 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrolysis of metal oxides in MgCl2 based molten salts with an inert graphite anode
Yating
Yuan
a,
Wei
Li
a,
Hualin
Chen
a,
Zhiyong
Wang
a,
Xianbo
Jin
*a and
George Z.
Chen
 b
b
aCollege of Chemistry and Molecular Sciences, Hubei Key Laboratory of Electrochemical Power Sources, Wuhan University, Wuhan, 430072, P. R. China. E-mail: xbjin@whu.edu.cn; Fax: +86 27 68756319; Tel: +86 27 68756319
bDepartment of Chemical and Environmental Engineering, Faculty of Engineering, The University of Nottingham, Nottingham, NG7 2RD, UK
First published on 22nd January 2016
Abstract
Electrolysis of solid metal oxides has been demonstrated in MgCl2–NaCl–KCl melt at 700 °C taking the electrolysis of Ta2O5 as an example. Both the cathodic and anodic processes have been investigated using cyclic voltammetry, and potentiostatic and constant voltage electrolysis, with the cathodic products analysed by XRD and SEM and the anodic products by GC. Fast electrolysis of Ta2O5 against a graphite anode has been realized at a cell voltage of 2 V, or a total overpotential of about 400 mV. The energy consumption was about 1 kW h kgTa−1 with a nearly 100% Ta recovery. The cathodic product was nanometer Ta powder with sizes of about 50 nm. The main anodic product was Cl2 gas, together with about 1 mol% O2 gas and trace amounts of CO. The graphite anode was found to be an excellent inert anode. These results promise an environmentally-friendly and energy efficient method for metal extraction by electrolysis of metal oxides in MgCl2 based molten salts.
Introduction
Metal materials are crucial to our society. Metals are mainly produced by pyrometallurgical methods such as carbothermic or metallothermic reductions. However, these processes usually involve multiple steps, and are energy intensive and environmentally unfriendly.1,2 At the end of the twentieth century, Chen et al.3 proposed the solid electrochemical reduction of TiO2 to produce Ti in CaCl2 molten salt, known as the FFC Cambridge process. Since then, this process has received much attention. Up till now, many metals, semi-metals and their alloys have been successfully prepared by this simple solid oxide reduction process.4,5 The FFC Cambridge process can be summarized by following reactions.Overall reaction:
| MOx → M + x/2O2 (inert anode) | (1a) |
| nMOx + xC → nM + xCOn (carbon anode, n = 1 or 2) | (1b) |
Cathodic reaction:
| MOx + 2xe → M + xO2− | (2) |
Anodic reaction:
| 2O2− → O2 + 2e (inert anode) | (3a) |
| nO2− + C → COn + 2ne (carbon anode) | (3b) |
In the above equations M represents the studied metal (e.g. Ti, Nb and Ta) or semi-metal (e.g. Si).
During electrolysis, the graphite anode is often used for its good electronic and mechanical properties. However, as shown by Reaction (3b), the anodic discharge of dissolved O2− ions in CaCl2, which come from the cathode by Reaction (2), will lead to anode consumption and consequently carbon emission. Moreover, the CO2 produced at the anode can react with O2− ions in the molten salt to form CO32− ions, which will move back to the cathode and discharge to carbon and O2− ions,6,7 leading to carbon contamination to the metal product, floating carbon on the surface of CaCl2 melt, as well as low current efficiency. Although many inert anodes have been developed for O2 evolution,8–12 they are yet pending for commercial verification. At present, similar to the electrolysis of dissolved Al2O3 in cryolite based molten fluorides, carbon anodes are still the choice for the electrolysis of solid metal oxides in molten CaCl2. To deal with the relevant problems, recently we have proposed a “disengaging strategy” by electrolysis of solid metal oxides in MgCl2 based molten salts, in which the solubility of O2− ions is insignificant to transport O2− to the anode.13 Alternatively, the released O2− ions stay in the cathode by forming MgO precipitation, and the anode reaction is thus changed to oxidation of Cl− ions to the Cl2 gas. This disengaging strategy makes the carbon anode effectively non-consumable, eliminating CO2 emission and all related problems. Our new process can be represented by Reactions (4–6).
Overall reaction:
| MOx + xMgCl2 = M + xMgO + xCl2 | (4) |
Cathodic reaction:
| MOx + xMgCl2 + 2xe → M + xMgO + 2xCl− | (5) |
Anodic reaction:
| 2xCl− → xCl2 + 2xe | (6) |
As indicated in Table 1, this new process, thermodynamically, can be applied to reduce most transitional metal oxides to respective metals, including some rare earth metals, such as uranium.
| Metal oxides | Decomposition voltage (V) |
|---|---|
| 2KCl = 2K + Cl2(g) | 3.566 |
| 2NaCl = 2Na + Cl2(g) | 3.329 |
| MgCl2 = Mg + Cl2(g) | 2.534 |
| 2MgCl2 + HfO2 = Hf + 2MgO + 2Cl2(g) | 2.388 |
| 2MgCl2 + UO2 = U + 2MgO + 2Cl2(g) | 2.335 |
| 2MgCl2 + ZrO2 = Zr + 2MgO + 2Cl2(g) | 2.317 |
| 2MgCl2 + TiO2 = Ti + 2MgO + 2Cl2(g) | 1.949 |
| 2MgCl2 + SiO2 = Si + 2MgO + 2Cl2(g) | 1.865 |
| 3MgCl2 + B2O3 = 2B + 3MgO + 3Cl2(g) | 1.731 |
| 5MgCl2 + Ta2O5 = 2Ta + 5MgO + 5Cl2(g) | 1.638 |
| 3MgCl2 + V2O3 = 2V + 3MgO + 3Cl2(g) | 1.616 |
| 5MgCl2 + Nb2O5 = 2Nb + 5MgO + 5Cl2(g) | 1.488 |
| 3MgCl2 + Cr2O3 = 2Cr + 3MgO + 3Cl2(g) | 1.478 |
| 3MgCl2 + Ga2O3 = 2Ga(l) + 3MgO + 3Cl2(g) | 1.298 |
| MgCl2 + ZnO = Zn(l) + MgO + Cl2(g) | 1.259 |
| 3MgCl2 + WO3 = W + 3MgO + 3Cl2(g) | 0.984 |
| 2MgCl2 + GeO2 = Ge + 2MgO + 2Cl2(g) | 0.977 |
| 2MgCl2 + SnO2 = Sn(l) + 2MgO + 2Cl2(g) | 0.944 |
| 3MgCl2 + Fe2O3 = 2Fe + 3MgO + 3Cl2(g) | 0.939 |
| 2MgCl2 + MnO2 = Mn + 2MgO + 2Cl2(g) | 0.855 |
| 3MgCl2 + MoO3 = Mo + 3MgO + 3Cl2(g) | 0.824 |
| 3MgCl2 + Sb2O3 = 2Sb(l) + 3MgO + 3Cl2(g) | 0.749 |
| MgCl2 + NiO = Ni + MgO + Cl2(g) | 0.743 |
| 4MgCl2 + Co3O4 = 3Co + 4MgO + 4Cl2(g) | 0.655 |
| 3MgCl2 + Bi2O3 = 2Bi(l) + 3MgO + 3Cl2(g) | 0.488 |
| MgCl2 + CuO = Cu + MgO + Cl2(g) | 0.316 |
Considering molten MgCl2 by itself has a relatively high vapor pressure, it is better to be used together with other chloride salts, such as NaCl and KCl. Using a mix of molten salts can also further decrease the solubility of MgO.14,15 The solubility of MgO in pure MgCl2 at 730 °C was reported to be about 0.36 mol%, which is fairly low in contrast with CaCl2 (>20 mol% at 850 °C) and LiCl (>10 mol% at 650 °C).16,17 However, if 70 mol% NaCl was added, the solubility of MgO can be significantly decreased to about 0.01 mol%. On the other hand, the solubility of MgO in equimolar NaCl–KCl at 730 °C is also less than 0.01 mol%.18 These data suggest that using the molten mixture of MgCl2–NaCl–KCl (MNK) can be more effective for disengaging the carbon anode from O2− ions.
In this work, we further demonstrate our new process by the electrolysis of Ta2O5 in the MNK melt at 700 °C. The mechanism of Ta2O5 reduction was investigated, and nanometer Ta powder (<50 nm) was produced at an energy consumption of about 1.0 kW h kgTa−1. The inert graphite anode was confirmed with the anodic gaseous products analysed in detail.
Experimental
Anhydrous MgCl2, NaCl, and KCl (analytical grade, Tianjin Guangfu Fine Chemical Research Institute, or Sinopharm Chemical Reagent Co., Ltd, China) were mixed in the eutectic ratio 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:2 (in mole ratio and 5 mol in total amount), and heated in a graphite or alumina crucible (inner diameter: 25–50 mm; height: 200–700 mm) in a programmable vertical furnace equipped with a sealable stainless steel retort (Wuhan Experimental Furnace Plant). The furnace temperature was slowly raised to and maintained at 400 °C for more than 12 h, and then to 700 °C until the salt mixture was fully molten. Pre-electrolysis of the molten salt was applied at 1.6 V to remove moisture and other redox-active impurities with the graphite crucible (or a graphite rod of 10 mm diameter in the alumina crucible) as the anode and a Mo wire (diameter: 2 mm) as the cathode. Pre-electrolysis lasted (ca. 10 h) until the current reached a low and stable background level.
:3:2 (in mole ratio and 5 mol in total amount), and heated in a graphite or alumina crucible (inner diameter: 25–50 mm; height: 200–700 mm) in a programmable vertical furnace equipped with a sealable stainless steel retort (Wuhan Experimental Furnace Plant). The furnace temperature was slowly raised to and maintained at 400 °C for more than 12 h, and then to 700 °C until the salt mixture was fully molten. Pre-electrolysis of the molten salt was applied at 1.6 V to remove moisture and other redox-active impurities with the graphite crucible (or a graphite rod of 10 mm diameter in the alumina crucible) as the anode and a Mo wire (diameter: 2 mm) as the cathode. Pre-electrolysis lasted (ca. 10 h) until the current reached a low and stable background level.
Cyclic voltammograms (CVs) were recorded using a Mo cavity electrode (MCE) loading with Ta2O5 powder, with the potential reference to a quartz sealed Ag/AgCl electrode. For constant potential, constant cell voltage and galvanostatic electrolysis, Ta2O5 powder (analytical grade; particle sizes: 300 nm; Zhuzhou Cemented Carbide Works Imp. & Exp. Company) was individually die-pressed into cylindrical pellets (10 MPa, 13 or 20 mm in diameter, 0.7–2 g: 1.1–1.7 mm in thickness, porosity: ∼50%), sintered in air at 1000 °C for 2 h, and sandwiched between two molybdenum (Mo) meshes to form an assembled cathode. After electrolysis for a designated time, the cathode was removed from the furnace, cooled in argon, washed in distilled water, or in dilute HCl (0.1 mol L−1) and water again, and then dried in a vacuum at 80 °C before further analyses. The CVs were recorded using CHI660A Electrochemical System (Shanghai Chenhua, China), while the electrolysis was controlled by a multichannel four-electrode potentiostat (Neware, China).
The products were characterised by X-ray diffraction spectroscopy (X-ray 6000 with Cu Kα1 radiation at λ = 1.5405 Å, Shimadzu, Japan), and a FEI Sirion Field Emission Gun SEM system. GC (FULI 9790II, with a thermal conductivity detector and argon serving as a carrier gas) was used for the analysis of the anodic gaseous products. Ion Chromatography (IC) analysis for Cl− ions was carried out using METROHM 881 Compact IC pro, with the ClO− ions reduced to Cl− ions by H2O2.
Results and discussion
Fig. 1 shows the cyclic voltammogram (CV) of Ta2O5 powders in MNK at 700 °C using the metallic cavity electrode (MCE). For comparison, a CV recorded on an empty MCE is superimposed. No significant reaction current can be observed on the blank CV before Mg deposition (C0) and dissolution, with an Mg/Mg2+ equilibrium potential of about −1.55 V vs. Ag/AgCl. The reduction current of Ta2O5 was found initially to be −0.4 V vs. Ag/AgCl. Preliminary comparison and analysis of the CVs of Ta2O5 in molten MNK and those previously reported in CaCl2 have revealed fairly similar main reduction features, showing mainly three reduction processes (C1–C3).19 However, the starting potential of C3 is unclear due to the overlap with C2.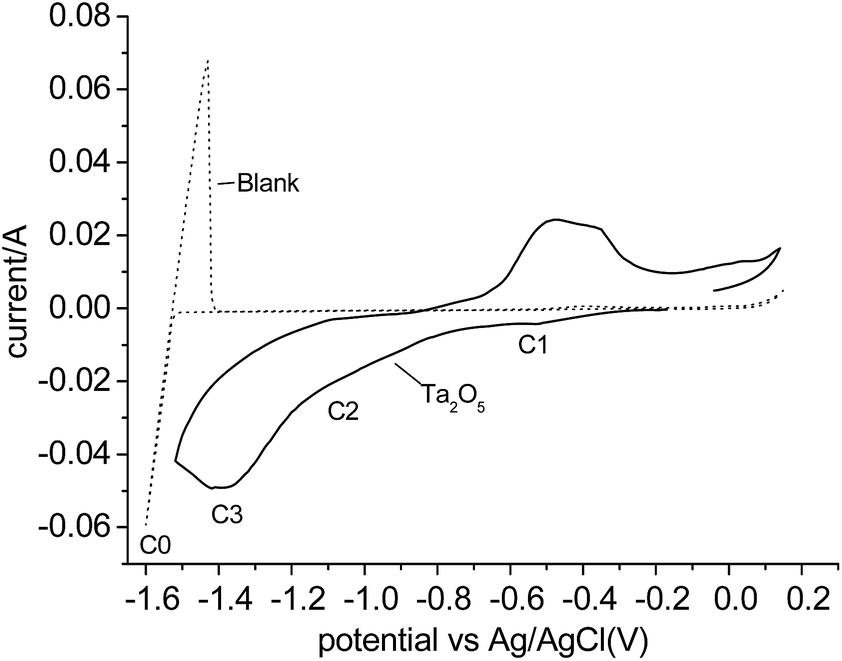 | ||
| Fig. 1 Cyclic voltammograms (CVs) of the blank MCE and the MCE loading with Ta2O5 powder in molten MgCl2–NaCl–KCl at 700 °C. | ||
According to Table 1, the reduction of Ta2O5 to Ta could occur at a potential about 0.9 V more positive than the deposition of Mg, thus, potentiostatic electrolysis of Ta2O5 was performed at −0.8 V vs. Ag/AgCl, a potential 0.75 V more positive than the Mg deposition potential so Ta was predicted to be produced. The current–time plot is shown in Fig. 2a. The reduction current rises initially to a peak suggesting the reduction followed the propagation of the three-phase interlines, which was generally observed during reduction of insulating oxides to metals.20
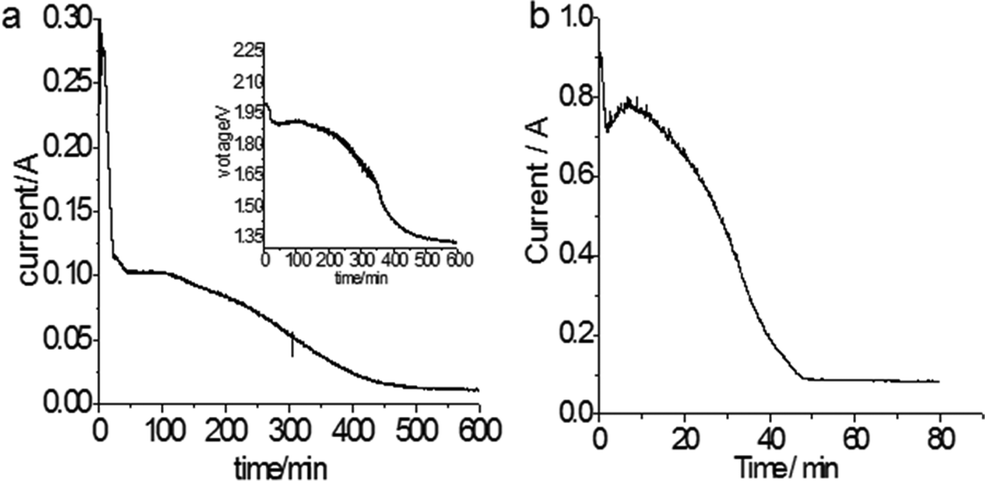 | ||
| Fig. 2 Current–time plots of (a) potentiostatic electrolysis of a Ta2O5 pellet in MNK at −0.8 V vs. Ag/AgCl and 700 °C. Insert: corresponding cell voltage profile. (b) Constant voltage electrolysis of a 0.7 g Ta2O5 pellet at 2.0 V and 700 °C. | ||
In line with the CV, Fig. 2a also shows several reduction processes. The first reduction displayed a high reduction current of about 0.25 A in the initial 30 minutes. The second reduction corresponded to the plateau current at about 0.1 A, which began to decrease at about 110 minutes, then a slope change at about 250 minutes could indicate a third reduction process. Finally, the current levelled off to background current after 500 minutes. To identify these reductions, XRD patterns of the electrolysis products after different times were scanned and displayed in Fig. 3. It can be seen that after 27 minutes, the Ta2O5 was almost completely reduced to TaO2. The electrolysis product at 300 minutes included Ta and Mg4Ta2O9. The appearance of Mg4Ta2O9 after TaO2 may suggest the possible disproportionation reaction of TaO2, possibly like its congener VO2, and the generation of an excess of MgO may have shifted the reaction to form the stabilized Mg4Ta2O9. The final electrolysis product after washing with water consisted of Ta and MgO. The presence of MgO in the cathode indicates its low solubility in the MNK melts as discussed above.
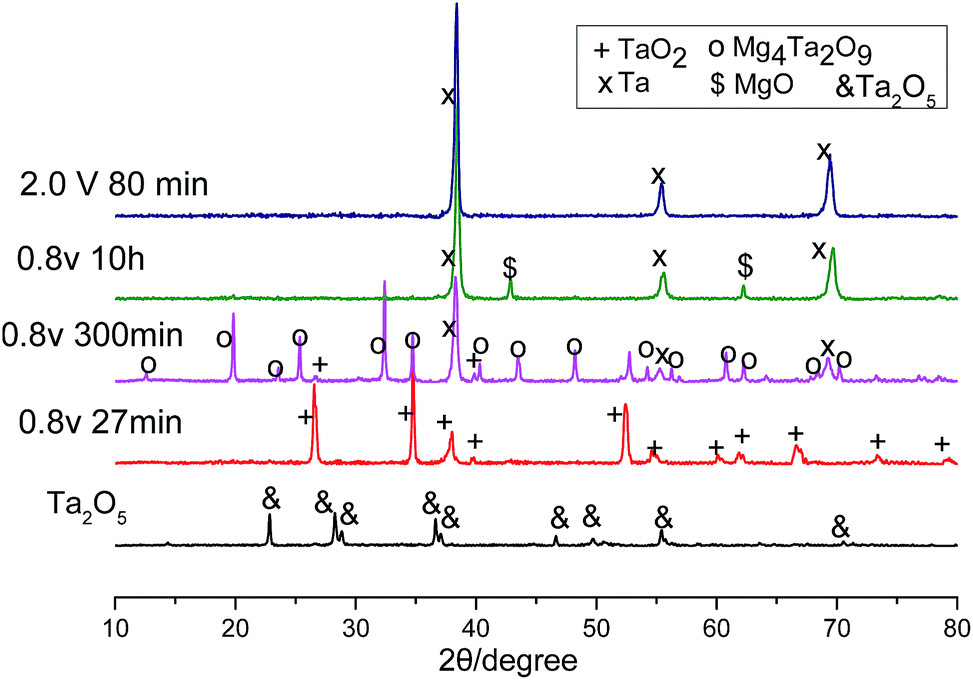 | ||
| Fig. 3 XRD patterns of the products collected at the indicated times of potentiostatic electrolysis (−0.8 V vs. Ag/AgCl, water washed only) and constant voltage electrolysis of Ta2O5 (2.0 V, water and acid washed) at 700 °C in MNK. | ||
The cell voltage (the inset in Fig. 2a) during the −0.8 V constant potential electrolysis was also monitored. In the main reduction stage of Ta2O5, the cell voltage was about 1.9 V, which was reasonably higher than the decomposition voltage ∼1.64 V of Ta2O5 in the MgCl2 based melt as calculated in Table 1. In addition, it can be seen that after the cell voltage decreased to about 1.64 V, it dropped suddenly. These may indicate that the electrolysis was mainly via the assumed Reaction (4) or specifically Reaction (7) for Ta2O5:
| 5MgCl2 + Ta2O5 = 2Ta + 5MgO + 5Cl2(g) | (7) |
Constant cell voltage electrolysis at 2.0 V was also carried out, and the current–time plot is shown in Fig. 2b. It can be seen that the reduction completed in about 50 minutes, and the dimensions of the cathode pellet remained almost unchanged before and after the reduction (Fig. 4a), suggesting the possibilities of both high electrolysis speed and high Ta recovery (nearly 100% in this work). Mass analysis was carefully carried out after the product was washed with water and 0.1 mol L−1 HCl respectively, and this confirmed that most of the MgO generated through Reaction (7) stayed in the cathode. It is interesting that although the porosity of the Ta2O5 pellet was only about 50% of that which is commonly used for electro-reduction in molten CaCl2, the influence of MgO accumulation in the oxide cathode was insignificant for the electro-reduction. However, it can be derived that the porosity of the Ta2O5 cathode needs to be higher than 30% to prevent the ion passages from becoming blocked.
 | ||
| Fig. 4 (a) Ta2O5 pellet collected after 80 min electrolysis in 700 °C MNK at 2.0 V. (b) Scanning Electron Microscopy (SEM) image of the Ta nanoparticles shown in (a) (after acid washing). | ||
On the other hand, although the cathode pellet taken out from the molten salt was perfectly strong, it dispersed to a powder after washing in water. After being further washed with acid, pure Ta was obtained, and the XRD pattern is shown in Fig. 3. The SEM images show very fine particles (∼50 nm) of the resulting Ta powder, which were smaller than those produced in molten CaCl2 at 900 °C, suggesting higher specific capacitance.19 This phenomenon may be partly associated with the relatively low molten salt temperature (700 °C) and also partly attributed to the MgO precipitated inside the pellet which prevented physical sintering of the Ta particles. The current efficiency was higher than 92%, with an energy consumption of about 1.3 kW h kgTa−1 for the electrolysis of 0.7 g Ta2O5 and 1 kW h kgTa−1 for the electrolysis of 2 g Ta2O5.
On the graphite anode, for verification of chlorine evolution, galvanostatic electrolysis of Ta2O5 at 1.5 A was performed in MNK. Since the MNK melt does contain some O2− ions, although with a concentration in the order of 10−4 in molar fraction, the discharge of O2− ions on graphite would occur. According to Scheme 1, the amounts of O2, CO2, CO and Cl2 in the anodic gaseous product were determined.
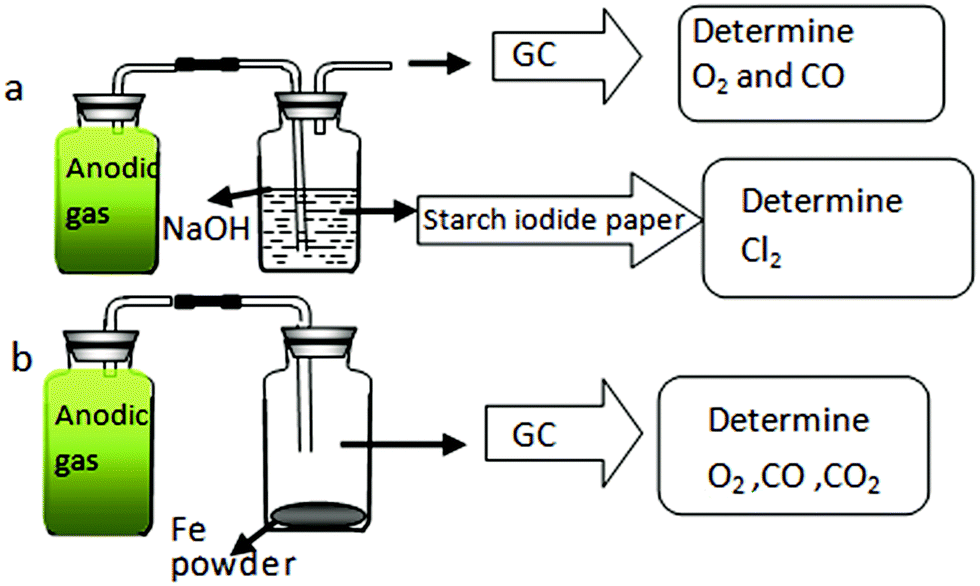 | ||
| Scheme 1 Illustration of the method for identification of anodic gas. | ||
First, about 500 mL of anodic gas was collected as shown in Fig. 5a. The colour of the anodic product was yellowish green. This is straightforward evidence for formation of Cl2 on the anode. After being absorbed by NaOH solution, a starch iodide test indicated the existence of ClO− in the solution, again convincing evidence for anodic Cl2 gas, which has been reacted with NaOH. In addition, ion chromatography (IC) analysis indicated that the Cl− and ClO− ion concentration in the NaOH solution approximately matched the absorbed gas volume (about 460 mL). About 40 mL colourless gas remained, and GC analysis (Fig. 6) indicated the existence of O2 and CO with concentrations in the total anodic gas of about 1.2 mol% and 0.2 mol%, respectively.
 | ||
| Fig. 5 (a) Bottles with 500 mL air (left) and anodic gas (right). (b) Photo of the colour change of the starch iodide paper after immersing into anodic gas bubbled through NaOH solution. | ||
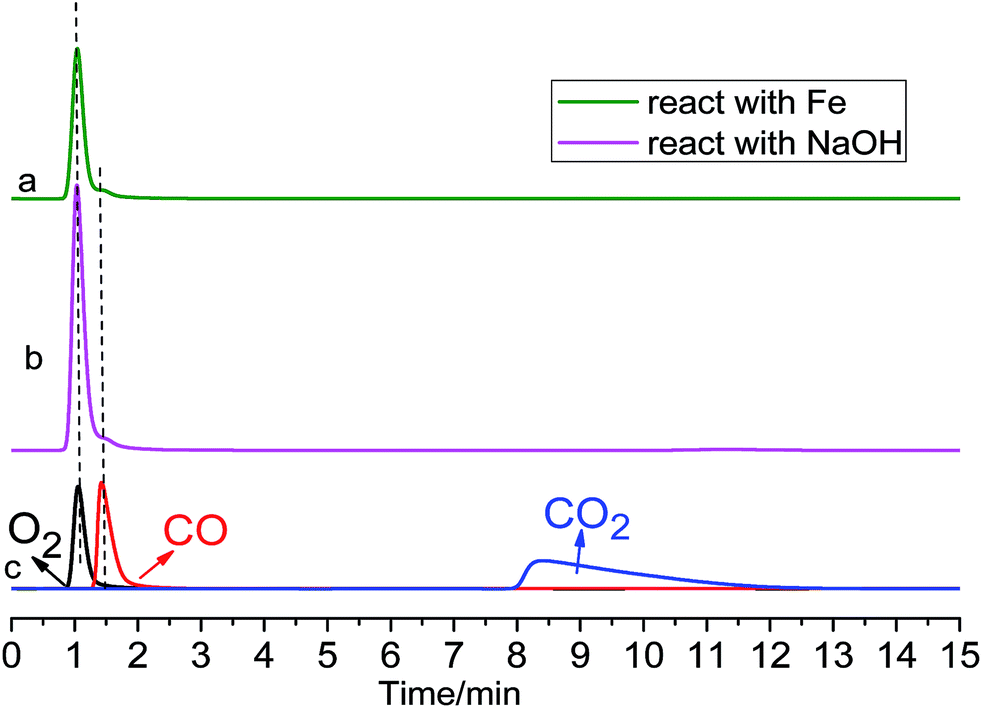 | ||
| Fig. 6 GC spectrogram of anodic gas. Line (a): after reacting with Fe powder. Line (b): after being absorbed by NaOH solution. Line (c): superimposed GC spectrograms of pure O2, CO and CO2. | ||
No CO2 was detected by GC, however, even if it was formed on a graphite anode, according to the Cl2 volume indicated by IC, the CO2 concentration would be very low. Similarly, it was reported that no CO2 gas was found as an anodic product during electrolysis of MgO in MgCl2–NdCl3 system at 700 °C.21 However, it seems strange that there was O2 as an anodic product without CO2 gas on a graphite anode. For confirmation, we designed another experiment. As shown in Scheme 1b, the anodic gaseous product was allowed to react with the reducing Fe powders thoroughly, and the remaining colourless gas was also detected by GC. According to Fig. 6, CO2 was in truth absent in the anodic product.
According to the above tests, the composition of the anodic gaseous product during galvanostatic electrolysis of Ta2O5 at 1.5 A consisted of mainly Cl2 gas, about 1 mol% O2 gas and trace amounts of CO gas, as summarized in Table 2. No CO2 was formed on the graphite anode.
| Compositions | Content (mol%) |
|---|---|
| Cl2 | 98.6 |
| O2 | 1.2 |
| CO | <0.2 |
| CO2 | None |
To determine the origination of anodic O2 gas, CVs of the graphite electrode in the MNK melt were recorded as shown in Fig. 7. According to Fig. 7, the evolution of chlorine (a0) was initiated at 1.1 V vs. Ag/AgCl. Coupled with the deposition potential of Mg (see Fig. 1a) on MCE, the decomposition voltage of MgCl2 was determined to be 2.65 V, which is fairly close to the thermodynamic value (2.534 V) calculated from HSC Chemistry 6. The measured value was slightly higher, which is reasonable considering that the molar fraction of MgCl2 in the MNK was only 0.5. The current corresponding to chlorine evolution increased rapidly with the positive scan, indicating that there is no kinetic difficulty on the discharge of Cl− ions on a graphite anode. Since Cl− ions are the main anions in the MNK melt, concentration polarization for the chlorine evolution is also negligible. On the contrary, in molten CaCl2, the anodic discharge of O2− ions on graphite is well known to be kinetically difficult both in terms of charge transfer and mass transfer.
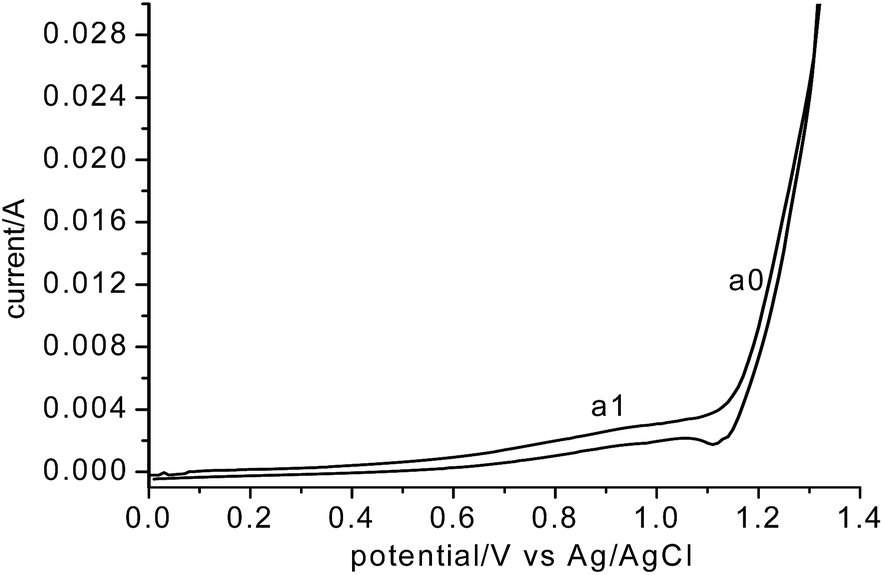 | ||
| Fig. 7 Cyclic voltammogram of graphite in eutectic MgCl2–NaCl–KCl (700 °C) vs. Ag/AgCl, scan rate 200 mV s−1. | ||
Before a0, there is an apparent anodic current at a1, which is very stable according to the multi-cycle scans and could be attributed to the discharge of O2− ions on the graphite anode. However, unlike the discharge of O2− on a carbon based anode in molten CaCl2, which exhibited complicated voltammograms involving formation of CO2, CO and O2 gases,22–24 here the CVs of O2− discharge in MNK featured a single reaction, probably the formation of O2 gas as mentioned above. To confirm it, potentiostatic electrolysis was performed at 1.0 V vs. Ag/AgCl against a Ta2O5 pellet cathode. The anodic current remained very stable at about 15 mA during the total 3 hour electrolysis. However, no anodic gas has automatically flowed out from the anodic gas chamber. The chamber was then sealed and the anodic gas was sampled at different times for GC analysis.
Fig. 8 shows the GC analysis results. As can be seen, after 0.5 h electrolysis, there was a small peak of O2 gas but no CO and CO2 gas was found. After about 1 h electrolysis, the O2 concentration increased significantly, and small peaks of CO and CO2 became just perceptible, and the O2:CO:CO2 ratio was about 8.2:1.3:0.5, which changed to 7.9:1.0:1.1. Considering initially there is only O2 evolution and the CO2 content became larger and larger, it is likely that the formation of CO2 is a result of a reaction between the graphite anode and the accumulated O2 in the anodic chamber. Combining the CV result and the GC analysis of the anodic gas during constant potential electrolysis at 1.0 V, it might be reasonable that the a1 current in Fig. 7 corresponds to the discharge of O2− ions to O2 gas on the graphite anode in the MNK melt at 700 °C. However, the small steady current (15 mA) at 1.0 V vs. Ag/AgCl was most likely to be the limited diffusion current of O2− ion discharge, which is only about 1.5% of the total current considering a 1.0 A g−1-Ta2O5 or higher current would be applied in the actual electrolysis.
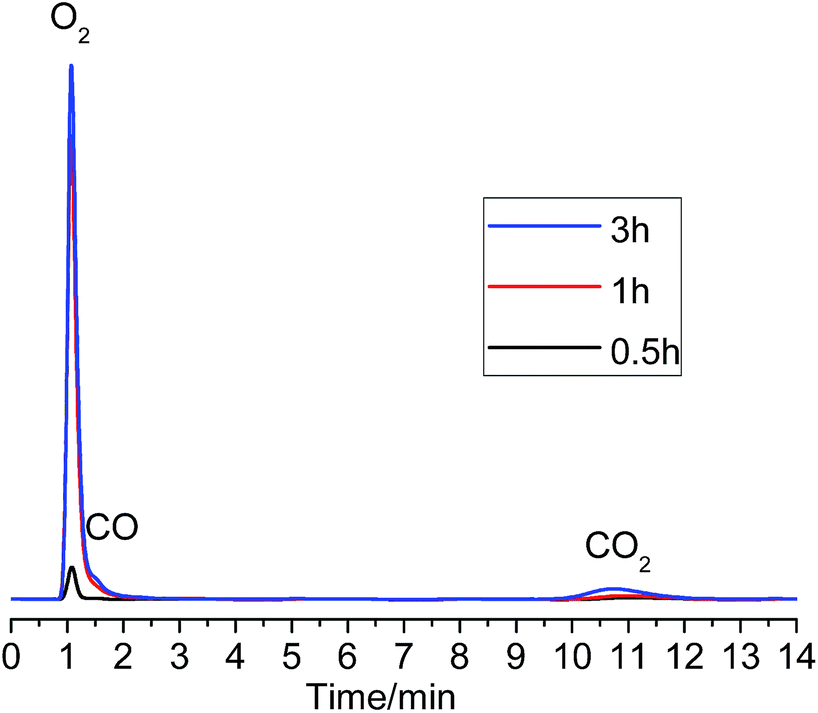 | ||
| Fig. 8 GC spectrograms of anodic gas produced by potentiostatic polarization of graphite in 700 °C MNK at 1.0 V vs. Ag/AgCl (time indicated). | ||
The optical images of the graphite anode before and after repeated electrolysis for ca. 100 h in the eutectic MNK melt were compared in Fig. 9. In line with the Cl2 and O2 gas evolution at the anode, the fine spiral machining marks of ca. 100 nm in width on the side of the graphite rod remained almost intact, even though these protruding parts are usually the most vulnerable sites to chemical or electrochemical attack. These observations suggested that the graphite anode is basically inert during the electrolysis of solid oxide cathode in the MgCl2 based molten salts. However, it is well-known that the anodic reaction of the solid oxide cathode electrolysis at a graphite electrode in CaCl2 was the discharge of the O2− to carbon oxides and carbonate ions, leading to significant erosion of the graphite anode, shuttle reactions of the oxygenated species (O2− and CO32−), and formation of a thick layer of floating carbon powder on the surface of the molten electrolyte. Since there was no corrosion to the graphite anode and consequently no carbon generated on the cathode due to the reduction of CO32− in a MgCl2 based electrolyte, no floating carbon was supposed to form on the surface of molten MNK. This was confirmed by the clear surface of MNK after 100 h electrolysis of Ta2O5 as shown in Fig. 9c.
 | ||
| Fig. 9 Optical photos of the graphite rod anode (dia. 2.0 cm) (a) before and (b) after electrolysis experiments for ca. 100 h in the eutectic MNK melt. (c) Photo of cooled MNK salts after 100 h electrolysis of Ta2O5. | ||
These results indicate that both the graphite anode and the MNK salt are ready for lengthy electrolysis. Of course, the consuming MgCl2 needs to be supplemented, for example, along with the metal oxide pellet. In addition, the Cl2 gas has many applications and can be collected as already commercially demonstrated in the chlor-alkali industry and the electrolysis of molten LiCl and MgCl2. It is also possible to use the Cl2 gas to regenerate MgCl2, noting the recent progress in developing H2–Cl2 fuel cells,25 which generate electricity and HCl while HCl is needed to convert MgO in the cathode back to MgCl2.
Conclusions
We have demonstrated the high speed, low energy consumption electrolysis of Ta2O5 in MgCl2–NaCl–KCl melt using an inert graphite anode. The reduction process was investigated by cyclic voltammetry, and potentiostatic and constant voltage electrolysis. The main intermediate products of the electro-reduction of Ta2O5 were identified to be TaO2 and Mg4Ta2O9. The final reduction product was nanometer Ta powder with sizes of about 50 nm. The anodic product included mainly Cl2 gas, plus about 1 mol% O2. There was only about 0.2 mol% CO in the anodic gas, and no CO2 was detected, suggesting that the graphite anode is fairly stable in our new process. The process should be applicable for the preparation of many other metals and alloys with high energy efficiency and low carbon emission.Acknowledgements
We thank the NSFC, Ministry of Education of China, the EPSRC, Guangdong Nat. Sci. Found., Nat. Key Fundamental R&D Program of China and Nat. Hi-Tech R&D Program of China for financial support (Grant No. 20773094, 21173161, 51204060, NCET-11-0397, EP/J000582/1, S2012040007501).References
- W. Kroll, Trans. Electrochem. Soc., 1940, 78, 35–47 CrossRef.
- T. Okabe, K. Nikami and K. Ono, Bulletin of the Iron and Steel Institute of Japan, 2002, 7, 39–45 CAS.
- G. Z. Chen, D. J. Fray and T. W. Farthing, Nature, 2000, 407, 361–364 CrossRef CAS PubMed.
- D. H. Wang, X. B. Jin and G. Z. Chen, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2008, 104, 189–234 RSC.
- A. M. Abdelkader, K. T. Kilby, A. Cox and D. J. Fray, Chem. Rev., 2013, 113, 2863–2886 CrossRef CAS PubMed.
- L. Cassayre, P. Palau, P. Chamelot and L. Massot, J. Chem. Eng. Data, 2010, 55, 4549–4560 CrossRef CAS.
- C. Schwandt and D. Fray, Electrochim. Acta, 2005, 51, 66–76 CrossRef CAS.
- D. R. Sadoway, JOM, 2001, 53, 34–35 CrossRef CAS.
- K. T. Kilby, S. Jiao and D. J. Fray, Electrochim. Acta, 2010, 55, 7126–7133 CrossRef CAS.
- C. Chen and X. Lu, Acta Metall. Sin. (Chin. Ed.), 2008, 44, 145 CAS.
- S.-W. Kim, E.-Y. Choi, W. Park, H. S. Im and J.-M. Hur, Electrochem. Commun., 2015, 55, 14–17 CrossRef CAS.
- S. Jiao, K.-N. P. Kumar, K. T. Kilby and D. J. Fray, Mater. Res. Bull., 2009, 44, 1738–1742 CrossRef CAS.
- W. Li, Y. T. Yuan, H. L. Chen, X. B. Jin, Z. Y. Wang and G. Z. Chen, Progress in Natural Science: Materials International, 2015, 11, 002 Search PubMed.
- S. Boghosian, A. Godø, H. Mediaas, W. Ravlo and T. Østvold, Acta Chem. Scand., 1991, 45, 145 CrossRef CAS.
- K. H. Sten, Electrochim. Acta, 1979, 24, 509 CrossRef.
- T. Usami, M. Kurata, T. Inoue, H. E. Sims, S. A. Beetham and J. A. Jenkins, J. Nucl. Mater., 2002, 300, 15 CrossRef CAS.
- D. A. Wenz, I. Johnson and R. D. Wolson, J. Chem. Eng. Data, 1969, 14, 250 CrossRef CAS.
- R. Combes, F. De Andrade, A. De Barros and H. Ferreira, Electrochim. Acta, 1980, 25, 371–374 CrossRef CAS.
- T. Wu, X. B. Jin, W. Xiao, X. H. Hu, D. H. Wang and G. Z. Chen, Chem. Mat., 2007, 19, 153–160 CrossRef CAS.
- G. Z. Chen, E. Gordo and D. J. Fray, Metall. Mater. Trans. B, 2004, 35, 223 CrossRef.
- K. Cathro, R. Deutscher and R. Sharma, J. Appl. Electrochem., 1997, 27, 404–413 CrossRef CAS.
- H. L. Chen, X. B. Jin, L. P. Yu and G. Z. Chen, J. Solid State Electrochem., 2014, 18, 3317 CrossRef CAS.
- M. Mohamedi, B. Børresen, G. M. Haarberg and R. Tunold, J. Electrochem. Soc., 1999, 146, 1472–1477 CrossRef CAS.
- R. Tunold, G. M. Haarberg, K. S. Osen, A. M. Martinez and E. Sandnes, ECS Trans., 2011, 35, 1–9 CAS.
- B. Huskinson, J. Rugolo, S. K. Mondal and M. J. Aziz, Energy Environ. Sci., 2012, 5, 8690 CAS.
| This journal is © The Royal Society of Chemistry 2016 |