
Investigation of cloud point extraction for the analysis of metallic nanoparticles in a soil matrix†
Hind
El Hadri
* and
Vincent A.
Hackley
 *
*
Materials Measurement Science Division, National Institute of Standards and Technology, 100 Bureau Drive, Gaithersburg, MD 20899-8520, USA. E-mail: h.el.hadri@hotmail.fr; vince.hackley@nist.gov
First published on 19th October 2016
Abstract
The characterization of manufactured nanoparticles (MNPs) in environmental samples is necessary to assess their behavior, fate and potential toxicity. Several techniques are available, but the limit of detection (LOD) is often too high for environmentally relevant concentrations. Therefore, pre-concentration of MNPs is an important component in the sample preparation step, in order to apply analytical tools with a LOD higher than the ng kg−1 level. The objective of this study was to explore cloud point extraction (CPE) as a viable method to pre-concentrate gold nanoparticles (AuNPs), as a model MNP, spiked into a soil extract matrix. To that end, different extraction conditions and surface coatings were evaluated in a simple matrix. The CPE method was then applied to soil extract samples spiked with AuNPs. Total gold, determined by inductively coupled plasma mass spectrometry (ICP-MS) following acid digestion, yielded a recovery greater than 90%. The first known application of single particle ICP-MS and asymmetric flow field-flow fractionation to evaluate the preservation of the AuNP physical state following CPE extraction is demonstrated.
Environmental significanceTo assess the impact of manufactured nanomaterials in the environment, one must first be able to detect, characterize and quantify them. This is extremely challenging due to their small size, very low environmental concentrations and the complexity of environmental matrices. The key finding of this work is that a surfactant-based process known as cloud point extraction can be optimized to concentrate and isolate metallic nanoparticles, while largely preserving their physical state. To clearly demonstrate this, two state-of-the-art analytical approaches were developed based on field flow fractionation and single-particle ICP-MS. The steps outlined here can be applied to a wide range of matrices, and therefore has broad significance for understanding the persistence of nanomaterials and their risk to the environment. |
1. Introduction
The increasing use of nanoscale materials in a wide variety of applications raises the potential problem of release into the environment and its associated effects.1–5 Quantification and characterization of manufactured nanoparticle (MNP) behavior in complex natural media, such as soils or sediments, is extremely challenging from a metrological perspective.2 The concentrations of MNPs found in the environment are frequently below the limit of detection (LOD) of commonly used analytical techniques. Therefore, pre-concentration of MNPs is often necessary in order to apply analytical tools with a LOD higher than the ng kg−1 level. However, the preservation of the original analyte (in both form and chemistry) is necessary to accurately assess the fate of MNPs in the environment. Indeed, modification of the sample by addition of chemicals can potentially alter MNP size, its interaction with natural colloids and its ecotoxicity. To mitigate these potential artifacts, effective and validated methods are needed to extract and pre-concentrate MNPs from complex environmental matrices.Commonly used methods for extraction and pre-concentration in aqueous media include pre-fractionation techniques such as centrifugation and ultrafiltration. Filtration is in theory a simple technique, however modification of MNPs (e.g., aggregation, poor recovery, etc.) can occur due to interaction with the filter membrane, cake formation or concentration polarization.6–8 Centrifugation is a technique with lower perturbation, but to pre-concentrate MNPs, it is sometimes necessary to apply a high g-force (as a function of the density and size), potentially leading to the formation of aggregates or shear forces that may otherwise alter weakly associated particles.9 Liquid–liquid or solid–liquid extractions can also be performed on the sample, but they are principally used for carbon-based MNPs (e.g., fullerenes).10,11 Single channel techniques such as field flow fractionation can also be utilized to pre-concentrate MNP samples; in this case, a large volume of sample is injected in the channel and the focusing step is extended.12,13 This method can be coupled online to appropriate detectors or concentrated fractions can be collected for off-line analysis. However, interactions can and do occur with the accumulation wall membrane due to the increased focusing time.14
Cloud point extraction (CPE) involves the addition of a surfactant at a concentration above its critical micelle concentration (CMC). If the solution temperature is increased above the cloud point temperature of the surfactant, micelles are formed via dehydration, and become less soluble in water. Two phases are obtained: viz. a surfactant-rich phase and a solvent (surfactant depleted) phase (the supernatant). Hydrophobic compounds are extracted with the surfactant micelles, while the specie selectivity between dissolved ions and NPs requires the addition of an additive.15–17 CPE has been used to extract and concentrate MNPs (e.g., AgNPs, AuNPs, CuONPs, carbon nanotubes) in model matrices and in more complex media such as natural waters16–21 or wastewater from treatment plants.22 Hartmann et al.18 observed that for several AgNP coatings, the extraction efficiency for CPE was in excess of 80%. Efficiency was significantly reduced only for protein-coated (bovine serum albumin) NPs. CPE was chosen for the present study in order to isolate AuNPs (as model MNPs) from soil extracts, due to its relative simplicity and cost effectiveness.
For quantification of MNPs after CPE, the most common techniques are ultraviolet-visible (UV-vis) spectroscopy, graphite furnace atomic absorption spectrometry (GFAAS) and inductively coupled plasma mass spectrometry (ICP-MS). The two latter techniques offer the advantage of very low LODs and high elemental specificity. Concerning MNP size determination, imaging techniques have been employed, such as transmission electron microscopy (TEM) and scanning EM combined with energy dispersive X-ray spectroscopy. While EM techniques are convenient and effective to visually assess the shape and the size of MNPs, sample preparation can be critical (e.g., the drying step can generate artifacts)23,24 and the results can be statistically limited. Single particle (sp) ICP-MS has recently evolved as a powerful analytical tool for the detection, sizing and determination of particle concentration for metallic or metal containing MNPs.25,26 This technique allows one to accurately measure the size and concentration of metal containing NPs at environmentally relevant concentrations (ng kg−1), limited primarily by the mass of individual particles (i.e., there is a lower size cut off for each element depending on sensitivity and the presence of the dissolved element in the background). Asymmetric flow field-flow fractionation (A4F) is a separation method widely used for the characterization of both natural colloids27,28 and MNPs.14,29,30 However, sensitivity is dependent on the detectors coupled to, or used in conjunction with, A4F. The use of ICP-MS as a detector provides low LODs, and its combination with CPE can provide an enhancement in analytical sensitivity where environmental samples are concerned.
In the present study, CPE was utilized to extract AuNPs from a model complex matrix (an agricultural soil extract). The primary objective was to optimize the CPE process relative to both recovery and preservation of the native MNP physical state, using state-of-the-art analytical tools, including hyphenated methods. Initially, different conditions for CPE were evaluated to pre-concentrate AuNPs (with different surface coating and size) in deionized (DI) water solutions. Several analytical techniques were used in order to determine the extraction efficiency and the size distribution of the AuNPs and natural colloids. For analysis of total Au content, ICP-MS was performed following acid digestion. The size distribution, along with the aggregation state, was evaluated using spICP-MS. A4F directly coupled to ICP-MS provided information on both the size and the elemental composition of the MNPs and natural colloids. In this study, A4F was used to characterize Au-spiked soil extract samples following treatment with CPE.
2. Materials and methods
2.1 Chemicals
High-purity (18 MΩ cm) DI water was obtained from a Type II biological grade water purification system (Aqua Solutions, Jasper, GA, USA)‡ and was used for all sample preparation and dilutions. Concentrated nitric, HNO3, (≥69%) and hydrochloric, HCl, (≥37%) acids were used in ICP-MS experiments for trace analysis (Fluka TraceSelect, Sigma-Aldrich, St. Louis, MO, USA). Citrate capped AuNPs with nominal diameters of 10 nm, 30 nm and 60 nm were obtained from Ted Pella (Redding, CA, USA); these will be denoted as BBI10, BBI30 and BBI60 after the original manufacturer BBI Solutions (Cardiff, UK). Additionally, NIST reference materials (RMs) with a nominal size of 30 nm and 60 nm, RM8012 (ref. 31) and RM8013 (ref. 32) (citrate capped AuNPs) respectively, were used in this study for calibration and traceability purposes. Moreover, branched polyethylenimine (bPEI, 25 kDa) and polyvinylpyrrolidone (PVP, 10 kDa) coated AuNPs with a nominal size of 30 nm were obtained from Nanocomposix (San Diego, CA, USA). NIST SRM 3121 Au and SRM 3140 Pt standard solutions were used to prepare dissolved elemental calibration standards. Two nonionic surfactants with different polyoxyethylene chain length: Triton X-114 (TX-114) and Triton X-100 (TX-100) were obtained from Sigma-Aldrich (St. Louis, MO, USA) for CPE experiments. Humic acid (Suwannee River Humic Acid Standard II, HA) was obtained from the International Humic Substances Society (IHSS, St. Paul, MN, USA). Citric acid (CA, 99.5%) and disodium ethylenediaminetetraacetic acid (EDTA) were obtained from Sigma-Aldrich and GFS Chemicals (Columbus, OH, USA), respectively. Ammonium nitrate (NH4NO3, 99%), TX-114 and sodium dodecyl sulfate (SDS), obtained from Sigma-Aldrich, were used in the mobile phase for A4F analyses; the mobile phase was first passed through a 0.2 μm regenerated cellulose filter prior to use. An agricultural soil (Nebraska SoNE-1) used in this study was obtained from the United States Geological Survey (USGS). SoNE-1 is a USGS in-house, non-certified reference material soil prepared specifically for the North American Soil Geochemical Landscapes Project.332.2 Total and single particle analyses by mass spectrometry
For the determination of total Au concentration and single particle analysis, a model 7700× ICP-MS from Agilent Technologies (Santa Clara, CA, USA) was used. The instrument was equipped with a concentric (Micromist) nebulizer and a refrigerated Scott chamber (2 °C). Before analysis, the ICP-MS was tuned using a multi-element standard solution from Agilent (1 μg L−1 each of 7Li, 89Y, 140Ce and 205Tl in 2% v/v HNO3) to ensure maximum sensitivity and minimum oxide (156CeO/140Ce) and doubly charged (70Ce++/140Ce+) level (<2%). Data were collected at m/z 197 for Au and m/z 195 for Pt. Dissolved Au calibration standards were prepared over a mass fraction range from (0.05 to 5) μg kg−1 either in diluted aqua regia (1.0% v/v 3 parts HCl/1 part HNO3, AR) or in DI water. Platinum was used as an internal standard for total analyses (spectrum analysis mode). For the spICP-MS analyses, the dwell time was set to 10 ms with a measurement time of 300 s for each sample (time resolved mode). The transport efficiency was determined daily by the use of standard dissolved Au solutions and AuNP reference materials (RM 8012 and RM 8013) according to a previously described method;34 the value averaged ≈6% during the course of this study. Between each sample analysis, the system was washed with DI water, 2% v/v HNO3 and/or 1% AR, as necessary to avoid contamination.2.3 A4F analyses
An Eclipse 3+ A4F system (Wyatt Technology, Santa Barbara, CA), equipped with a 1200 series UV-vis absorbance diode array detector (Agilent Technologies) and a multi-angle laser light scattering (MALS) detector (DAWN HELEOS, Wyatt Technology) was used as a separation and analysis system. The A4F nominal (trapezoidal) channel height was established using a spacer of 250 μm (with dimensions of 26.5 cm length and narrowing width from 2.1 to 0.6 cm). This spacer permits the fractionation of a wide size range in a reasonable analysis time;35 for soil extracts, colloids range in size from tens of nm to 500 nm. Polyethersulfone (PES) 10 kDa membranes were purchased from Wyatt Technology and used as the accumulation wall. Mobile phase flow was generated using an 1100 series isocratic pump (Agilent Technologies) equipped with a degasser (Gastorr TG-14, Flom Co., Ltd., Tokyo, Japan). All injections were performed with an Agilent Technologies 1260 ALS series autosampler using a 100 μL injection volume. Data from the A4F detectors was processed using Astra 6.1.4.25 (Wyatt Technology) and OpenLab (Agilent Technologies) software. A4F recovery was determined as the ratio between UV peak areas of the sample injected with and without application of the cross-flow field. The radius of gyration (rG) was determined using the (first order) Berry formalism to analyze MALS data at different angles.36 Hyphenation of A4F with ICP-MS to analyze AuNPs and soil colloids was performed using a collision cell, with He as collision gas, to reduce interferences. The isotopes 197Au, 27Al, 55Mn and 57Fe were monitored. Under the conditions of this experiment, 57Fe, though less abundant than 56Fe, nevertheless produced a cleaner signal with less interferences and similar signal-to-background at the levels encountered here.2.4 Sample preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) have been used previously to extract and concentrate NPs,16,19,37,38 and were therefore chosen for the present study. The final percentage of surfactant in the solution before heating and centrifugation was 0.1% w/w (i.e., 1.60 mmol L−1 and 1.86 mmol L−1, for TX-100 and TX-114, respectively); this surfactant percentage is much higher than the reported CMC of (0.2–0.3) mmol L−1.39–41 To about 10 mL of a sample (AuNPs in DI water or in soil extract at a concentration ranging from 10 μg kg−1 to 500 μg kg−1), 200 μL of 5% w/w surfactant was added. The addition of Triton surfactant to the sample can be accompanied by the addition of salt (NaCl or CaCl2) and/or acid (HNO3, CA or EDTA). These additives were chosen based on previous studies.16,19,42,43 Generally the added volume was 100 μL to avoid a dilution effect. All reactants were added to the sample with a pipette and measured gravimetrically. Table 1 shows the CPE conditions used in this set of experiments. Salt is added to decrease the Coulombic repulsion and acid is added to increase the extraction efficiency by shifting the pH closer to the point of zero charge for citrate coated AuNPs.19
:1) have been used previously to extract and concentrate NPs,16,19,37,38 and were therefore chosen for the present study. The final percentage of surfactant in the solution before heating and centrifugation was 0.1% w/w (i.e., 1.60 mmol L−1 and 1.86 mmol L−1, for TX-100 and TX-114, respectively); this surfactant percentage is much higher than the reported CMC of (0.2–0.3) mmol L−1.39–41 To about 10 mL of a sample (AuNPs in DI water or in soil extract at a concentration ranging from 10 μg kg−1 to 500 μg kg−1), 200 μL of 5% w/w surfactant was added. The addition of Triton surfactant to the sample can be accompanied by the addition of salt (NaCl or CaCl2) and/or acid (HNO3, CA or EDTA). These additives were chosen based on previous studies.16,19,42,43 Generally the added volume was 100 μL to avoid a dilution effect. All reactants were added to the sample with a pipette and measured gravimetrically. Table 1 shows the CPE conditions used in this set of experiments. Salt is added to decrease the Coulombic repulsion and acid is added to increase the extraction efficiency by shifting the pH closer to the point of zero charge for citrate coated AuNPs.19
| Sample name | Acid (pH) | Salt (final concentration) |
|---|---|---|
| a Used in section 3.1.3, influence of size and coating on CPE efficiency. b pH not available, but all TX-114 solutions with inert salt have similar pH. c pH of EDTA solution alone. | ||
| TX-114 alone | None (6.4) | None |
| TX-114 + NaCl | None (6.2) | NaCl (2 mmol L−1) |
| TX-114 + HNO3 | HNO3 0.1 M (3.0) | None |
| TX-114 + CaCl2 | None (≈6.2)b | CaCl2 (2 mmol L−1) |
| TX-114 + HNO3 + NaCl | HNO3 0.1 M (3.0) | NaCl (2 mmol L−1) |
| TX-114 + CAa | Citric acid (3.2) | None |
| TX-114 + EDTAa | EDTA (4.5)c | None |
Samples were shaken vigorously and then heated for 30 min at 40 °C or 70 °C in a water bath, for TX-114 or TX-100, respectively, to induce cloud formation. Phase separation was completed by centrifugation at 4000 rpm (2060g) for 10 min using a Beckman J2-HC centrifuge (Beckmann Coulter Life Sciences, Indianapolis IN, USA) with a J20.1 fixed angle rotor (15 mL tube volume). The supernatant and the surfactant-rich phase extract were separated and the extract was diluted 2 to 3 times in DI water prior to digestion. Typically, CPE samples are diluted in alcohol (usually ethanol);19 however, the autocatalytic oxidation between concentrated (≈70%) nitric acid and ethanol is potentially explosive and can generate toxic gas (NO2), even if the hazard is reduced with aqua regia (AR).44,45 Moreover, in order to preserve maximal sample integrity, dilution in water is preferable. Indeed, the nature of the initial media (aqueous) is conserved in this way.
An aliquot of the supernatant and surfactant-rich phases (extract) were each digested with AR. Undiluted AR was added to the surfactant-rich phase and left at room temperature for a few hours in a fume hood. The solution was then diluted to the final volume with DI water to yield an AR concentration of 1% to 2%. For spICP-MS analyses, samples were diluted several times in DI water to obtain a concentration of about 5 ng kg−1 and 40 ng kg−1, for 30 nm and 60 nm, respectively. For each size, these concentrations yielded a particle concentration of about 15000 mL−1.
In addition to citrate, surface coatings evaluated in this study include nonionic PVP and cationic bPEI. Additionally, AuNPs were mixed with natural organic matter (humic acid, HA) to assess its influence on the CPE process. HA (at a concentration of 2 mg kg−1) was added to citrate capped AuNPs (BBI30), prior to CPE in order to modify the surface in a manner that mimics environmental exposure, and mixed 2 h with a bench-top rotator. The HA solution pH was at the natural value near 4 or was adjusted to pH 8 with dropwise addition of NaOH.
2.5 Uncertainty statement
Unless otherwise noted, all uncertainty intervals for the extraction efficiency values reported in this work represent one standard deviation about the mean, s, based on 2 to 10 measurements performed under repeatability conditions, where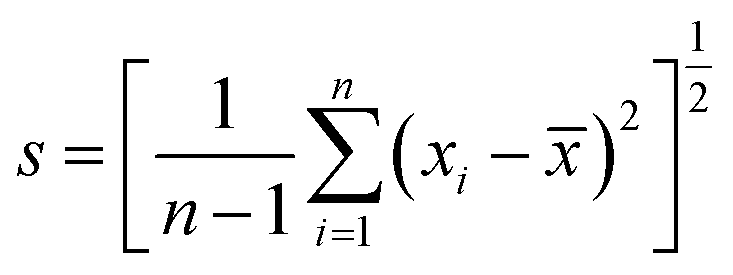 , n is the number of replicate measurements and
, n is the number of replicate measurements and ![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) is the mean value. For the mass-based diameter determined from spICP-MS data using a Gaussian model, the uncertainty is the standard deviation of the means obtained by Gaussian fitting. The LOD and the limit of quantification (LOQ) for the ICP-MS instrument coupled to A4F is defined as the signal background plus 3s or 10s, respectively.
is the mean value. For the mass-based diameter determined from spICP-MS data using a Gaussian model, the uncertainty is the standard deviation of the means obtained by Gaussian fitting. The LOD and the limit of quantification (LOQ) for the ICP-MS instrument coupled to A4F is defined as the signal background plus 3s or 10s, respectively.
3. Results and discussion
3.1 Cloud point extraction optimization in a simple matrix (DI water)
The initial objective was to optimize CPE conditions using a simple matrix (DI water) in order to select the most relevant conditions to evaluate with more complex matrices (soil extracts). Both extraction efficiency and size distribution were assessed for AuNPs with different coatings, core size and concentration.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1).
Fig. 1 presents Au recovery determined by ICP-MS (after digestion) using different surfactant compositions. From the extraction efficiencies shown, we conclude that single component TX-114 is the most appropriate surfactant composition for AuNP extraction by CPE in a simple aqueous solution (DI water). It is worth noting that TX-114 contains a shorter polyoxyethylene chain (n = 7–8) compared with TX-100 (n = 9–10) and has a lower cloud point temperature (22 °C vs. 65 °C). This is important, because following the heating step, centrifugation is performed at a temperature that is lower than the cloud point temperature for TX-100 or the TX-mixture (maximum temperature in the centrifuge is 30 °C). The results therefore imply a re-dissolution of the micelles in the aqueous phase, and consequently a decrease in the extraction efficiency. Notably, the addition of inert salt (NaCl) to the TX-mixture increased the extraction efficiency, most likely by lowering the cloud point temperature.49–51 However, recoveries remain low relative to pure TX-114 as shown in Fig. 1. Additionally, a relatively large amount of Au was detected in the supernatant of both mixtures. Overall, TX-114 was selected as the CPE surfactant for the remainder of the study.
:1).
Fig. 1 presents Au recovery determined by ICP-MS (after digestion) using different surfactant compositions. From the extraction efficiencies shown, we conclude that single component TX-114 is the most appropriate surfactant composition for AuNP extraction by CPE in a simple aqueous solution (DI water). It is worth noting that TX-114 contains a shorter polyoxyethylene chain (n = 7–8) compared with TX-100 (n = 9–10) and has a lower cloud point temperature (22 °C vs. 65 °C). This is important, because following the heating step, centrifugation is performed at a temperature that is lower than the cloud point temperature for TX-100 or the TX-mixture (maximum temperature in the centrifuge is 30 °C). The results therefore imply a re-dissolution of the micelles in the aqueous phase, and consequently a decrease in the extraction efficiency. Notably, the addition of inert salt (NaCl) to the TX-mixture increased the extraction efficiency, most likely by lowering the cloud point temperature.49–51 However, recoveries remain low relative to pure TX-114 as shown in Fig. 1. Additionally, a relatively large amount of Au was detected in the supernatant of both mixtures. Overall, TX-114 was selected as the CPE surfactant for the remainder of the study.
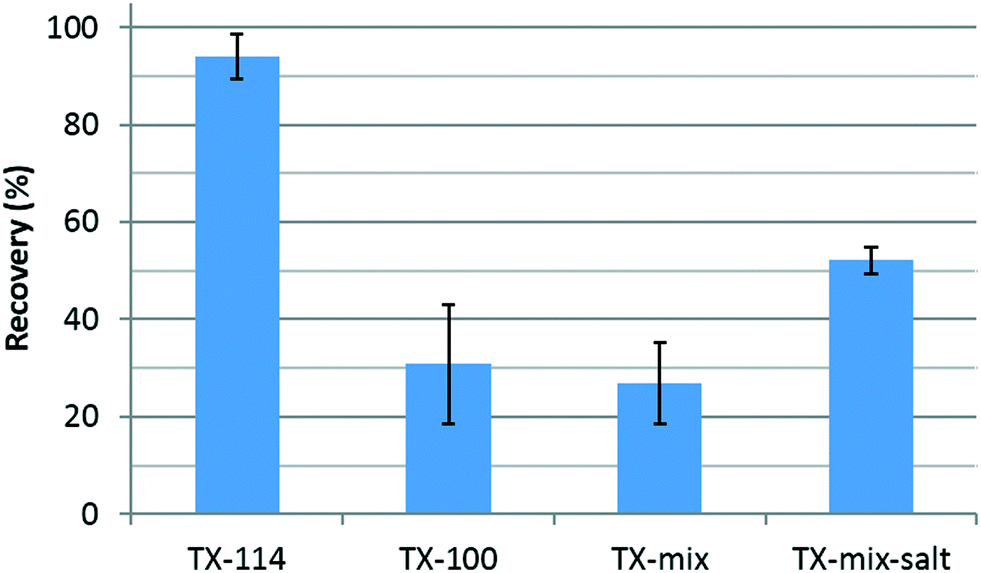 | ||
| Fig. 1 Au recovery from DI water solutions after CPE for different surfactant compositions, using BBI60 at an initial concentration of 100 μg kg−1. The salt is NaCl. Error bars represent one standard deviation of the replicates (n = 3). | ||
TX-114 was evaluated at three concentrations (0.1%, 0.2% and 0.5%), all of which are above the CMC. The recoveries were similar regardless of the concentration (data not shown).
3.1.2.1 Extraction efficiency in DI water. Fig. 2 shows the extraction efficiency (recovery) for BBI60 AuNP suspensions under different conditions. Regardless of the conditions, the recovery is consistently above 80%. The average enrichment factor was about 10×. Consequently, all conditions provide satisfactory extraction efficiencies relative to previously reported results.52,53
 | ||
| Fig. 2 CPE recoveries of BBI60 AuNPs, at an initial nominal concentration of 100 μg kg−1, from the simple matrix (DI water) determined after digestion of the extract with AR and analysis by ICP-MS (the number of sample preparation replicates are from left to right 11, 5, 4, 2 and 4). Supernatants are also presented here to assess the conservation of the mass. Error bars represent one standard deviation of the replicates. | ||
3.1.2.2 Size distribution as a function of CPE conditions. Here we assess the conservation of the size distribution for AuNPs following extraction. For this purpose, spICP-MS analyses were performed on the CPE extracts. Fig. 3 presents the size distributions of nominally 60 nm AuNPs after CPE using different conditions of extraction. It appears that AuNPs extracted with TX-114 alone (Fig. 3a) and TX-114 + NaCl (Fig. 3b) preserve the original size distribution with a mean size of 60.3 ± 0.3 nm and 60.1 ± 0.3 nm, respectively, obtained from Gaussian fitting. These results are statistically equivalent to the size distribution determined for the native BBI60 AuNPs measured without CPE under the same conditions, which yielded a mean size of 59.9 ± 0.1 nm (see Fig. S1 in the ESI†). Conversely, the extraction with TX-114 + HNO3 (Fig. 3c) and TX-114 + CaCl2 (Fig. 3d) yield a wider size distribution with a population around 60 nm and at least one other population centered above 70 nm. The sizes determined from spICP-MS data are based on a calculation where the particles are assumed to be perfect spheres with the density of bulk gold.34 In this case, if the particles form aggregates, the dimers would have a mass 2× higher and the spherical equivalent size would be roughly 75 nm. In the same way, a trimer of 60 nm AuNPs determined by spICP-MS would have a spherical equivalent diameter of about 86 nm. Fig. 3c and d clearly show a population around 75 nm, which likely corresponds to dimers. Above 80 nm the particle frequencies may be due to formation of trimers and more massive aggregates (from the mass of each particle event, it can be deduced that 4 particles correspond to 95 nm, 5 particles to 104 nm, etc.).
 | ||
| Fig. 3 Size distribution of BBI60 AuNPs from spICP-MS analyses for a) TX-114 only, b) TX-114 + NaCl, c) TX-114 + HNO3 and d) TX-114 + CaCl2. | ||
It is worth noting that spICP-MS has a slightly greater capacity to differentiate monomers from discrete clusters with low particle number, due to the mass-based measurement. By comparison, the hydrodynamic diameter (i.e., as measured by dynamic light scattering) for a dimer and trimer of spheres is 1.2D1 and 1.35D1, respectively, where D1 is the diameter of the monomer; thus, for 60 nm AuNPs, the hydrodynamic diameter of the dimer and trimer is 72 nm and 81 nm, respectively.
For CPE using HNO3 or CaCl2, the size distributions are not reproducible. In certain cases the aggregation state is predominant and the frequency for the monomer decreases substantially. Because aggregation is a kinetic phenomenon, it is reasonable to expect greater variance between experiments when aggregation is present. Moreover, the increase of aggregation may imply a lower recovery, when it is determined by spICP-MS compared with total analysis by ICP-MS; the more massive aggregates may settle in the autosampler vial prior to analysis, or the large size may inhibit complete ionization in the plasma. To quantify the aggregation state of the samples after CPE, an aggregation number parameter was used. The first aggregation number (AN1) was determined as the ratio of the mass sum of the dimer (mdimer) over the mass sum of the monomer (mmono):
 | (1) |
For larger aggregates (containing three or more AuNPs) the aggregation number (AN2) is defined by:
 | (2) |
More specifically, from the mass distribution determined by spICP-MS, the monomer peak can be deduced (see Fig. S2†). The summation ∑mmono corresponds to the sum of the masses in the entire monomer peak. The dimer peak is obtained by multiplying the maximum of the first (monomer) peak by two. All frequencies over the dimer peak are collectively lumped together as trimer-plus. To avoid counting events corresponding to coincidence (i.e., signal from two monomers occurring in one dwell time increment) during the analysis, the number of particles is determined in such a way that the particle number is below 500 min−1, where no coincidence (AN1+2 < 0.1) was observed for the control sample (BBI60 without CPE).
Fig. 4 compares the aggregation numbers for the different extraction conditions. ANs are below 0.2 for TX-114 alone and TX-114 + NaCl. AN1s for the other conditions are close to 0.5 and AN2s range from 20 ± 17 for TX-114 + HNO3 to 47 ± 20 for TX-114 + HNO3 + NaCl. The uncertainties determined as one standard deviation of the replicates of sample preparation show a wide variability for the size distributions and confirm the instability of the AuNPs extracted under these conditions (i.e., addition to TX-114 of HNO3, HNO3 + NaCl or CaCl2). In the same way, samples of AuNPs were analyzed without performing CPE (control samples), while ANs were determined in the same manner: AN1 = 0.08 ± 0.02 and AN2 = 0.02 ± 0.02. By comparing with samples extracted with TX-114 alone and TX-114 + NaCl, the AN1s are similar. AN2s are slightly lower for TX-114 alone at 0.06 ± 0.03. For TX-114 + NaCl, AN2 is equal to 0.11 ± 0.8, which is 5× higher than the native AuNP control sample; however, the uncertainty shows a large variability. Thus, AuNPs extracted with TX-114 alone and TX-114 + NaCl did not show a significant change in size distribution following CPE.
 | ||
| Fig. 4 Aggregation number (AN) determined for several conditions of extraction using BBI60 AuNPs. The insert is an enlargement of the graph. Error bars represent one standard deviation of the replicates (the number of sample preparation replicates were, from left to right, 9, 9, 9, 7 and 4). | ||
Overall, it is reasonable to conclude that the use of TX-114 alone or TX-114 + NaCl, as the extraction formulation, presents the best stability and conservation of size after CPE in a simple matrix for citrate-capped AuNPs. Previous studies, employing TEM to determine the size of the AuNPs13 and AgNPs,16 showed that the size was unaffected by CPE, even in the presence of acid. The redisolution of the surfactant-rich phase sample in alcohol after CPE may explain the observed difference with the current study (disaggregation). Moreover, these TEM analyses can only confirm that the core size of the particles is preserved (i.e., no change of the monomer size or change in shape), whereas aggregation events are difficult at best to quantify by TEM, due to drying artifacts. Therefore, spICP-MS is a more sensitive technique to evaluate the aggregation rate and state in situ.
Subsequently, the 30 nm size AuNPs were chosen to evaluate the effect of surface coating on CPE. In addition to TX-114 alone and TX-114 + NaCl, CA and EDTA were used as additives (see Table 1). Fig. 5 compares the extraction efficiency for different AuNP coatings used in this study. We observe that HA modification decreased the extraction efficiency to <30% for TX-114 alone. There is a small improvement for pH 8 with 22% extraction efficiency compared to 13% at pH 4. Ghouas et al. (2012) showed that extraction of HA by a nonionic surfactant is influenced by the presence of electrolytes and acids.55 The solubility of HA in water decreases with increasing ionic strength or decreasing pH.56 Therefore, factors such as decrease of repulsive interactions (at higher ionic strength) and protonation of HA to the neutral molecular form (at acidic pH) in the water/surfactant media may enhance the interaction of HA-coated AuNPs with the micellar aggregates and thus improve extraction efficiency.55,57 Similarly, in the present study, the recovery of PVP-coated AuNPs is less than 20% when TX-114 alone is used for extraction. Recovery for the HA treated AuNPs is improved by the addition of NaCl, CA and EDTA (see visual observation, Fig. S5†). The bPEI coating does not significantly reduce extraction efficiency compared with citrate, when TX-114 alone is used. However, when NaCl, CA or EDTA are added into the mixture, a decrease of the extraction efficiency is observed. It should be noted that bPEI contains a mixture of primary, secondary and tertiary amines,58 and except under strongly acidic conditions, carries a high positive charge density. Therefore, changes in surface charge (screening effects) may be the origin for the observed decrease in extraction efficiency when salt or acid is added to the bPEI-AuNP suspension. Additionally, the hydration of bPEI-coated AuNPs is greater (more water of hydration) compared to negatively charged AuNPs, and this could also play a role in determining extraction efficiency.
 | ||
| Fig. 5 Influence of surface coating on CPE recovery. BBI30 with a nominal core size of 30 nm was used at a concentration of 100 μg kg−1. AuNPs were mixed with HA at pH 4 and pH 8. Error bars represent one standard deviation of the replicates (for HA pH 4 n = 3; for HA pH 8 n = 3; for PVP n = 5, 2, 4 and 3 for TX-114 alone, TX-114 + NaCl, TX-114 + CA and TX-114 + EDTA, respectively; for bPEI n = 5 for TX-114 alone and 3 for the other additives; for citrate n = 10, 8, 4 and 4 for TX-114 alone, TX-114 + NaCl, TX-114 + CA and TX-114 + EDTA, respectively). | ||
According to the literature, if a compound is hydrophobic, it is more likely to be trapped in the micelles.59 Therefore, PVP-AuNPs and HA-AuNPs should be easily extracted because they are more hydrophobic due to the carbon chain and the aromaticity, respectively.60 However, it was observed that for TX-114 alone, citrate coating was extracted more efficiently. Compared to PVP (zeta-potential = −17.4 ± 2.9 mV, likely due to residual citrate) and HA (zeta-potential = −35.5 ± 1.5 mV), the citrate coating (zeta-potential = −24.0 ± 1.2 mV) has a very low molar mass. The size of the coating may also influence extraction. Therefore it appears that extraction efficiency is not simply a question of hydrophobicity, otherwise PVP and HA should have a higher measured efficiency.
Additionally, spICP-MS analyses for citrate coated AuNPs after the addition of CA exhibit a high aggregation rate, no doubt as a result of charge screening. For HA, PVP and bPEI coated surfaces, the size distribution remains largely unchanged when CA and EDTA (except for bPEI) are present in the solution. These coatings appear to stabilize the AuNPs, preventing significant aggregation from occurring during CPE (Fig. S4†).
3.2 Optimization of A4F methodology in a simple matrix (DI water)
Following CPE, samples have a high surfactant content. After dilution in water, the concentration of the supernatant reaches a maximum value of 0.5% TX-114. The presence of the surfactant in the sample may induce different interactions between NPs in the sample and the A4F membrane (accumulation wall); the elution process, and consequently the retention time, can be affected by these interactions. To assess these potential effects, different mobile phase compositions were tested: viz. 0.5 mmol L−1 NH4NO3, 0.5 mmol L−1 NH4NO3 + TX-114 at different concentrations and 0.4 mmol L−1 NH4NO3 + 0.1 mmol L−1 SDS. AuNP stock solutions were diluted in DI water and in a solution of 0.5% of TX-114.The nonionic surfactant (TX-114) was formulated in the mobile phase at several concentrations: viz. below, equal to, and above the CMC (0.2 mmol L−1). When a concentration of TX-114 equal to or above 0.2 mmol L−1 was used, the response of UV and MALS detectors was noisy and presented a high baseline signal intensity, likely due to the incipient formation of micelles.61,62 Below the CMC (TX-114 set at 0.001% or 0.02 mmol L−1), the background is greatly improved and returns to normal operating conditions. However, for the AuNP samples tested here (BBI30 and BBI60), two peaks were observed with a total recovery below 20%. Moreover, replicate analyses showed that the fractograms were not reproducible, as the signal of the analyzed NPs in subsequent runs decreased relative to the initial run. The low recovery may be due to passivation of the membrane by the nonionic surfactant. Thus, the electrostatic repulsion between membrane and the negatively charged particles is reduced. We have concluded that TX-114 is not a compatible surfactant additive in the mobile phase for A4F fractionation, at least under the conditions of the present study.
An anionic surfactant (SDS) at 0.1 mmol L−1 was used in combination with 0.4 mmol L−1 NH4NO3 to improve the anionic character of the PES membrane and potentially reduce passivation of the membrane by TX-114.62,63 The fractogram obtained, using a channel flow rate of 0.5 mL min−1 and a cross flow rate of 0.3 mL min−1 (see Fig. S6†), for BBI60 in 0.5% TX-114, exhibits two peaks that are not baseline resolved. One part of the population is not retained in the channel and elutes in the void peak (elution time of the unretained species at around 1 min) with a relative abundance of 63% ± 2%. It is due presumably to a high level of repulsion with the membrane and may be accelerated by the presence of micelles. The second population elutes at around 6 min.
The use of a mobile phase containing only a simple salt (0.5 mmol L−1 NH4NO3) was also considered. In the first step, BBI AuNPs (30 nm and 60 nm) were diluted in water and injected in order to establish a point of comparison. After two injections of blanks containing 0.5% TX-114, AuNPs diluted in 0.5% TX-114 were injected into the A4F system. Fig. 6 presents fractograms (UV-vis trace at 520 nm) for BBI30 and BBI60 AuNPs diluted into the two different media. For samples fractionated in TX-114 relative to DI water, the observations are as follows: 1) appearance of a small peak just after the void time at ≈1 min, 2) a wider main peak for both sizes in TX-114 and 3) a shift of the main peak toward faster (i.e., lower) retention times (by about 10%). The first population peak (retention time, tR = 1.3 min) may contain a small amount of AuNPs that are not retained due to the surrounding surfactant (absorption at 520 nm). Indeed, some AuNPs may be trapped in the residual TX-114 micelles and thus the nonionic surfactant might degrade the particle dispersion;62 therefore, due to repulsion with the membrane they are eluted faster. This hypothesis is confirmed by the analysis of blanks (sample of 0.5% TX-114 with no AuNPs, Fig. 6), which showed lower signal intensity at this retention time. Therefore, this peak is not due to the surfactant alone, but contains a population of AuNPs.
 | ||
| Fig. 6 A4F fractograms for citrate capped AuNPs (BBI30, dashed line and BBI60, solid line) diluted in DI water and diluted in 0.5% TX-114. | ||
The AuNPs were analyzed several times. Fig. S7† shows the fractograms of the replicates for a nominal size of 30 nm (see Fig. S7a†) and 60 nm (see Fig. S7b†). Importantly, it is observed that the retention times are repeatable for both sizes: about 3.9 min for 30 nm and 6.2 min for 60 nm. Additionally, the main peak narrows after the first run and the first peak near tR = 1 min has decreased. The resolution ( , with w the peak width at half height) thus becomes comparable with the AuNPs diluted in water, 0.67 ± 0.04 and 0.76 ± 0.04, for 30 nm and 60 nm, respectively. The resolution is acceptable even though it is <1 (baseline resolution would be indicated by a value of 1.5 or larger). Indeed, the goal is to obtain good separation in a reasonable analysis time for the entire size range of colloids present (up to 450 nm).
, with w the peak width at half height) thus becomes comparable with the AuNPs diluted in water, 0.67 ± 0.04 and 0.76 ± 0.04, for 30 nm and 60 nm, respectively. The resolution is acceptable even though it is <1 (baseline resolution would be indicated by a value of 1.5 or larger). Indeed, the goal is to obtain good separation in a reasonable analysis time for the entire size range of colloids present (up to 450 nm).
The experiments conducted here suggest that a mobile phase of 0.5 mmol L−1 NH4NO3 is the more appropriate condition for extracted samples containing a certain percentage (0.5%) of nonionic surfactant (TX-114), providing that preliminary runs are performed in order to condition the membrane for this type of sample. AuNPs diluted in DI water were also analyzed after conditioning. The signal intensity decreased significantly, indicating a substantial loss of material on the membrane surface. Thus, after the conditioning of the membrane, only sample containing TX-114 can be analyzed.
The coupling of A4F with ICP-MS (high sensitivity element-specific detector) is particularly useful when the AuNP concentration in the samples is relatively low (below 500 μg kg−1). The retention times are similar at low (200 μg kg−1) and high (13000 μg kg−1) concentrations (see Fig. S8†). Moreover, the ICP-MS signal for 200 μg kg−1 is above the LOQ (about 3 times higher) and offers better sensitivity compared to MALS or UV-vis (see Fig. S8b†).
3.3 CPE on soil extracts
Following CPE experiments in DI water, optimized conditions were applied to soil extract samples (obtained as described in section 2.4.2.). Thus, the extraction efficiency and the size distribution were assessed in the same way as the simple matrix, using spICP-MS (for AuNPs) and A4F-UV-MALS-ICP-MS (for natural colloids). | ||
| Fig. 7 Comparison of Au recovery after CPE for soil extract samples and simple MHW. Error bars represent one standard deviation of the replicates (n = 3). BBI60 AuNPs were initially dosed in the sample at a concentration of 500 μg kg−1. | ||
The recoveries for the soil extracts containing AuNPs are very high (over 90%) and comparable to the efficiency of extraction of AuNPs without soil colloids (MHW only). This result is in accordance with observations made in a simple matrix in section 3.1.3.
In order to test if the extraction efficiency of PVP coated AuNPs remains low when mixed with a soil extract matrix, PVP30 was spiked into the soil extract and CPE was performed using TX-114 alone. The recovery was 112% ± 5% (duplicate samples; data not shown). That is to say, high recovery is obtained. Thus, the presence of natural colloids can apparently improve the extraction efficiency by changing the physico-chemical properties of the solution (pH, ionic strength) and/or the surface characteristics of the NPs. We conclude that PVP-coated AuNPs can bind to the natural colloids such that they are co-extracted during the CPE process.
 | ||
| Fig. 8 Size distributions obtained from spICP-MS for BBI60 AuNPs. a) Soil extract after CPE with TX-114 alone, b) soil extract after CPE with TX-114 + NaCl, c) soil extract without CPE and d) MHW after CPE with TX-114 alone. The size distributions presented here are the collective sum of triplicate samples for a), b) and c). | ||
A second population around 75 nm is also observed in Fig. 8. As for the simple matrix, this peak corresponds to the aggregation of AuNPs and more particularly to the formation of dimers, as discussed previously. Aggregation numbers were calculated for the soil samples before and after CPE and are presented in Fig. 9.
 | ||
| Fig. 9 Aggregation number determined for soil extracts (SE) with and without CPE and a sample of AuNPs prepared in MHW (no soil) after CPE. BBI60 AuNPs were used. Columns from left to right correspond with the samples in Fig. 8(a) to (d), respectively. AN1 and AN2 are presented after subtraction of AN determined for AuNPs in DI water (referred to here as a blank, blk). Error bars represent one standard deviation of the replicates (n = 3). | ||
The observed aggregation may be due to interaction with components within the complex media. The extracts contain soil colloids (extracted in MHW) and other species (salts, organic matter, etc.) that may induce homoaggregation. Additionally, heteroaggregation with the natural colloids must also be considered as a possible outcome. The analysis of the soil extracts without CPE has also exhibited aggregation phenomena, demonstrating that this effect is not due to the CPE process itself. Additionally, CPE of AuNPs in MHW can also induce aggregation, because of the salts (monovalent and divalent) present in the MHW. Indeed, in Fig. 8d, for AuNPs prepared in MHW alone, aggregation is also observable (AN1 + AN2 = 0.55 ± 0.06).
The same observation applies to the effect of AuNP concentration (10 and 100 μg kg−1). The size distributions obtained by spICP-MS analysis indicates that they are largely preserved and not altered by the CPE process, independent of concentration (Fig. S9†).
A4F analyses of soil extracts dosed with BBI60 and treated by CPE are presented in Fig. 10. UV-vis, MALS and ICP-MS signals for Au and the major elements (Al, Fe and Mn) were monitored. The radius of gyration, rG, shows that the size varies between 30 nm and 250 nm over the main population range, indicating that effective fractionation is achieved. The fractograms show that Au is mainly present at the retention time corresponding to the principal population of AuNPs around 6 min. The natural colloids are represented by the broad peak at longer retention times apparent in MALS, UV and ICP-MS traces. If the Au signal is expanded (see Fig. S10†), it appears that a small quantity of the AuNPs are co-eluted with the broad (natural colloid) peak. Thus, AuNPs may be homoaggregated and/or bound (heteroaggregated) to the natural colloids present in the soil; both processes would yield longer retention times and broadened peaks. Similar results were previously observed in the literature where AgNPs and AuNPs interacted with different particulate components of the natural samples.64,65 For 30 nm AuNPs, Meisterjahn et al.65 observed that the retention time of AuNPs spiked in natural colloids is similar to the pure dispersion, but the peak presented an asymmetric and broadened shape.
 | ||
| Fig. 10 A4F fractograms of a soil extract with BBI60 AuNPs after CPE (TX-114 alone), Vp = 0.5 mL min−1 and Vc = 0.3 mL min−1. MALS signal at 90° is reported. The ×20 and ×5 parameters in the legend represent the scaling factor. | ||
4. Conclusions
We systematically investigated and optimized cloud point extraction or CPE for AuNPs using a simple aqueous matrix, then applied the method to an agricultural soil extract composed of the sub-0.45 μm fraction (colloids + dissolved species). The results quantitatively confirm the feasibility of using CPE with TX-114 surfactant on soil extract samples spiked with surface modified AuNPs, as a first step toward the development of a robust validated methodology for detecting, characterizing and quantifying manufactured metallic NPs in soils. The AuNPs are successfully pre-concentrated and the size distribution of the sample is preserved (as uniquely determined by single particle ICP-MS and asymmetric flow field-flow fractionation or A4F), under optimized conditions. Furthermore, enrichment of the analyte improves the selective detection and quantification of metallic MNPs by hyphenated A4F-ICP-MS. The use of other additives (HNO3, NaCl, CA and EDTA) to potentially enhance extraction show that acidification of the mixture improves extraction efficiency, but causes an important aggregation event for citrate coated AuNPs. Nevertheless, it is important to preserve, as much as possible, sample integrity. The influence of the MNP surface coating on the extraction efficiency in a simple matrix is significant, especially when TX-114 is used alone. However, in a complex matrix (soil extract) the inherent ionic strength and the presence of other colloids enhances the recovery for PVP and HA coated AuNPs without further additives. We achieve an average optimized enrichment factor of about 10×, but this can be improved by increasing the initial volume (i.e., a container volume of 50 mL instead of 10 mL with an appropriate sized centrifuge rotor) or by decreasing the volume of TX-114. In conclusion, enrichment by an optimized CPE method is shown to be feasible, reproducible, non-perturbing and improves detection and quantification of the MNP analyte. On-going research efforts focus on expanding this methodology to a wider range of MNPs (e.g., silver NPs) and additional environmental matrices (e.g., soils, sediments, industrial and residential sludge).Acknowledgements
The authors wish to express their gratitude to Stephen Wilson, Coordinator for the U.S. Geological Survey, Reference Materials Project, for kindly providing USGS reference soil for these studies. We acknowledge the nanoEHS initiative at NIST, coordinated by Debra Kaiser in the Materials Measurement Laboratory, for financial and programmatic support of this project and the postdoctoral research associateship of HEH. The authors would like to thank Christopher M. Sims, Arnab K. Mukherjee and R. David Holbrook for their helpful reviews of the manuscript.References
- M. E. Vance, T. Kuiken, E. P. Vejerano, S. P. McGinnis, M. F. Hochella Jr, D. Rejeski and M. S. Hull, Beilstein J. Nanotechnol., 2015, 6, 1769–1780 CrossRef CAS PubMed.
- V. H. Grassian, A. J. Haes, I. A. Mudunkotuwa, P. Demokritou, A. B. Kane, C. J. Murphy, J. E. Hutchison, J. A. Isaacs, Y.-S. Jun, B. Karn, S. I. Khondaker, S. C. Larsen, B. L. T. Lau, J. M. Pettibone, O. A. Sadik, N. B. Saleh and C. Teague, Environ. Sci.: Nano, 2016, 3, 15–27 RSC.
- F. v. d. Kammer, S. Legros, T. Hofmann, E. H. Larsen and K. Loeschner, TrAC, Trends Anal. Chem., 2011, 30, 425–436 CrossRef.
- F. Laborda, E. Bolea, G. Cepriá, M. T. Gómez, M. S. Jiménez, J. Pérez-Arantegui and J. R. Castillo, Anal. Chim. Acta, 2016, 904, 10–32 CrossRef CAS PubMed.
- S. M. Majedi and H. K. Lee, TrAC, Trends Anal. Chem., 2016, 75, 183–196 CrossRef CAS.
- M. Hassellöv, J. W. Readman, J. F. Ranville and K. Tiede, Ecotoxicology, 2008, 17, 344–361 CrossRef PubMed.
- E. Alayemieka, S.-H. Lee and J.-I. Oh, Environ. Eng. Res., 2006, 11, 201–207 CrossRef.
- D. Zirkler, F. Lang and M. Kaupenjohann, Colloids Surf., A, 2012, 399, 35–40 CrossRef CAS.
- M. Roca, N. H. Pandya, S. Nath and A. J. Haes, Langmuir, 2010, 26, 2035–2041 CrossRef CAS PubMed.
- D. Bouchard and X. Ma, J. Chromatogr. A, 2008, 1203, 153–159 CrossRef CAS PubMed.
- K. L. Chen and M. Elimelech, Environ. Sci. Technol., 2008, 42, 7607–7614 CrossRef CAS PubMed.
- M. Baalousha, F. V. D. Kammer, M. Motelica-Heino and P. Le Coustumer, J. Chromatogr. A, 2005, 1093, 156–166 CrossRef CAS PubMed.
- T. K. Mudalige, H. Qu and S. W. Linder, Anal. Chem., 2015, 87, 7395–7401 CrossRef CAS PubMed.
- J. Gigault, J. M. Pettibone, C. Schmitt and V. A. Hackley, Anal. Chim. Acta, 2014, 809, 9–24 CrossRef CAS PubMed.
- M. A. Mesquita da Silva, V. L. Azzolin Frescura and A. J. Curtius, Spectrochim. Acta, Part B, 2000, 55, 803–813 CrossRef.
- G. Hartmann and M. Schuster, Anal. Chim. Acta, 2013, 761, 27–33 CrossRef CAS PubMed.
- J.-B. Chao, J.-F. Liu, S.-J. Yu, Y.-D. Feng, Z.-Q. Tan, R. Liu and Y.-G. Yin, Anal. Chem., 2011, 83, 6875–6882 CrossRef CAS PubMed.
- G. Hartmann, T. Baumgartner and M. Schuster, Anal. Chem., 2014, 86, 790–796 CrossRef CAS PubMed.
- J.-F. Liu, J.-B. Chao, R. Liu, Z.-Q. Tan, Y.-G. Yin, Y. Wu and G.-B. Jiang, Anal. Chem., 2009, 81, 6496–6502 CrossRef CAS.
- S. M. Majedi, B. C. Kelly and H. K. Lee, Anal. Chim. Acta, 2014, 814, 39–48 CrossRef CAS PubMed.
- G. Z. Tsogas, D. L. Giokas and A. G. Vlessidis, Anal. Chem., 2014, 86, 3484–3492 CrossRef CAS PubMed.
- L. Li, G. Hartmann, M. Döblinger and M. Schuster, Environ. Sci. Technol., 2013, 47, 7317–7323 CAS.
- R. F. Domingos, M. A. Baalousha, Y. Ju-Nam, M. M. Reid, N. Tufenkji, J. R. Lead, G. G. Leppard and K. J. Wilkinson, Environ. Sci. Technol., 2009, 43, 7277–7284 CrossRef CAS PubMed.
- G. G. Leppard, Curr. Nanosci., 2008, 4, 278–301 CrossRef CAS.
- R. B. Reed, C. P. Higgins, P. Westerhoff, S. Tadjiki and J. F. Ranville, J. Anal. At. Spectrom., 2012, 27, 1093–1100 RSC.
- J. Liu, K. E. Murphy, R. I. MacCuspie and M. R. Winchester, Anal. Chem., 2014, 86, 3405–3414 CrossRef CAS PubMed.
- F. v. d. Kammer and U. Förstner, Water Sci. Technol., 1998, 37, 173–180 CrossRef CAS.
- H. El Hadri, J. Gigault, P. Chéry, M. Potin-Gautier and G. Lespes, Anal. Bioanal. Chem., 2013, 406, 1639–1649 CrossRef PubMed.
- M. Bouby, H. Geckeis and F. W. Geyer, Anal. Bioanal. Chem., 2008, 392, 1447–1457 CrossRef CAS PubMed.
- M. Baalousha, B. Stolpe and J. R. Lead, J. Chromatogr. A, 2011, 1218, 4078–4103 CrossRef CAS PubMed.
- NIST Reference Material 8012 Gold Nanoparticles, Nominal 30 nm Diameter, National Institute of Standards and Technology, Report of Investigation, 2015 Search PubMed.
- NIST Reference Material 8013 Gold Nanoparticles, Nominal 60 nm Diameter, National Institute of Standards and Technology, Report of Investigation, 2015 Search PubMed.
- D. B. Smith, L. G. Woodruff, R. M. O'Leary, W. F. Cannon, R. G. Garrett, J. E. Kilburn and M. B. Goldhaber, Appl. Geochem., 2009, 24, 1357–1368 CrossRef CAS.
- H. E. Pace, N. J. Rogers, C. Jarolimek, V. A. Coleman, C. P. Higgins and J. F. Ranville, Anal. Chem., 2011, 83, 9361–9369 CrossRef CAS PubMed.
- K.-G. Wahlund, J. Chromatogr. A, 2013, 1287, 97–112 CrossRef CAS PubMed.
- M. Andersson, B. Wittgren and K.-G. Wahlund, Anal. Chem., 2003, 75, 4279–4291 CrossRef CAS PubMed.
- M. F. Nazar, S. S. Shah, J. Eastoe, A. M. Khan and A. Shah, J. Colloid Interface Sci., 2011, 363, 490–496 CrossRef CAS PubMed.
- Z. Sosa Ferrera, C. Padrón Sanz, C. Mahugo Santana and J. J. Santana Rodríguez, TrAC, Trends Anal. Chem., 2004, 23, 469–479 CrossRef CAS.
- S. Goheen and R. Matsor, J. Am. Oil Chem. Soc., 1989, 66, 994–997 CrossRef CAS.
- D. F. Lowe, C. L. Oubre and C. H. Ward, Surfactants and Cosolvents for NAPL Remediation A Technology Practices Manual, Taylor & Francis, 1999 Search PubMed.
- I. Baranowska, Handbook of Trace Analysis: Fundamentals and Applications, Springer International Publishing, 2015 Search PubMed.
- G. Hartmann, C. Hutterer and M. Schuster, J. Anal. At. Spectrom., 2013, 28, 567–572 RSC.
- S. M. Majedi, H. K. Lee and B. C. Kelly, Anal. Chem., 2012, 84, 6546–6552 CrossRef CAS PubMed.
- E. Camera, G. Modena and B. Zotti, Propellants, Explos., Pyrotech., 1983, 8, 70–73 CrossRef CAS.
- B. A. A. van Woezik, Runaway and thermally safe operation of a nitric acid oxidation in a semi-batch reactor, PhD thesis, University of Twente, Enschede, 2000 Search PubMed.
- Liquid-Solid partitioning as a function of liquid-to-solid ratio in solid materials using a parallel batch procedure-Method 1316, US Environmental Protection Agency, 2012 Search PubMed.
- Methods for measuring the acute toxicity of effluents and receiving waters to freshwater and marine organisms, US Environmental Protection Agency, 5th edn, 2002 Search PubMed.
- L. J. Gimbert, P. M. Haygarth, R. Beckett and P. J. Worsfold, Environ. Sci. Technol., 2005, 39, 1731–1735 CrossRef CAS PubMed.
- B. Fricke, Anal. Biochem., 1993, 212, 154–159 CrossRef CAS PubMed.
- L. P. Malpiedi, B. B. Nerli, D. S. P. Abdalla and A. Pessoa, Biotechnol. Prog., 2014, 30, 554–561 CrossRef CAS PubMed.
- C. C. Nascentes and M. A. Z. Arruda, Talanta, 2003, 61, 759–768 CrossRef CAS PubMed.
- AOAC Guidelines for Single Laboratory Validation of Chemical Methods for Dietary Supplements and Botanicals, A. O. A. C. International, Gaithersburg, MD, 2002 Search PubMed.
- C. J. Murphy, J. D. MacNeil and S. G. Capar, J. AOAC Int., 2013, 96, 190–203 CrossRef CAS PubMed.
- Y. T. He, J. Wan and T. Tokunaga, J. Nanopart. Res., 2008, 10, 321–332 CrossRef CAS.
- H. Ghouas, B. Haddou, M. Kameche, Z. Derriche and C. Gourdon, J. Hazard. Mater., 2012, 205–206, 171–178 CrossRef CAS PubMed.
- H. Kipton, J. Powell and R. M. Town, Anal. Chim. Acta, 1992, 267, 47–54 CrossRef CAS.
- D. L. Giokas, J. Antelo, E. K. Paleologos, F. Arce and M. I. Karayannis, J. Environ. Monit., 2002, 4, 505–510 RSC.
- M. Jager, S. Schubert, S. Ochrimenko, D. Fischer and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 4755–4767 RSC.
- A. S. Yazdi, TrAC, Trends Anal. Chem., 2011, 30, 918–929 CrossRef CAS.
- Y. Xiao and M. R. Wiesner, J. Hazard. Mater., 2012, 215, 146–151 CrossRef PubMed.
- S. Dubascoux, F. Von Der Kammer, I. Le Hécho, M. P. Gautier and G. Lespes, J. Chromatogr. A, 2008, 1206, 160–165 CrossRef CAS PubMed.
- M. Moon, Bull. Korean Chem. Soc., 1995, 16, 613–619 CAS.
- E. Bolea, J. Jiménez-Lamana, F. Laborda and J. R. Castillo, Anal. Bioanal. Chem., 2011, 401, 2723–2732 CrossRef CAS PubMed.
- J. Gigault and V. A. Hackley, Anal. Chim. Acta, 2013, 763, 57–66 CrossRef CAS PubMed.
- B. Meisterjahn, E. Neubauer, F. v. d. Kammer, D. Hennecke and T. Hofmann, J. Chromatogr. A, 2014, 1372, 204–211 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6en00322b |
| ‡ The identification of any commercial product or trade name does not imply endorsement or recommendation by the National Institute of Standards and Technology. |
| This journal is © The Royal Society of Chemistry 2017 |