 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cutting the cost of carbon capture: a case for carbon capture and utilization†
Lennart
Joos
a,
Johanna M.
Huck
b,
Veronique
Van Speybroeck
a and
Berend
Smit
*bcd
aCenter for Molecular Modeling, Ghent University, B-9052 Zwijnaarde, Belgium. E-mail: veronique.vanspeybroeck@ugent.be
bDepartment of Chemical and Biomolecular Engineering, University of California, Berkeley, CA 94720-1462, USA
cDepartment of Chemistry, University of California, Berkeley, CA 94720- 1462, USA
dLaboratory of Molecular Simulation, Institut des Sciences et Ingénierie Chimiques, Valais, Rue d'Industrie 17, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1950 Sion, Switzerland. E-mail: berend.smit@epfl.ch
First published on 11th March 2016
Abstract
A significant part of the cost for carbon capture and storage (CCS) is related to the compression of captured CO2 to its supercritical state, at 150 bar and typically 99% purity. These stringent conditions may however not always be necessary for specific cases of carbon capture and utilization (CCU). In this manuscript, we investigate how much the parasitic energy of an adsorbent-based carbon capture process may be lowered by utilizing CO2 at 1 bar and adapting the final purity requirement for CO2 from 99% to 70% or 50%. We compare different CO2 sources: the flue gases of coal-fired or natural gas-fired power plants and ambient air. We evaluate the carbon capture performance of over 60 nanoporous materials and determine the influence of the initial and final CO2 purity on the parasitic energy of the carbon capture process. Moreover, we demonstrate the underlying principles of the parasitic energy minimization in more detail using the commercially available NaX zeolite. Finally, the calculated utilization cost of CO2 is compared with the reported prices for CO2 and published costs for CCS.
1 Introduction
Nowadays, there are no technological hurdles to the industrial deployment of post-combustion carbon capture. However, the high energy penalty of the process and the associated financial costs hamper the actual use of the technology. Drastically lowering the price of this process is therefore a requisite for the competitiveness of carbon capture as a viable technology to reduce CO2 emissions.1–5The most mature technology for carbon capture and storage (CCS) to separate CO2 from the post-combustion flue gases is amine scrubbing, a two-step process in which CO2 is first chemically bound to the amines and later recovered in a pure form by ‘stripping’ the CO2 from the amines at high temperatures.6,7 A commonly used amine in this process is monoethanolamine (MEA). Heating the dilute aqueous amine mixture imposes a severe energy penalty on the process. Alternative technologies are therefore proposed, such as adsorption in nanoporous materials. A variety of classes of nanoporous materials exists with high CO2/N2 selectivity, good CO2 uptake, as well as a less energy demanding regeneration than the amine solutions.8–10 Among them are metal–organic frameworks (MOFs),11,12 zeolitic imidazolate frameworks (ZIFs),13,14 porous polymer networks (PPNs),15–17 and zeolites.18–21
There are several possibilities to lower the cost of post-combustion carbon capture with nanoporous materials, which will be outlined in the following subsections. In the first place, ongoing advances in material research and computational screening methods can help to design a new material with potentially better CCS properties. Secondly, novel ways to operate the carbon capture process can also reduce the energy consumption. Thirdly, changing the operating conditions of the carbon capture process also provides opportunities to save energy. The latter will be the focus of this manuscript.
1.1 Alternative materials
Due to rational material synthesis approaches there has been an explosion of new materials in recent years. Since their discovery in the 1990's, some 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Metal–Organic Frameworks (MOFs) have been synthesized, and countless new ones as well as specific modifications are designed every year.23 Moreover, the importance of zeolites in the petrochemical industry has spurred new developments in the field of zeolite synthesis as well.24,25
000 Metal–Organic Frameworks (MOFs) have been synthesized, and countless new ones as well as specific modifications are designed every year.23 Moreover, the importance of zeolites in the petrochemical industry has spurred new developments in the field of zeolite synthesis as well.24,25
To compare nanoporous materials among each other, Bae and Snurr presented five criteria to evaluate CCS performance: CO2 uptake, CO2/N2 selectivity, CO2 working capacity, regenerability of the material and a sorbent selection parameter.26 Some of the authors of this manuscript proposed to use one metric instead, the “parasitic energy”, i.e. the energy output of the power plant immediately consumed by the CCS process.27,28 This metric can easily be related to the industrial application of CCS, as it straightforwardly identifies the material with the lowest energy requirement for CCS.
However, experimentally determining any CCS metric for thousands of possible candidates would be nearly impossible. Therefore, high-throughput computational screenings of databases with existing as well as hypothetical materials have become very attractive tools. They allow us to evaluate millions of materials for their CCS performance and guide experimental efforts in the direction of the most promising materials. With these computer screenings, it is not only possible to assess the boundaries of the material space for their carbon capture performance, but also get molecular insights on the best performing materials. A screening of nanoporous materials from different material families identified that adsorbent-based carbon capture can yield a parasitic energy 30% lower than that of the amine scrubbing process, but that further improvements are not to be expected.27,28 Finally, going beyond brute force computational screenings requires machine learning techniques, which have already been applied for gas adsorption in nanoporous materials.12,29,30
1.2 Alternative technologies
Although the discovery of new materials may gradually improve carbon capture performance, previously mentioned screening studies showed that this approach has its limits. Therefore, it is worth taking a second look at the carbon capture process, shown in Fig. 1. In conventional nanoporous materials-based carbon capture process, the exhaust gases of the power plant are first cooled and CO2 is adsorbed at low temperatures to maximize CO2 uptake. Once the adsorption bed is saturated with CO2, the nanoporous material is regenerated either by heating the material or by applying a vacuum. Either way, a parasitic energy penalty is imposed on the process. Moreover, H2O in the exhaust gases will compete for the same adsorption sites as CO2, thereby lowering the CO2 uptake and increasing the parasitic energy.31 | ||
| Fig. 1 Layout of a coal-fired power plant. (A) represents a power plant retrofitted with conventional carbon capture technology based on CO2 adsorption on a nanoporous material. The exhaust gases are first cooled and CO2 is subsequently adsorbed. Regeneration of the bed requires a parasitic energy from the power plant, which is even higher when H2O is present. (B) is a power plant retrofitted with the proposed High-temperature Adsorption and Low-temperature Desorption (HALD) technology, whereby adsorption takes place at high temperatures and the material is regenerated by cooling the adsorbent while saturating it with H2O.22 | ||
In the newly proposed “High-temperature Adsorption and Low-temperature Desorption” (HALD) set-up, the temperature-dependent competitive adsorption of CO2 and H2O is exploited to overcome the high energy requirement.22,32 At high temperatures, the competitive adsorption of CO2 and H2O is in favor of CO2, so adsorption at higher temperatures (without first cooling the exhaust gases) improves the selectivity towards CO2. As the competition is in favor of H2O at low temperatures, CO2 can be desorbed at low temperatures, by cooling and saturating the absorbent with H2O. When the regenerated zeolite – void of CO2 but full with H2O – is brought into contact again with the hot exhaust gases, the waste energy of the flue gas desorbs the H2O from the material and restores the competitive advantage of CO2. This temperature swing process utilizes the residual heat of the exhaust gases to overcome the competition with H2O and capture the CO2 from this CO2/N2/H2O mixture and hence does not require the input of energy for regeneration. The upside-down HALD alternative to traditional CCS methods thereby opens new perspectives to reduce the energy penalty of CCS.
1.3 Alternative operating conditions
For the application of carbon capture and storage (CCS), the captured CO2 has to be of high purity (99%) before it can be compressed to a supercritical state at around 150 bar.33 Only in these conditions can CO2 efficiently be stored in geological formations. The high purity requirement and the compression to high pressure constitute a large part of the energy penalty for CCS. However, the stringent conditions may not always be necessary for specific cases of carbon capture and utilization (CCU). As a result, the price per tonne CO2 may actually be significantly lower, shifting the economics of CCU into a more favorable direction.In Table 1, a few examples of CCU are given. From left to right, the examples are organized with decreasing CO2 purity for the application, 99%, 70% or 50%. From top to bottom, the CO2 source is indicated: coal-fired power plants, with 14% CO2 in their flue gases, gas turbine exhaust gases, with about 4% CO2 flue gases, and air, with CO2 levels of about 400 ppm.
| In/out | 99% | 70% | 50% |
|---|---|---|---|
| COAL | EOR | Carbstone | Algae |
| NG | Fire extinguisher | Welding/molding | Greenhouses |
| AIR | CCS | Fumigation | Bubbles in drinks |
An application in which 50% CO2 could suffice, but for which large amounts of CO2 are needed, is the cultivation of algae for the production of biofuels.34 Increased CO2 levels are used to heighten the crop production in greenhouses, although for this application smaller quantities suffice, provided by a gas turbine or a biomass plant for instance.35,36 Finally, for the carbonation of drinks, local CO2 production directly from the air could be an option. An interesting example for 70% CO2 is the production of “Carbstone”.37 In a chemical process, finely ground CaO and MgO, waste products from the metallurgical industry, react with CO2 to produce CaCO3/MgCO3 bricks and tiles. This is an example where CO2 is both permanently sequestered in a chemically stable solid product, as well as utilized for a useful application (CCUS). Smaller amounts of CO2 could be used as an inert gas in welding,38 or for specific molding techniques, in which CO2 was found to increase the hardness.39 Finally, locally producing small amounts of CO2 could also be practical for fumigation, for instance of bed bugs.40 Some cases however do require the use of very pure, 99% CO2. Enhanced Oil Recovery (EOR) for instance, injects CO2 in near-depleted oil wells under supercritical conditions.41 The market for this application is in the order of millions of tonnes of CO2 per year, and therefore large coal-fired power plants are needed to fill that demand. For fire extinguishers, pure CO2 is also necessary, but much smaller CO2 sources can be utilized.42 Finally, direct air capture could serve as a solution of last resort, when the climate turns really bad and the CO2 level in the atmosphere has to be suppressed on a very short term, direct air capture devices can be used for carbon capture and storage, directly from the atmosphere.43,44
2 Goal
The central question in this manuscript is how much the parasitic energy of carbon capture can be lowered by utilizing CO2 at 1 bar and adapting the final purity requirements for CO2 from different CO2 sources. First, we investigate the influence of the final purity requirement for the case of coal-fired power plant flue gases. Then, we extend this analysis to other sources of CO2, the flue gases of natural gas-fired power plants and ambient air. Afterwards, we go into more detail on the purity of the desorbed mixture. Then, the properties of NaX, a commercially available material, and the minimization of the parasitic energy are studied in more detail. Finally, we will compare the calculated CCU cost with reported costs for CCS and available prices for CO2.3 Methodology
The parasitic energy of the carbon capture process stems from the regeneration of the nanoporous material, either by heating the material, by applying a vacuum or a combination of the two. In the first place, the material can be heated up in order to trigger CO2 desorption. This thermal heating energy, Qthermal, not only incorporates the sensible heat requirement to heat the material but also the required desorption heat of CO2. The latter heat term is required to undo the binding of the CO2 to the nanoporous material in order to proceed with the storage process. Accordingly, the thermal energy requirement per mass CO2 is given by: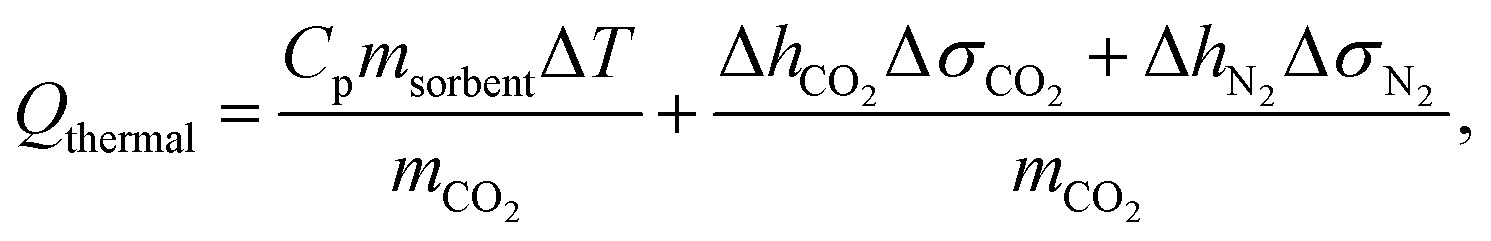 | (1) |
The second contribution to the parasitic energy is related to the compression of the desorbed CO2 to either 1 bar, for utilization, or to 150 bar, for transportation and storage. The compression energy is mainly determined by the CO2 purity and the desorption pressure. The pumping work, Wcomp, is estimated by means of the NIST REFPROP database46 and a linear regression based on this data. The isentropic efficiency of the pump is assumed to be 85% for gas below the supercritical point and 90% above it.
For the overall parasitic energy, we assume that heat is delivered by steam from the power plant and that the compressors are driven by the produced electricity directly. To calculate the total parasitic energy, the loss in the power plant's production, the compression work can be used directly, whereas for the heat, a typical turbine efficiency of 75%47 and the Carnot efficiency η to convert thermal energy into electrical work have to be taken into account to translate the heat loss in an output loss:
| Eparasitic = (0.75QthermalηCarnot) + Wcomp. | (2) |
This total energy requirement can be minimized by varying the final operation conditions of the desorption process, i.e. temperature and/or pressure. These desorption processes are referred to as Temperature–Pressure Swing Adsorption (TPSA), Temperature Swing Adsorption (TSA) or Pressure Swing Adsorption (PSA), depending which parameters are varied or fixed, in each case. TPSA operates at changing temperature and pressure, whereas TSA is characterized by constant pressure (set to pdes = 1 atm; this equals adsorption pressure) and PSA features a fixed temperature (Tdes = 333 K). The considered desorption conditions range from 0.01 atm < pdes < 3 atm and 333 K < Tdes < 473 K for pressure and temperature, respectively.
The database includes five classes of nanoporous materials, which are partially of hypothetical nature. These comprise metal–organic frameworks (MOFs),48–61 zeolitic imidazolate frameworks (ZIFs),62–64 porous polymer networks (PPNs),65 zeolites20 and cation exchanged zeolites (CEZs).18 Moreover, two classes of hypothetical materials are considered: HMOFs – analogs of the well-studied MOF-5 (ref. 66) – and hypothetical ZIF structures. The class of CEZs is represented by Linde type A and type X zeolites where the Na-cations are partially exchanged by the alkaline earth metals magnesium and calcium. Alongside the fully coordinated MOFs, UMCM-1 and MOF-177, we also included MOFs with open metal sites, like the series M-MOF-74, CuBTC, and CuBTTri.11,26,67–69 Of special interest in recent years are also porous polymer networks (PPNs) tethered with different polyamines.70,71
A full description of the methodology, the assumptions and the considered materials can be found in ref. 28.
4 Results & discussion
4.1 Minimum energy
Before we dig deeper into the performance of the different materials, it is worth looking at this problem from an elementary thermodynamical viewpoint.72,73 It is possible to calculate the minimum work to separate CO2 from a two-component gas mixture, Wsepmin, using the molar entropy of an ideal gas mixture containing a mole fraction x of CO2, according to| sim(x) = −R[xlnx + (1 − x)ln(1 − x)] | (3) |
| −Wsepmin = −Tsep[nemsim(xem) + ncapsim(xcap) − nfluesim(xflue)], | (4) |
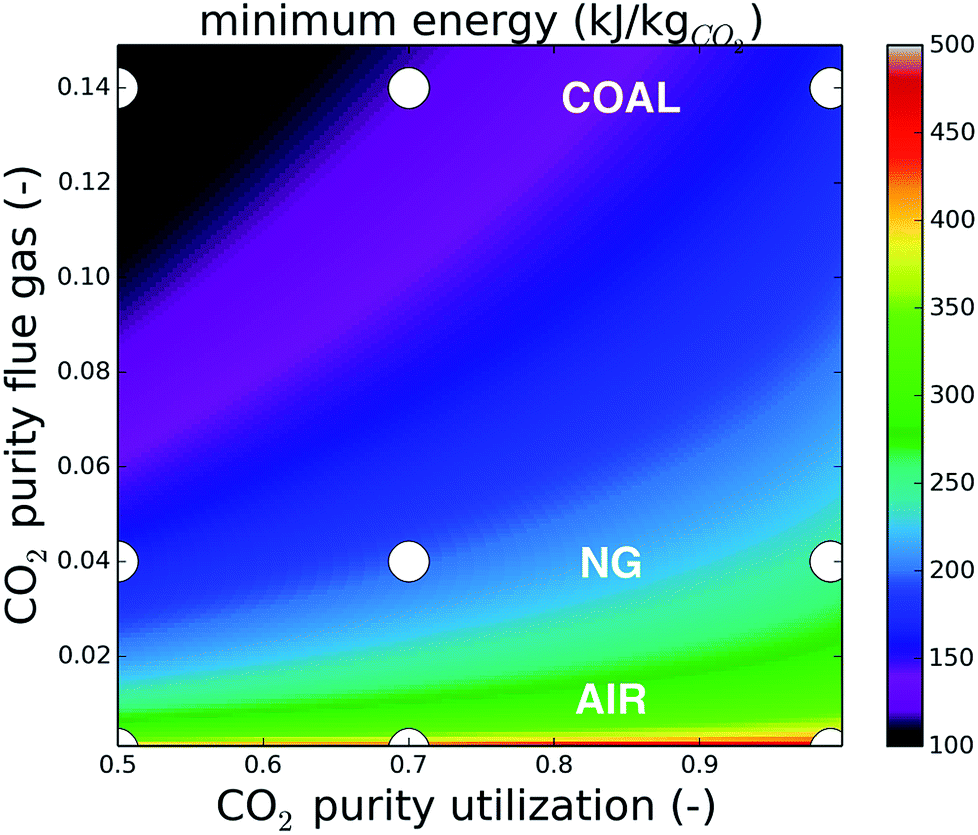 | ||
| Fig. 2 The minimum energy (in kJ kgCO2−1) to separate CO2 from a binary mixture with an initial content (y-axis) to a final composition (x-axis), at 313 K and 1 bar. The points relevant in this manuscript are shown in white. The adsorption temperature in the case of direct air capture is 288 K in this manuscript, which lowers the minimum energy compared to the values in this figure. | ||
| CO2 source | Final composition | |||
|---|---|---|---|---|
| 99% | 70% | 50% | ||
| COAL | 14% | 168 | 119 | 89 |
| NG | 4% | 245 | 197 | 166 |
| AIR | 0.04% | 477 | 432 | 404 |
Direct air capture is situated in a very unfavorable region of this energy landscape and relative improvements are relatively small. For coal on the other hand, almost 50% minimum energy can be saved by lowering the purity of the final mixture from 99% to 50%. The energy requirement for upgrading the natural gas flue gas from 4% to 50% is almost identical to upgrading coal flue gas from 14% to 99%. This observation already opens perspectives to lower the cost of carbon capture from natural-gas fired power plants, given that applications for CO2 at 50% purity can be found.
4.2 Influence of the purity requirement
Now, let us return to the selection of 62 nanoporous materials to consider carbon capture from the flue gas of coal-fired power plants. This flue gas has the highest CO2 content and is therefore the most practical CO2 source for post-combustion carbon capture. Fig. 3 shows the parasitic energy to capture CO2 from coal flue gas as a function of the CO2 Henry coefficient (kH,CO2) at 300 K, for the original requirement of CCS, with CO2 at 99% purity and compressed to 150 bar and for the cases of 99%, 70% and 50% purity at 1 bar. First of all, the compression work from 1 to 150 bar comprises a large part of the parasitic energy and is hence an important part of the cost for CCS. Fig. S1 in the ESI† shows that this contribution to the parasitic energy exceeds 55%, especially for the best performing materials. Furthermore, lowering the purity requirement from 99% down to 70 or 50% further reduces the parasitic energy, although the relative improvements are much smaller than the parasitic energy reduction when omitting the 1 to 150 bar compression work.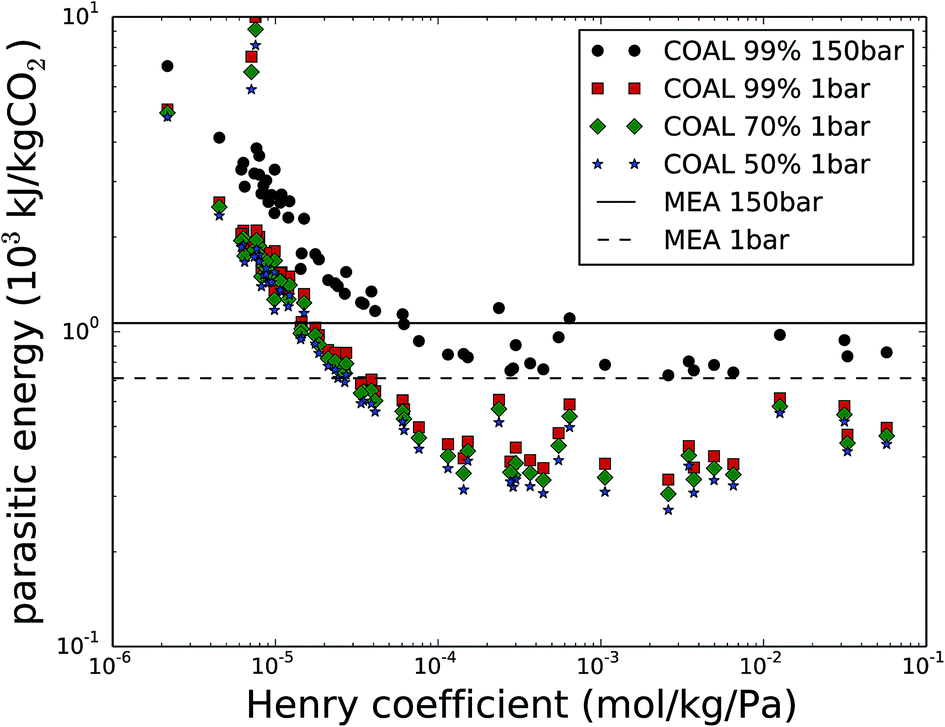 | ||
| Fig. 3 The parasitic energy to capture CO2 from coal flue gas as a function of the CO2 Henry coefficient (kH,CO2) at 300 K. The original parasitic energy is given in black, with CO2 at 99% purity and compressed to 150 bar. The cases for 99%, 70% and 50% purity at 1 bar are shown in red, green and blue respectively. Current MEA technology is marked as a solid line whereas the heating requirement of the MEA technology (excluding the compression from 1 to 150 bar) is shown as the dashed line. | ||
The parasitic energy of the amine technology with monoethanolamine (MEA), 1065 kJ kgCO2−1, is indicated with a solid line. Please note that for the MEA technology, the 353 kJ kgCO2−1 compression work from 1 bar to 150 bar can be avoided, leaving the heating energy as the only contribution to the parasitic energy (indicated with a dashed line in Fig. 3). This corresponds to 712 kJ kgCO2−1, 33% lower than the original case. The purity of the final mixture however cannot be altered for MEA, as only CO2 reacts with the amines and is therefore recovered at 100%. Within the scope of lowering the energy requirement of carbon capture for certain cases, of carbon utilization, solid adsorbents therefore provide larger margins for energy improvements than amines.
Table 3 shows the parasitic energy of the best performing materials for carbon capture from coal-fired power plants, for different final requirements. Mg-MOF-74 is the best candidate regardless of the final purity requirement. Not compressing the CO2 to 150 bar saves 388 kJ kgCO2−1 and lowers the parasitic energy with 54%. The net compression work is slightly higher than for MEA, as the CO2 is not 100% pure when using Mg-MOF-74, so some N2 has to be compressed as well. Further lowering the purity requirement decreases the parasitic energy from 339 to 271 kJ kgCO2−1, an additional improvement of 20%. This is however much lower than the contribution of the 1–150 bar compression work.
| COAL | |||||||
|---|---|---|---|---|---|---|---|
| 150 bar, 99% | 1 bar, 99% | 1 bar, 70% | 1 bar, 50% | ||||
| Mg-MOF-74 | 727 | Mg-MOF-74 | 339 | Mg-MOF-74 | 305 | Mg-MOF-74 | 271 |
| PPN-6-CH2-TETA | 742 | MgX | 369 | MgX | 338 | MgX | 306 |
| mmen-CuBTTri | 752 | NaX | 370 | NaX | 340 | NaX | 308 |
| NaX | 754 | PPN-6-CH2-TETA | 380 | CaX | 344 | CaX | 310 |
| MgX | 760 | CaX | 381 | NaA | 350 | Zn-MOF-74 | 315 |
The order of the other top-performing materials shifts slightly, depending on the imposed final conditions. The polyamine-tethered porous polymer network PPN-6-CH2-TETA74 disappears from the selection at 70% and 50%. The same is true for the amine-functionalized metal–organic framework mmen-CuBTTri. Finally, for all considered cation-exchanged X-type zeolite (Mg, Na, Ca), the parasitic energy is very similar across the considered cases. The cation-exchanged zeolites are of high interest, as they combine their good carbon capture properties with commercial availability on the tonne scale. This is especially true for zeolite NaX, which is often used as a reference material for solid-adsorbent carbon capture.31,75–77 Finally, as the difference in carbon capture performance between the readily available NaX, and the yet-to-be commercialized Mg-MOF-74 is small, there is no reason to wait for the industrial production of novel materials in order to do carbon capture with solid adsorbents.
4.3 Influence of the carbon source
Extending the analysis to other sources of CO2, such as the flue gas of natural gas-fired power plants (with typically 4% CO2 content) and ambient air (with CO2 levels of 400 ppm), yields Fig. 4a. The 1–150 bar compression work is omitted in all cases and focus is on the imposed final purity. Fig. 4b shows the relative improvement between the parasitic energy for 50% versus 99% imposed purity for coal flue gases, natural gas flue gases and ambient air.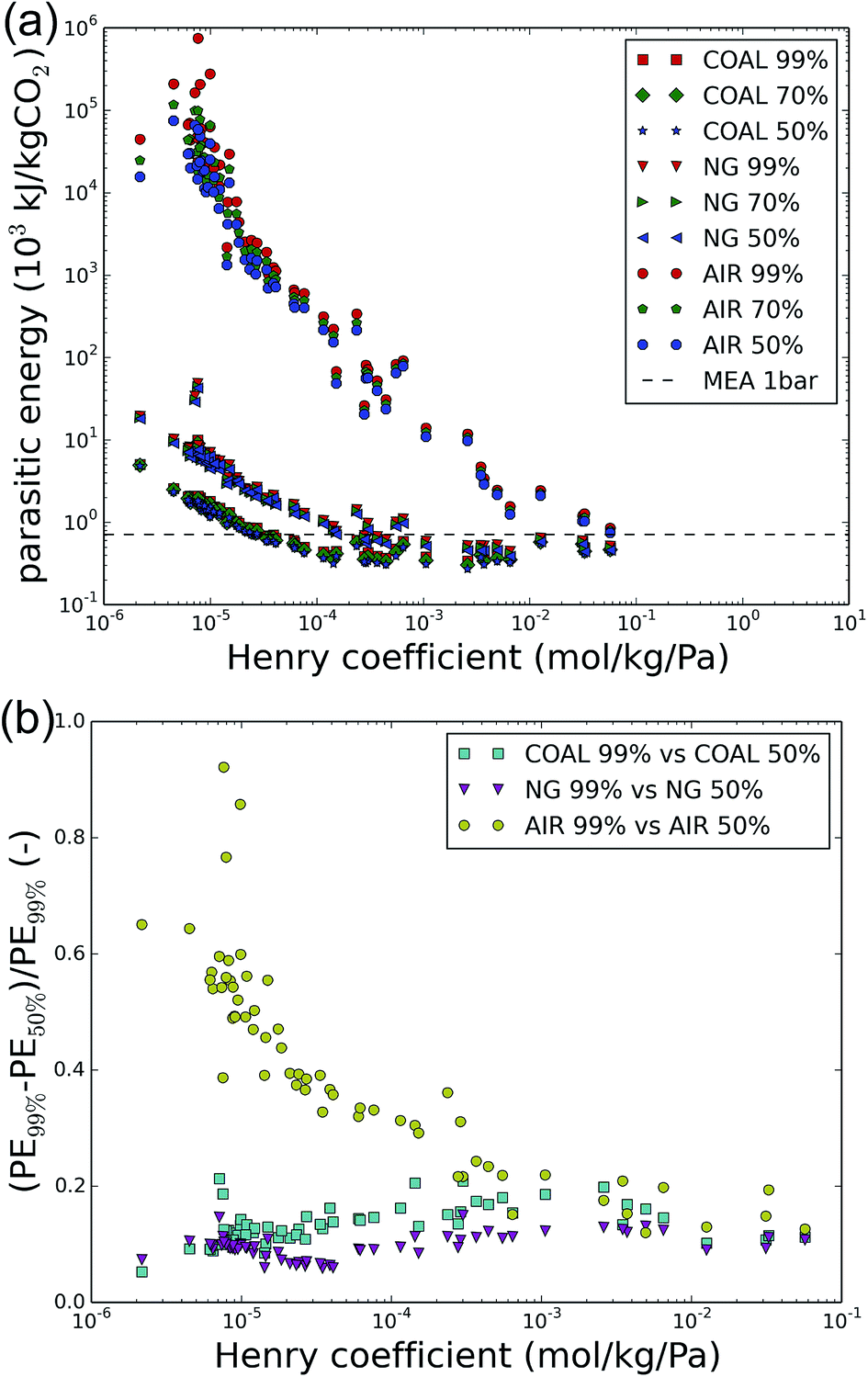 | ||
| Fig. 4 (a) The parasitic energy to capture CO2 from a coal-fired power plant (COAL), a natural gas-fired power plant (NG) or directly from air (AIR). Final purity requirements of 50, 70 and 99% are shown and 1 bar is maintained across all cases. (b) The relative gain in parasitic energy (PE) between the parasitic energy for 50% versus 99% imposed purity for coal flue gases, natural gas flue gases and ambient air. | ||
As expected from the minimum energy calculations in Table 2, the parasitic energy for direct air capture is higher than that for carbon capture from natural gas flue gases or coal flue gases. Moreover, throughout the carbon sources, lower final purity requirements decrease the parasitic energy. The largest gains are found for direct air capture, for which a lot of the poorly performing materials exhibit relative improvements of 20–60% between 99% and 50% purity. This observation does not correspond to the conclusions from the minimum energy calculations, which predicted much lower improvements for direct air capture. Therefore, these large improvements are likely related to improvements in the process, rather than in the thermodynamics of air capture. For coal and natural gas, 10–20% energy can typically be saved, especially for the best performing materials.
Table 4 lists the five best performing materials for carbon capture from the flue gas of natural gas-fired power plants and directly from air, for the different final requirements. For natural gas flue gases, the functionalized PPN-6 materials have the lowest parasitic energy requirements. The top performer is polyamine-tethered porous polymer network PPN-6-CH2-TETA, which was already mentioned among the five best candidates for carbon capture from coal flue gases. The metal–organic material (MOM) zinc hexafluorosilicate (SiF6−2), SIFSIX-3-Zn, is highly selective for CO2.78,79 In cases of lower purity, Mg-MOF-74 again acts as a promising candidate. And finally, the commercially available zeolite NaX is present in the top-5 materials for all purities. As opposed to the case of coal flue gases however, its performance is drastically lower than its competitors. For CO2 capture at 99% purity and 150 bar, NaX has a parasitic energy of 925 kJ kgCO2−1 whereas for PPN-6-CH2-TETA, this is only 807 kJ kgCO2−1.
| 150 bar, 99% | 1 bar, 99% | 1 bar, 70% | 1 bar, 50% | ||||
|---|---|---|---|---|---|---|---|
| NG | |||||||
| PPN-6-CH2-TETA | 807 | PPN-6-CH2-TETA | 444 | PPN-6-CH2-TETA | 416 | PPN-6-CH2-TETA | 389 |
| PPN-6-CH2-TAEA | 858 | PPN-6-CH2-TAEA | 494 | PPN-6-CH2-TAEA | 466 | PPN-6-CH2-TAEA | 439 |
| PPN-6-CH2-DETA | 880 | PPN-6-CH2-DETA | 515 | Mg-MOF-74 | 486 | Mg-MOF-74 | 454 |
| SIFSIX-3-Zn | 907 | NaX | 518 | PPN-6-CH2-DETA | 487 | SIFSIX-3-Zn | 455 |
| NaX | 925 | SIFSIX-3-Zn | 520 | NaX | 488 | NaX | 455 |
|
|||||||
| AIR | |||||||
| PPN-6-CH2-DETA | 1215 | PPN-6-CH2-DETA | 854 | PPN-6-CH2-DETA | 797 | PPN-6-CH2-DETA | 746 |
| SIFSIX-3-Cu | 1617 | SIFSIX-3-Cu | 1206 | SIFSIX-3-Cu | 1112 | SIFSIX-3-Cu | 1026 |
| PPN-6-CH2-TAEA | 1645 | PPN-6-CH2-TAEA | 1281 | PPN-6-CH2-TAEA | 1145 | PPN-6-CH2-TAEA | 1033 |
| PPN-6-CH2-TETA | 1948 | PPN-6-CH2-TETA | 1564 | PPN-6-CH2-TETA | 1398 | PPN-6-CH2-TETA | 1254 |
| ZIF-36-CAG | 3240 | ZIF-36-CAG | 2439 | ZIF-36-CAG | 2248 | ZIF-36-CAG | 2122 |
For CO2 capture directly from air, again the functionalized PPN-6 materials are suitable candidates, with PPN-6-CH2-DETA as the material with the lowest parasitic energy. The differences in parasitic energy, even among the five best performers, are much larger than in the case for capture from coal or natural gas flue gases. Due to this wider spacing, the relative order of the materials does not change with changing purity requirements. Copper hexafluorosilicate (SiF6−2), SIFSIX-3-Cu, is the second best material. And the zeolitic imidazolate framework ZIF-36-CAG also appears in this list. NaX is in the seventh place, with a parasitic energy of almost four times that of PPN-6-CH2-DETA (e.g. 2889 kJ kgCO2−1 for NaX versus 746 kJ kgCO2−1 for PPN-6-CH2-DETA in the case of 50% pure CO2).
In Table 5, the parasitic energies of the best performing materials for each source of CO2 are compared across the imposed purities. From Tables 3 and 4, we concluded that for CO2 capture from coal-fired power plants, Mg-MOF-74 had the optimal properties, for natural gas-fired power plants, this was PPN-6-CH2-TETA and for direct air capture PPN-6-CH2-DETA. Lowering the final purity requirement from 99% to 50% lowers the parasitic energy with almost 20% for Mg-MOF-74, whereas for PPN-6-CH2-TETA and PPN-6-CH2-DETA, the possible improvement is only 12%. Interestingly, for direct air capture, the parasitic energy requirement when purifying CO2 from 400 ppm to 50% (746 kJ kgCO2−1) is only marginally higher than the original parasitic energy requirement for CO2 at 150 bar and 99% purity from coal flue gas (727 kJ kgCO2−1). This indicates that direct air capture could be an attractive method to win CO2 for specific cases of carbon utilization.
| 150 bar, 99% | 1 bar, 99% | 1 bar, 70% | 1 bar, 50% | |
|---|---|---|---|---|
| COAL | 727 | 339 | 305 | 271 |
| NG | 807 | 444 | 416 | 389 |
| AIR | 1215 | 854 | 797 | 746 |
4.4 Importance of the final purity
In the previous sections, the CO2 purity was a key factor in the discussion. The initial purity of CO2 is determined by the composition of the flue gas or the air, and is therefore fixed by the carbon source. For the final purity of CO2, we assumed that this is imposed by the operation of the carbon capture process. However, Fig. 5 reveals that this is not necessarily the case. In this plot, the results from Fig. 3 are color coded based on the final purity, after optimization of the parasitic energy. For a coal-fired power plant flue gas and with final purities set to 99%, 70% and 50%, only the best performing materials actually attain purities higher than 90%. Moreover, even when the purity requirement is gradually lowered (circles, squares and triangles at the same Henry coefficient), this barely influences the final purity. This indicates that the final purity of the recovered gas mixture is in fact imposed by the material properties (e.g. Henry coefficient), rather than by the operating conditions.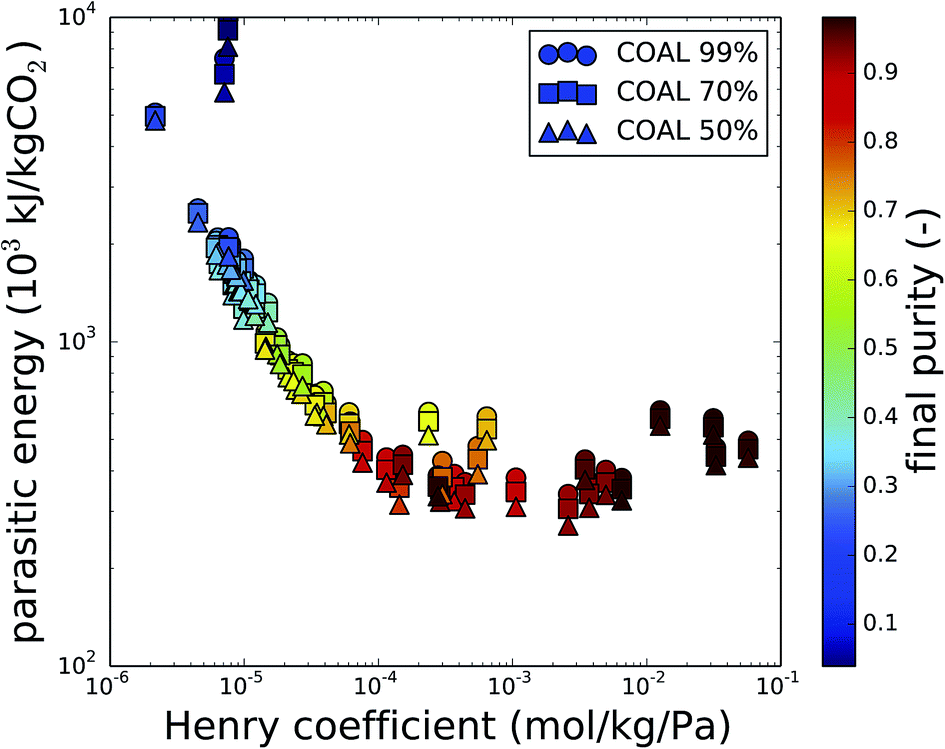 | ||
| Fig. 5 The parasitic energy to capture CO2 from coal flue gas as a function of the CO2 Henry coefficient (kH,CO2) at 300 K, for CO2 at 99%, 70% or 50% and 1 bar. The color code indicates the purity of the final gas mixture and shows that the purity requirement of 99% is only obtained for the best performing materials. | ||
Fig. 6 shows the final purity as a function of the CO2/N2 selectivity of the material at adsorption conditions. A correlation exists between the CO2 selectivity, a material property, and the final CO2 purity, an outcome of the carbon capture process optimization. For coal flue gases, there is no significant difference between the different cases of imposed purity, which is a process parameter. Fig. S2 and S3 in the ESI† show that the points for natural gas flue gases and direct air capture are slightly further apart. In any case, the CO2/N2 selectivity of the used material influences the final purity more than the imposed purity does.
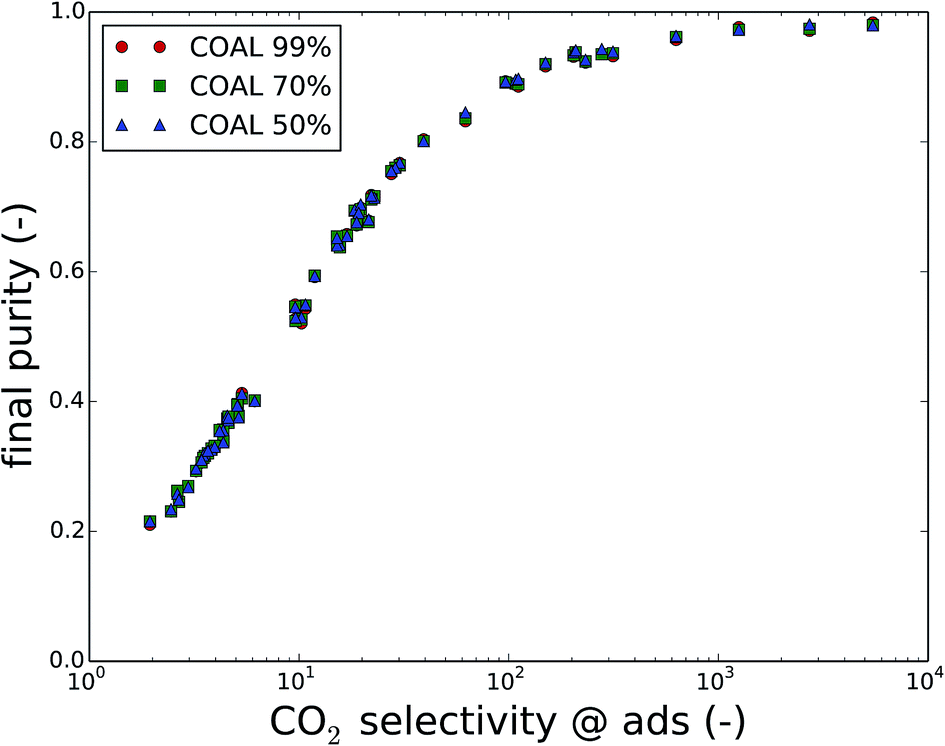 | ||
| Fig. 6 The final purity as a function of the CO2/N2 selectivity of the material at adsorption conditions for carbon capture from coal flue gases at different imposed purities (99%, 70%, 50%). Note that the selectivity is a material property, the imposed purity an adaptable process parameter and the final purity an outcome of the process optimization. | ||
Moreover, a couple of interesting correlations with the purity are shown in Fig. 7. For clarity, the points are limited to the flue gases of coal-fired power plants. In the ESI,† the correlations are also given for natural gas flue gases and air. In Fig. 7a, the parasitic energy is plotted as a function of the final purity. Generally, the parasitic energy of a material decreases with increasing final purity. At very high final purity (>95%) however, the parasitic energies increase again, suggesting that the last percents are the most difficult to obtain. As was already clear from previous plots, the imposed purity also has an influence on the parasitic energy. For air capture (See Fig. S4 in the ESI†), most materials fail to meet the purity requirements, and hence result in parasitic energies of up to 4 orders of magnitude higher. Fig. 7b reveals an even clearer correlation between the final purity and the compression energy. Also here, both the imposed and the final purity have an effect on the compression energy. Therefore, it pays off to use a highly selective CO2 adsorbent and lower the purity requirements later, rather than using a less selective material CO2 which would meet the goals. This is due to the compression work of the additional N2 in the capture gas stream, as can be explained by Fig. 7c. The pressure at which CO2 is recovered varies the most at high purity, dropping significantly across the imposed final purity, whereas at low purity, there is almost no improvement possible. Finally, in Fig. 7d, the parasitic energy across the different cases is compared with the original parasitic energy requirement for CO2 at 99% and 150 bar shows that improvements of almost 60% are possible, especially when using coal flue gases as the carbon source. For direct air capture, the possible improvement of most materials is very low, but there are also some promising outliers at high purity (see Fig. S4 in ESI†).
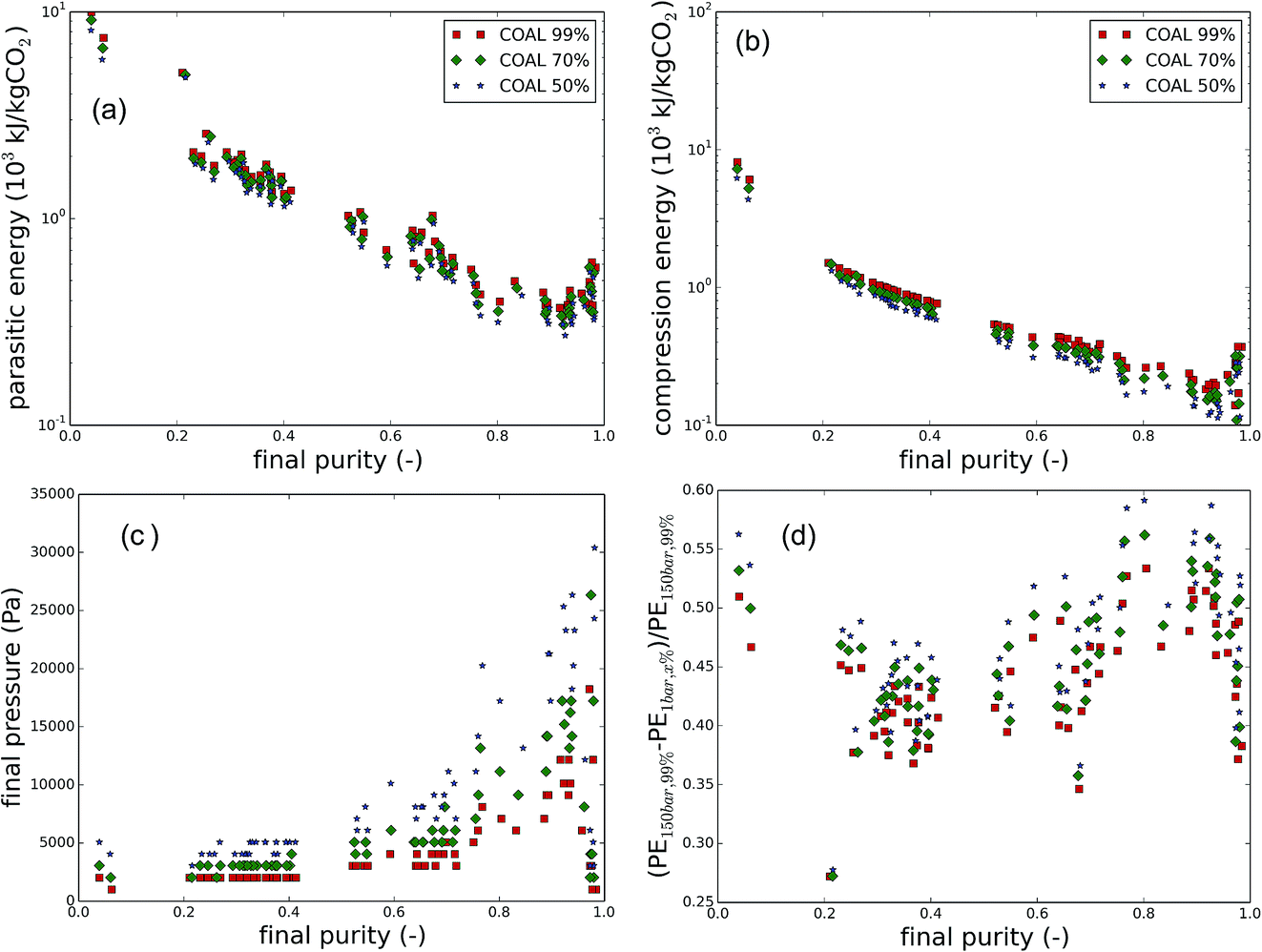 | ||
| Fig. 7 (a) Parasitic energy as a function of the final purity, (b) the compression part of the parasitic energy as a function of the final purity, (c) the final pressure as a function of the final purity, (d) improvement of the parasitic energy, compared to the reference case of CO2 at 150 bar and 99% imposed purity, all as a function of the final purity. | ||
These plots demonstrate that on the one hand, for one specific material, the parasitic energy is reduced when lower purity requirements are imposed. On the other hand, across the different materials, the trend is that the higher the purity of the final mixture, the lower the parasitic energy requirement. Although the purity is mainly imposed by the CO2/N2 selectivity of the material, which is fixed, there is some margin to tune the desorption pressure in order to lower the compression work of the regeneration. Therefore, the best strategy is to choose a highly selective material, which will hence yield a high final purity, ensuring a low parasitic energy among competing materials. Subsequently, the carbon capture process can be operated at a lower imposed purity, thereby reducing the parasitic energy.
Finally, when the desired purity is not met, a second adsorption step can be introduced. This step will be less energy-demanding, but will increase the parasitic energy of the overall process. This is an interesting extension for future work.
4.5 Case study: zeolite NaX
Zeolites are the work horses of the petrochemical industry, with widespread uses in catalysis and adsorption. Moreover, they have excellent regenerability and high stability in a wide range of environments. The commercial availability of zeolite NaX (or 13X) in particular, is truly unique among the considered materials.80 Only a handful of MOFs have been commercialized so far, with prices that are many times that of zeolites.81 The other materials discussed in this manuscript are even further away from industrial applications.Fig. 8 shows the Langmuir isotherms of CO2 and N2 in NaX, for carbon capture from coal flue gases (a) and directly from air (b) respectively. The numerical values for the desorption pressure, temperature and purity, as well as the compression, heating and parasitic energy are given in Table 6. For coal flue gases, the CO2 loading during adsorption conditions is high among its competitors: almost 150 gCO2 kgNaX−1, as is its selectivity (CO2/N2) with 314, resulting in a relatively low N2 uptake. Optimizing the parasitic energy includes a trade-off between pulling a stronger vacuum, and hence evacuating more CO2 from the material in one step, and keeping a higher desorption pressure, but requiring more energy for heating the material. The result of this optimization is the ideal amount of CO2 recovered in one step, the difference in the CO2 loading at adsorption and the CO2 loading at desorption. This amount remains more or less constant across the imposed final purities. Lowering the final purity requirement means that the desorption sweep stream contains less CO2 and that therefore, the driving force for CO2 to leave the framework increases. As a result, the desorption isotherm moves towards higher total pressures, and hence, lower pressures are needed to evacuate the same amount of CO2.
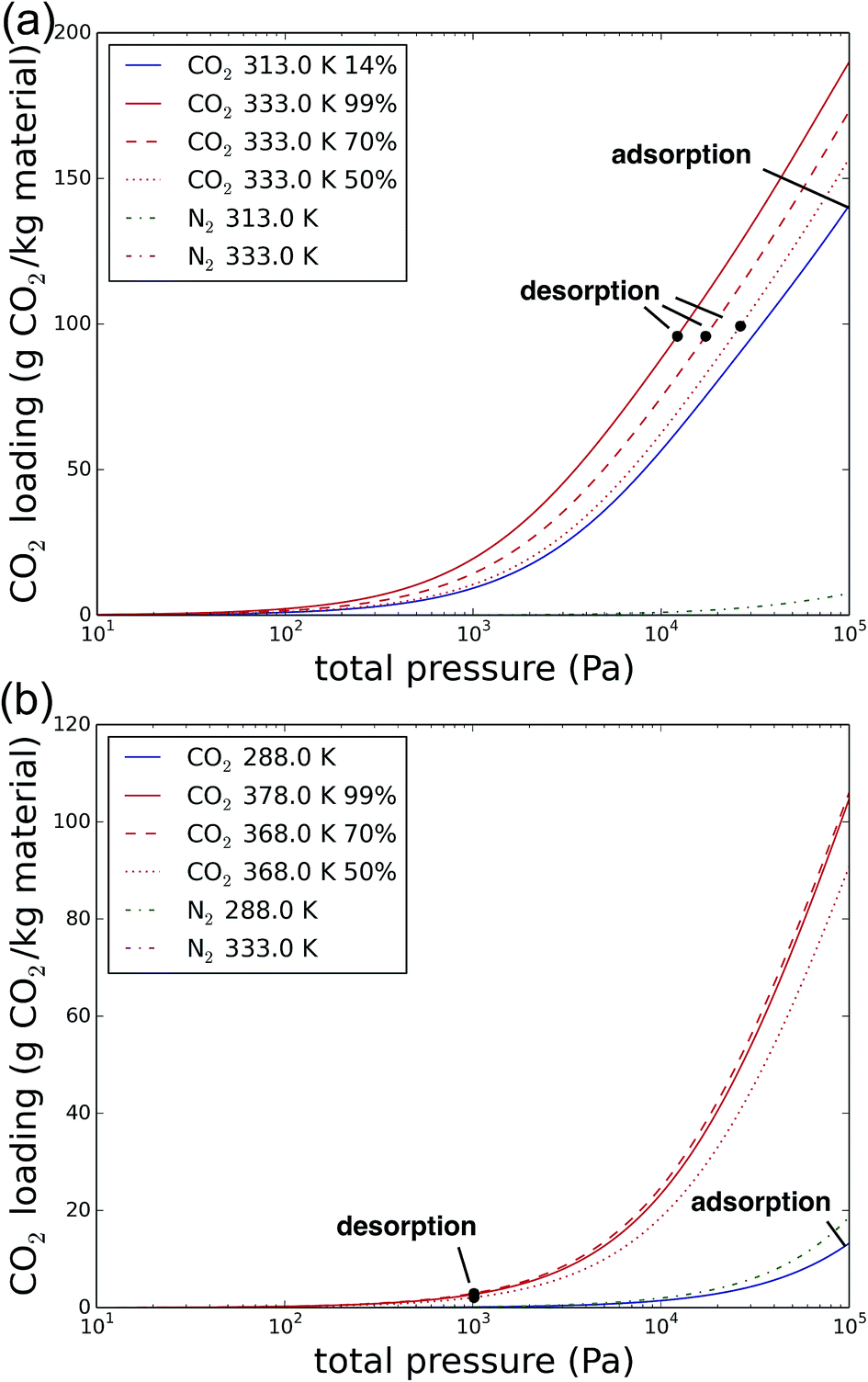 | ||
| Fig. 8 The CO2 and N2 isotherms of NaX for carbon capture from coal-fired power plant flue gases (a) and direct air capture (b), respectively. The adsorption and different desorption conditions indicated are depending on the imposed purity of the CO2. | ||
| NaX | Fin P (Pa) | Fin T (K) | Fin PUR (–) | CE1–150 bar (kJ kgCO2−1) | PE (kJ kgCO2−1) | CE (kJ kgCO2−1) | HE (kJ kgCO2−1) | CE/PE (–) | CO2 prod (kg mmaterial−3) |
|---|---|---|---|---|---|---|---|---|---|
| COAL 99% | 12159 |
333 | 0.932 | 383 | 370 | 180 | 191 | 0.49 | 42.0 |
| COAL 70% | 17225 |
333 | 0.937 | 381 | 340 | 149 | 190 | 0.44 | 42.1 |
| COAL 50% | 26345 |
333 | 0.939 | 380 | 308 | 113 | 194 | 0.37 | 38.9 |
| NG 99% | 3040 | 333 | 0.886 | 406 | 518 | 313 | 205 | 0.60 | 42.7 |
| NG 70% | 4053 | 333 | 0.891 | 404 | 488 | 286 | 203 | 0.59 | 44.4 |
| NG 50% | 6080 | 333 | 0.889 | 405 | 455 | 250 | 205 | 0.55 | 42.5 |
| AIR 99% | 1013 | 378 | 0.338 | 1158 | 3412 | 1102 | 2309 | 0.32 | 8.7 |
| AIR 70% | 1013 | 368 | 0.333 | 1176 | 3103 | 1118 | 1984 | 0.36 | 8.5 |
| AIR 50% | 1013 | 368 | 0.353 | 1106 | 2889 | 1054 | 1835 | 0.36 | 9.2 |
For direct air capture, Fig. 8b, the performance of NaX is suboptimal. In this case, the desorption pressure reached the lower limit of 1013 Pa for all cases. Therefore, the temperature has to be increased in order to recover enough CO2. However, the CO2 working capacity is still poor (only 20% of that for coal flue gases) and the final purity is below any of the imposed purities (around 34%). It is however possible to imagine carbon utilization at 30%, for instance to improve crop growth in greenhouses.35,36
5 Cost from a sequestration and utilization viewpoint
As for any other chemical compound, there is a market for CO2. As opposed to any other market however, the CO2 supply however exceeds its demand with several orders of magnitude, as illustrated in Table 7. On the supply side, the 36 billion metric tons of CO2 emitted in 2015. On the demand side, some 80 million metric tons of CO2, most of which were used for Enhanced Oil Recovery (EOR). Currently, the CO2 used for EOR is delivered from natural CO2 reservoirs, rather than from carbon capture in power plants.82 Moreover, the recovered oil will in turn be converted into CO2 emissions, giving rise to a net increase of CO2 emissions to the atmosphere. Carbon capture and utilization for EOR will only be economically viable if the price of the carbon capture technology is lower than the price to extract CO2 from natural reservoirs.Market mechanisms alone will therefore not have a significant impact in order to lower CO2 emissions to the atmosphere. That is why a growing number of countries, regions and cities are putting a price on CO2 emissions, either through taxes or cap-and-trade mechanisms. Taxes straightforwardly provide incentives for polluters to reduce emissions, as it requires them to pay a fixed amount for each ton of CO2 emitted. Cap-and-trade mechanisms require polluters to buy units of allowed emissions, which can be traded on the market, like any other commodity. Last year, this global market traded 7 billion metric tons of CO2, a significant portion of the total emissions. At first sight, the total market for CCS is two orders of magnitude larger than the market for CCU. However, it only pays off to capture and sequester CO2 emissions if the price to emit one ton of CO2 is higher than the cost of a technology that avoids the emission of one ton of CO2.
The trading prices of CO2 in Europe and California were around 8 EUR and 12 USD per metric ton of CO2 in 2015, whereas reported CCS costs are generally a few times higher (see Table 8). Moreover, on average, CO2 emissions are priced at less than 10 USD per ton.83 As a result, there is currently no market for CCS. With the current CO2 trading prices, it is cheaper to dump CO2 into the atmosphere and pay for it, than to avoid the emissions using CCS. In view of carbon utilization, it is not straightforward to find prices for bulk CO2, as they are highly dependent on the source, and especially the distance from the source to the utilization site. We found a relatively low price for CO2 from natural reservoirs, 15–19 USD per ton, but if new pipelines have to be build, this price will increase rapidly. Likewise, some chemical plants produce CO2 with high purity and low costs (5–25 USD per ton), for instance from ammonia synthesis, but if the CO2 is not used in the proximity of the refinery, the cost will go up drastically. Moreover, when food-grade CO2 is required, the price will also be higher than that of industrial grade, etc.
| ton CO2−1 | Currency | Year | Ref. | |
|---|---|---|---|---|
| Carbon trading price | ||||
| EU | 8 | EUR | 2015 | 84 |
| California | 12 | USD | 2015 | 85 |
|
||||
| CCS cost | ||||
| McKinsey | 30–45 | EUR | 2008 | 2 |
| BCG | 45 | EUR | 2008 | 3 |
| IEA (coal) | 55 | USD | 2011 | 5 |
| IEA (NG) | 80 | USD | 2011 | 5 |
| IPCC (coal) | 9–44 | USD | 2005 | 86 |
| IPCC (NG) | 16–68 | USD | 2005 | 86 |
|
||||
| CO 2 production price | ||||
| Industrial grade | 51 | USD | 2010 | 87 |
| Natural reservoirs | 15–19 | USD | 2011 | 82 |
| Concentrated sources | 20 | USD | 2011 | 82 |
| Refinery | 5–25 | USD | 2005 | 88 |
| Consumer market | 90–100 | USD | 2005 | 88 |
|
||||
| Social cost of carbon | ||||
| Interagency working group | 43 | USD | 2013 | 89 |
The central question in this manuscript is whether utilization at ambient pressure and lower purity further shift the economics in a more favorable direction. Since the parasitic energy is defined as the electricity loss of the power plant, the values in kJ kgCO2−1 can be converted to currency, by assuming an average electricity price. In 2015, the price for electricity for industrial customers was on average 0.12 EUR per kW per h in the European Union90 and 0.07 USD in the United States of America.91 Note that there are large differences across these regions, and that no distinction is made between the relative costs of the different fuels (coal, natural gas, nuclear, renewable). Table 9 shows the monetary cost for the different carbon capture scenarios discussed above. These values do not include the capital cost, or the cost of transportation and sequestration, which is around 10 EUR per ton of CO2.
| 150 bar, 99% | 1 bar, 99% | 1 bar, 70% | 1 bar, 50% | |
|---|---|---|---|---|
| EUR | ||||
| COAL | 24 | 11 | 10 | 9 |
| NG | 27 | 15 | 14 | 13 |
| AIR | 41 | 28 | 27 | 25 |
|
||||
| USD | ||||
| COAL | 14 | 7 | 6 | 5 |
| NG | 16 | 9 | 8 | 8 |
| AIR | 24 | 17 | 15 | 14 |
The cost of 24 EUR per ton in Table 9 for carbon capture from the flue gas of coal-fired power plants is in agreement with the CCS costs reported in Table 8 when subtracting the transportation, storage and capital cost. The price of 14 USD is also relatively close to the 15–19 USD for CO2 from natural CO2 reservoirs, indicating that captured CO2 from coal flue gases could possibly be used for EOR. Secondly, in Table 9, the difference between coal-fired power plants and natural gas-fired power plants is much smaller than what was expected from the policy reports in Table 8. For CO2 capture directly from the air, we did not find reference prices in the literature, but the values in Table 9 are likely on the low side.
Most interestingly, not compressing the capture CO2 from 1 bar to 150 bar saves around 13 EUR or 7 USD per ton of CO2. Lowering the purity requirement from 99% to 70% or 50% can save 1–2 EUR or 1–2 USD, a modest improvement. The CO2 price of 9 EUR or 5 USD per ton from coal flue gases is very interesting for applications where 1 bar CO2 at 50% purity is desired. Finally, an interagency working group determined the US social cost of CO2 at 43 USD per ton of CO2 in 2020 assuming a 3% discount rate.89,92 At this socially desirable CO2 price, sequestration and utilization of CO2 would be economically viable.
6 Conclusions
Before CO2 can be sequestered in underground reservoirs, it has to be purified to 99% and subsequently compressed to 150 bars, making up a large part of the cost for carbon capture and storage. For some examples of carbon capture and utilization however, compression to 150 bar is superfluous and the final purity requirement may be lower than 99%. The use of CO2 to increase crop growth in greenhouses, algae growth for biofuel production or “Carbstone” are a few notable examples. In this manuscript, we investigate how far the parasitic energy – and therefore costs – of carbon capture can be lowered by considering less stringent final conditions. To find reliable trends, we screen a selection of over 60 materials, both synthesized and hypothetical, from a variety of material families.First of all, we find that the compression work from 1 to 150 bar comprises up to 55% of the parasitic energy for CCS. Therefore, avoiding this compression work in a CCU application is by far the biggest possible improvement we identified. Moreover, lowering the purity requirement from 99% down to 50% further lowers the parasitic energy up to 20%. This stems from the fact that CO2 can be recovered at a less strong vacuum, which requires less compression work.
When comparing different materials, two effects are at play. Across different materials, the higher the purity of the final mixture, the lower the parasitic energy requirement. For a single material however, the parasitic energy is lowered when lower purity requirements are imposed. There is a systematic distinction between the imposed purity and the final purity. We found that the CO2/N2 selectivity of the material, a fixed material property, and not the imposed final purity, one of the carbon capture process conditions, has the largest influence on the final CO2 purity, a result of the parasitic energy minimization. When a lower purity is imposed, the final purity does not change significantly, but the desorption pressure is higher and the compression work of the regeneration is hence lower. Therefore, a highly selective material yields a high final purity, and the carbon capture process can subsequently be operated at a lower imposed purity, thereby lowering the parasitic energy.
As a result, the best performing materials remain the same across the considered final conditions, although the relative order may somewhat shift. For carbon capture from coal flue gases, natural gas flue gases and air, these materials are Mg-MOF-74, PPN-6-CH2-TETA, and PPN-6-CH2-DETA, respectively. Zeolite NaX combines its commercial availability with good performance, especially for coal flue gases and natural gas flue gases.
Interestingly, omitting the 1–150 bar compression and lowering the purity requirement reduces the parasitic energy of direct air capture almost to the parasitic energy to recover CO2 at 99% purity and 150 bar from coal flue gases. Direct air capture could hence be attractive for specific cases of carbon utilization. Bear in mind that changing the purity requirement is not possible with the MEA technology, as 100% pure CO2 is stripped from the amines in the regeneration step. This underlines the extra potential of solid adsorbent carbon capture in the case of CCU.
As a conclusion, when CO2 is not sequestered at 150 bar and 99% purity, but instead utilized at ambient pressure and lower purities, the parasitic energy and associated cost of carbon capture may be reduced by almost 60%. This drastically lowers the cost of carbon capture for utilization purposes. Moreover, the calculated price of CO2 capture from coal flue gases, for utilization at 1 bar and 50% purity (9 EUR or 5 USD respectively), is competitive with CO2 from natural reservoirs or refineries. This makes a strong case for carbon capture and utilization.
Acknowledgements
L. J. and V. V. S. acknowledge the Flemish Fund for Scientific Research (FWO12/ASP/151) and BELSPO in the frame of IAP/7/05 for financial support. V. V. S. acknowledges funding from the European Research Council under the European Community's Seventh Framework Programme FP7 (2007–2013) ERC grant agreement number 240483, of the European Union's Horizon 2020 research and innovative programme under consolidator ERC grant agreement number 647755-DYNPOR (2015–2020). B. S. was supported by the Center for Gas Separations Relevant to Clean Energy Technologies, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, and Office of Basic Energy Sciences, under Award Number DE-SC0001015. J. M. H. was supported by the Deutsche Forschungsgemeinschaft (DFG, priority program SPP 1570).References
- A. S. Bhown and B. C. Freeman, Environ. Sci. Technol., 2011, 45, 8624–8632 CrossRef CAS PubMed.
- McKinsey&Company, Carbon Capture & Storage: Assessing the Economics, 2008, http://www.globalccsinstitute.com/publications/carbon-capture-storage-assessing-economics Search PubMed.
- Boston Consulting Group, Carbon Capture and Storage: A Solution to the Problem of Carbon Emissions, 2008, https://www.bcg.com/documents/file15263.pdfs Search PubMed.
- British Petroleum, BP Energy Outlook 2035, 2015, https://www.bp.com/energyoutlook Search PubMed.
- International Energy Agency, World Energy Outlook 2013 factsheet, 2013, http://www.worldenergyoutlook.org/resources/factsheets/ Search PubMed.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- G. Rochelle, E. Chen, S. Freeman, D. Van Wagener, Q. Xu and A. Voice, Chem. Eng. J., 2011, 171, 725–733 CrossRef CAS.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- S. Choi, J. Drese and C. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS PubMed.
- G. Sneddon, A. Greenaway and H. H. P. Yiu, Adv. Energy Mater., 2014, 4, 1301873 CrossRef.
- A. O. Yazaydin, R. Q. Snurr, T.-H. Park, K. Koh, J. Liu, M. D. LeVan, A. I. Benin, P. Jakubczak, M. Lanuza, D. B. Galloway, J. J. Low and R. R. Willis, J. Am. Chem. Soc., 2009, 131, 18198–18199 CrossRef CAS PubMed.
- M. Fernandez, P. G. Boyd, T. D. Daff, M. Z. Aghaji and T. K. Woo, J. Phys. Chem. Lett., 2014, 5, 3056–3060 CrossRef CAS PubMed.
- N. T. T. Nguyen, H. Furukawa, F. Gándara, H. T. Nguyen, K. E. Cordova and O. M. Yaghi, Angew. Chem., Int. Ed., 2014, 53, 10645–10648 CrossRef CAS PubMed.
- B. Wang, A. P. Cote, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Nature, 2008, 453, 207–211 CrossRef CAS PubMed.
- D. Yuan, W. Lu, D. Zhao and H.-C. Zhou, Adv. Mater., 2011, 23, 3723–3725 CrossRef CAS PubMed.
- W. Lu, D. Yuan, D. Zhao, C. I. Schilling, O. Plietzsch, T. Muller, S. Bräse, J. Guenther, J. Blümel, R. Krishna, Z. Li and H.-C. Zhou, Chem. Mater., 2010, 22, 5964–5972 CrossRef CAS.
- W. Lu, W. M. Verdegaal, J. Yu, P. B. Balbuena, H.-K. Jeong and H.-C. Zhou, Energy Environ. Sci., 2013, 6, 3559 CAS.
- T.-H. Bae, M. R. Hudson, J. A. Mason, W. L. Queen, J. J. Dutton, K. Sumida, K. J. Micklash, S. S. Kaye, C. M. Brown and J. R. Long, Energy Environ. Sci., 2013, 6, 128–138 CAS.
- J. Kim, M. Abouelnasr, L.-C. Lin and B. Smit, J. Am. Chem. Soc., 2013, 135, 7545–7552 CrossRef CAS PubMed.
- M. W. Deem, R. Pophale, P. A. Cheeseman and D. J. Earl, J. Phys. Chem. C, 2009, 113, 21353–21360 CAS.
- M. M. F. Hasan, E. L. First and C. A. Floudas, Phys. Chem. Chem. Phys., 2013, 15, 17601–17618 RSC.
- L. Joos, K. Lejaeghere, J. Huck, V. Van Speybroeck and B. Smit, Energy Environ. Sci., 2015, 8, 2480–2491 CAS.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- V. Van Speybroeck, K. Hemelsoet, L. Joos, M. Waroquier, R. G. Bell and C. R. A. Catlow, Chem. Soc. Rev., 2015, 44, 7044–7111 RSC.
- J. Li, A. Corma and J. Yu, Chem. Soc. Rev., 2015, 44, 7112–7127 RSC.
- Y.-S. Bae and R. Q. Snurr, Angew. Chem., Int. Ed., 2011, 50, 11586–11596 CrossRef CAS PubMed.
- L.-C. Lin, A. H. Berger, R. L. Martin, J. Kim, J. A. Swisher, K. Jariwala, C. H. Rycroft, A. S. Bhown, M. Deem, M. Haranczyk and B. Smit, Nat. Mater., 2012, 11, 633–641 CrossRef CAS PubMed.
- J. M. Huck, L.-C. Lin, A. H. Berger, M. N. Shahrak, R. L. Martin, A. S. Bhown, M. Haranczyk, K. Reuter and B. Smit, Energy Environ. Sci., 2014, 7, 4132–4146 CAS.
- C. Simon, R. Mercado, S. Schnell, B. Smit and M. Haranczyk, Chem. Mater., 2015, 27, 4459–4475 CrossRef CAS.
- C. Simon, J. Kim, D. Gomez-Gualdron, J. Camp, Y. Chung, R. Martin, R. Mercado, M. Deem, D. Gunter, M. Haranczyk, D. Sholl, R. Snurr and B. Smit, Energy Environ. Sci., 2015, 8, 1190–1199 CAS.
- L. Joos, J. A. Swisher and B. Smit, Langmuir, 2013, 29, 15936–15942 CrossRef CAS PubMed.
- S. Heylen, L. Joos, T. N. Parac-Vogt, V. Van Speybroeck, C. E. A. Kirschhock and J. A. Martens, Angew. Chem., Int. Ed., 2012, 51, 11010–11013 CrossRef CAS PubMed.
- B. Smit, J. R. Reimer, C. M. Oldenburg and I. C. Bourg, Introduction to Carbon Capture and Sequestration, Imperial College Press, London, 1st edn, 2014 Search PubMed.
- S. A. Scott, M. P. Davey, J. S. Dennis, I. Horst, C. J. Howe, D. J. Lea-Smith and A. G. Smith, Curr. Opin. Biotechnol., 2010, 21, 277–286 CrossRef CAS PubMed.
- S. A. Prior, G. B. Runion, S. C. Marble, H. H. Rogers, C. H. Gilliam and H. A. Torbert, HortScience, 2011, 46, 158–162 CAS.
- H. H. Rogers, G. Runion and S. V. Krupa, Environ. Pollut., 1994, 83, 155–189 CrossRef CAS PubMed.
- C. Meyer, M. G. Wichmann and T. S. Spengler, J. Econ. Bus., 2016, 1–36 Search PubMed.
- D. H. Abbott and C. E. Albright, J. Laser Appl., 1994, 6, 69 CrossRef CAS.
- W. Klemm and R. Berger, Cem. Concr. Res., 1972, 2, 567–576 CrossRef CAS.
- C. Wang, L. Lü and M. Xu, J. Med. Entomol., 2012, 49, 1076–1083 CrossRef CAS PubMed.
- E. N. T. George Douglas Hobson, Introduction to Petroleum Geology, Scientific Press Limited, 1st edn, 1975 Search PubMed.
- E. Davidson, Process of extinguishing fires, US Pat., 1,010,870, 1911 Search PubMed.
- Q. Wang, J. Luo, Z. Zhong and A. Borgna, Energy Environ. Sci., 2011, 4, 42–55 CAS.
- A. Kumar, D. G. Madden, M. Lusi, K.-J. Chen, E. A. Daniels, T. Curtin, J. J. Perry and M. J. Zaworotko, Angew. Chem., Int. Ed., 2015, 54, 14372–14377 CrossRef CAS PubMed.
- A. L. Myers and J. M. Prausnitz, AIChE J., 1965, 11, 121–127 CrossRef CAS.
- E. W. Lemmon, M. L. Huber and M. O. McLinden, NIST Reference Fluid Thermodynamic and Transport Properties Database (REFPROP): Version9.0, 2010, http://www.nist.gov/srd/nist23.cfm Search PubMed.
- S. A. Freeman, R. Dugas, D. H. Van Wagener, T. Nguyen and G. T. Rochelle, Int. J. Greenhouse Gas Control, 2010, 4, 119–124 CrossRef CAS.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- J. M. Simmons, H. Wu, W. Zhou and T. Yildirim, Energy Environ. Sci., 2011, 4, 2177 CAS.
- J. A. Mason, K. Sumida, Z. R. Herm, R. Krishna and J. R. Long, Energy Environ. Sci., 2011, 4, 3030 CAS.
- T. M. McDonald, D. M. D'Alessandro, R. Krishna and J. R. Long, Chem. Sci., 2011, 2, 2022 RSC.
- P. Aprea, D. Caputo, N. Gargiulo, F. Iucolano and F. Pepe, J. Chem. Eng. Data, 2010, 55, 3655–3661 CrossRef CAS.
- P. Chowdhury, S. Mekala, F. Dreisbach and S. Gumma, Microporous Mesoporous Mater., 2012, 152, 246–252 CrossRef CAS.
- P. D. C. Dietzel, R. Blom and H. Fjellvåg, Eur. J. Inorg. Chem., 2008, 2008, 3624–3632 CrossRef.
- P. D. C. Dietzel, V. Besikiotis and R. Blom, J. Mater. Chem., 2009, 19, 7362 RSC.
- L. Grajciar, A. D. Wiersum, P. L. Llewellyn, J.-S. Chang and P. Nachtigall, J. Phys. Chem. C, 2011, 115, 17925–17933 CAS.
- Z. Liang, M. Marshall and A. L. Chaffee, Energy Fuels, 2009, 23, 2785–2789 CrossRef CAS.
- B. Mu, P. M. Schoenecker and K. S. Walton, J. Phys. Chem. C, 2010, 114, 6464–6471 CAS.
- Z. R. Herm, J. A. Swisher, B. Smit, R. Krishna and J. R. Long, J. Am. Chem. Soc., 2011, 133, 5664–5667 CrossRef CAS PubMed.
- R. Banerjee, H. Furukawa, D. Britt, C. Knobler, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 3875–3877 CrossRef CAS PubMed.
- H. Amrouche, S. Aguado, J. Pérez-Pellitero, C. Chizallet, F. Siperstein, D. Farrusseng, N. Bats and C. Nieto-Draghi, J. Phys. Chem. C, 2011, 115, 16425–16432 CAS.
- H. Huang, W. Zhang, D. Liu, B. Liu, G. Chen and C. Zhong, Chem. Eng. Sci., 2011, 66, 6297–6305 CrossRef CAS.
- D. Yuan, W. Lu, D. Zhao and H.-C. Zhou, Adv. Mater., 2011, 23, 3723–3725 CrossRef CAS PubMed.
- R. L. Martin, L.-C. Lin, K. Jariwala, B. Smit and M. Haranczyk, J. Phys. Chem. C, 2013, 117, 12159–12167 CAS.
- P. Bai, M. Tsapatsis and J. I. Siepmann, Langmuir, 2012, 28, 15566–15576 CrossRef CAS PubMed.
- J. W. Yoon, Y.-K. Seo, Y. K. Hwang, J.-S. Chang, H. Leclerc, S. Wuttke, P. Bazin, A. Vimont, M. Daturi, E. Bloch, P. L. Llewellyn, C. Serre, P. Horcajada, J.-M. Grenèche, A. E. Rodrigues and G. Férey, Angew. Chem., Int. Ed., 2010, 49, 5949–5952 CrossRef CAS PubMed.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308–2322 RSC.
- W. Lu, D. Yuan, J. Sculley, D. Zhao, R. Krishna and H.-C. Zhou, J. Am. Chem. Soc., 2011, 133, 18126–18129 CrossRef CAS PubMed.
- W. Lu, J. P. Sculley, D. Yuan, R. Krishna, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 7480–7484 CrossRef CAS PubMed.
- K. S. Lackner, Energy, 2013, 50, 38–46 CrossRef CAS.
- Direct Air Capture of CO2 with Chemicals: A Technology Assessment for the APS Panel on Public Affairs, 2011, https://www.aps.org/policy/reports/assessments/upload/dac2011.pdf.
- W. Lu, J. P. Sculley, D. Yuan, R. Krishna, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 7480–7484 CrossRef CAS PubMed.
- J. Mérel, M. Clausse and F. Meunier, Environ. Prog., 2006, 25, 327–333 CrossRef.
- D. Ko, R. Siriwardane and L. T. Biegler, Ind. Eng. Chem. Res., 2003, 42, 339–348 CrossRef CAS.
- K. T. Chue, J. N. Kim, Y. J. Yoo, S. H. Cho and R. T. Yang, Ind. Eng. Chem. Res., 1995, 34, 591–598 CrossRef CAS.
- P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS PubMed.
- O. Shekhah, Y. Belmabkhout, Z. Chen, V. Guillerm, A. Cairns, K. Adil and M. Eddaoudi, Nat. Commun., 2014, 5, 4228 CAS.
- Transparency Market Research, Global Synthetic Zeolite Market, http://www.transparencymarketresearch.com/pressrelease/synthetic-zeolites-market.htm.
- B. Yilmaz, N. Trukhan and U. Müller, Chin. J. Catal., 2012, 33, 3–10 CrossRef CAS.
- Accelerating the Uptake of CCS: Industrial Use of Capture Carbon Dioxide, 2011, https://www.hub.globalccsinstitute.com/publications/.
- State and Trends of Carbon Pricing, 2015, http://www.worldbank.org/content/dam/Worldbank/document/Climate/State-and-Trend-Report-2015.pdf.
- European Emission Allowances—Global Environmental Exchange, 2015, https://www.eex.com/en/market-data/emission-allowances/spot-market/european-emission-allowances.
- California Carbon Dashboard, 2015, http://www.calcarbondash.org.
- IPCC Special Report on Carbon Capture and Storage, 2005 Search PubMed.
- Carbon Dioxide Enhanced Oil Recovery – Untapped Domestic Energy Supply and Long Term Carbon Storage Solution, 2010, https://www.netl.doe.gov/file/%20library/research/oil-gas/small_CO2_EOR_Primer.pdf.
- Carbon Dioxide Apps Are Key In Ethanol Project Developments, 2011, http://www.ethanolproducer.com/articles Search PubMed.
- Social Cost of Carbon for Regulatory Impact Analysis? Under Executive Order 12866, 2013, http://www.1.usa.gov/18ftAsH.
- Energy price statistics, 2015, http://www.eceuropa.eu/eurostat/statistics-explained/index.php/Energy_price_statistics.
- Electric Power Monthly, 2015, http://www.eia.gov/electricity/monthly/pdf/epm.pdf.
- A. Fraas, R. Lutter, S. Dudley, T. Gayer, J. Graham, J. F. Shogren and W. K. Viscusi, Science, 2016, 351, 569 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6fd00031b |
| This journal is © The Royal Society of Chemistry 2016 |