
Hesperetin and its sulfate and glucuronide metabolites inhibit TNF-α induced human aortic endothelial cell migration and decrease plasminogen activator inhibitor-1 (PAI-1) levels†
Juan Antonio
Giménez-Bastida
,
Antonio
González-Sarrías
*,
Fernando
Vallejo
,
Juan Carlos
Espín
and
Francisco A.
Tomás-Barberán
Research Group on Quality, Safety and Bioactivity of Plant Foods, Dept. Food Science and Technology, CEBAS-CSIC, P.O. Box 164, 30100 Campus de Espinardo, Murcia, Spain. E-mail: agsarrias@cebas.csic.es; Fax: +34 968 396213; Tel: +34 968 396249
First published on 29th September 2015
Abstract
Epidemiological, clinical and preclinical studies have reported the protection offered by citrus consumption, mainly orange, against cardiovascular diseases, which is primarily mediated by the antiatherogenic and vasculoprotective effects of the flavanone hesperetin-7-O-rutinoside (hesperidin). However, flavanone aglycones or glycosides are not present in the bloodstream but their derived phase-II metabolites could be the actual bioactive molecules. To date, only a few studies have explored the effects of circulating hesperetin-derived metabolites (glucuronides and sulfates) on endothelial cells. Herein, we describe for the first time the effects of hesperetin 3′-O-glucuronide, hesperetin 7-O-glucuronide, hesperetin 3′-O-sulfate, hesperetin 7-O-sulfate and hesperetin on human aortic endothelial cell (HAEC) migration upon pro-inflammatory stimuli as an essential step to angiogenesis. Hesperetin and its derived metabolites, at physiologically relevant concentrations (1–10 μM), significantly attenuated cell migration in the presence of the pro-inflammatory cytokine TNF-α (50 ng mL−1), which was accompanied and perhaps mediated by a significant decrease in the levels of the thrombogenic plasminogen activator inhibitor-1 (PAI-1). However, hesperetin metabolites did not counteract the TNF-α-induced production of pro-inflammatory interleukin-6 (IL-6) and IL-8. We also study here for the first time, the metabolism of hesperetin and its derived metabolites by HAEC with and without a pro-inflammatory stimulus. All these results reinforce the concept according to which circulating phase-II hesperetin metabolites are critical molecules contributing to the cardioprotective effects upon consumption of citrus fruits such as orange.
1. Introduction
Atherosclerosis is the primary cause for cardiovascular diseases (CVDs) in western countries.1 Atherosclerosis is a chronic inflammatory process characterized by lipid accumulation, monocyte infiltration and foam cell formation which lead to the development of the atherosclerotic plaque. Growth of the plaques generates hypoxia and promotes neovascularization.2 Formation of new capillaries from endothelial cells is an important mechanism involved in atherosclerosis. Angiogenesis is a complex process that typically involves endothelial cell proliferation, migration and alignment to form tubular structures.3,4 Many different molecules are involved in the regulation of endothelial cell migration and proliferation. TNF-α is a pro-inflammatory cytokine that regulates endothelial cell migration as well as promotes the secretion of multiple interleukins, including IL-6 and IL-8, and other molecules involved in the regulation of cell migration such as PAI-1.4–6 Inhibition of TNF-α-induced arterial inflammation and the subsequent endothelial proliferation and migration by natural compounds has a high value in the treatment of atherosclerosis.7An important number of epidemiological studies have reported a positive effect of flavonoid-containing food consumption on CVDs.8–11 Citrus fruits have acquired an increasing interest due to their beneficial effects. It has been reported that the consumption of orange juice and orange, grapefruit juice and blackcurrant juice has been associated with a protective effect on the cardiovascular system.12–17 This beneficial effect has been partially associated with the flavanone content. The most abundant component in sweet orange juices, regardless of the variety, is by far hesperidin (20–59 mg per 100 mL), followed by narirutin (1.6–8.4 mg per 100 mL). These are all flavanone-O-glycosides, which account for most of the flavonoid content in orange juice.18 In this regard, few preclinical studies (endothelial cell and animal models) have shown that citrus flavanone extracts as well as hesperidin, naringin, and naringenin, among others can exert antiatherogenic and vasculoprotective effects by modulating the expression of molecules related to inflammation, preventing the formation of foam cells, reducing the area of atherosclerotic plaques, improving the lipid profile, inhibiting the adhesion of monocytes to the endothelium and modulating cell migration.19–25 However, most of these studies assayed higher concentrations than those reported in vivo and endothelial cells are not exposed to aglycones or glycosides because they are not present in the bloodstream. Furthermore, the actual compounds responsible for the effects observed are not yet fully known.
Hesperidin metabolism mainly occurs in the large intestine where hesperetin is released after rhamnosidase catalysis by the gut microbiota. This involves the delayed absorption and conjugation of hesperetin, with a time to peak plasma concentration of about 4–7 h in contrast to flavonoid glucosides such as onion quercetin glucosides that are absorbed predominantly in the small intestine (a time to peak plasma concentration of about 1 h).26–29 Upon consumption of orange, the main circulating metabolites are hesperetin sulfates (3′- and 7-O-sulfate), and hesperetin glucuronides (3′- and 7-O-glucuronide). In addition, low hesperetin sulfoglucuronide concentration levels can be also found.30,31
To the best of our knowledge, hitherto only a few studies have assayed the metabolites of hesperetin (glucuronides and sulfates) using endothelial cells. These studies reported that naringenin and hesperetin metabolites (glucuronides and sulfates) were effective, similar to their aglycones, in ameliorating monocyte adhesion to endothelial cells and modulating the expression of proteins associated with inflammation and suppressing induced inflammation.19,32 However, the effects of flavanone metabolites on endothelial cell migration and proliferation, and the important steps of atherosclerosis and angiogenesis, have not been reported so far.
Taking together all of the above, the aim of the present study was to determine the effects of hesperetin and its in vivo circulating metabolites, i.e. hesperetin 3′-O-glucuronide, hesperetin 7-O-glucuronide, hesperetin 3′-O-sulfate and hesperetin 7-O-sulfate on the migration and proliferation of human aortic endothelial cells (HAECs) upon pro-inflammatory (TNF-α) signals. The metabolism of hesperetin and its derived metabolites by HAECs, as well as the role of key actors in angiogenesis and atherosclerosis development (PAI-1, IL-6 and IL-8) will be also explored.
2. Materials and methods
2.1. Materials and reagents
Hesperetin and its sulfate and glucuronide metabolites (Fig. 1) were chemically synthesized by Villapharma SL (Parque Tecnológico de Fuente Álamo, Murcia, Spain). Human recombinant TNF-α and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) were purchased from Sigma Aldrich (St Louis, USA). Phosphate Buffered Saline (PBS) was from Fisher Scientific (USA). Dimethylsulfoxide (DMSO), diethyl-ether and HPLC reagents, formic acid and acetonitrile (ACN), were obtained from Panreac (Barcelona, Spain). Ultrapure Millipore water was used throughout the study. | ||
| Fig. 1 Chemical structures of hesperetin, hesperetin 3′-O-glucuronide, hesperetin 7-O-glucuronide, hesperetin 3′-O-sulfate and hesperetin 7-O-sulfate. | ||
2.2. Cell lines and cell culture conditions
Human aortic endothelial cells (HAECs) were cultured in endothelial cell growth medium (ECGM) and sub-cultured using the recommended subculture reagent kit. Cells and media were obtained from the European Collection of Cell Cultures (Salisbury, UK). Cells were routinely seeded at a density of 104 cells per cm2 in 25 cm2 flasks and maintained at 37 °C under a humidified atmosphere at 5% of CO2. The culture medium was changed every other day until the cells reached >80% confluence. Passages between 2 and 6 were used for all the experiments.2.3. MTT assay
HAECs were seeded into 96-well plates at a density of 3200 cells per well and were maintained under appropriate culture conditions until reaching 60–70% confluence. The medium was then replaced and the cells were exposed for 24 h to an experimental medium containing hesperetin aglycone or its metabolites (glucuronides and sulfates) at 10 μM, both in the presence or absence of TNF-α (50 ng mL−1). After treatments, a mixture of 50 μL of MTT (5 mg mL−1) and 200 μL of fresh medium was added to PBS-washed cells, and then incubated at 37 °C for 4 hours. Next, the MTT reaction agent was removed and 100 μL of DMSO was added to the cell culture plates and incubated at 37 °C for 15 minutes to solubilize the formazan. The optical density (OD) of the coloured solution was measured at 570 nm using a multimode microplate reader (FLUOstar Omega, BMG Labtech, Ortenberg, Germany). The final results of the treated cells are expressed as percentages of those values obtained for control cells (containing 0.5% DMSO). Data are presented as the mean ± standard deviation (SD) of three separate experiments (n = 12 wells per experiment).2.4. Measurement of cytokines by enzyme-linked immunosorbent assay (ELISA)
Confluent HAECs were cultured at an initial density of 104 cells per cm2 (96-well plates) and were grown up to confluence. The culture medium was changed every other day. After reaching confluence, the cells were treated with TNF-α (50 ng mL−1) and each of the tested metabolites (aglycones, glucuronides and sulfates) for 24 h. After treatment, cell culture supernatants were collected and frozen at −80 °C until further analysis. IL-8, IL-6, and PAI-1 were analysed using commercially available ELISA kits from Peprotech (Rocky Hill, NJ, USA). The minimum detection level was 8 pg mL−1 for IL-8 and 23 pg mL−1 for IL-6 and PAI-1. The concentration of each molecule was related to the intensity of the colour measured by using a microplate reader (Infinite M200, Tecan, Grodig, Austria). This assay was performed four times (n = 6 wells per experiment) and the values are presented as the mean ± SD.2.5. Migration assay
HAECs were cultured in 24-well plates at an initial density of 104 cells per cm2. After reaching confluence, an empty area was created with a sterile pipette tip. Media, dislodged cells and debris were aspirated, and the cells were washed with Hank's buffered salt solution (twice) and exposed to the culture medium containing each of the tested compounds (10, 1, 0.1 μM) in the presence or absence of the pro-inflammatory cytokine TNF-α (50 ng mL−1). Control cells were exposed to an equivalent amount of DMSO (0.5% v/v).The scratched area (μm2) generated was considered as time 0. Different photos (2–3) were captured along the scraped area on each well (2 wells per treatment) at the beginning (time 0), and after 12 and 16 hours using a contrast inverted microscope and a charge-coupled device (CCD) camera attached to the microscope. The ability of the cells to migrate in the presence of the tested compounds was calculated by measuring the uncovered surface (scratched area in μm2) in each picture at time 0, 12 and 16 hours. The data are expressed as the percentage of the surface covered by the cells at 12 and 16 hours compared to the surface measured at time 0 (100% of uncovered surface) as described in ESI Fig. 1.† Data are presented as the mean ± SD representative of 3 independent experiments. Each experiment was performed in duplicate (n = 2 wells per treatment).
2.6. Determination of metabolites in cell media of HAECs
To determine if HAECs were able to metabolize hesperetin and its derived metabolites, cell culture supernatants were collected at the end of cell treatment and kept at −80 °C. ACN (100 μL) was added per 100 μL of culture media, vortexed and centrifuged at 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 435g for 10 min. The supernatant was then concentrated in a Speedvac® concentrator (Savant SPD 121P) and the residue was re-dissolved in 50 μL of MeOH and filtered (0.45 μm) before analysis by UPLC-Triple Quadrupole mass spectrometry (UPLC-QqQ-MS) using the same conditions described below.
435g for 10 min. The supernatant was then concentrated in a Speedvac® concentrator (Savant SPD 121P) and the residue was re-dissolved in 50 μL of MeOH and filtered (0.45 μm) before analysis by UPLC-Triple Quadrupole mass spectrometry (UPLC-QqQ-MS) using the same conditions described below.
2.7. Analysis of hesperetin metabolites by UPLC-QqQ-MS
Determination of the phase II metabolites of hesperetin and its derived metabolites in cell media was identified and quantified by using a UPLC 1290 Infinity Series (Agilent Technologies, Waldbronn, Germany) equipped with a triple quadrupole mass spectrometer (6460 Jet Stream Series, Agilent Technologies). A Poroshell 120 C18 column (100 × 3.0 mm i.d., 2.7 μm) (Agilent, Palo Alto, CA) was used at room temperature, and the injected volume was 3 μL. Gradient elution was carried out using water/formic acid (99:1, v/v) and acetonitrile at a constant flow rate of 0.5 mL min−1. The gradient used was [t (min), % acetonitrile v:v]: (0, 1), (10, 40), (12, 90) and (15, 1). The optimum mass spectrometry parameters for detection of hesperetin aglycone, hesperetin 7- and 3′-O-glucuronide and hesperetin 7- and 3′-O-sulfate (50 μM) were optimized, connecting the column directly to the ‘Jet Stream’.
Source parameters: capillary voltage −3500 V, charging potential −500 V, nebulizer pressure 40 (psi), auxiliary gas heated to 275 °C and introduced at a flow rate of pure nitrogen 9 L min−1. The multiple reaction monitoring (MRM) method monitored five transitions for each analysis: hesperetin, m/z 301 → 164; hesperetin glucuronide, m/z 477 → 301; hesperetin diglucuronide, m/z 653 → (477) → 301; hesperetin sulfoglucuronide, m/z 557 → (477) → 301; hesperetin sulfate, m/z 381 → 301 with a dwell time for each transition of 8 ms. Concentrations of hesperetin diglucuronide and hesperetin sulfoglucuronide metabolites were tentatively estimated by using synthesized hesperetin glucuronide calibration curves (20, 10, 5, 2.5, 1 μM).
Limits of quantification (LOQs) were 80, 50/80 and 40/50 nM for hesperetin, hesperetin 7- and 3′-O-glucuronides and hesperetin 7- and 3′-O-sulfates, respectively. Limits of detection (LODs) were 30, 20/30 and 15/20 nM respectively. The intraday repeatability of the UPLC-QqQ method was assessed from 10 consecutive chromatographic runs using a standard solution with 2.5 μM of every standard in MeOH:0.1% (v/v) formic acid. The inter-day repeatability of the method was assessed by analysing the same standard solution for 2 consecutive days. The relative standard deviation (RSD) for the peak area was in the range of 0.5–4.7% in the intraday test and 1.3–3.5% in the case of the inter-day test.
2.8. Statistical analyses
The results of all the experiments are presented as mean values ± SD. For variance analysis, Levene's test was performed. For normally distributed data, one-way ANOVA followed by Dunnett's post-hoc test was used. The results with p values <0.05 were considered statistically significant.3. Results
3.1. Effect of hesperetin and its metabolites on cell proliferation of HAECs in the presence or absence of TNF-α (50 ng mL−1)
Hesperetin and its conjugated metabolites at 10 μM, alone or in combination with TNF-α (50 ng mL−1), did not significantly affect cell growth or cause cytotoxicity as measured by the MTT assay at 24 h (Fig. 2). Once this was confirmed, concentrations of 10, 1 and 0.1 μM were assayed in subsequent experiments.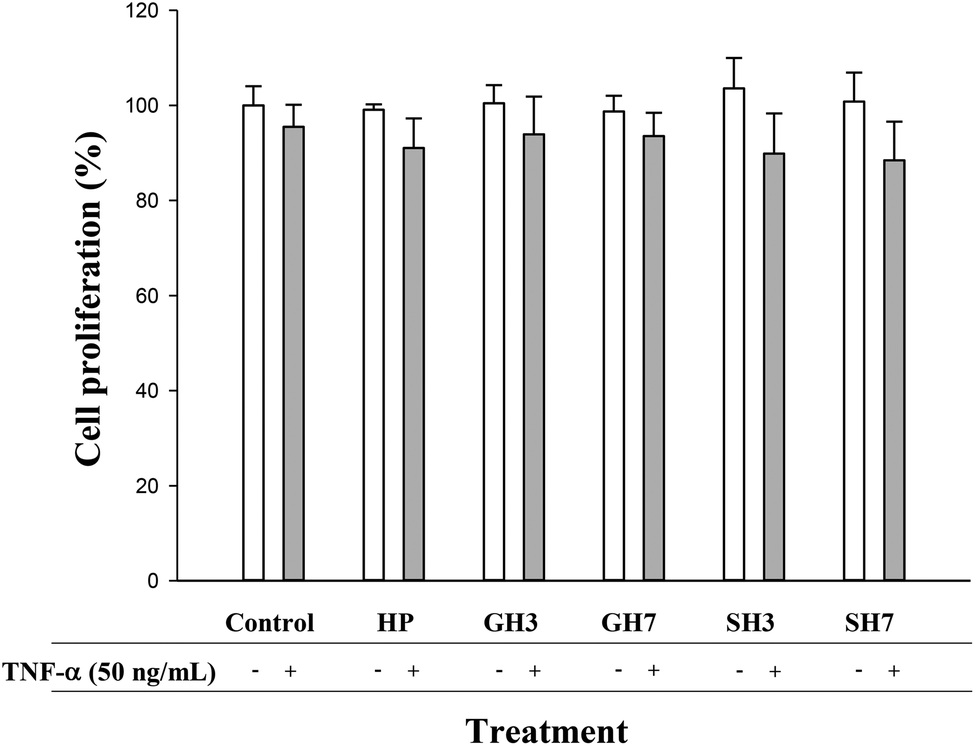 | ||
| Fig. 2 Effect of hesperetin (HP), hesperetin 3′-O-glucuronide (GH3), hesperetin 7-O-glucuronide (GH7), hesperetin 3′-O-sulfate (SH3) and hesperetin 7-O-sulfate (SH7) (10 μM) on cell proliferation (%) in HAECs in the presence or absence of TNF-α (50 ng mL−1) at 24 h. Values (%) are expressed as mean ± SD (n = 3). | ||
3.2. Effect of hesperetin and its metabolites on IL-6, IL-8 and PAI-1
TNF-α (50 ng mL−1) treatment for 24 h induced significant (p < 0.05) levels of IL-6, IL-8 and PAI-1. Hesperetin glucuronides and sulfates had no effect on the TNF-α-induced secretion of IL-6 and IL-8. A slight effect was exerted by the aglycone hesperetin at 10 μM, which decreased the concentration of IL-6 (8%) and IL-8 (10%), although this reduction was not statistically significant (Fig. 3). | ||
| Fig. 3 Effect of hesperetin (HP), hesperetin 3′-O-glucuronide (GH3), hesperetin 7-O-glucuronide (GH7), hesperetin 3′-O-sulfate (SH3) and hesperetin 7-O-sulfate (SH7) (10 μM, 1 μM, and 0.1 μM) on TNF-α-induced IL-8 (A) and IL-6 (B) levels in HAECs at 24 h. Values are expressed as mean ± SD (n = 4 replicates; 3 wells per replicate). ap < 0.05 vs. control group (DMSO). | ||
Hesperetin decreased the level of PAI-1 in the presence of TNF-α at concentrations ranging from 0.1 to 10 μM (Fig. 4). The maximum reduction observed was 15% when the cells were exposed to both TNF-α and hesperetin (10 μM). This effect was less significant when the cells were exposed to the metabolites (glucuronides and sulfates). Hesperetin 3′-O-glucuronide and hesperetin 7-O-sulfate (10 μM) reduced the concentration of PAI-1 by 17.6% and 11.2% (p < 0.05), respectively. However, hesperetin 7-O-glucuronide and hesperetin 3′-O-sulfate did not exert significant effects at the concentrations tested.
 | ||
| Fig. 4 Effect of hesperetin (HP), hesperetin 3′-O-glucuronide (GH3), hesperetin 7-O-glucuronide (GH7), hesperetin 3′-O-sulfate (SH3) and hesperetin 7-O-sulfate (SH7) (10 μM, 1 μM, and 0.1 μM) on TNF-α-induced PAI-1 levels in HAECs at 24 h. Values are expressed as mean ± SD (n = 4 replicates; 3 wells per replicate). ap < 0.05 vs. control group (DMSO); bp < 0.05 vs. TNF-α treatment. | ||
3.3. Effect of hesperetin and circulating metabolites on the migration of TNF-α-activated HAECs
HAECs were co-treated with TNF-α (50 ng mL−1) plus each metabolite for 12 and 16 h, and the migration of cells was investigated (Fig. 5A). As expected, TNF-α (50 ng mL−1) increased the capacity of the cells to migrate after 12 h (89.6% induction; p < 0.05). However, the co-incubation with both hesperetin and its derived metabolites, at a concentration of 10 μM, ameliorated significantly (p < 0.05) the effect of TNF-α, hesperetin and hesperetin 7-O-sulfate (71.7 and 61.6% of inhibition, respectively) being the compounds that exerted the most notable effect. In addition, no significant differences in the migration were found on co-incubation with any tested compound at the highest dose (10 μM) compared to control cells (Fig. 5A; ESI Fig. S2†). At a concentration of 1 μM, hesperetin metabolites reduced the migration in the range of 24 to 40% compared to TNF-α alone, although these effects were not statistically significant. However, hesperetin aglycone (1 μM) significantly reduced cell migration (47.7%; p < 0.05). No effects were observed when the cells were exposed to TNF-α and the tested compounds at a concentration of 0.1 μM for 12 hours (data not shown). | ||
| Fig. 5 Effect of hesperetin (HP), hesperetin 3′-O-glucuronide (GH3), hesperetin 7-O-glucuronide (GH7), hesperetin 3′-O-sulfate (SH3) and hesperetin 7-O-sulfate (SH7) on TNF-α-stimulated migration (50 ng mL−1) of HAECs for 12 hours (A) and in the absence of TNF-α for 12 hours (B). The results are expressed as mean ± SD of three different experiments (n = 2 wells per experiment). ap < 0.05 vs. control group (DMSO); bp < 0.05 vs. TNF-α treatment. | ||
At longer times of exposition (16 h), the cells exposed to TNF-α, alone or in combination with hesperetin metabolites, showed an increased capacity to migrate compared to untreated cells. Only co-incubation with hesperetin aglycone at 10 μM reduced this effect (83.2% of inhibition; p < 0.05) (ESI Fig. S2†).
We next evaluated the effect on cell migration in the absence of TNF-α. Fig. 5B shows that none of the compounds exerted any statistically significant effect on the migration compared to control cells. In addition, hesperetin aglycone and hesperetin 7-O-sulfate at 10 μM caused a slight and not statistically significant reduction of cell migration after 12 hours compared to control cells (Fig. 5B).
3.4. Hesperetin metabolism by HAEC cells
The metabolism of hesperetin and its derivatives (10 μM) by HAECs was evaluated by analysing the presence of metabolites excreted into the medium using a UPLC-QqQ-MS equipment after 12 and 24 h of incubation. In addition, the cell metabolism of these metabolites was explored under normal and inflammation (TNF-α 50 ng mL−1) conditions. All metabolites were very stable in the cell media (in the presence or absence of HAECs) without significant degradation during the assay time (results not shown) under both normal and inflammation conditions. The analysis of cell media during the incubation with hesperetin aglycone (10 μM) in HAECs showed a decrease of around 30 and 55% at 12 and 24 h, respectively, concomitant with the accumulation of both hesperetin 3′-O-sulfate and hesperetin 7-O-sulfate (8–10 and 20–34% after 12 and 24 h, respectively) (Fig. 6B). Fig. 6A shows the extracted ion chromatograms (EIC). In contrast, no conjugated or deconjugated metabolites were detected in the cell media after incubation of both glucuronide or sulfate hesperetin conjugates (data not shown). Finally, no differences were found in the hesperetin metabolism under normal or inflammation conditions at the tested times. | ||
| Fig. 6 Cell metabolism of hesperetin in HAECs. Extracted ion chromatograms (EIC) (A) and concentrations (B) of hesperetin (HP) and/or hesperetin 3′-O-sulfate (SH3) and hesperetin 7-O-sulfate (SH7) (μM) at 0, 12 and 24 h. Data are expressed as mean ± SD values (n = 3). | ||
3. Discussion
Inflammation plays a crucial role in the development of atherosclerosis by promoting the secretion of cytokines and chemoattractant molecules, recruiting immune cells and triggering the expression of adhesion molecules.33 Despite the fact that prevention of atherosclerosis by dietary compounds including citrus flavanones has attracted increasing interest from food scientists; the number of studies investigating the effect of flavanones on endothelial cells is scarce. As reviewed by Chanet et al. (2012),34 citrus flavanones such as naringenin, naringin, hesperidin and hesperetin have been reported to exert antiatherogenic effects in preclinical studies. Thus, previous studies reported that hesperidin, naringin, and naringenin downregulated the TNF-α-induced expression of VCAM-1 in human umbilical vein endothelial cells (HUVECs), diminished the adhesion of U937 monocytes to HUVECs stimulated with TNF-α as well as mitigated induced oxidative stress and inflammation.20,35,36 However, although these studies may indicate that flavanones can ameliorate the progression of atherosclerosis, cells of the vascular system are not exposed to the glycosides or aglycones of flavanones, and therefore, the physiological relevance is limited.Hesperidin deconjugation is known to be catalyzed by different fecal microbial enzymes (α-rhamnosidases, β-glucosidases, and β-glucuronidases) to yield the corresponding aglycone hesperetin that is absorbed from the colon and transformed by the phase II enzymes into its glucuronide and sulfate derivatives.37–39 A high variability in flavanone bioavailability has been described in plasma with concentrations ranging from 0.04 to 6 μM.34 To date, the number of studies investigating the effect of flavanone metabolites (at concentrations detected in vivo) on mechanisms associated with atherosclerosis development is still limited. The glucuronides and sulfates of the flavanones, at concentrations that can be achieved in vivo (from 0.5 to 10 μM), have been reported to reduce the adhesion of monocytes to the endothelial cells and modulate the expression of genes related to inflammation,32 which can contribute to the understanding of the beneficial effects of flavanones in cardiovascular protection. However, the effects of these compounds on other mechanisms such as migration and proliferation, critical processes involved in angiogenesis and atherosclerosis,40 have not yet been investigated.
Our results indicated for the first time that hesperetin and its derived metabolites reduced the TNF-α-induced migration of human aortic endothelial cells (HAECs), used as a model of the human vascular system, while sparing the proliferative capacity of these cells. These results are in agreement with previous studies conducted on other types of endothelial cells such as human umbilical vein endothelial cells (HUVECs) widely used as cell migration models. HUVECs are derived from immune-naive foetal tissue and show significant functional differences from the adult vascular endothelium,41 while HAECs are derived from a big vessel, aorta, providing an excellent model system to study all aspects of cardiovascular function and disease including atherosclerosis.42 Thus, hesperetin was reported to inhibit significantly the VEGF-induced migration of HUVECs, although at high (non-physiological) concentrations (25–100 μM).25 Another study showed that treatment with a flavanone-rich orange extract (∼60 μM total flavanones) inhibited the migration of TNF-α-induced colon fibroblast CCD-18Co cells.21
Increased levels of IL-6 have been shown to induce migration of vascular smooth muscle cells,43,44 whereas a high concentration of IL-8 has been associated with induction of migration and proliferation in rat aortic cells.45 According to these previous studies, our data revealed that although TNF-α increased the release of IL-6 and IL-8 to the culture medium, none of the investigated flavanones (conjugated metabolites or aglycone) exerted any effect on the levels of these cytokines, indicating that the inhibitory effect of hesperetin and its circulating metabolites on migration could be mediated by molecules other than IL-6 and IL-8.
In this regard, the role of PAI-1 was also explored. PAI-1 is a thrombogenic protein involved in a wide range of cardiovascular diseases as well as in cell migration.46 Indeed, the inhibition of this molecule is a potential strategy in the prevention of atherosclerosis and CVDs.47 Our data showed that hesperetin aglycone was most efficient in reducing the release of PAI-1 into the culture medium, which matched with the highest inhibitory effect on cell migration. In addition, hesperetin 7-O-glucuronide was also able to reduce the concentration of PAI-1 in TNF-α-stimulated HAECs at concentrations ranging from 1 to 10 μM, whereas the concentrations of 10 μM of hesperetin 3′-O-glucuronide and hesperetin 7-O-sulfate were needed to observe a statistical reduction. Our data are in agreement with previous studies that reported the hypotensive, vasodilatory and anti-inflammatory activities in vitro and in vivo of hesperetin,19,48 as well the reduction of TNF-α-induced adhesion of monocytes THP-1 to HUVECs.32 Moreover, these studies suggested that the biological activity of the hesperetin metabolites tested might depend on the position of the conjugation, and to a lesser extent on the type of substituent (glucuronide or sulfate), although contradictory results have been found so far. Thus, hesperetin 7-O-glucuronide, similar to hesperetin aglycone, was reported to be more effective than hesperetin 3′-O-glucuronide as hypotensive, vasodilatory and anti-inflammatory in primary endothelial cells isolated from Sprague-Dawley rat aortas,19 as well as in HUVECs.48 In contrast, among the hesperetin metabolites, hesperetin 3′-O-glucuronide and sulfate (2 μM) reduced the TNF-α-induced adhesion of monocytes THP-1 to HUVECs and modulated the expression of genes related to inflammation, whereas hesperetin 7-O-glucuronide had no effect.32
Two recent studies have reported the cell metabolism (glucuronidation, sulfation and methylation) of the flavanol epicatechin,49 and isoflavones such as genistein and daidzein in HUVECs.50 In the present study, the uptake and metabolism of hesperetin by HAECs was also investigated. Our data showed that hesperetin was metabolized into both hesperetin 7-O-sulfate and hesperetin 3′-O-sulfate reaching over 50% of total metabolites after 24 h of incubation. Although the conversion of hesperetin into glucuronidated and sulfated metabolites has been reported in cell models of the gastrointestinal tract,39,51 to the best of our knowledge, the metabolism of hesperetin and its derived circulating metabolites by HAECs has not been reported so far.
Overall, our results suggest that the effects obtained by hesperetin on cell migration as well as on PAI-1 levels might be, at least partly, also as a result of the action of sulfate hesperetin conjugates together with hesperetin. Therefore, further research is needed to ascertain possible synergistic or additive effects between hesperetin and its derived metabolites.
Finally, with all due caution when extrapolating in vitro models to in vivo scenarios, our results (based on specific assay conditions such as 12–16 h of exposure time and 1–10 μM dose) suggest that a long term consumption of hesperidin-containing foods could provide certain blood circulating levels (low μM concentrations) of their main derived metabolites, which could interact with the aortic tissue and exert atheroprotective effects.
5. Conclusions
Our results show for the first time that hesperetin and its main circulating plasma metabolites, at physiologically relevant concentrations, efficiently attenuated the migration of TNF-α-inflamed HAECs, which was accompanied and perhaps mediated by a significant decrease in PAI-1 levels. In addition, we also described for the first time, the metabolism (sulfation) of hesperetin and its related metabolites by HAECs, independently of the inflammatory status. Further in vivo investigations are warranted to explore the possible role of hesperetin metabolites as well as other flavanone metabolites as potential atheroprotective compounds.Abbreviations
| ACN | Acetonitrile |
| CVDs | Cardiovascular diseases |
| DMSO | Dimethyl sulfoxide |
| ECGM | Endothelial cell growth medium |
| EIC | Extracted ion chromatograms |
| GH3 | Hesperetin 3′-O-glucuronide |
| GH7 | Hesperetin 7-O-glucuronide |
| HAECs | Human aortic endothelial cells |
| HP | Hesperetin |
| HUVECs | Human umbilical vein endothelial cells |
| IL | Interleukin |
| MeOH | Methanol |
| MTT | 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PBS | Phosphate buffered saline |
| S.D. | Standard deviation |
| SH3 | Hesperetin 3′-O-sulfate |
| SH7 | Hesperetin 7-O-sulfate |
| TNF-α | Tumor necrosis factor alpha |
| UPLC-QqQ-MS | UPLC-Triple Quadrupole mass spectrometry |
| VCAM-1 | Vascular cell adhesion molecule 1 |
Conflict of interest
All authors declare no conflict of interests.Acknowledgements
This research has been supported by the Project CICYT-AGL2011-22447 (MINECO, Spain) and project 201370E068 (CSIC). AGS is holder of a ‘Juan de la Cierva’ contract from MINECO (Spain).References
- D. S. Celermajer, C. K. Chow, E. Marijon, N. M. Anstey and K. S. Woo, J. Am. Coll. Cardiol., 2012, 60, 1207–1216 CrossRef PubMed.
- A. S. Jaipersad, G. Y. Lip, S. Silverman and E. Shantsila, J. Am. Coll. Cardiol., 2014, 63, 1–11 CrossRef CAS PubMed.
- W. Risau, Nature, 1997, 386, 671–674 CrossRef CAS PubMed.
- L. Lamalice, F. Le Boeuf and J. Huot, Circ. Res., 2007, 100, 782–794 CrossRef CAS PubMed.
- Z. Szekanecz, M. R. Shah, L. A. Harlow, W. H. Pearce and A. E. Koch, Pathobiology, 1994, 62, 134–139 CrossRef CAS PubMed.
- J. A. Giménez-Bastida, M. Larrosa, A. González-Sarrías, F. Tomás-Barberán, J. C. Espín and M. T. García-Conesa, J. Agric. Food Chem., 2012, 60, 8866–8876 CrossRef PubMed.
- J. Xiao, J. Song, V. Hodara, A. Ford, X. L. Wang, Q. Shi, L. Chen and J. L. Vandeberg, J. Diabetes Res., 2013, 2013, 185172 Search PubMed.
- J. Mursu, S. Voutilainen, T. Nurmi, T. P. Tuomainen, S. Kurl and J. T. Salonen, Br. J. Nutr., 2008, 100, 890–895 CrossRef CAS PubMed.
- L. Hooper, P. A. Kroon, E. B. Rimm, J. S. Cohn, I. Harvey, K. A. Le Cornu, J. J. Ryder, W. L. Hall and A. Cassidy, Am. J. Clin. Nutr., 2008, 88, 38–50 CAS.
- A. Cassidy, E. B. Rimm, E. J. O'Reilly, G. Logroscino, C. Kay, S. E. Chiuve and K. M. Rexrode, Stroke, 2012, 43, 946–951 CrossRef CAS PubMed.
- D. Grassi, G. Desideri, P. Di Giosia, M. De Feo, E. Fellini, P. Cheli, L. Ferri and C. Ferri, Am. J. Clin. Nutr., 2013, 98, 1660S–1666S CrossRef CAS PubMed.
- C. Dalgård, F. Nielsen, J. D. Morrow, H. Enghusen-Poulsen, T. Jonung, M. Hørder and M. P. de Maat, Br. J. Nutr., 2009, 101, 263–269 CrossRef PubMed.
- C. Morand, C. Dubray, D. Milenkovic, D. Lioger, J. F. Martin, A. Scalbert and A. Mazur, Am. J. Clin. Nutr., 2011, 93, 73–80 CrossRef CAS PubMed.
- S. Foroudi, A. S. Potter, A. Stamatikos, B. S. Patil and F. Deyhim, J. Med. Food, 2014, 17, 612–617 CrossRef CAS PubMed.
- P. J. Curtis, J. Potter, P. A. Kroon, P. Wilson, K. Dhatariya, M. Sampson and A. Cassidy, Am. J. Clin. Nutr., 2013, 97, 936–942 CrossRef CAS PubMed.
- M. Y. Schär, P. J. Curtis, S. Hazim, L. M. Ostertag, C. D. Kay, J. F. Potter and A. Cassidy, Am. J. Clin. Nutr., 2015, 101, 931–938 CrossRef PubMed.
- J. Constans, C. Bennetau-Pelissero, J. F. Martin, E. Rock, A. Mazur, A. Bedel, C. Morand and A. M. Bérard, Clin. Nutr., 2014 DOI:10.1016/j.clnu.2014.12.016.
- F. Tomas-Barberán and M. Clifford, J. Sci. Food Agric., 2000, 80, 1073–1080 CrossRef.
- M. Yamamoto, H. Jokura, K. Hashizume, H. Ominami, Y. Shibuya, A. Suzuki, T. Hase and A. Shimotoyodome, Food Funct., 2013, 4, 1346–1351 CAS.
- I. T. Nizamutdinova, J. J. Jeong, G. H. Xu, S. H. Lee, S. S. Kang, Y. S. Kim, K. C. Chang and H. J. Kim, Int. Immunopharmacol., 2008, 8, 670–678 CrossRef CAS PubMed.
- J. A. Giménez-Bastida, M. Martínez-Florensa, J. C. Espín, F. A. Tomás-Barberán and M. T. García-Conesa, J. Agric. Food Chem., 2009, 57, 9305–9315 CrossRef PubMed.
- S. Rizza, R. Muniyappa, M. Iantorno, J. A. Kim, H. Chen, P. Pullikotil, N. Senese, M. Tesauro, D. Lauro, C. Cardillo and M. J. Quon, J. Clin. Endocrinol. Metab., 2011, 96, E782–E792 CrossRef CAS PubMed.
- A. Chanet, D. Milenkovic, C. Deval, M. Potier, J. Constans, A. Mazur, C. Bennetau-Pelissero, C. Morand and A. M. Bérard, J. Nutr. Biochem., 2012, 23, 469–477 CrossRef CAS PubMed.
- J. M. Assini, E. E. Mulvihill, B. G. Sutherland, D. E. Telford, C. G. Sawyez, S. L. Felder, S. Chhoker, J. Y. Edwards, R. Gros and M. W. Huff, J. Lipid Res., 2013, 54, 711–724 CrossRef CAS PubMed.
- G. D. Kim, Prev. Nutr. Food Sci., 2014, 19, 299–306 CrossRef PubMed.
- P. C. Hollman, M. N. Bijsman, Y. van Gameren, E. P. Cnossen, J. H. de Vries and M. B. Katan, Free Radical Res., 1999, 31, 569–573 CrossRef CAS PubMed.
- C. Felgines, S. Talavéra, M. P. Gonthier, O. Texier, A. Scalbert, J. L. Lamaison and C. Rémésy, J. Nutr., 2003, 133, 1296–1301 CAS.
- W. Hollands, G. M. Brett, J. R. Dainty, B. Teucher and P. A. Kroon, Mol. Nutr. Food Res., 2008, 52, 1097–1105 CAS.
- A. J. Day, J. M. Gee, M. S. DuPont, I. T. Johnson and G. Williamson, Biochem. Pharmacol., 2003, 65, 1199–1206 CrossRef CAS.
- M. Tomás-Navarro, F. Vallejo, F. Borrego and F. A. Tomás-Barberán, J. Agric. Food Chem., 2014, 62, 9458–9462 CrossRef PubMed.
- M. Tomás-Navarro, F. Vallejo, E. Sentandreu, J. L. Navarro and F. A. Tomás-Barberán, J. Agric. Food Chem., 2014, 62, 24–27 CrossRef PubMed.
- A. Chanet, D. Milenkovic, S. Claude, J. A. Maier, M. Kamran Khan, N. Rakotomanomana, S. Shinkaruk, A. M. Bérard, C. Bennetau-Pelissero, A. Mazur and C. Morand, Br. J. Nutr., 2013, 110, 587–598 CrossRef CAS PubMed.
- C. Weber and H. Noels, Nat. Med., 2011, 17, 1410–1422 CrossRef CAS PubMed.
- A. Chanet, D. Milenkovic, C. Manach, A. Mazur and C. Morand, J. Agric. Food Chem., 2012, 60, 8809–8822 CrossRef CAS PubMed.
- J. S. Choi, Y. J. Choi, S. H. Park, J. S. Kang and Y. H. Kang, J. Nutr., 2004, 134, 1013–1019 CAS.
- I. Orhan, S. Nabavi, M. Daglia, G. Tenore, K. Mansouri and S. Nabavi, Curr. Pharm. Biotechnol., 2015, 16, 245–251 CAS.
- G. M. Brett, W. Hollands, P. W. Needs, B. Teucher, J. R. Dainty, B. D. Davis, J. S. Brodbelt and P. A. Kroon, Br. J. Nutr., 2009, 101, 664–675 CrossRef CAS PubMed.
- F. Vallejo, M. Larrosa, E. Escudero, M. P. Zafrilla, B. Cerdá, J. Boza, M. T. García-Conesa, J. C. Espín and F. A. Tomás-Barberán, J. Agric. Food Chem., 2010, 58, 6516–6524 CrossRef CAS PubMed.
- W. Brand, M. G. Boersma, H. Bik, E. F. Hoek-van den Hil, J. Vervoort, D. Barron, W. Meinl, H. Glatt, G. Williamson, P. J. van Bladeren and I. M. Rietjens, Drug Metab. Dispos., 2010, 38, 617–625 CrossRef CAS PubMed.
- M. Fosbrink, F. Niculescu, V. Rus, M. L. Shin and H. Rus, J. Biol. Chem., 2006, 281, 19009–19018 CrossRef CAS PubMed.
- P. H. Tan, C. Chan, S. A. Xue, R. Dong, B. Ananthesayanan, M. Manunta, C. Kerouedan, N. J. Cheshire, J. H. Wolfe, D. O. Haskard, K. M. Taylor and A. J. George, Atherosclerosis, 2004, 173, 171–183 CrossRef CAS PubMed.
- Y. Chen, D. Li, Y. Xu, Y. Zhang, L. Tao, S. Li, Y. Jiang and X. Shen, J. Evidence-Based Complementary Altern. Med., 2014, 2014, 956824 Search PubMed.
- Z. Wang and W. H. Newman, J. Surg. Res., 2003, 111, 261–266 CrossRef CAS.
- M. V. Autieri, ISRN Vasc. Med., 2012, 2012, 17 Search PubMed.
- Y. Qin, F. Fan, Y. Zhao, Y. Cui, X. Wei, K. Kohama, J. R. Gordon, F. Li and Y. Gao, IUBMB Life, 2013, 65, 67–75 CrossRef CAS PubMed.
- V. A. Ploplis, Curr. Drug Targets, 2011, 12, 1782–1789 CrossRef CAS.
- A. Rouch, C. Vanucci-Bacque, F. Bedos-Belval and M. Baltas, Eur. J. Med. Chem., 2015, 92, 619–636 CrossRef CAS PubMed.
- H. Takumi, H. Nakamura, T. Simizu, R. Harada, T. Kometani, T. Nadamoto, R. Mukai, K. Murota, Y. Kawai and J. Terao, Food Funct., 2012, 3, 389–398 CAS.
- A. Rodriguez-Mateos, N. Toro-Funes, T. Cifuentes-Gomez, M. Cortese-Krott, C. Heiss and J. P. Spencer, Arch. Biochem. Biophys., 2014, 559, 17–23 CrossRef CAS PubMed.
- N. Toro-Funes, F. J. Morales-Gutiérrez, M. T. Veciana-Nogués, M. C. Vidal-Carou, J. P. Spencer and A. Rodriguez-Mateos, Food Funct., 2015, 6, 98–108 Search PubMed.
- W. Brand, P. A. van der Wel, M. J. Rein, D. Barron, G. Williamson, P. J. van Bladeren and I. M. Rietjens, Drug Metab. Dispos., 2008, 36, 1794–1802 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5fo00771b |
| This journal is © The Royal Society of Chemistry 2016 |