
Hydroxytyrosol and tyrosol sulfate metabolites protect against the oxidized cholesterol pro-oxidant effect in Caco-2 human enterocyte-like cells†
Angela
Atzeri
a,
Ricardo
Lucas
b,
Alessandra
Incani
a,
Pablo
Peñalver
c,
Alberto
Zafra-Gómez
d,
M. Paola
Melis
a,
Roberto
Pizzala
e,
Juan C.
Morales
*bc and
Monica
Deiana
*a
aDipartimento di Scienze Biomediche, Unità di Patologia Sperimentale, Università degli Studi di Cagliari, Cittadella Universitaria SS 554, 09042 Monserrato, Cagliari, Italy. E-mail: mdeiana@unica.it; Fax: +39 070 6754032; Tel: +39 070 6754126
bDepartment of Bioorganic Chemistry, Instituto de Investigaciones Químicas, CSIC-Universidad de Sevilla, 49 Americo Vespucio, 41092 Sevilla, Spain
cInstitute of Parasitology and Biomedicine López Neyra, Avenida del Conocimiento, 17, 18016 Armilla-Granada, Spain. E-mail: jcmorales@ipb.csic.es; Fax: +34 95 8181632; Tel: +34 95 8181644
dDepartment of Analytical Chemistry, Faculty of Sciences, University of Granada, C/Severo Ochoa, s/n, 18001 Granada, Spain
eDipartimento di Medicina Molecolare, Università degli Studi di Pavia, Via Ferrata 9, 27100 Pavia, Italy
First published on 8th October 2015
Abstract
The aim of this study was to investigate the ability of the sulfate metabolites of hydroxytyrosol (HT) and tyrosol (TYR) to act as antioxidants counteracting the pro-oxidant effect of oxidized cholesterol in intestinal cells. For this purpose, we synthesized sulfate metabolites of HT and TYR using a chemical methodology and examined their antioxidant activity in Caco-2 monolayers in comparison with the parent compounds. Exposure to oxidized cholesterol led to ROS production, oxidative damage, as indicated by the MDA increase, a decrease of reduced glutathione concentration and an enhancement of glutathione peroxidase activity. All the tested compounds were able to counteract the oxidizing action of oxidized cholesterol; HT and TYR sulfate metabolites showed an efficiency in protecting intestinal cells comparable to that of the parent compounds, strengthening the assumption that the potential beneficial effect of the parent compounds is retained, although extensive metabolisation occurs, the resulting metabolites being able to exert a biological action themselves.
Introduction
Polyphenols present in extra virgin olive oil, the principal fat component of the Mediterranean diet, have been demonstrated to exert many potentially beneficial biological effects due to, at least in part, their ability to protect against oxidative stress mediated damage.1,2 Hydroxytyrosol (3,4-dihydroxyphenylethanol; HT) and tyrosol (4-hydroxyphenylethanol; TYR) (Fig. 1) are the most biologically active phenolic alcohols present in the oil, where they exist in simple forms or as conjugates (such as oleuropein); they are structurally identical except that HT has an extra hydroxyl group at the meta position. | ||
| Fig. 1 Structure of hydroxytyrosol (HT), tyrosol (TYR) and their sulfate metabolites 3–5. | ||
The literature on phenolic absorption and metabolism after olive oil ingestion is extensive;3,4 however, there are still several issues to be clarified. Some complex olive oil polyphenols, among the secoiridoids, are relatively stable under gastric conditions and reach the intestine where they may be directly absorbed or metabolised under absorption;5 most of the complex olive oil polyphenols however seem to undergo gastrointestinal biotransformation (see de Bock for an excellent review),6 increasing the relative amount of simple phenols, mainly TYR and HT, entering the small and large intestine.7 At this location the concentration of simple phenols may be quite high, in the high μM range,7 and they might exert direct protective effects by scavenging reactive species and/or preventing their formation.8 However, in the process of crossing enterocytes olive oil phenolic compounds are subjected to classical phase I/II biotransformation and to an important first pass metabolism, resulting in almost undetectable concentrations of free HT and TYR in body fluids.9
Sulfated and glucuronidated HT and TYR are the predominant metabolites found in human plasma and urine,10,11 and they have also been shown to concentrate in the intestinal epithelium, since glucuronidation and sulfation are the major pathways of phase II xenobiotic metabolism in the human intestine.12,13 The potential health benefits of HT, TYR and their derivatives are likely to be due to both parental compounds and their phase I and phase II major metabolites. Glucuronidated metabolites of HT showed a more efficient radical scavenging potency than HT itself11 and the ability to protect renal cells14 and erythrocytes from oxidative injury was also better than the parent compound.15 However no data are available to date concerning the antioxidant activity of sulfate metabolites of HT and TYR.
In this study we investigated the possible protective effect of the sulfate metabolites of TYR and HT, in comparison with the parent compounds, against the oxidative damage to intestinal mucosa due to oxidized cholesterol exposure in the human colon adenocarcinoma cell line, Caco-2. After confluence, these cells spontaneously undergo full differentiation in vitro with enterocyte-like features.16 Caco-2 cells have been recognized as a suitable model for evaluating the effect of nutrient components, for both normal dietary constituents and toxicants, as oxidizing agents.17 Large amounts of lipid oxidation products become available, of both exogenous and endogenous origin, at the level of the intestinal mucosa. Dietary oxysterols, derived from cholesterol degradation and oxidation after prolonged storage or cooking of foods rich in cholesterol, have recently been shown to contribute to the onset and further development of oxidative stress and inflammation related intestinal diseases.18,19 The major oxysterols found in food may contribute to the oxidative unbalance of the intestinal epithelium by inducing the generation of reactive oxygen species (ROS).18,20 Our purpose was to investigate the ability of sulfate metabolites of HT and TYR to act as antioxidants counteracting the oxysterol induced changes of the cellular redox state and compare them with their parent compounds. For this purpose, we synthesized sulfate metabolites of HT and TYR using a chemical methodology and evaluated their uptake/stability in Caco-2 monolayers. Cytotoxicity, production of MDA and ROS species, and levels of glutathione and glutathione peroxidase activity were measured to assess the protective effect of the sulfate metabolites in comparison with their parent phenolic compounds.
Materials and methods
Preparation of oxidized cholesterol
Oxidized cholesterol was prepared essentially as previously described.21 In brief, 2 ml aliquots of pure cholesterol (Sigma Aldrich, St. Louis, MO) solution (5 mg ml−1 in EtOH) were dried down into round-bottomed glass test tubes under vacuum and heated in a 140 °C oil bath in air for 3 h. The oxidized mixture was separated and analysed by GC-MS for the determination of the relative oxysterols. Briefly, 2 μL of the mixture was diluted in 2 mL of EtOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 dilution). After evaporating under a nitrogen stream, 50 μL of pyridine (Sigma Aldrich) and 50 μL of N,O-bis(trimethylsilyl)trifluoroacetamide with trimethylchlorosilane (BSTFA:TMCS; 99:1) (Sigma Aldrich) were added, mixed and kept at 60 °C for 45 min. The derivatized sample (1.5 μL) was injected into an Agilent (Waldbronn, DE) GC-MS in splitless mode and the column temperature was programmed starting from 70 °C to 250 °C (10 °C per minute), after up to 290 °C (5 °C min−1) maintained for 10 min, ending at 300 °C (2 °C min−1) for 5 min (duration about 45 min total). The helium flow was set at 1.3 mL min−1. The specific parameters of the instrument were as published by Calderon-Santiago et al.22 Spectra were acquired in scan mode.
:1000 dilution). After evaporating under a nitrogen stream, 50 μL of pyridine (Sigma Aldrich) and 50 μL of N,O-bis(trimethylsilyl)trifluoroacetamide with trimethylchlorosilane (BSTFA:TMCS; 99:1) (Sigma Aldrich) were added, mixed and kept at 60 °C for 45 min. The derivatized sample (1.5 μL) was injected into an Agilent (Waldbronn, DE) GC-MS in splitless mode and the column temperature was programmed starting from 70 °C to 250 °C (10 °C per minute), after up to 290 °C (5 °C min−1) maintained for 10 min, ending at 300 °C (2 °C min−1) for 5 min (duration about 45 min total). The helium flow was set at 1.3 mL min−1. The specific parameters of the instrument were as published by Calderon-Santiago et al.22 Spectra were acquired in scan mode.
GS-MS analysis demonstrated that the mixture contained 58.9% of cholesterol, 13.5% of 7-ketocholesterol, 9.54% of 7β-hydroxycholesterol, 5.6% of 6β-hydroxycholesterol, 5.87% of 7α-hydroxycholesterol, 4.5% of cholesta-4,6-dien-3-ol and 2.1% of 3-keto-4-cholestene.
Synthesis of sulfate metabolites
:1) to obtain the corresponding triethylammonium salt as a white powder (94–98% yield).
:MeOH, 10:1) showed the formation of a major product and complete consumption of the initial material. Solvents were removed and the crude extract was purified by using Sephadex LH-20 (CH2Cl2:MeOH, 1:1) to afford 7 (92 mg, 98%) as a white powder. 1H-NMR (300 MHz, D2O) δ: 6.72, 7.18 (2d, 4H, J = 8.7 Hz, H arom), 4.24 (t, 2H, CH2OAc), 3.10 (q, 6H, –CH2CH3), 2.88 (t, 2H, CH2Ar), 2.19 (t, 2H, J = 7.5 Hz, CH2), 1.45 (m, 2H, CH2), 1.18 (t, 9H, CH2CH3), 0.74 (t, 3H, CH3); 13C-NMR (125 MHz, D2O) δ: 177.1 (CO), 150.0, 136.2, 130.3 (2 × CH arom), 121.3 (2 × CH arom), 65.4 (CH2OAc), 46.5, 36.0 (CH2Ar), 34.0, 18.3, 12.6, 8.0 (CH3). ESI-HRMS (ES−) Calcd for C12H15O6S (M − H) 287.0589, Found: 287.0594.
:MeOH (from 100:0 to 70:30). Fractions containing the desired product were concentrated and freeze-dried affording compound 5 as a white solid (68 mg, 94%). 1H-NMR (400 MHz, D2O) δ: 7.18, 6.86 (2d, 4H, J = 8.4 Hz, H arom), 3.78, 2.78 (2t, 4H, J = 6.7 Hz, CH2OH, CH2Ar); 13C-NMR (75 MHz, D2O) δ: 157 (Cq), 130.3 (2 × CH arom), 129.8 (Cq), 117.1 (2 × CH arom), 63.0 (CH2OH), 36.9 (CH2Ar). ESI-HRMS (ES−) Calcd for C8H9O5S (M − H) 217.0171, Found: 217.0171.
:ethyl acetate 3:1) at that point indicated that the reaction was complete. The pale yellow reaction mixture was diluted with EtOAc (100 mL), cast into a separatory funnel, and washed with water (2 × 50 mL) and brine (50 mL), and the organic phase was dried (Na2SO4). Filtration and concentration in a vacuum afforded the crude extract that was purified by flash column chromatography (hexane:ethyl acetate from 15:1 to 10:1) to afford 9 and 10 (314 mg, 90%, powder) like a regioisomeric mixture in the ratio of ∼1:1. 1H NMR (400 MHz, CDCl3) δ 6.88 (d, 1H, J = 8.1 Hz, H arom), 6.83 (s, 1H, H arom), 6.77 (d, 1H, J = 8.4 Hz, H arom), 6.73 (d, 1H, J = 8.4 Hz, H arom), 6.71 (s, 1H, H arom), 6.62 (d, 1H, J = 8.1 Hz, H arom), 5.52, 5.45 (2s, 2H, 2 × OH), 4.26 (t, 2H, J = 6.7 Hz, CH2OAc), 4.24 (t, 2H, J = 6.4 Hz, CH2OAc), 2.86 (t, 2H, J = 6.7 Hz, CH2Ar), 2.84 (t, 2H, J = 6.4 Hz, CH2Ar), 2.07–2.05 (2s, 6H, CH3C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1.05, 1.03 (2s, 18H, C(CH3)3 × 2), 0.30, 0.29 (2s, 12H, –Si(CH3)2 × 2); 13C-NMR (125 MHz, CDCl3) δ: 171.1, 171.0 (CO), 147.1, 145.9, 142.3, 141.0, 131.8, 129.6 (Cq arom), 122.4, 120.2, 118.6, 117.7, 115.4, 114.8 (CH arom), 65.2, 65.0 (CH2OAc), 34.5 (2 × CH2Ar), 25.7 (C(CH3)3), 21.0, 20.9 (CH3CO), 18.2 (C(CH3)3), −4.2 (Si(CH3)2); HRMS (ES+) Calcd for C16H26O4NaSi (M + Na) 333.1498, Found: 333.1508.
:MeOH 10:1) showed the formation of a major product and complete consumption of the starting material. Solvents were removed and the crude was purified by using Sephadex LH-20 in a solvent mixture of CH2Cl2:MeOH 1:1 to afford 11 and 12 (231 mg, 94%, powder) like a regioisomeric mixture in the ratio ∼1:1. 1H-NMR (400 MHz, CDCl3) δ: 7.50 (d, 1H, J = 7.9 Hz, H arom), 7.46 (s, 1H, H arom), 6.82 (d, 1H, J = 8.2 Hz, H arom), 6.78 (d, 1H, J = 8.2 Hz, H arom), 6.73 (d, 1H, J = 7.9 Hz, H arom), 6.72 (s, 1H, H arom), 4.20 (t, 4H, J = 7.08 Hz, CH2OAc), 3.10–3.00 (dq, 12H, CH2CH3), 2.83 (t, 2H, J = 7.1 Hz, CH2Ar), 2.82 (t, 2H, J = 7.05 Hz, CH2Ar), 2.04, 2.03 (2s, 6H, CH3CO), 1.26 (t, 18H, CH2CH3), 1.00, 0.99 (2s, 18H, C(CH3)3 × 2), 0.21, 0.20 (2s, 12H, –Si(CH3)2 × 2); 13C-NMR (125 MHz, CDCl3) δ: 171.0, 170.9 (CO), 146.8, 145.6, 143.9, 142.8, 134.0, 130.7 (Cq arom), 124.5, 122.4, 121.9, 121.8, 121.6, 121.0 (CH arom), 65.1 (CH2OAc), 46.3 (CH2CH3), 34.5 (CH2Ar), 25.7 (C(CH3)3), 21.0 (CH3CO), 18.8 (C(CH3)3), 8.8 (CH2CH3), −4.2 (Si(CH3)2); ESI-HRMS (ES−) Calcd for C16H25O7SiS (M − H) 389.1090, Found: 389.1092.
:MeOH (from 100:0 to 70:30). Fractions containing the desired product were concentrated and freeze-dried affording compounds 3 and 4 (115 mg, 90%, white powder) like a regioisomeric mixture in the ratio ∼1:1. 1H-NMR (300 MHz, D2O) δ: 7.21 (d, 2H, J = 8.0 Hz, H arom), 7.17 (s, 1H, H arom), 6.98 (d, 1H, J = 8.5 Hz, H arom), 6.88 (d, 1H, J = 8.5 Hz, H arom), 6.80 (s, 1H, H arom), 6.70 (d, 1H, J = 8.0 Hz, H arom), 3.76–3.69 (m, 4H, CH2OAc), 2.73–2.70 (m, 4H, CH2Ar); 13C-NMR (125 MHz, D2O) δ: 149.4, 147.4, 139.0, 138.4, 137.8, 130.7, 127.6, 123.0, 122.6, 120.0, 118.2, 117.6, 62.6, 62.4 (CH2OAc), 37.4, 36.9 (CH2Ar). HRMS-ESI (ES−) Calcd for C8H9O6S (M − H) 233.0120, Found: 233.0126.
O), 1.05, 1.03 (2s, 18H, C(CH3)3 × 2), 0.30, 0.29 (2s, 12H, –Si(CH3)2 × 2); 13C-NMR (125 MHz, CDCl3) δ: 171.1, 171.0 (CO), 147.1, 145.9, 142.3, 141.0, 131.8, 129.6 (Cq arom), 122.4, 120.2, 118.6, 117.7, 115.4, 114.8 (CH arom), 65.2, 65.0 (CH2OAc), 34.5 (2 × CH2Ar), 25.7 (C(CH3)3), 21.0, 20.9 (CH3CO), 18.2 (C(CH3)3), −4.2 (Si(CH3)2); HRMS (ES+) Calcd for C16H26O4NaSi (M + Na) 333.1498, Found: 333.1508.
:MeOH 10:1) showed the formation of a major product and complete consumption of the starting material. Solvents were removed and the crude was purified by using Sephadex LH-20 in a solvent mixture of CH2Cl2:MeOH 1:1 to afford 11 and 12 (231 mg, 94%, powder) like a regioisomeric mixture in the ratio ∼1:1. 1H-NMR (400 MHz, CDCl3) δ: 7.50 (d, 1H, J = 7.9 Hz, H arom), 7.46 (s, 1H, H arom), 6.82 (d, 1H, J = 8.2 Hz, H arom), 6.78 (d, 1H, J = 8.2 Hz, H arom), 6.73 (d, 1H, J = 7.9 Hz, H arom), 6.72 (s, 1H, H arom), 4.20 (t, 4H, J = 7.08 Hz, CH2OAc), 3.10–3.00 (dq, 12H, CH2CH3), 2.83 (t, 2H, J = 7.1 Hz, CH2Ar), 2.82 (t, 2H, J = 7.05 Hz, CH2Ar), 2.04, 2.03 (2s, 6H, CH3CO), 1.26 (t, 18H, CH2CH3), 1.00, 0.99 (2s, 18H, C(CH3)3 × 2), 0.21, 0.20 (2s, 12H, –Si(CH3)2 × 2); 13C-NMR (125 MHz, CDCl3) δ: 171.0, 170.9 (CO), 146.8, 145.6, 143.9, 142.8, 134.0, 130.7 (Cq arom), 124.5, 122.4, 121.9, 121.8, 121.6, 121.0 (CH arom), 65.1 (CH2OAc), 46.3 (CH2CH3), 34.5 (CH2Ar), 25.7 (C(CH3)3), 21.0 (CH3CO), 18.8 (C(CH3)3), 8.8 (CH2CH3), −4.2 (Si(CH3)2); ESI-HRMS (ES−) Calcd for C16H25O7SiS (M − H) 389.1090, Found: 389.1092.
:MeOH (from 100:0 to 70:30). Fractions containing the desired product were concentrated and freeze-dried affording compounds 3 and 4 (115 mg, 90%, white powder) like a regioisomeric mixture in the ratio ∼1:1. 1H-NMR (300 MHz, D2O) δ: 7.21 (d, 2H, J = 8.0 Hz, H arom), 7.17 (s, 1H, H arom), 6.98 (d, 1H, J = 8.5 Hz, H arom), 6.88 (d, 1H, J = 8.5 Hz, H arom), 6.80 (s, 1H, H arom), 6.70 (d, 1H, J = 8.0 Hz, H arom), 3.76–3.69 (m, 4H, CH2OAc), 2.73–2.70 (m, 4H, CH2Ar); 13C-NMR (125 MHz, D2O) δ: 149.4, 147.4, 139.0, 138.4, 137.8, 130.7, 127.6, 123.0, 122.6, 120.0, 118.2, 117.6, 62.6, 62.4 (CH2OAc), 37.4, 36.9 (CH2Ar). HRMS-ESI (ES−) Calcd for C8H9O6S (M − H) 233.0120, Found: 233.0126.
Cell culture
Cell culture materials were purchased from Invitrogen (Milano, Italy). Caco-2 cells were obtained from the European Collection of Cell Cultures (ECACC, Salisbury UK) and grown in Dulbecco's modified Eagle's medium (DMEM), supplemented with 2.5% of heat-inactivated bovine serum and 100 U mL−1 penicillin and 100 μg mL−1 streptomycin, at 37 °C under a humidified atmosphere of 95% air and 5% CO2. For experimental studies Caco-2 cells, at passage 45–60, were plated at a density of about 1 × 105 mL−1 and used 21 days post seeding.Cytotoxic activity
The cytotoxic effect caused by the exposure to the increasing concentrations of oxidized cholesterol and the protective effect of HT, TYR, and sulfate metabolites were assessed on Caco-2 cells, seeded in 24-well plates, by the neutral red method.25 Cells were exposed to oxidized cholesterol (0–175 μg mL−1) in complete medium and incubated for 24 h. In order to assess the protective effect of the phenolic compounds the cells were pretreated with the tested compounds (2.5–10 μM in water solution, 30 min) prior to oxidized cholesterol exposure (100 μg mL−1 for 24 h). After incubation, the medium was removed; a neutral red solution (Sigma Aldrich) (0.33% in medium) was then added to the wells to determine the cell viability. After 30 min of incubation, the neutral red solution was carefully removed and the cells were quickly rinsed with PBS. The incorporated dye was then solubilised in neutral red solubilisation solution, acetic acid/water/ethanol (1/49/50, v/v/v), and the absorbance was measured at 540 nm.Determination of MDA
MDA levels were determined in the medium of treated cells by the TBARS test with HPLC quantification, using the method described by Templar et al.,26 with some modifications. Briefly, 100 μL of 10% trichloroacetic acid (TCA, Sigma Aldrich) was added to 400 μL of the medium, and the samples were mixed and left at room temperature. After 20 min, 200 μL of 2-thiobarbituric acid (TBA, Sigma Aldrich) (0.6%) were added; samples were incubated at 90 °C for 45 min and then centrifuged at 5000g for 15 min at 4 °C. Aliquots of the supernatant were injected into an Agilent 1100 HPLC system (Agilent Technologies, Santa Clara, CA) equipped with a diode-array detector (HPLC–DAD) and separation was achieved using a Varian (Middelburg, The Netherlands) Inertsil 5 ODS-2, 150 × 4.6 mm column; the mobile phase used was a mixture of KH2PO4 50 mM pH 7/MeOH (65/35, v/v) at a constant flow rate of 1 mL min−1. The formation of the MDA–TBA adduct was revealed measuring its absorbance at 532 nm. A standard curve was prepared using a 1,1,3,3-tetraethoxypropane (TEP, Sigma Aldrich) solution in PBS (0.05–10 μM).ROS production in Caco-2 cells
Intracellular ROS production was monitored by adding 2′,7′-dichlorodihydrofluorescein diacetate (H2-DCF-DA, Sigma Aldrich), according to Dinicola et al.,27 in Caco-2 cells exposed to oxidized cholesterol (75 μg mL−1) in complete medium and incubated for 0–180 min and in cells pretreated with the tested phenolic compounds (5–25 μM in water solution, 30 min), prior to exposure to oxidized cholesterol (75 μg mL−1 for 30 min), with some modifications. After incubation the culture medium was replaced with PBS and cells were loaded with 10 μM H2-DCF-DA for 30 min. After incubation, PBS and H2-DCF-DA were removed and the cells were washed twice. The increase in cell fluorescence was measured at excitation and emission wavelengths of 485 and 530 nm, respectively, using an Infinite F200 auto microplate reader (Tecan, Salzburg, Austria), at 25 °C.Glutathione (GSH) level and glutathione peroxidase (GPx) activity
To assess the changes in the levels of GSH and GPx activity, the cells were seeded in plate dishes and exposed to oxidized cholesterol (75 μg mL−1) in complete medium for 0–24 h. In order to assess the effect of HT, TYR, HT-S and TYR-S, the cells were pretreated with the tested compounds (5–25 μM in water solution, 30 min) prior to exposure to oxidized cholesterol 75 μg mL−1 for 30 min to determine the GSH level and for 18 h to measure the GPx activity. At the end of the incubation time, cells were washed with PBS, and subsequently scraped into 500 μL of 5% metaphosphoric acid. Samples were then sonicated and centrifuged at 10000g for 20 min at 4 °C; supernatants were collected and used to determine the GSH level and GPx activity, using the Glutathione Assay Kit and the Glutathione peroxidase Assay Kit (Cayman Chemical Company, Ann Arbor, USA) according to the manufacturer's instructions.
Statistical analysis
Data are expressed as means ± S.D (n = 12 for each sample/condition). The statistical evaluation of the results was performed by analysis of variance (ANOVA) followed by a Bonferroni post-hoc test using GraphPad InStat version 3.05 (GraphPad Software, San Diego, CA, USA).Results
Synthesis of HT and TYR sulfate metabolites has been carried out using a protection–deprotection strategy together with the use of microwaves in the critical sulfation step. In the case of TYR, the primary alcohol was acyl-protected using immobilized lipase Novozym 435® and vinyl butyrate in tert-butyl methyl ether (98% yield) (Scheme 1). Next, microwave-assisted sulfation was performed by treatment with the SO3·NMe3 complex and triethylamine in acetonitrile at 100 °C (98% yield). The final deprotection with K2CO3 in MeOH afforded tyrosol sulfate 5 in good yield (94%).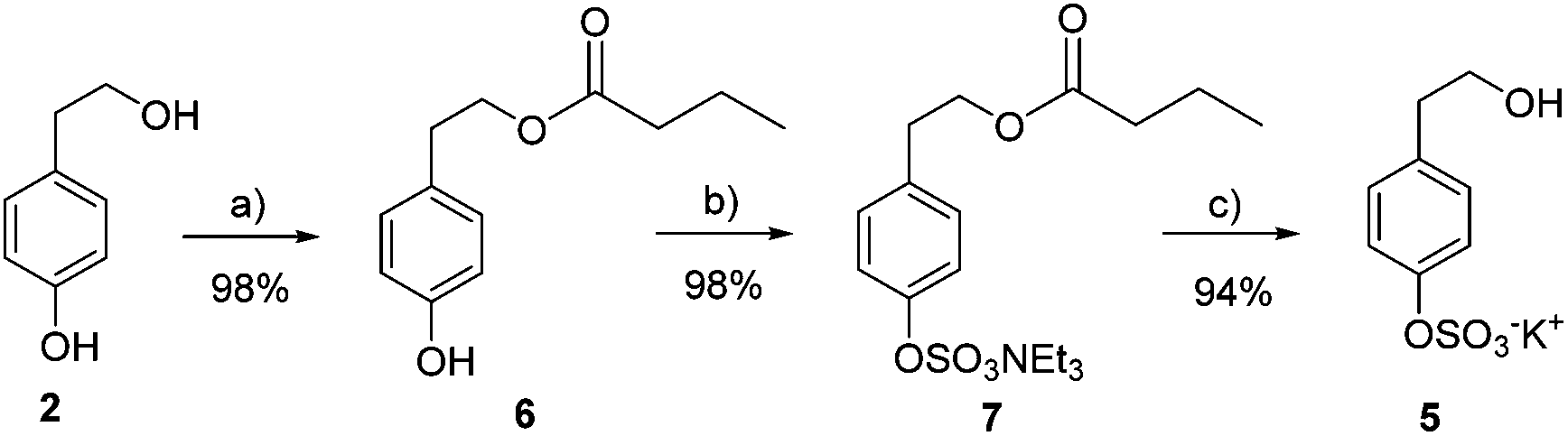 | ||
| Scheme 1 Preparation of tyrosol sulfate 5. (a) Novozym435, vinyl butyrate, TBME; (b) SO3·NMe3, TEA, CH3CN, 100 °C, 20 min, MW; (c) K2CO3, MeOH. | ||
Mono-sulfated hydroxytyrosol derivatives 3 and 4 were prepared as a regioisomeric mixture using a synthetic strategy similar to that of TYR. In this case, hydroxytyrosol acetate 8 was mono-silyl protected to avoid disulfated products difficult to separate from the mono-sulfated derivatives (Scheme 2).
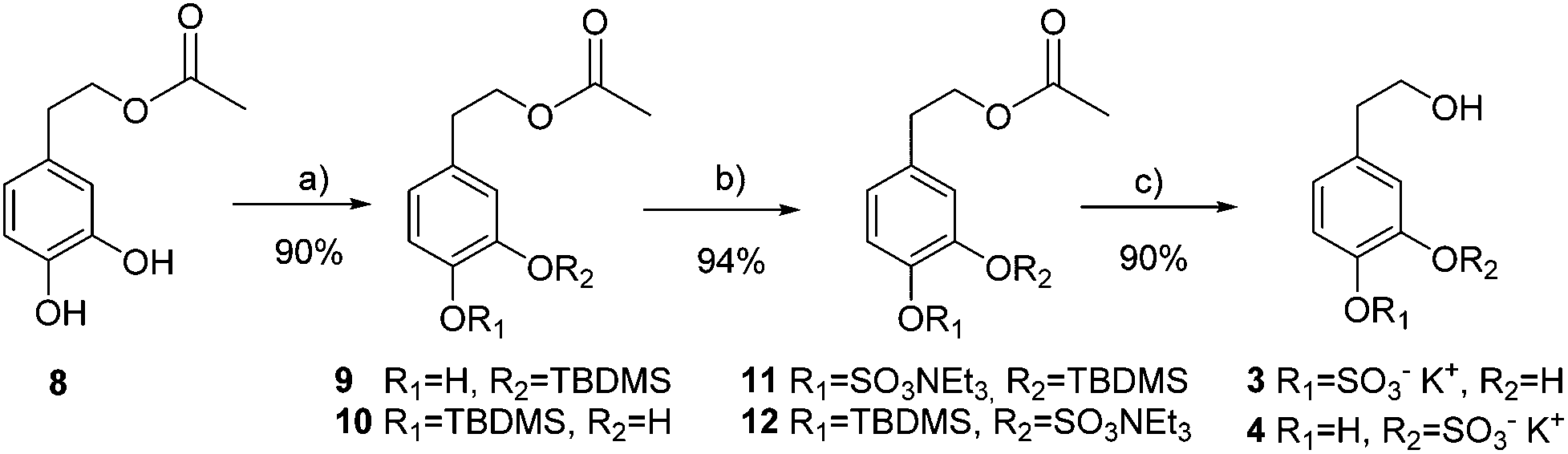 | ||
| Scheme 2 Preparation of hydroxytyrosol sulfate metabolites 3 and 4. (a) TBDMSOTf, DIPEA, CH2Cl2; (b) SO3·NMe3, TEA, CH3CN, 100 °C, 20 min, MW; (c) KF, K2CO3, MeOH. | ||
Random TBDMS-protection of HT acetate 8 and subsequent chromatographic separation afforded a 1:1 regioisomeric mixture of the two possible mono-phenolic compounds 9 and 10. The same sulfation reaction conditions used before were applied to give 94% yield of a 1:1 mixture of isomers 11 and 12. Finally, acetyl and silyl deprotection was carried out in one step using KF and K2CO3 in MeOH to obtain a mixture of mono-sulfated hydroxytyrosol derivatives 3 and 4 (90% yield).
To investigate the potential protective effect of the HT and TYR sulfate metabolites, in comparison with their parent compounds, against the pro-oxidant effect of oxidized cholesterol in intestinal cells, Caco-2 monolayers were treated for 24 h with a mixture obtained from the oxidation of cholesterol at 140 °C for 3 h. Under these oxidizing conditions half of the initial cholesterol was turned into oxidation products (reported in the Materials and methods section). Treatment with the increasing concentration of the mixture induced production of MDA and cell death (Fig. 2), indicating the presence of oxidative cell injury.
 | ||
| Fig. 2 Values of MDA (A) and cell viability (B) measured in Caco-2 cells after 24 h incubation with different concentrations of oxidized cholesterol. * = p < 0.05 versus control. | ||
Pretreatment with the phenolic compounds significantly inhibited the increase of MDA (Fig. 3) in Caco-2 cells treated with 75 μg mL−1 of the oxidizing mixture, the highest amount able to induce a significant production of MDA but not cell death. MDA reduction was significant from 2.5 μM for HT-S and from 5 μM for HT; TYR and its sulfate metabolite exerted the same efficacy starting from 2.5 μM.
 | ||
| Fig. 3 Values of MDA measured in Caco-2 cells after 24 h incubation with 75 μg mL−1 oxidized cholesterol and pretreated (30 min) with TYR, HT, HT sulfate metabolites 3–4 (HT-S) and TYR sulfate metabolite 5 (TYR-S) (2.5–10 μM). * = p < 0.05 versus control, a = p < 0.05 versus oxidized cholesterol treated (Ox). | ||
Next we examined the ability of HT, TYR and their corresponding sulfate metabolites to protect cells against death in Caco-2 cells treated with 100 μg mL−1 of oxidized cholesterol, the lowest amount able to induce a significant cell death (Fig. 4). We observed that HT preserved cell viability from the concentration of 5 μM whereas HT sulfates 3–4 showed protection at the 10 μM concentration. In the case of TYR and its sulfate derivatives, both improved cell viability at 10 μM concentration.
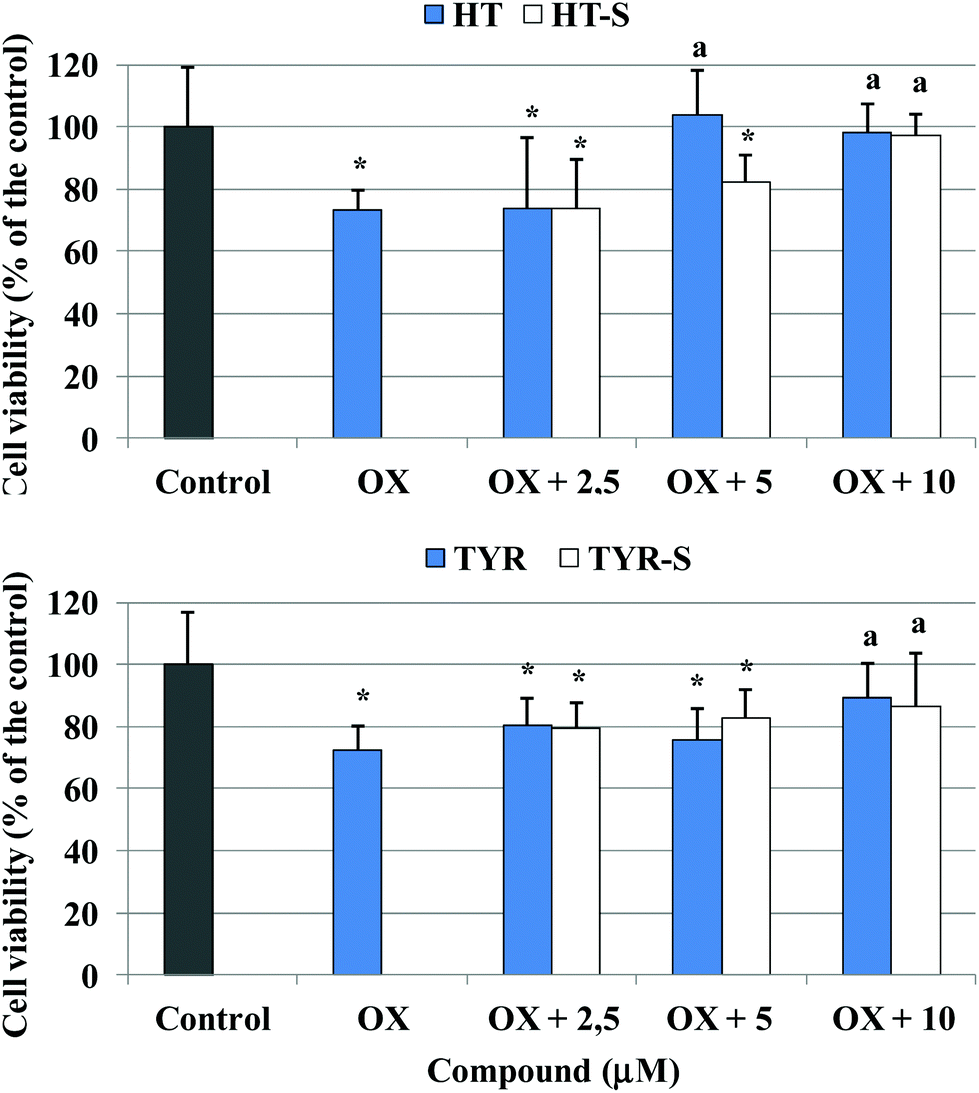 | ||
| Fig. 4 Cell viability measured in Caco-2 cells after 24 h incubation with 100 μg mL−1 oxidized cholesterol and pretreated (30 min) with TYR, HT or their sulfate metabolites (2.5–10 μM). * = p < 0.05 versus control, a = p < 0. 05 versus oxidized cholesterol treated (Ox). | ||
The oxidizing action of the mixture of oxidized cholesterol was then investigated monitoring the alteration of the cellular redox status with time: after 30 min of incubation a significant production of ROS was observed in the cells treated with the oxidized cholesterol in comparison with the control (Fig. 5A). Next, we measured ROS production at 30 min, after pretreatment with the different phenolic compounds (5, 10, 25 μM). We observed that ROS production was significantly lower from the concentration of 10 μM for all the tested compounds, except for TYR-S (Fig. 5B).
 | ||
| Fig. 5 ROS level, expressed as % of the control samples, in Caco-2 cells, treated with 75 μg mL−1 oxidized cholesterol for different incubation times (A) or pretreated (30 min) with TYR, HT or their sulfate metabolites (5–25 μM) and treated with oxidized cholesterol for 30 min (B), exposed to 2′,7′-dichlorodihydrofluorescein diacetate H2-DCF-DA (10 μM) for 30 min. * = p < 0.05 versus control, a = p < 0.05 versus oxidized cholesterol treated (Ox), ° = p < 0.05 versus 0 min. | ||
After 30 min of incubation with the oxidized cholesterol, ROS production was associated with a significant reduction of Caco-2 cellular GSH, around 40% of the initial value, as shown in Fig. 6A. The level of GSH increased with time, reaching the control value at 3/6 h, to decrease again thereafter. Pretreatment with both HT and TYR preserved the control level of GSH at all the tested concentrations and their sulfate metabolites exerted a comparable efficacy (Fig. 6B). Interestingly, none of the tested compounds was able to alter the GSH level when incubated alone (25 μM) under the experimental conditions used (p > 0.5 versus control).
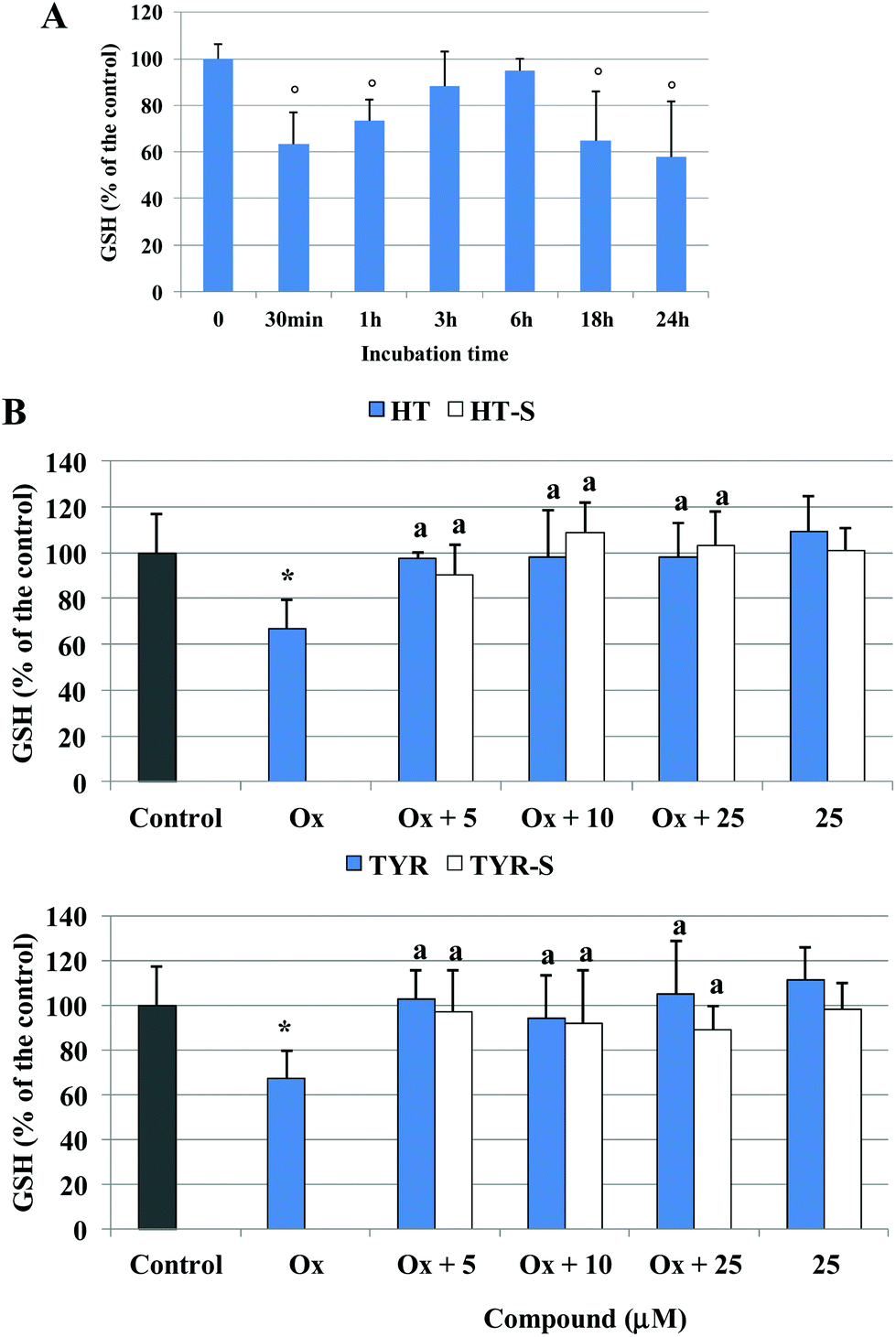 | ||
| Fig. 6 GSH level, expressed as % of the control samples, in Caco-2 cells, treated with 75 μg mL−1 oxidized cholesterol for different incubation times (A) or pretreated (30 min) with TYR, HT or their sulfate metabolites (5–25 μM) and treated with oxidized cholesterol for 30 min (B). * = p < 0.05 versus control, a = p < 0.05 versus oxidized cholesterol treated (Ox), ° = p < 0.05 versus 0 min. | ||
With respect to the considered parameters, we did not observe any concentration dependent protective effect of the tested phenolic compounds, nor significant differences among the parent compounds and metabolites.
A significant increase of GPx activity, around 50% above the control level, was also observed in Caco-2 cells treated with the oxidized cholesterol after 18 h of incubation (Fig. 7A). HT (25 μM) induced an increase, although not significant, of the basal GPx activity; however, once incubated together with the oxidized cholesterol HT reduced the increase of the enzyme activity from 10 μM with respect to the oxidized samples (Fig. 7B). HT sulfate metabolites 3 and 4 did not alter the control level but exerted the same activity of the parent compound for the oxidized samples.
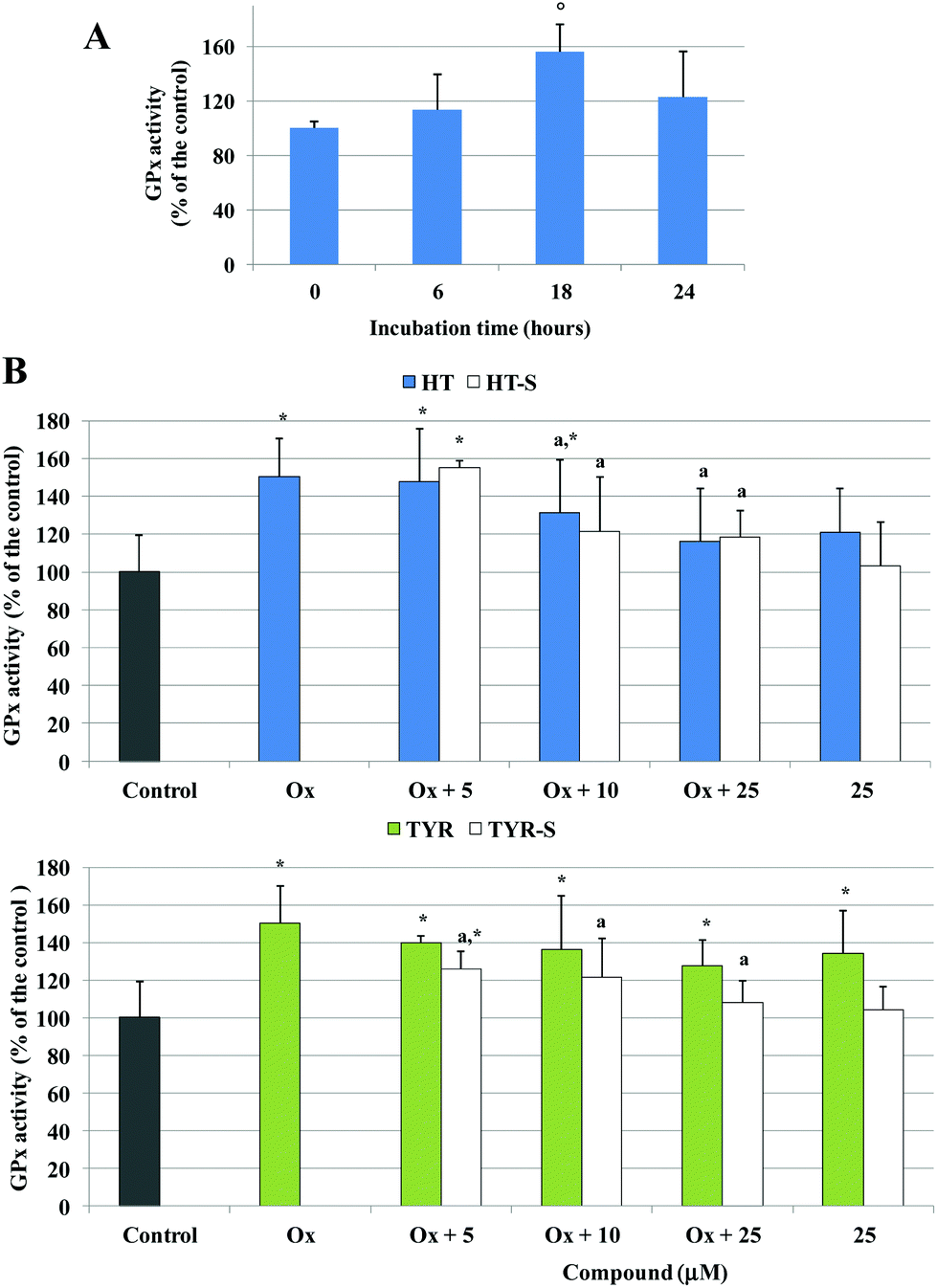 | ||
| Fig. 7 GPx activity, expressed as % of the control samples, in Caco-2 cells, treated with 75 μg mL−1 oxidized cholesterol for different incubation times (A) or pretreated (30 min) with TYR, HT or their sulfate metabolites (5–25 μM) and treated with oxidized cholesterol for 18 h (B). * = p < 0.05 versus control, a = p < 0.05 versus oxidized cholesterol treated (Ox), ° = p < 0.05 versus 0 h. | ||
In the presence of TYR, GPx activity was significantly above the control level; a small effect was observed in the pretreated samples with respect to the oxidized ones. In contrast, Caco-2 cells pretreated with all the tested concentrations of TYR sulphate 5 resulted in an enzyme activity significantly lower with respect to the oxidized samples, and the enzyme basal activity was not influenced (Fig. 7B).
Finally, we evaluated the stability and uptake of HT, TYR and their sulfate metabolites from 30 min to 24 h of incubation in the Caco-2 cultures. UPLC-MS analyses (see the ESI†) revealed a partial loss of the compounds in the culture medium, maybe due to the action of the fetal bovine serum present or due to the intrinsic stability to air oxidation of each compound. Analysing medium and supernatants (from the lysed cells) we observed that all the tested compounds entered the cells from 30 min of incubation and undergo an extensive metabolisation, giving rise to a pool of metabolites, among which we detected mainly sulfate and methyl-sulfate metabolites.
Discussion
It is known that polyphenols undergo strong phase I and phase II metabolism, in which they are hydrolyzed (phase I) and later conjugated (phase II) into their glucuronidated, methylated and sulfated forms in order to be absorbed and excreted. Recently, such metabolites are being taken into consideration to provide in vivo relevance for any activity.Sulfate metabolites of phenolic compounds, such as resveratrol,28–30 quercetin, epicatechin and catechin,31–33 have been shown to exert a biological activity comparable to that of the parent compound. Quercetin-3-O-sulfate has been shown to retain an antioxidant effect, significantly inhibiting the Cu2+-induced oxidation of human LDL.34 The free radical scavenging activity of a 3-sulfate metabolite of resveratrol was comparable to that of the parent compound, when measuring the ability to quench the DPPH radical.28
In this work we have studied the potential protecting activity of HT and TYR sulfate metabolites in Caco-2 human endothelial intestinal cells where oxidative stress was induced by oxidized cholesterol. Although the liver is the major organ for metabolism of many drugs and xenobiotics, the small intestine also contributes substantially to it by several pathways involving phase I and phase II reactions. At the same time, the concentration of sulfate metabolites in the intestinal epithelium may be high, as glucuronidation and sulfation are major pathways of phase II xenobiotic metabolism in the human intestine,12,13 and might contribute to preserve the intestinal epithelium integrity against pro-oxidant dietary agents.
We selected oxidized cholesterol for our oxidative stress cell model since cholesterol and its oxidation products have recently been shown to potentially interfere with the homeostasis of the intestinal epithelium,18,20 due to their pro-oxidant and pro-inflammatory properties. Oxysterols are more polar and more readily diffusible through cell membranes, and have consistently been shown to be more reactive than unoxidized cholesterol, possessing marked pro-inflammatory and cytotoxic effects in a number of cells and tissues.35,36 Under our oxidizing conditions, about 50% of the initial cholesterol was turned into oxidation products, mainly 7-ketocholesterol and some of the hydroxycholesterol isomers commonly found in cholesterol rich foodstuffs.37 It has been reported that dietary cholesterol and some oxysterols of dietary origin are able to lead differentiated Caco-2 cells to both necrotic and apoptotic death, depending on the experimental conditions.18,38–40
In the present study, oxidized cholesterol toxic effects were investigated in terms of alteration of the cellular redox state and oxidative damage. Oxidized cholesterol exposure resulted in a significant production of ROS, in accordance with the results reported by Biasi et al.,18,41 this likely being the cause of oxidative damage and death. ROS production in Caco-2 cells treated with specific oxysterols has been reported to be due, at least in part, to the activation of NADPH oxidase.18 Under our experimental conditions ROS production was inversely correlated with the decrease of GSH level. GSH is the main non-enzymatic antioxidant defence within the cell, reducing different peroxides, hydroperoxides, and radicals (alkyl, alkoxyl, peroxyl, etc.). It is usually assumed that GSH depletion reflects intracellular oxidation whereas an increase in GSH concentration could be expected to prepare the cell against a potential oxidative insult.42,43
In our oxidative stress model, in cells pretreated with HT, we observed an inhibition in the formation of ROS and also prevention of the GSH level decrease. The HT protective effect could predominantly be attributed to its capacity of acting as a free radical scavenger, as already documented for the same cells treated with other oxidizing agents, TBH8 and acrylamide.44 The well known free radical scavenging and metal chelating activities are mostly due to the HT ortho-diphenolic structure, whose high antioxidant activity may be explained by the high electron donating effect of the second hydroxyl group.
Despite the lack of the catechol group in their structure, HT sulfate metabolites (3–4) were as effective as HT itself. It is interesting to note however that, as shown by our uptake studies (see the ESI†), both HT and its sulfate metabolite enter the cells and are partially converted one into the other, thus what we observe is in part the resultant of the activity of both compounds.
At the same time, TYR and its sulfate metabolite 5 display very similar protecting ability against oxidation in our cell model, and TYR possesses only one OH phenolic group whereas TYR sulfate does not have any hydroxyl substituent in its aromatic ring.
In the case of TYR, it has been shown to have scavenging effects on ONOO−,45 O2− (ref. 46) and peroxyl radicals.8
It is important to remark that, in addition to the antioxidant capacity of natural polyphenols by directly scavenging extracellular/intracellular ROS, several phenolic compounds have been shown to provide a parallel protection by enhancing the level of GSH and the activity of a number of related protective enzymes.47 Among these enzymes, GPx catalyses the reduction of peroxides and is suggested to act as a barrier against hydroperoxide attack.48 Under our experimental conditions, none of the tested compounds altered the basal level of GSH, but TYR was able to significantly increase the activity of GPx after 18 h of incubation. At that time point, in Caco-2 cells exposed to the oxidized cholesterol we detected a significant increase of the GPx activity, indicating a positive response of the cell defence system to face an oxidative insult.42,43 Thus, pre-treatment with HT and HT sulfates prevented the increase in the activity of GPx induced by oxidative stress, either by direct action on the enzyme activity or via the decreasing of ROS production and consequently in cell damaging peroxide species.
The same result was found in the presence of TYR sulfate metabolites. GPx activity in Caco-2 cells was significantly enhanced with respect to the control in all the samples pretreated with TYR, in the presence or absence of oxidized cholesterol. This outcome is the first evidence of an inductive effect on the GPx activity of TYR, which has been shown to preserve the GSH level under oxidative stress conditions without altering the GPx basal level in Caco-249 and macrophage-like (J774 A1)50 cell lines. Our data are in agreement with that previously published with HT in HepG243,51 and in Caco-244 cells. Similarly, flavanols, such as epicatechin-3-gallate and procyanidin B2, induce a significant increase in several antioxidant-related enzyme activities, accompanied by a decrease in ROS production not affecting the GSH content in Caco-2 cells.52
When Caco-2 cells were treated with oxidized cholesterol for 24 h, a significant increase of MDA production was observed, together with a decrease in cell viability, indicating a lipo-peroxidative injury to the cells. Pretreatment with all the phenolic compounds and the corresponding sulfate metabolites inhibited MDA formation at all the tested concentrations. At 24 h of incubation, regardless of the phenolic compound initially present in the incubation mixture (parent compound or sulfate metabolite), we found a pool of compounds (see the ESI†), mainly sulfate and methyl-sulfate metabolites, in agreement with a study by Rubio et al.,13 due to the intense metabolising activity of Caco-2 cells, all of which probably contribute to the final effect.
None of the compounds however was equally efficient in preventing cell death. This result suggests that the alteration of the cellular redox status not only led to a direct oxidative injury, but likely modulated signalling pathways involved in cell survival, as demonstrated in differentiated Caco-2 cells, where both unoxidized cholesterol as well as an oxysterol mixture were able to induce apoptosis as a consequence of enhanced ROS production.18
The actual contribution of phenolic metabolites to the biological activity of the parent compounds is still quite controversial. In fact, Khymenets et al.53 reported the lack of antioxidant activity for HT glucuronate metabolites compared with that of the parent compounds, suggesting that they are not chief contributors to the antioxidant effects provided by olive oil consumption. In contrast, Paiva-Martins et al.15 recently described the protective effects of HT glucuronides on erythrocyte oxidative-induced hemolysis to a similar extent to that observed for HT itself. These results suggested that HT metabolites may play an important role in the protective activity of olive oil phenolic compounds.
Conclusions
In conclusion, our data provide evidence of the ability of sulfate metabolites of HT and TYR to protect intestinal cells against the pro-oxidant effect of oxidized cholesterol, with an efficiency comparable to that of the parent compounds. Our data strengthen the assumption that the potential beneficial effect of the parent compounds is retained, if not enhanced, even when extensive metabolisation occurs, being the resulting metabolites able to exert a biological action themselves. Sulfate metabolites may be present in the intestinal epithelium, as a result of deconjugation/conjugation processes; thus, they may significantly contribute to the protective activity exerted by olive oil simple phenols against the oxidation process triggered by lipid peroxides on the intestinal mucosa.Abbreviations
| HT | Hydroxytyrosol |
| TYR | Tyrosol |
| HT-S | HT sulfate metabolites 3–4 |
| TYR-S | TYR sulfate metabolite 5 |
| MDA | Malonyldialdehyde |
| NR | Neutral red |
| ROS | Reactive oxygen species |
| GSH | Glutathione |
| GPx | Glutathione peroxidase |
Acknowledgements
We gratefully acknowledge financial support from the Regione Autonoma della Sardegna (LR7-CRP-25300), Fondazione Banco di Sardegna and Junta de Andalucía (CVI-5007).References
- S. Martin-Pelaez, M. I. Covas, M. Fito, A. Kusar and I. Pravst, Mol. Nutr. Food Res., 2013, 57, 760–771 CAS.
- F. Visioli and E. Bernardini, Curr. Pharm. Des., 2011, 17, 786–804 CrossRef CAS.
- M. N. Vissers, P. L. Zock, A. J. Roodenburg, R. Leenen and M. B. Katan, J. Nutr., 2002, 132, 409–417 CAS.
- L. Rubio, R. M. Valls, A. Macia, A. Pedret, M. Giralt, M. P. Romero, R. de la Torre, M. I. Covas, R. Sola and M. J. Motilva, Food Chem., 2012, 135, 2922–2929 CrossRef CAS PubMed.
- J. Pinto, F. Paiva-Martins, G. Corona, E. S. Debnam, M. Jose Oruna-Concha, D. Vauzour, M. H. Gordon and J. P. Spencer, Br. J. Nutr., 2011, 105, 1607–1618 CrossRef CAS PubMed.
- M. de Bock, E. B. Thorstensen, J. G. Derraik, H. V. Henderson, P. L. Hofman and W. S. Cutfield, Mol. Nutr. Food Res., 2013, 57, 2079–2085 Search PubMed.
- G. Corona, X. Tzounis, M. A. Dessi, M. Deiana, E. S. Debnam, F. Visioli and J. P. E. Spencer, Free Radical Res., 2006, 40, 647–658 CrossRef CAS PubMed.
- M. Deiana, G. Corona, A. Incani, D. Loru, A. Rosa, A. Atzeri, M. P. Melis and M. A. Dessi, Food Chem. Toxicol., 2010, 48, 3008–3016 CrossRef CAS PubMed.
- R. de la Torre, Inflammopharmacology, 2008, 16, 245–247 CrossRef CAS PubMed.
- E. Miro-Casas, M. I. Covas, M. Farre, M. Fito, J. Ortuno, T. Weinbrenner, P. Roset and R. de la Torre, Clin. Chem., 2003, 49, 945–952 CAS.
- K. L. Tuck, P. J. Hayball and I. Stupans, J. Agric. Food Chem., 2002, 50, 2404–2409 CrossRef CAS PubMed.
- J. H. Lin, M. Chiba and T. A. Baillie, Pharmacol. Rev., 1999, 51, 135–158 CAS.
- L. Rubio, A. Macia, A. Castell-Auvi, M. Pinent, M. T. Blay, A. Ardevol, M. P. Romero and M. J. Motilva, Food Chem., 2014, 149, 277–284 CrossRef CAS PubMed.
- M. Deiana, A. Incani, A. Rosa, A. Atzeri, D. Loru, B. Cabboi, M. P. Melis, R. Lucas, J. C. Morales and M. A. Dessi, Chem. – Biol. Interact., 2011, 193, 232–239 CrossRef CAS PubMed.
- F. Paiva-Martins, A. Silva, V. Almeida, M. Carvalheira, C. Serra, J. E. Rodrigues-Borges, J. Fernandes, L. Belo and A. Santos-Silva, J. Agric. Food Chem., 2013, 61, 6636–6642 CrossRef CAS PubMed.
- M. Pinto, S. Robine-Leon, M.-D. Appay, M. Kedinger, N. Triadou, E. Dussaulxs, B. Lacrox, P. Simon-Assmann, K. Haffen, J. Fogh and A. Zweibaum, Biol. Cell, 1983, 47, 323–330 Search PubMed.
- Q. Li, Y. Sai, Y. Kato, I. Tamai and A. Tsuji, Pharmacol. Res., 2003, 20, 1119–1124 CrossRef CAS.
- F. Biasi, C. Mascia, M. Astegiano, E. Chiarpotto, M. Nano, B. Vizio, G. Leonarduzzi and G. Poli, Free Radicals Biol. Med., 2009, 47, 1731–1741 CrossRef CAS PubMed.
- F. Biasi, C. Mascia and G. Poli, Carcinogenesis, 2008, 29, 890–894 CrossRef CAS PubMed.
- C. Mascia, M. Maina, E. Chiarpotto, G. Leonarduzzi, G. Poli and F. Biasi, Free Radicals Biol. Med., 2010, 49, 2049–2057 CrossRef CAS PubMed.
- A. Rosa, M. Deiana, A. Atzeri, G. Corona, A. Incani, M. P. Melis, G. Appendino and M. A. Dessi, Chem. – Biol. Interact., 2007, 165, 117–126 CrossRef CAS PubMed.
- M. Calderon-Santiago, A. Peralbo-Molina, F. Priego-Capote and M. D. L. de Castro, Eur. J. Lipid Sci. Technol., 2012, 114, 687–694 CrossRef CAS PubMed.
- S. Grasso, L. Siracusa, C. Spatafora, M. Renis and C. Tringali, Bioorg. Chem., 2007, 35, 137–152 CrossRef CAS PubMed.
- R. Lucas, F. Comelles, D. Alcantara, O. S. Maldonado, M. Curcuroze, J. L. Parra and J. C. Morales, J. Agric. Food Chem., 2010, 58, 8021–8026 CrossRef CAS PubMed.
- R. Fautz, B. Husein and C. Hechenberger, Mutat. Res., 1991, 253, 173–179 CAS.
- J. Templar, S. P. Kon, T. P. Milligan, D. J. Newman and M. J. Raftery, Nephrol. Dial. Transplant., 1999, 14, 946–951 CrossRef CAS PubMed.
- S. Dinicola, M. A. Mariggio, C. Morabito, S. Guarnieri, A. Cucina, A. Pasqualato, F. D'Anselmi, S. Proietti, P. Coluccia and M. Bizzarri, Br. J. Nutr., 2013, 110, 797–809 CrossRef CAS PubMed.
- J. Hoshino, E. J. Park, T. P. Kondratyuk, L. Marler, J. M. Pezzuto, R. B. van Breemen, S. Mo, Y. Li and M. Cushman, J. Med. Chem., 2010, 53, 5033–5043 CrossRef CAS PubMed.
- V. Aires, E. Limagne, A. K. Cotte, N. Latruffe, F. Ghiringhelli and D. Delmas, Mol. Nutr. Food Res., 2013, 57, 1170–1181 CAS.
- R. Ruotolo, L. Calani, E. Fietta, F. Brighenti, A. Crozier, C. Meda, A. Maggi, S. Ottonello and D. Del Rio, Nutr. Metab. Cardiovasc. Dis., 2013, 23, 1086–1092 CrossRef CAS PubMed.
- J. Terao, J. Med. Invest., 1999, 46, 159–168 CAS.
- T. Koga and M. Meydani, Am. J. Clin. Nutr., 2001, 73, 941–948 CAS.
- S. Tribolo, F. Lodi, C. Connor, S. Suri, V. G. Wilson, M. A. Taylor, P. W. Needs, P. A. Kroon and D. A. Hughes, Atherosclerosis, 2008, 197, 50–56 CrossRef CAS PubMed.
- C. Manach, C. Morand, V. Crespy, C. Demigne, O. Texier, F. Regerat and C. Remesy, FEBS Lett., 1998, 426, 331–336 CrossRef CAS.
- A. Vejux, L. Malvitte and G. Lizard, Braz. J. Med. Biol. Res., 2008, 41, 545–556 CrossRef CAS PubMed.
- G. Leonarduzzi, P. Gamba, S. Gargiulo, B. Sottero, A. Kadl, F. Biasi, E. Chiarpotto, N. Leitinger, G. Vendemiale, G. Serviddio and G. Poli, Aging Cell, 2008, 7, 375–382 CrossRef CAS PubMed.
- J. Plat, J. A. Nichols and R. P. Mensink, J. Lipid Res., 2005, 46, 2468–2476 CrossRef CAS PubMed.
- A. J. O'Sullivan, Y. C. O'Callaghan, J. A. Woods and N. M. O'Brien, J. Appl. Toxicol., 2003, 23, 191–197 CrossRef PubMed.
- E. Ryan, J. Chopra, F. McCarthy, A. R. Maguire and N. M. O'Brien, Br. J. Nutr., 2005, 94, 443–451 CrossRef CAS.
- S. Roussi, A. Winter, F. Gosse, D. Werner, X. Zhang, E. Marchioni, P. Geoffroy, M. Miesch and F. Raul, Cell Death Differ., 2005, 12, 128–135 CrossRef CAS PubMed.
- F. Biasi, T. Guina, M. Maina, B. Cabboi, M. Deiana, C. I. Tuberoso, S. Calfapietra, E. Chiarpotto, B. Sottero, P. Gamba, S. Gargiulo, V. Brunetto, G. Testa, M. A. Dessi, G. Poli and G. Leonarduzzi, Biochem. Pharmacol., 2013, 86, 138–145 CrossRef CAS PubMed.
- M. Alia, R. Mateos, S. Ramos, E. Lecumberri, L. Bravo and L. Goya, Eur. J. Nutr., 2006, 45, 19–28 CrossRef CAS PubMed.
- L. Goya, R. Mateos and L. Bravo, Eur. J. Nutr., 2007, 46, 70–78 CrossRef CAS PubMed.
- I. Rodriguez-Ramiro, M. A. Martin, S. Ramos, L. Bravo and L. Goya, Toxicology, 2011, 288, 43–48 CrossRef CAS PubMed.
- R. de la Puerta, M. E. Martinez Dominguez, V. Ruiz-Gutierrez, J. A. Flavill and J. R. Hoult, Life Sci., 2001, 69, 1213–1222 CrossRef CAS.
- A. A. Bertelli, M. Migliori, V. Panichi, B. Longoni, N. Origlia, A. Ferretti, M. G. Cuttano and L. Giovannini, Ann. N. Y. Acad. Sci., 2002, 957, 295–301 CrossRef CAS PubMed.
- R. Masella, R. Di Benedetto, R. Vari, C. Filesi and C. Giovannini, J. Nutr. Biochem., 2005, 16, 577–586 CrossRef CAS PubMed.
- X. G. Lei, W. H. Cheng and J. P. McClung, Annu. Rev. Nutr., 2007, 27, 41–61 CrossRef CAS PubMed.
- C. Giovannini, E. Straface, D. Modesti, E. Coni, A. Cantafora, M. De Vincenzi, W. Malorni and R. Masella, J. Nutr., 1999, 129, 1269–1277 CAS.
- R. Di Benedetto, R. Vari, B. Scazzocchio, C. Filesi, C. Santangelo, C. Giovannini, P. Matarrese, M. D'Archivio and R. Masella, Nutr. Metab. Cardiovasc. Dis., 2007, 17, 535–545 CrossRef CAS PubMed.
- M. A. Martin, S. Ramos, A. B. Granado-Serrano, I. Rodriguez-Ramiro, M. Trujillo, L. Bravo and L. Goya, Mol. Nutr. Food Res., 2010, 54, 956–966 CAS.
- I. Rodriguez-Ramiro, M. A. Martin, S. Ramos, L. Bravo and L. Goya, Eur. J. Nutr., 2011, 50, 313–322 CrossRef CAS PubMed.
- O. Khymenets, M. Fito, S. Tourino, D. Munoz-Aguayo, M. Pujadas, J. L. Torres, J. Joglar, M. Farre, M. I. Covas and R. de la Torre, Drug Metab. Dispos., 2010, 38, 1417–1421 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR and 13C-NMR spectra of compounds 3–5, 7, 9–12 and TYR, HT and sulfate metabolites’ stability/uptake in Caco-2 cultures. See DOI: 10.1039/c5fo00074b |
| This journal is © The Royal Society of Chemistry 2016 |