 CrossRef CAS PubMed.
- C. D. Ferris, S. R. Jaffrey, A. Sawa, M. Takahashi, S. D. Brady, R. K. Barrow, S. A. Tysoe, H. Wolosker, D. E. Baranano, S. Dore, K. D. Poss and S. H. Snyder, Nat. Cell Biol., 1999, 1, 152–157 CrossRef CAS PubMed.
- G. Y. Suh, Y. Jin, A. K. Yi, X. M. Wang and A. M. Choi, Am. J. Respir. Cell Mol. Biol., 2006, 35, 220–226 CrossRef CAS PubMed.
- L. Baird and A. T. Dinkova-Kostova, Arch. Toxicol., 2011, 85, 241–272 CrossRef CAS PubMed.
- M. Ye, Q. Wang, W. Zhang, Z. Li, Y. Wang and R. Hu, Biochem. Cell Biol., 2014, 92, 337–348 CrossRef CAS PubMed.
- T. W. Kensler, N. Wakabayashi and S. Biswal, Annu. Rev. Pharmacol. Toxicol., 2007, 47, 89–116 CrossRef CAS PubMed.
- C. H. Park, S. H. Choi, J. W. Koo, J. H. Seo, H. S. Kim, S. J. Jeong and Y. H. Suh, J. Neurosci. Res., 2002, 70, 484–492 CrossRef CAS PubMed.
- H. L. Yuan, B. Li, J. Xu, Y. Wang, Y. He, Y. Zheng and X. M. Wang, CNS Neurosci. Ther, 2012, 18, 584–590 CrossRef CAS PubMed.
- H. Zhang, T. Han, L. Zhang, C. H. Yu, D. G. Wan, K. Rahman, L. P. Qin and C. Peng, Phytomedicine, 2008, 15, 587–594 CrossRef CAS PubMed.
- H. Lv, C. Zhu, Y. Liao, Y. Gao, G. Lu, W. Zhong, Y. Zheng, W. Chen and X. Ci, Respir. Physiol. Neurobiol., 2015, 216, 43–51 CrossRef CAS PubMed.
- G. S. Drummond and A. Kappas, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 6466–6470 CrossRef CAS.
- A. Murakami and H. Ohigashi, Int. J. Cancer, 2007, 121, 2357–2363 CrossRef CAS PubMed.
- S. Moncada, J. R. Soc. Med., 1999, 92, 164–169 CAS.
- T. Lawrence, D. A. Willoughby and D. W. Gilroy, Nat. Rev. Immunol., 2002, 2, 787–795 CrossRef CAS PubMed.
- H. L. Pahl, Oncogene, 1999, 18, 6853–6866 CrossRef CAS PubMed.
- E. Cario, I. M. Rosenberg, S. L. Brandwein, P. L. Beck, H. C. Reinecker and D. K. Podolsky, J. Immunol., 2000, 164, 966–972 CrossRef CAS.
- Y. J. Surh, K. S. Chun, H. H. Cha, S. S. Han, Y. S. Keum, K. K. Park and S. S. Lee, Mutat. Res., 2001, 480–481, 243–268 CrossRef CAS.
- M. Guha and N. Mackman, Cell Signalling, 2001, 13, 85–94 CrossRef CAS.
- B. Zhang, L. Yan, P. Zhou, Z. Dong, S. Feng, K. Liu and Z. Gong, Int. Immunopharmacol., 2013, 15, 289–295 CrossRef CAS PubMed.
- P. Chakraborty, G. Saraswat and S. N. Kabir, Toxicol. Appl. Pharmacol., 2014, 277, 95–107 CrossRef CAS PubMed.
- Y. J. Surh and H. K. Na, Genes Nutr., 2008, 2, 313–317 CrossRef CAS PubMed.
- J. S. Lee and Y. J. Surh, Cancer Lett., 2005, 224, 171–184 CrossRef CAS PubMed.
- J. M. Lee and J. A. Johnson, J. Biochem. Mol. Biol., 2004, 37, 139–143 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.