
Protective effects of trigonelline against indomethacin-induced gastric ulcer in rats and potential underlying mechanisms
Paulrayer
Antonisamy†
a,
Mariadhas Valan
Arasu†
b,
Muniappan
Dhanasekaran
c,
Ki Choon
Choi
d,
Adithan
Aravinthan
a,
Nam Soo
Kim
a,
Chang-Won
Kang
a and
Jong-Hoon
Kim
*a
aCollege of Veterinary Medicine, Biosafety Research Institute, Chonbuk National University, 54596 79 Gobong-ro, Iksan-city, Jeollabuk-Do, Republic of Korea. E-mail: jhkim1@jbnu.ac.kr; Fax: +82 63 850 0910; Tel: +82 63 850 0952
bDepartment of Botany and Microbiology, Addiriyah Chair for Environmental Studies, College of Science, King Saud University, P.O. Box 2455, Riyadh 11451, Saudi Arabia
cDivision of Ethnopharmacology, Entomology Research Institute, Loyola College, Chennai 600 034, Tamil Nadu, India
dGrassland and forage division, National Institute of Animal Science, RDA, Seonghwan-Eup, Cheonan-Si, Chungnam 330-801, Republic of Korea
First published on 12th October 2015
Abstract
The present study was undertaken to explore gastroprotective effects of trigonelline (TRG) and to determine the potential mechanisms involved in this action. In order to evaluate the gastroprotective efficiency of TRG, an indomethacin-induced ulcer model has been applied. Antioxidants, cytokines, adhesion markers and apoptosis levels have been analyzed for the biochemical mechanism involved in TRG activity. TRG (45 mg kg−1) pretreated rats significantly inhibited gastric lesions by 81.71%. Indomethacin administration raises the levels of leukotriene B4 (LTB4), lipid peroxidation and myeloperoxidase (MPO) with the significant declines of prostaglandin E2 (PGE2), superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GSH-px) levels. Conversely, TRG (45 mg kg−1) pretreated animals showed significant rises in PGE2 and antioxidant levels along with substantial reductions in LTB4, lipid peroxidation and MPO levels. Indomethacin-induced rats also exhibited considerable increases of pro-inflammatory cytokines including interleukin-6 (IL-6), interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), and interferon-γ (IFN-γ) levels and decreases of anti-inflammatory cytokines such as interleukin-10 (IL-10) and interleukin-4 (IL-4), but these imbalances were normalized through treatment of TRG. The protective activity of TRG against indomethacin-induced gastric ulcer has been ascribed to three important mechanisms: (1) anti-inflammatory; (2) antioxidant; (3) anti-apoptotic pathways.
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs), such as indomethacin and ketoprofen, are generally used to alleviate swelling and pain caused by inflammatory diseases including rheumatoid arthritis and osteoarthritis. Despite their benefits of an anti-inflammatory nature, these drugs may cause peptic ulcers.1,2 The major causes of peptic ulcers include gastric acid, pepsin, bile salts, NSAIDs, Helicobacter pylori infection, consumption of alcohol and tobacco. An earlier report stated that the NSAID users have a greater threat of peptic ulcers than those with Helicobacter pylori infection.3 Thus, it is imperative to search for novel compounds that may help to prevent ulceration of the gastrointestinal tract. NSAID gastropathy is considered a “silent epidemic” and, therefore, has been an area of intense research. Among the commonly used NSAIDs, indomethacin possesses the highest ulcerogenic potential for humans.4,5 Inhibition of cyclooxygenases (COXs) and the associated reduced prostaglandin (PG) synthesis were previously believed to be the major reasons for gastric pathogenesis caused by NSAIDs including indomethacin.6–8 However, accumulated evidence suggests that other COX-independent factors also play equally important roles in the process.9–11 In order to prevent and treat gastric ulcers, indigenous healers and herbalists traditionally used phytogenic agents. In recent decades, gastroprotection using medicinal plant products as possible therapeutic alternatives has become a subject of active scientific investigations.12Trigonelline (TRG) is a pyridine alkaloid, commonly found in Trigonella foenum-graecum L. (fenugreek) seeds and coffee beans.13,14 TRG as a coffee ingredient is one of the most often consumed alkaloids. The anti-diabetic properties of TRG and its beneficial influence on lipid profile have been proven.15,16 TRG attenuates adipocyte differentiation and lipid accumulation in 3T3-L1 cells.17 Also, this alkaloid has been taken into consideration as a potential neuroprotective agent, especially in Alzheimer's disease (AD). Previous reports exhibit that TRG shows memory improvement in β-amyloid-induced memory impairment in rats and in neurite outgrowth in rats and humans.14,18 It has also shown antioxidant properties.19 TRG has antioxidant effectiveness in cell-free systems and human colon cell lines.20 However, the role of TRG against acute gastric ulcers induced by indomethacin remains unidentified. The objective of this study was to assess the effects of TRG against indomethacin-induced gastric damage in rats and its potential gastroprotective mechanism.
Materials and methods
Animals
A total of 102 male Sprague–Dawley (SD) rats (200–220 g) were used for this experiment. For the dose selection study 48 animals were used (8 groups with 6 rats each). For a study on the role of different antagonists in TRG produced gastroprotectivity, 54 animals were used (9 groups with 6 rats each). Animals were kept at a precise temperature of 23 ± 2 °C, a relative humidity of 65–80% and exposed to 12 h dark–light cycles (lights on at 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :00) and fed with a standard pellet diet (Samyang, Daejeon, Republic of Korea) and tap water ad libitum. Animals were maintained in accordance with the guidelines delivered by the National Institute of Health for the Care and Use of Laboratory Animals (NIH Publication 80-23, revised in 1996). All the in vivo studies were performed in accordance with the Ethics Committee norms (permit number CBNU-2014-92) established by the Institutional Animal Care and Use Committee at Chonbuk National University (Jeonju, Republic of Korea).
:00) and fed with a standard pellet diet (Samyang, Daejeon, Republic of Korea) and tap water ad libitum. Animals were maintained in accordance with the guidelines delivered by the National Institute of Health for the Care and Use of Laboratory Animals (NIH Publication 80-23, revised in 1996). All the in vivo studies were performed in accordance with the Ethics Committee norms (permit number CBNU-2014-92) established by the Institutional Animal Care and Use Committee at Chonbuk National University (Jeonju, Republic of Korea).
Drugs and chemicals
Trigonelline (TRG), indomethacin, omeprazole, SC560, celecoxib, Nω-nitro-L-arginine methyl ester (L-NAME), N-ethylmaleimide (NEM), yohimbine, glibenclamide and ELISA kits for SOD (superoxide dismutase) caspase-3 were purchased from Sigma Chemical Co. (St. Louis, MO, USA). The apoptosis assay kit was acquired from Boehringer Mannheim. ELISA kits for ICAM-1, VCAM-1, E-selectin, P-selectin, prostaglandin E2 (PGE2) and leukotriene B4 (LTB4) were obtained from R&D Systems (Minneapolis, MN, USA). ELISA kits for TNF-α, IL-6 and IL-10 were from BioLegend (San Diego, CA, USA). ELISA kits for hepatocyte growth factor (HGF), vascular endothelial growth factor (VEGF), epidermal growth factor (EGF) and the transcription factor kit were obtained from Cayman Chemical (Ann Arbor, USA). Antibodies for COX-1 and β-actin were from Sigma-Aldrich. Antibodies for eNOS, iNOS, IL-10 and TNF-α were obtained from Cell Signaling Technology Inc. (Beverly, MA, USA). All other chemicals used were of analytical reagent grade.Dose selection
To determine the lowermost effective dose of TRG, gastric ulcers were induced by indomethacin after TRG treatment. In this analysis, forty eight (48) SD rats were used, divided into eight (8) groups containing six (n = 6) rats each. Rats were starved for 24 hours and accommodated in cages. They were only allowed free access to drinking water. Drugs were dissolved in 0.5% CMC (carboxymethyl cellulose) as the vehicle, and administered orally via orogastric-intubations. An experimental gastric ulcer was induced based on a previous method described by Antonisamy et al. (2014)21 using indomethacin as the ulcerogen.Group 1 (normal control) received 0.5 mL of 0.5% CMC.
Group 2 (ulcer control) received 0.5 mL of 0.5% CMC.
Group 3 (positive control) received 0.5 mL of 0.5% CMC of the standard drug omeprazole (40 mg kg−1) based on the previous report.21
Groups 4, 5, 6, 7 and 8 received 0.5 mL of 0.5% CMC of the TRG at a dosage of 15, 30, 45, 60 and 75 mg kg−1 respectively.
After 30 min, Group 1 received 0.5 mL of 0.5% CMC. Groups 2–8 received indomethacin (20 mg kg−1 orally). Animals were sacrificed under anesthesia with ketamine/xylazine (0.5 mL of 100 mg per mL ketamine combined with 0.05 mL of 20 mg per mL xylazine) at a dosage of 0.55 mL per 100 g body weight and six (6) hours later indomethacin administration and the ulcer score were macroscopically examined according to a previous method.22
Role of different antagonists on TRG produced gastroprotectivity
Rats were assigned to nine (9) groups, each comprising of six rats (n = 6). The treatment groups and experimental protocol are detailed below:Group 1 (normal control) received 0.5 mL of 0.5% CMC.
Group 2 (ulcer control) received 0.5 mL of 0.5% CMC.
Group 3 (TRG treatment) received TRG (45 mg kg−1 p.o.)
Group 4 (SC560 + TRG treatment) received SC560 (5 mg kg−1 p.o.) and TRG (45 mg kg−1 p.o.)
Group 5 (celecoxib + TRG treatment) received celecoxib (3.5 mg kg−1 p.o.) and TRG (45 mg kg−1 p.o.)
Group 6 (YO + TRG treatment) received YO (2 mg kg−1 i.p.) and TRG (45 mg kg−1 p.o).
Group 7 (L-NAME + TRG treatment) received L-NAME (50 mg kg−1 i.p.) and TRG (45 mg kg−1 p.o).
Group 8 (NEM + TRG treatment) received NEM (10 mg kg−1 s.c.) and TRG (45 mg kg−1 p.o).
Group 9 (GLIB + TRG treatment) received GLIB (5 mg kg−1 p.o.) and TRG (45 mg kg−1 p.o).
All drugs were administered using 0.5% CMC as the vehicle. After 30 min, each group of animals except the normal group received 20 mg per kg of indomethacin. Selective COX-1 inhibitor (SC560), COX-2 inhibitor (celecoxib), α2-receptor antagonist (yohimbine), nonselective nitric oxide synthase (NOS) inhibitor (L-NAME), endogenous sulfhydryl antagonist (NEM), and K+ATP channel antagonist (glibenclamide) were administered to rats 30 min before TRG treatment and 1 h prior to indomethacin induction. Six hours later, the animals were killed under anesthesia with ketamine/xylazine at a dosage of 0.55 mL per 100 g body weight and the stomach was surgically removed, opened along the greater curvature and the lesions were macroscopically examined according to the ulcer score described by a previous method.22 Concisely, ulcers are either circular (assessed on the basis of diameter) or linear (assessed on the basis of length). Deep circular ulcers more than 8 mm = 10; 7–8 mm = 8; 6–7 mm = 7; 5–6 mm = 6; 4–5 mm = 5; 3–4 mm = 4; 2–3 mm = 3; 1–2 mm = 2 and 0–1 mm = 1. The deep linear ulcers more than 10 mm in length = 6 and linear ulcer less than 10 mm in length = 3. The score for each single lesion was then summed up for the determination of the ulcer index (mm).
The percentage inhibition was calculated through the method described by Demirbilek et al. (2004):23 (UI nontreated − UI treated)/UI nontreated) × 100.
Gastric tissue homogenate preparation
Immediately after the animals were killed, gastric mucosa was removed from rats and washed carefully with ice-cold saline. Using a homogenizer a small fragment of each stomach was homogenized (10% w/v) in ice cold PBS (0.1 mol l−1) containing mammalian protease inhibitor cocktail. The homogenates were centrifuged at 10000g at 4 °C for 15 min. A pure supernatant was used to quantify the biochemical markers.
Determination of the effect of TRG on biochemical markers
MPO activity was determined as previously described.24 The absorbance was read at 650 nm. MPO activity was expressed as mU per 100 mg of wet tissue.The SOD activity was evaluated based on the manufacturer's instructions. The absorbance was read at 560 nm.25 The results were expressed as U per mg of proteins.
Catalase activity was measured using the method described by Aebi (1984).26 Absorbance was measured at 510 nm. The results were expressed as U per g of tissue.
Lipid peroxidation was determined by estimating the level of thiobarbituric acid reactive substances (TBARS) measured as malondialdehyde (MDA), according to the method of Mihara and Uchiyama.27 The absorbance was measured at 535 nm. The results were expressed as nmol of MDA per g of tissue.
Glutathione peroxidase (GPx) activity was spectrophotometrically determined based on a previous method.28 The absorbance was measured at 340 nm. The results were expressed as units per g of wet tissue.
PGE2 and LTB4 assays were performed according to the manufacturer's instructions. Results were expressed as ng per g of wet tissue (for PGE2) and pg per g of wet tissue (for LTB4).
Measurements of apoptosis and caspase-3 were performed according to the manufacturer's instructions. Levels were expressed as μM pNA per min per g of wet tissue.
For apoptotic measurements, the gastric mucosal cells were collected from the stomachs of freshly dissected rats for a quantitative analysis of apoptosis. Gastric mucosal cells were collected and incubated in the lysis buffer and centrifuged; the supernatant having the cytoplasmic histone-associated DNA fragments was reacted with an immobilized anti-histone antibody in the microtitrator wells. After the wells were washed, the retained complex was reacted with anti-DNA peroxidase and probed with ABTS [2,2′-azinobis (3-ethylbenzthiazolinesulfonic acid)] reagent for spectrophotometric quantification.29 The apoptosis level was expressed as U per mg of protein.
The levels of P-selectin, E-selectin, VCAM-1 and ICAM-1 in serum samples were estimated using ELISA kits according to the manufacturer's protocol. The values were expressed as ng ml−1 (for ICAM-1, VCAM-1 and P-selectin) and pg per ml for E-selectin.
The levels of VEGF, EGF and HGF in the gastric tissue were estimated using commercially available ELISA kits. The values were expressed as ng per g of wet tissue.
The levels of pro-inflammatory cytokines such as TNF-α, IFN-γ, IL-1β, IL-6, and anti-inflammatory cytokines including IL-10 and IL-4 were evaluated based on the manufacturer's instructions. The values were expressed as pg per mg of proteins.
Gastric mucosal NOS activity was measured by previous methods.30,31 The difference between absorption at 401 and 421 nm was frequently detected with a dual wavelength recording spectrophotometer at 37 °C. The induced NOS (iNOS) level was calculated by subtraction of the constitutive NOS (cNOS) level from the total NOS (tNOS) level.
The NO content was quantified by measuring nitrite/nitrate concentrations using the Griess assay.32 The absorbance was measured at 540 nm. The results were expressed as μmol per g of tissue.
Preparation of nuclear fraction and determination of the effect of TRG on transcription factor
Stomach tissues were homogenized in ice cold PBS, and centrifuged at 3000g for 5 min. The resulting supernatants were discarded. Precipitates were washed twice by ice cold PBS and then re-suspended in buffer A (10 mM HEPES buffer, pH 7.9, containing 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol (DTT), 50 mM NaF, 30 mM β glycerophosphate, 1 mM Na3VO4 and 1 mM phenylmethylsulfonyl fluoride (PMSF) and 10% NP-40), the resulting homogenates were centrifuged at 15000g for 15 min after an incubation period of 15 min on ice and strong shocked for 45 s. The pellets were washed three times with buffer A, then resuspended in buffer B (20 mM HEPES buffer, pH 7.9, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT and 1 mM PMSF) and shocked at 4 °C for 30 min, then centrifuged at 15000g for 15 min. The resulting supernatants were considered as nuclear extracts, and frozen at −80 °C for measurements of NF-κB. NF-κB p65 and NF-κB p50 subunits were detected by ELISA following the manufacturer's instructions. The optical density (OD) was measured at 655 nm. Percentage inhibition was calculated by the following formula: (OD of control − OD of treated/OD of control × 100).
Western blot analysis
Western blot analysis was carried out to detect the expression of various target proteins. Gastric mucosal samples were homogenized in RIPA lysis buffer. The total protein content in the supernatant was assayed by BCA protein assay reagent (Pierce, Rockford, IL, USA). Equal amounts of proteins (20 μg) were loaded for 10% sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis and then electro transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA, USA). The membranes were incubated with a 1:1000 dilution of the respective primary antibodies, followed by a 1:2000 dilution of a horseradish peroxide-conjugated secondary antibody. Protein bands were visualized by ECL (GE Healthcare, Pittsburgh, PA, USA). ImageJ analysis software was used to quantify the density of each band.
Determination of the effect of TRG on microvascular permeability
In this experiment rats were allocated to five different groups, having six animals each. Prior to experimentation rats were fasted for 24 h and permitted free access to water. The treatment groups and the experimental protocol are given below:Group 1 (normal control) received 0.5 mL of 0.5% CMC.
Group 2 (ulcer control) received 0.5 mL of 0.5% CMC.
Group 3 (TRG treatment) received TRG (45 mg kg−1).
Group 4 (SC560 + TRG treatment) received SC560 + TRG (30 mg kg−1 + 45 mg kg−1).
Group 5 (celecoxib + TRG treatment) received celecoxib + TRG (30 mg kg−1 + 45 mg kg−1).
All drugs were suspended in 0.5% CMC and treated by oral administration 1 h before ulcer induction using indomethacin. Microvascular permeability was assessed 6 h after indomethacin treatment through measuring the amount of extravasated Evan's blue dye in mucosa based on a previous method.33 Briefly, each rat received 1 mL of 1% (w/v) Evan's blue in sterile saline through intravenous injection 30 min before being sacrificed. Under ether anesthesia, rats were killed by bleeding from the descending aorta, their stomachs were removed, and gastric mucosa was scraped off and immersed in distilled water. The dye was extracted with formamide and quantified spectrophotometrically at 620 nm, and results were expressed as μg per mg of proteins.
Statistical analysis
All results were expressed as mean ± S.D. (standard deviation). Data were analysed for normal distribution using a Kolmogorov–Smirnov test. Normally distributed data were analysed with one way ANOVA using Tukey's post hoc test; otherwise the Kruskal–Wallis test was used. If the Kruskal–Wallis test for analysis of variance was significant, the Mann–Whitney U-test was used for comparison between two selected groups. Statistical significance was accepted at a P value less than 0.05.Results
Macroscopic reflection indicated that pretreatment of TRG (Fig. 1C) or omeprazole (Fig. 1D) considerably reduced gastric mucosal injury compared to the indomethacin-induced ulcer control group, where elongated bands of hemorrhages have been observed in gastric mucosa (Fig. 1B). However, the normal group displays undamaged stomachs without any injuries (Fig. 1A).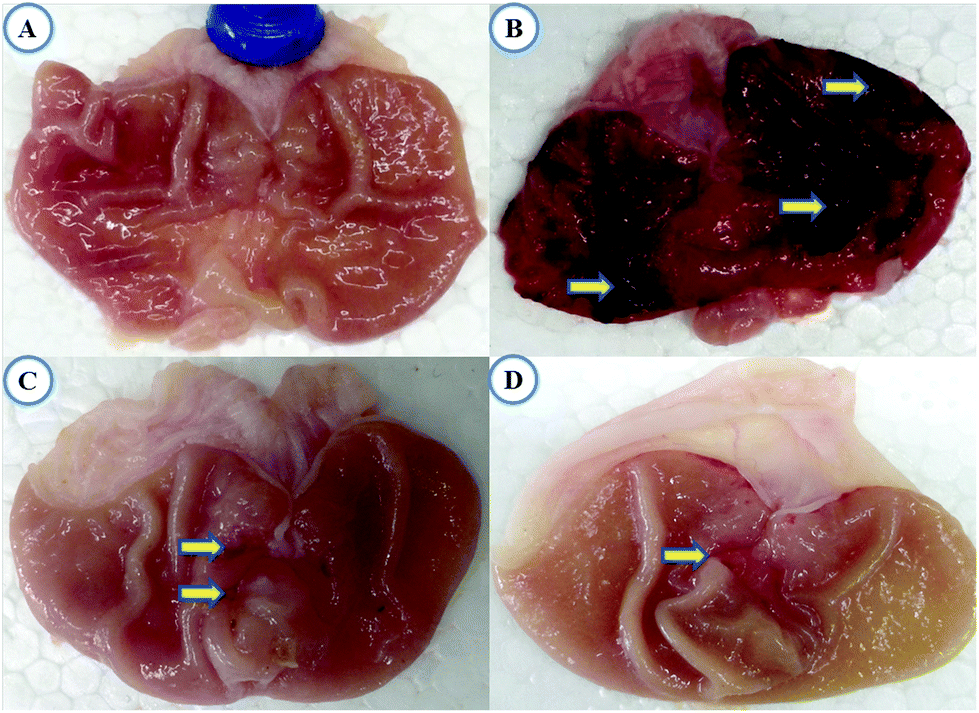 | ||
| Fig. 1 Macroscopic appearance of the gastric mucosa. (A) Normal group, (B) indomethacin-induced ulcer group, (C) TRG (45 mg kg−1) pretreated group, and (D) OMP (40 mg kg−1) pretreated group. Indomethacin-induced severe injuries to the gastric mucosa that appear as elongated bands of hemorrhage (yellow arrow). Note: OMP (omeprazole); TRG (trigonelline). | ||
Fig. 2 represents the effective dose determination. In comparison with the indomethacin treated group, TRG at 45 mg kg−1 provided a significant gastroprotective effect, inhibiting the gastric ulcer by 81.71%, which does not vary statistically from upper doses such as 60 and 75 mg kg−1. Therefore, 45 mg kg−1 was selected as the lowermost effective dose of TRG.
 | ||
| Fig. 2 Effect of TRG (15, 30, 45, 60 and 75 mg kg−1) on the indomethacin-induced ulcer index in rats. Values are mean ± SD (n = 6). †P < 0.05 compares IND with all the groups; nsP < 0.05 compare TRG 45 mg kg−1 with TRG 60 and 75 mg kg−1. Note: OMP (omeprazole); TRG (trigonelline); IND (indomethacin). | ||
Levels of mucosal SOD, CAT, GSH-px and PGE2 were reduced in the indomethacin group 3.36, 2.62, 1.84 and 1.94 fold respectively as compared to normal rats. These levels were reverted by TRG (45 mg kg−1) pretreatment that was 3.15 (SOD), 2.31 (CAT), 1.86 (GSH-px) and 1.69 (PGE2) fold. On the other hand, MDA, MPO and LTB4 levels were significantly increased in the indomethacin group as compared with the normal group 2.17, 3.94 and 1.52 fold respectively. However, pretreatment of TRG (45 mg kg−1) significantly reduced MDA (2.07 fold), MPO (3.50 fold) and LTB4 (1.58 fold) levels compared to the indomethacin group (Fig. 3A–C).
 | ||
| Fig. 3 (A) Effect of TRG (45 mg kg−1) on gastric SOD, CAT and GSH-Px levels, (B) MDA and MPO levels, (C) PGE2, and LTB4 levels in indomethacin-induced ulcerated rats. Values are mean ± SD (n = 6). †P < 0.05 compares IND with all the groups. Note: TRG (trigonelline); IND (indomethacin). | ||
TNF-α (14.78 fold), IFN-γ (3.16 fold), IL-1β (2.25 fold) and IL-6 (7.98 fold) levels were significantly increased and IL-4 (6.05 fold) and IL-10 (1.91 fold) levels were considerably reduced in the indomethacin group as compared to the normal group. Pretreatment of TRG (45 mg kg−1) significantly reduced TNF-α, IFN-γ, IL-1β, and IL-6 levels 12.47, 2.34, 2.03 and 7.11 fold respectively as compared to the indomethacin-induced ulcerated group (Fig. 4A and B).
 | ||
| Fig. 4 (A) Effect of TRG (45 mg kg−1) on gastric TNF-α and IFN-γ levels, (B) IL-6 and IL-1β levels, (C) IL-10 and IL-4 levels in indomethacin-induced ulcerated rats. Values are mean ± SD (n = 6). †P < 0.05 compares IND with all the groups. Note: TRG (trigonelline); IND (indomethacin). | ||
iNOS (12.62 fold) and TNF-α (4.18 fold) protein expression levels were significantly increased and eNOS (3.88 fold), COX-1 (17.15 fold) and IL-10 (1.56 fold) levels were considerably reduced in the indomethacin group as compared to the normal group. Pretreatment of TRG (45 mg kg−1) significantly reduced iNOS and TNF-α levels 3.46 and 4.59 fold respectively. The levels of eNOS (5.13 fold), COX-1 (16.92 fold) and IL-10 (2.84 fold) were significantly increased in the TRG pretreated group compared to the indomethacin-induced ulcerated group (Fig. 5).
 | ||
| Fig. 5 Effect of TRG (45 mg kg−1) on the protein expression level of eNOS, iNOS, COX-1, IL-10 and TNF-α in gastric mucosa. Levels of proteins of interest were normalized to the level of β-actin. Data are expressed as mean ± SD (†P < 0.05 when compared to the IND group). Note: TRG (trigonelline); IND (indomethacin). | ||
ICAM-1, VCAM-1, P-selectin and E-selectin levels were considerably elevated following the administration of indomethacin reaching 4.50, 2.40, 4.60 and 3.79 fold respectively as compared to normal rats. The TRG (45 mg kg−1) pretreated group showed significantly reduced levels of ICAM-1 (2.59 fold), VCAM-1 (1.97 fold), P-selectin (2.14 fold) and E-selectin (2.30 fold) as compared to the indomethacin-induced ulcerated group (Fig. 6A and B).
 | ||
| Fig. 6 (A) Effect of TRG (45 mg kg−1) on gastric ICAM-1 and VCAM-1 levels, (B) E-selectin and P-selectin levels, (C) VEGF, HGF and EGF levels, (D) cNOS, iNOS, tNOS and NO levels in indomethacin-induced ulcerated rats. Values are mean ± SD (n = 6). †P < 0.05 compares IND with all the groups. Note: TRG (trigonelline); IND (indomethacin). | ||
Indomethacin-induced ulcerated rats show significant increases of iNOS (11.33 fold) and NO (2.52 fold) levels, as compared with normal rats. However, the TRG (45 mg kg−1) pretreated animals show a 5.66 and 1.86 fold decline in the levels of iNOS and NO respectively as compared to the indomethacin group (Fig. 6C and D).
Levels of apoptosis (6.24 fold), caspase-3 (10.27 fold), NF-κB p65 (22.33 fold) and NF-κB p50 (14.20 fold) were significantly elevated in the indomethacin-induced ulcerated group compared to normal animals. However, pretreatment of TRG (45 mg kg−1) significantly reduced apoptosis, caspase-3, NF-κB p65 and NF-κB p50 levels 5.63, 3.52, 2.91 and 3.73 fold respectively (Fig. 7A and B).
 | ||
| Fig. 7 (A) Effect of TRG (45 mg kg−1) on caspase-3 and apoptosis levels, (B) p50 and p65 levels, (C) gastric microvascular permeability levels in indomethacin-induced ulcerated rats. Values are mean ± SD (n = 6). †P < 0.05 compare IND with all the groups; *P < 0.05 compare TRG (45 mg kg−1) + IND with SC560 + TRG (45 mg kg−1) + IND and celecoxib + TRG (45 mg kg−1) + IND. Note: TRG (trigonelline); IND (indomethacin). | ||
The microvascular permeability level was significantly increased in the indomethacin group compared to the normal group. Pretreatment of TRG (45 mg kg−1) reduced the level of microvascular permeability (77.17%). However, administration of SC560 decreased the percentage of inhibition of microvascular permeability from 77.17% to 3.62%; whereas, treatment of celecoxib did not affect the TRG activity against microvascular permeability (Fig. 7C).
Administration of SC560, L-NAME, and NEM significantly reduced the ulcer index inhibition percentage elicited by TRG (45 mg kg−1) from 81.71% to −6.52%, 3.52%, and −0.87% respectively. But, the treatment of celecoxib, yohimbine and glibenclamide did not affect the ulcer protective activity of TRG (Fig. 8).
 | ||
| Fig. 8 Effect of SC560 (COX-I specific inhibitor), celecoxib (COX-II specific inhibitor), YO (α2-receptors antagonist), L-NAME (NOS inhibitor), NEM (endogenous sulfhydryl antagonist) and GLIB (K+ATP channels antagonist) on ulcer protective effects of TRG (45 mg kg−1) against indomethacin-induced ulcer. Values are mean ± SD (n = 6). †P < 0.05 compare IND with all the groups; *P < 0.05 compare TRG (45 mg kg−1) + IND with SC560 + TRG (45 mg kg−1) + IND or celecoxib + TRG (45 mg kg−1) + IND or YO + TRG (45 mg kg−1) + IND or L-NAME + TRG (45 mg kg−1) + IND or NEM + TRG (45 mg kg−1) + IND or GLIB + TRG (45 mg kg−1) + IND. Note: TRG (trigonelline); IND (indomethacin); L-NAME (Nω-nitro-L-arginine methyl ester); NEM (N-ethylmaleimide); YO (yohimbine); GLIB (glibenclamide). | ||
Discussion
Our knowledge about NSAID-induced ulceration has progressed significantly from a basic understanding in the past 10 years. However, these advancements in knowledge have not translated into widespread applications in a clinical setting. This study will address the gastroprotective activity of TRG against indomethacin-induced gastric ulcer along with the underlying mechanisms.Prostaglandins (PGs) have a crucial role in the preservation of physiological processes including mucosal blood flow, angiogenesis, and mucus and bicarbonate secretions.34 PGs were synthesized by cyclooxygenase-I (COX-I) and cyclooxygenase-II (COX-II) isozymes. Since indomethacin is a non-specific COX inhibitor, it causes gastric ulceration and intensifies gastric ulcers already present in humans and rodents through suppression of PG synthesis.8 Consistent with these results, our investigational outcomes revealed that exposure of indomethacin significantly reduced gastric mucosal PGE2 levels as compared to normal rats. However, pretreatment of TRG (45 mg kg−1) significantly increased PGE2 levels as compared to indomethacin-induced ulcerated rats. In this study that the gastroprotective activity of TRG has been reverted by SC560 (COX-I selective inhibitor) and not by celecoxib (COX-II selective inhibitor) indicates the involvement of COX-I synthesized PGs in TRG affording gastroprotection. This observation was consistent with previous reports.21,35
Activated neutrophils produce myeloperoxidase (MPO), cytokines, reactive oxygen species (ROS) and reactive nitrogen species (RNS) which have been responsible for oxidative stress in gastric endothelial cells. Since MPO has prominently been produced by neutrophils, infiltration of neutrophils into the endothelium was identified via quantification of the MPO level. Previous experiments stated that the MPO activity was elevated on indomethacin-induced gastric ulcer.21,35 Taken together, the present data indicate that TRG significantly reduced MPO activity as compared to that in indomethacin-induced ulcerated animals.
Previous experiments explicate an increase in pro-inflammatory cytokines and a decline of anti-inflammatory cytokines during a gastric ulcer.21,35 Consistent with these findings, this work revealed that administration of indomethacin significantly increased pro-inflammatory cytokines (TNF-α, IFN-γ, IL-1β and IL-6) and decreased anti-inflammatory cytokines (IL-10 and IL-4) level; however, pretreatment of TRG significantly reverted these markers to the normal level. Among the pro-inflammatory cytokines TNF-α possesses multiple pathophysiological roles in gastric ulcer including activation of NF-κB, apoptosis, iNOS and neutrophil infiltration. Similarly, IL-6 is another important pro-inflammatory cytokine, activating PMNs at the inflammatory site, and triggering the oxidative pathway responsible for tissue damage during gastric ulcer.34,36 Nonetheless, IL-10 is a vital anti-inflammatory and immunosuppressive cytokine able to inhibit TNF-α production. That TRG considerably inhibits TNF-α and IL-6 along with augmenting IL-10 elucidates its anti-inflammatory nature.
Neutrophilic PMNs deal with vascular endothelium in vastly coordinated manners that consist of leukocyte rolling, arrest, firm adhesion, and diapedesis. This interaction occurs under high shear stresses within venules and depends on multiple families of adhesion molecules including ICAM-1, VCAM-1, P-selectin and E-selectin.37 Adhesion molecules facilitated the transendothelial migration of neutrophils into the site of gastric tissue injury. In this study, levels of adhesion molecules were considerably elevated in indomethacin-induced ulcerated animals. However, TRG pretreatment reduces the pathological levels of these adhesion molecules to normal. This result was in agreement with previous studies.34,38
The lipid peroxidation level in gastric tissue was measured by determining the quantity of MDA; this investigation may convey the level of gastric tissue injury.39 Our findings show significant increases of MDA in the indomethacin-induced group, however, pretreatment of TRG significantly prevented MDA production. Superoxide dismutase (SOD) converted superoxide anions (O2−) to hydrogen peroxide which in turn is detoxified by glutathione peroxidase (GSH-px) and catalase (CAT). These enzymes constitute an endogenous antioxidant system, and prevent cell damage induced by ROS.40 In this study, TRG significantly increased the levels of SOD, GSH-px and CAT compared with the indomethacin-induced ulcerated group which suggests its endogenous antioxidant stimulatory potential against indomethacin-induced ulcers.
NF-κB is believed to play a pivotal role in the inducible expression of many genes, including TNF-α, IL-1β, IL-6, iNOS and adhesion molecules.41,42 In this study, indomethacin-induced ulcerated animals showed significant increases of both p50 and p65 subunits, nonetheless, pretreatment of TRG considerably decreased these pathophysiological markers to the normal level. This result was in agreement with previous reports.43,44
Nitric oxide (NO) plays an important role in controlling numerous components of mucosal defense, including increased gastric mucus secretion, blood flow, and reduced neutrophil adhesion.39,40 A previous report shows considerable increases of iNOS in indomethacin-induced ulcerated rats than in normal rats.35 In agreement with this finding, the present results revealed significant increases of iNOS and NO in the indomethacin group compared to normal rats. However, pretreatment of TRG significantly reverted the levels of iNOS and NO compared to the indomethacin group. A previous study indicated that L-NAME, a nonspecific NOS inhibitor, augmented indomethacin-induced gastric injury in rats;45 consistent with this finding, the present work revealed that pretreatment of L-NAME considerably decreased the ulcer protective efficiency of TRG and simultaneously increased the ulcer index.
VEGF as a growth factor elicits endothelial proliferation, migration and ulcer healing via stimulation of angiogenesis.21 Similarly, HGF supports the angiogenesis process by multiple mechanisms including COX activation and increases EGF expression that is essential for the acceleration of ulcer healing by stimulating cell migration and proliferation in epithelial cell monolayers, repairing tissue, and diminishing gastric acid secretion.46 The current study shows that indomethacin administration significantly decreased the mucosal VEGF, HGF, and EGF levels compared to normal rats. However, pretreatment of TRG considerably augmented growth factor levels. These outcomes are in agreement with previous studies.21,35
In indomethacin-induced ulcers, apoptosis is another vital pathophysiological pathway. In the present study, TRG showed a substantial decline of caspase-3 and apoptosis in indomethacin-induced ulcer. Previous results explore that apoptosis was stimulated by lipid peroxidation and TNF-α and inhibited by PGE2.40 The present study shows that TRG significantly increased the production of PGE2 and reduced lipid peroxidation and TNF-α levels explicate the possibility that the decline of apoptosis and caspase-3 in the TRG-pretreated group is due to the augmentation of PGE2 and inhibition of lipid peroxidation and TNF-α.
Intestinal permeability is thought to be a central and essential mechanism for translating the biochemical/cellular events of NSAIDs into tissue reaction in the small bowel.47 Indeed, elevated microvascular permeability was observed in indomethacin-induced animals. However, this condition has been reduced by TRG treatment. However, treatment of SC560 significantly affects the activity of TRG against vascular permeability. However, treatment of celecoxib did not alter TRG activity. These observations are in agreement with previous reports.35,48
Nonprotein endogenous NP-SH compounds bind with free radicals generated by ulcerogens including indomethacin and finally detoxify them. NP-SH compounds are also able to control mucus production, and the recycling of antioxidants.49,50 Our results show significant inhibition of the protective effects of TRG after NEM treatment in comparison with the TRG treated animals, indicating that the gastroprotective effects of TRG are at least partly mediated by NP-SH compounds. In the stomach physiological functions such as gastric blood flow regulation, acid secretion, and stomach contractility have been mediated by the opening of K+ATP channels, a class of ligand-gated proteins.39 In this study, the gastroprotective mechanism of TRG was K+ATP-channel independent, since its protective activity was not affected by pretreatment with glibenclamide, a potent antagonist of these channels. Presynaptic α2-receptors regulate different activities in the gastrointestinal tract including the regulation of gastric acid secretion.51 Pretreatment of yohimbine (α2-receptor antagonist) could not block the protective effects of TRG against indomethacin-induced ulcers, indicating that α2-receptors were not involved in the gastroprotective effects of TRG.
Conclusions
To sum up, this is the first report to determine the defensive effects of TRG against the indomethacin-induced gastric ulcer model in rats. Overall pieces of evidence were depicted in this study which showed that TRG vividly overcomes oxidative stress, cytokine imbalance, inflammation and apoptosis through augmenting the activities of antioxidant enzymes, preventing the production of inflammatory markers, and inhibition of microvascular permeability and anti-apoptotic activities. From these total observations, we concluded that TRG has a solid preventive potential against gastric ulcers induced by indomethacin.Conflict of interest
The author(s) declare(s) that they have no conflicts of interest to disclose.Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2012R1A1A4A01011658). The authors would like to extend their sincere appreciation to the Deanship of Scientific Research at King Saud University for its funding of this research through the Research Group project no. (RG-1435-071).References
- D. E. Furst, R. W. Ulrich and S. Prakash, Basic and clinical pharmacology, McGraw Hill, United States, 12th edn, 2012 Search PubMed.
- M. A. Morsy and M. A. El-Moselhy, Pharmacology, 2013, 91, 267 CrossRef CAS PubMed.
- C. Musumba, D. M. Pritchard and M. Pirmohamed, Aliment. Pharmacol. Ther., 2009, 30, 517 CrossRef CAS PubMed.
- A. A. Asmari, S. A. Omani, M. A. Otaibi, A. A. Abdulaaly, I. Elfaki, K. A. Yahya and M. Arshaduddin, Int. J. Clin. Exp. Med., 2014, 7, 586 Search PubMed.
- W. I. E. Hinojosa, M. A. Quiroz, I. R. Alvarez, P. E. Castaneda, M. L. Villarreal and A. C. Taketa, J. Ethnopharmacol., 2014, 155, 1156 CrossRef PubMed.
- S. K. Yadav, B. Adhikary, S. Chand, B. Maity, S. K. Bandyopadhyay and S. Chattopadhyay, Free Radicals Biol. Med., 2012, 52, 1175 CrossRef CAS PubMed.
- K. Takeuchi, World J. Gastroenterol., 2012, 18, 2147 CrossRef CAS PubMed.
- F. Halter, A. S. Tarnawski, A. Schmassmann and B. M. Peskar, Gut, 2001, 49, 443 CrossRef CAS.
- T. Yoshikawa, Y. Naito, A. Kishi, T. Tomii, T. Kaneko, S. Iinuma, H. Ichikawa, M. Yasuda, S. Takahashi and M. Kondo, Gut, 1993, 34, 732 CrossRef CAS.
- S. Fiorucci, E. Antonelli and A. Morelli, Dig. Liver Dis., 2001, 31, S35 CrossRef.
- J. L. Wallace, Curr. Opin. Pharmacol., 2005, 5, 573 CrossRef CAS PubMed.
- F. Borrelli and A. A. Izzo, Phytother. Res., 2000, 14, 581 CrossRef CAS.
- S. Shailajan, S. Menon, A. Singh, M. Mhatre and N. Sayed, Pharm. Methods, 2011, 2, 157 CrossRef PubMed.
- C. Tohda, T. Kuboyama and K. Komatsu, Neurosignals, 2005, 14, 34 CrossRef CAS PubMed.
- O. Yoshinari and K. Igarashi, Curr. Med. Chem., 2010, 17, 2196 CrossRef CAS.
- J. Zhou, S. Zhou and S. Zeng, Fundam. Clin. Pharmacol., 2013, 27, 279 CrossRef CAS PubMed.
- S. Ilavenil, M. V. Arasu, L. Jeong-Chae, D. H. Kim, S. G. Roh, H. S. Park, G. J. Choi, V. Mayakrishnan and K. C. Choi, Phytomedicine, 2014, 21, 758 CrossRef CAS PubMed.
- C. Tohda, N. Nakamura, K. Komatsu and M. Hattori, Biol. Pharm. Bull., 1999, 22, 679 CAS.
- S. Acharya, K. Acharya, S. Paul and S. Basu, Can. J. Plant Sci., 2011, 105, 91 Search PubMed.
- T. Bakuradze, R. Lang, T. Hofmann, H. Stiebitz, G. Bytof, I. Lantz, M. Baum, G. Eisenbrand and C. Janzowski, Mol. Nutr. Food Res., 2010, 54, 1734 CAS.
- P. Antonisamy, M. Dhanasekaran, S. Ignacimuthu, V. Duraipandiyan, J. D. Balthazar, P. Agastian and J. H. Kim, Phytomedicine, 2014, 21, 966 CrossRef CAS PubMed.
- U. Valcavi, R. Caponi, A. Brambilla, M. Palmira, F. Minoja, F. Bernini, R. Musanti and R. Fumagalli, Arzneim. Forsch., 1982, 32, 657 CAS.
- S. Demirbilek, I. Gürses, N. Sezgin, A. Karaman and N. Gürbüz, J. Pediatr. Surg., 2004, 9, 57 CrossRef PubMed.
- K. M. Mullane, R. Kraemer and B. Smith, J. Pharmacol. Methods, 1985, 14, 157 CrossRef CAS.
- C. C. Winterbourn, R. E. Hawkins, M. Brian and R. W. Carrell, J. Lab. Clin. Med., 1975, 85, 337 CAS.
- H. Aebi, Methods Enzymol., 1984, 105, 121 CAS.
- M. Mihara and M. Uchiyama, Anal. Biochem., 1978, 86, 271 CrossRef CAS.
- D. Paglia and W. Valentine, J. Lab. Clin. Med., 1967, 70, 158 CAS.
- B. L. Slomiany and A. Slomiany, IUBMB Life, 2004, 56, 41 CrossRef CAS PubMed.
- M. Feelisch and E. A. Noack, Eur. J. Pharmacol., 1987, 139, 19 CrossRef CAS.
- R. G. Knowles, M. Merrett, M. Salter and S. Moncada, Biochem. J., 1990, 270, 833 CrossRef CAS.
- K. M. Miranda, M. G. Espey and D. A. Wink, Nitric Oxide, 2001, 5, 62 CrossRef CAS PubMed.
- C. L. Chander, A. R. Moore, F. M. Desa, D. Howat and D. A. Willoughby, J. Pharm. Pharmacol., 1988, 40, 745 CrossRef CAS PubMed.
- B. Adhikary, S. K. Yadav, S. K. Bandyopadhyay and S. Chattopadhyay, Food Funct., 2011, 2, 338 CAS.
- P. Antonisamy, P. Kannan, A. Aravinthan, V. Duraipandiyan, M. V. Arasu, S. Ignacimuthu, N. A. Al-Dhabi and J. H. Kim, Sci. World J., 2014, 1 CrossRef PubMed.
- A. L. Rozza, F. Meira de Faria, A. R. Souza Brito and C. H. Pellizzon, PLoS One, 2014, 9, e86686 Search PubMed.
- S. Chakraborty, S. K. Yadav, M. Subramanian, M. Iwaoka and S. Chattopadhyay, Biochim. Biophys. Acta, 2014, 1840, 3385 CrossRef CAS PubMed.
- D. Thong-Ngam, S. Choochuai, S. Patumraj, M. Chayanupatkul and N. Klaikeaw, World J. Gastroenterol., 2012, 18, 1479 CrossRef CAS PubMed.
- P. Antonisamy, V. Duraipandiyan, A. Aravinthan, N. A. Al-Dhabi, S. Ignacimuthu, K. C. Choi and J. H. Kim, Eur. J. Pharmacol., 2015, 750, 167 CrossRef CAS PubMed.
- P. Antonisamy, P. Subash-Babu, A. A. Alshatwi, A. Aravinthan, S. Ignacimuthu, K. C. Choi and J. H. Kim, Chem.–Biol. Interact., 2014, 224, 157 CrossRef CAS PubMed.
- U. Raju, R. Lu, F. Noel, G. J. Gumin and P. J. Tofilon, J. Biol. Chem., 1997, 272, 24624 CrossRef CAS PubMed.
- D. Zhou, T. Yu, G. Chen, S. A. Brown, Z. Yu, M. P. Mattson and J. S. Thompson, Int. J. Radiat. Biol., 2001, 77, 763 CrossRef CAS PubMed.
- C. Linard, C. Marquette, J. Mathieu, A. Pennequin, D. Clarençon and D. Mathé, Int. J. Radiat. Oncol., Biol., Phys., 2004, 58, 427 CrossRef CAS PubMed.
- K. Uehara, S. Miura, T. Takeuchi, T. Taki, M. Nakashita, M. Adachi, T. Inamura, T. Ogawa, Y. Akiba, H. Suzuki, H. Nagata and H. Ishii, J. Pharmacol. Exp. Ther., 2003, 305, 232 CrossRef CAS PubMed.
- M. M. Khattab, M. Z. Gad and D. Abdallah, Pharmacol. Res., 2001, 43, 463 CrossRef CAS PubMed.
- T. Brzozowski, P. C. Konturek, S. J. Konturek, R. Pajdo, D. Schuppan, D. Drozdowicz, A. Ptak, M. Pawlik, T. Nakamura and E. G. Hahn, J. Physiol. Pharmacol., 2000, 51, 751 CAS.
- I. Bjarnason and K. Takeuchi, J. Gastroenterol., 2009, 44, S23 CrossRef PubMed.
- A. Tanaka, S. Hase, T. Miyazawa, R. Ohno and K. Takeuchi, J. Pharmacol. Exp. Ther., 2002, 303, 1248 CrossRef CAS PubMed.
- A. L. Rozza, M. M. Tde, H. Kushima, A. Tanimoto, M. O. Marques, T. M. Bauab, C. A. Hiruma-Lima and C. H. Pellizzon, Chem.–Biol. Interact., 2011, 189, 82 CrossRef CAS PubMed.
- P. Antonisamy, V. Duraipandiyan, S. Ignacimuthu and J. H. Kim, South Ind. J. Biol. Sci., 2015, 1, 34 Search PubMed.
- K. Gyires, K. Müllner, S. Fürst and A. Z. Rónai, J. Physiol. (Paris), Suppl., 2000, 94, 117 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |