
Effect of humic acid on the kinetics of silver nanoparticle sulfidation†
Basilius
Thalmann
ab,
Andreas
Voegelin
a,
Eberhard
Morgenroth
ab and
Ralf
Kaegi
*a
aEawag, Swiss Federal Institute of Aquatic Science and Technology, Überlandstrasse 133, CH-8600 Dübendorf, Switzerland. E-mail: ralf.kaegi@eawag.ch
bETH Zürich, Institute of Environmental Engineering, CH-8093 Zürich, Switzerland
First published on 15th December 2015
Abstract
The sulfidation of metallic silver nanoparticles (AgNP) observed in urban wastewater systems and in natural waters reduces their toxicity by several orders of magnitude. However, the reaction rate of this transformation is only poorly understood and the influence of humic acid (HA) on AgNP sulfidation has not been studied to date. We therefore investigate the sulfidation kinetics of AgNP reacted with bisulfide (HS−) in the absence and presence of HA and evaluate different kinetic models to describe the observed reaction kinetics. Citrate-stabilized AgNP of different sizes (20–200 nm) were reacted with an excess of HS− in the absence of HA as well as at HA concentrations ranging from 50 to 1000 mg L−1. The extent of AgNP sulfidation after the selected reaction times was determined by X-ray absorption spectroscopy (XAS). The overall sulfidation rate increased with decreasing AgNP size and increasing HA concentration. The sulfidation rate of the AgNP was best described by a diffusion-limited solid state reaction model (parabolic rate law). The corresponding half-lives of the AgNP ranged from minutes to hours. The increase of the sulfidation rate with increasing HA concentration may be explained by the adsorption of HA onto the AgNP surface facilitating the access of HS− to the particle surface. The results from analytical transmission electron microscopy suggest that the AgNP were sulfidized asymmetrically in the absence of HA. In the presence of HA, the initially formed concentric core–shell Ag0–Ag2S structures developed into hollow Ag2S nanoparticles with increasing reaction time, possibly via the Kirkendall effect.
Nano impactSilver nanoparticles (AgNP) are of ecotoxicological concern. The sulfidation of AgNP observed in urban wastewater systems and in wetlands reduces their toxicity by several orders of magnitudes. However, the sulfidation rate and the influence of humic acid (HA) on the sulfidation are still poorly understood. We show that the sulfidation rate increases with decreasing AgNP diameter and increasing HA concentrations. We reveal that the sulfidation of AgNP leads to the initial formation of the Ag2S–Ag0 core–shell structures. Outward diffusion of Ag from the metallic core results in the formation of hollow Ag2S NP by the Kirkendall effect. |
Introduction
Silver nanoparticles (AgNP) are widely applied to consumer products (e.g., textiles and cosmetics),1 with the intention to slowly release antimicrobially active Ag+.2,3 During the use and the washing of such products, AgNP can detach from the host material4–6 and may reach the aquatic environment causing environmental concern.7 Mass flow analysis revealed that the largest fraction of AgNP released will be discharged into urban wastewater systems8 and about 5% will pass the wastewater treatment plants and reach surface waters.9Released AgNP will interact with various constituents such as natural organic matter (NOM) present in the aquatic environment. Humic acid (HA), a part of NOM, was shown to increase the colloidal stability of AgNP due to the attachment of HA onto the particles' surface.10 Additionally, the adsorption of HA onto the AgNP surface can reduce the toxicity of the AgNP.11
The environmentally most important AgNP transformations are the dissolution of AgNP and the formation of Ag2S, AgCl or Ag complexes with organics. In the presence of HA and light, the formation of smaller AgNP has been reported.12–14 Dissolution is dependent on the size and the coating of the AgNP in addition to pH and dissolved molecular oxygen.15–18 In chloride-rich environments, the dissolution of AgNP is followed by the precipitation of AgCl or the formation of soluble chloride complexes.19 In the presence of sulfides, the formation of Ag2S is dominant due to its low solubility product.20 In sulfidation experiments, HS− reacting with AgNP decreased faster with decreasing particle diameter and in the presence of HA, but these qualitative observations were not explored in more detail.21
In urban wastewater, the sulfidation of AgNP to Ag2S starts in the sewer system22,23 and continues in the wastewater treatment plants due to elevated HS− concentrations.9,24–27 Within typical hydraulic residence times in the urban wastewater system of ∼24 h, metallic AgNP get almost completely sulfidized. The sulfidation of AgNP has also been reported in freshwater wetland sediments.28 We recently showed that AgNP sulfidation also occurs in oxic surface waters by reaction with metal sulfides such as ZnS and CuS with lower thermodynamic stability than Ag2S.29 The toxicity of very poorly soluble Ag2S is several orders of magnitude lower than the toxicity of soluble Ag salts and the oxidation of Ag2S in oxic waters proceeds rather slowly.19,30–32 Furthermore, substantial amounts of HA are present in wastewater, as up to 85 mg of organic C per g of sewage sludge has been reported.33 Despite the overarching importance of the sulfidation of AgNP and the ubiquitous presence of HA, the dependence of the sulfidation rates on the AgNP size and the HA concentrations is only poorly understood.21,29,34–36
The objectives of this study were, therefore, to investigate the sulfidation kinetics and the mechanism of AgNP reacting with HS− under oxic conditions and to determine the influence of HA on the sulfidation rates and mechanisms. We investigated the effects of AgNP (nominal) size (20, 40, 100 and 200 nm) and HA concentrations (0, 50, 250, 1000 mgHA L−1) on the sulfidation rates. The reaction progress was monitored by measuring the Ag speciation at the selected reaction times using X-ray absorption spectroscopy (XAS). We evaluated four different kinetic models to best describe the sulfidation kinetics. Further insights into the reaction mechanism were obtained by transmission electron microscopy (TEM) analysis of partially and fully sulfidized AgNP.
Materials and methods
Starting materials
Citrate-stabilized AgNP (20, 40, 100 and 200 nm in diameter; Nanocomposix, 1000 mgAg L−1, BioPure, CA, USA) were used for all experiments. The particles were characterized using transmission electron microscopy (TEM), dynamic light scattering (DLS), phase analysis light scattering (PALS) and UV/vis spectroscopy. The zeta potential was calculated from the electrophoretic mobility (PALS) using the Smoluchowski approximation. DLS and electrophoretic mobility measurements were performed using a Zetasizer (NanoZS, Malvern Instruments, UK). The plasmon resonance was recorded using a UV/vis spectrometer (Cary 100, Varian). The results are given in the ESI† and are in good agreement with the data provided by the supplier (Fig. S1 and S2, Table S1†).For the sulfidation experiments, 56.4 mg of sodium hydrogen sulfide (NaHS, Alfa Aesar) was dissolved in 10 mL of deionized water (doubly deionized (DDI) water was used in all experiments; Millipore, 18.2 MΩ cm) to obtain a bisulfide (HS−) stock solution of ∼100 mM. The HS− concentration (100.4 mM) was determined iodometrically (according to Eaton, et al.37). In brief, 1 mL of Lugol's solution was mixed with 50 mL of water and 20 μL of the stock solution followed by the addition of 1 mL of starch solution (10 g L−1). This solution was titrated with thiosulfate (3.26 mM) until it became colorless.
Humic acid (HA, Sigma Aldrich, lot: #STBD5313V, technical grade) was used as received. A stock solution was produced by dissolving 70 mg of HA in 50 mL of DDI water. The iron concentration of the HA was 254 mmolFe kgHA−1 (measured by inductively coupled plasma optical emission spectroscopy (ICP-OES) after acid digestion of 100 μL of HA solution with 1 mL of HNO3 (Ultrapure 60%, Merck, DE) and 100 μL of H2O2 (30%) in an UltraClave3 (MLS GmbH)).
Methods
The interaction of HA with HS− was studied by reacting specific amounts of HS− (1–4.5 mM) with a buffered solution (pH = 7.5, 50 mM HEPES) containing 250 mgHA L−1 in 1.5 mL Eppendorf tubes with minimal head space. An aliquot of 100 μL was used to determine the initial HS− concentration in every vial. The tubes were closed and sealed with Parafilm and the HA was separated by centrifugation (18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, 60 min, Mikro 220R, Hettich Zentrifugen) at the end of the experiments. Additional experiments were conducted with 250 mgHA L−1 and 2.3 mM HS− without centrifugation for the same time period (60 min). The HS− concentration in the supernatant/bulk solution was determined according to the methylene blue method (4500-S2−), as it is very selective to S2−.37 In brief, 100 μL of the sample was mixed with 0.9 mL of DDI water and 0.1 mL of N,N-dimethylbenzene-1,4-diamine (337.5 mg (Sigma-Aldrich, CH) dissolved in 50 mL of 49% H2SO4, 50 mM). This solution was reacted with 30 μL of an iron(III) chloride solution (9 M, FeCl3,Sigma-Aldrich, CH) for 4 min, followed by the addition of 0.32 mL of diammonium hydrogen phosphate (3.8 M, (NH3)2HPO4, Sigma-Aldrich, CH). The absorption peak maximum at 605 nm of the stoichiometrically formed methylene blue dye was measured using a spectrophotometer and the concentration was calculated using the extinction coefficient ε = 132000 cm M−1.38
000g, 60 min, Mikro 220R, Hettich Zentrifugen) at the end of the experiments. Additional experiments were conducted with 250 mgHA L−1 and 2.3 mM HS− without centrifugation for the same time period (60 min). The HS− concentration in the supernatant/bulk solution was determined according to the methylene blue method (4500-S2−), as it is very selective to S2−.37 In brief, 100 μL of the sample was mixed with 0.9 mL of DDI water and 0.1 mL of N,N-dimethylbenzene-1,4-diamine (337.5 mg (Sigma-Aldrich, CH) dissolved in 50 mL of 49% H2SO4, 50 mM). This solution was reacted with 30 μL of an iron(III) chloride solution (9 M, FeCl3,Sigma-Aldrich, CH) for 4 min, followed by the addition of 0.32 mL of diammonium hydrogen phosphate (3.8 M, (NH3)2HPO4, Sigma-Aldrich, CH). The absorption peak maximum at 605 nm of the stoichiometrically formed methylene blue dye was measured using a spectrophotometer and the concentration was calculated using the extinction coefficient ε = 132000 cm M−1.38
514 eV) at the Dutch Belgian Beamline (DUBBLE, BM01B) at the European Synchrotron Radiation Facility (ESRF, Grenoble, France). A closed-cycle He-cryostat adjusted to 80 K (DUBBLE) was used to cool the samples. The reference samples Ag0 (metallic foil) and Ag2S (acanthite) were measured in transmission mode. The frozen samples were measured using a 9-element monolithic Ge fluorescence detector (Canberra, CT, USA).
Athena39 was used to process the X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra and to quantify the metallic and the sulfidic fractions by linear combination fitting (LCF). The E0 was set to 25514 eV and a first-order polynomial fit to the data from 25414 to 25454 eV was subtracted from the raw data. A second-order polynomial fit to the data from 25554 to 25814 eV was used to normalize the edge-jump at E0 to unity and to flatten the spectrum. LCF analysis of the XANES spectra was performed over the energy-range from 25494 to 25814 eV, and LCF analysis of the EXAFS spectra was performed over the k-range from 2.5 to 9 Å−1. The individual fractions of Ag0 and Ag2S were constrained to values between 0 and 1; the sum was not constrained. The fractions of Ag0 derived from the LCF analysis of the XANES and EXAFS data were averaged and are reported together with the respective standard deviations.
Results and discussion
Sulfidation kinetics of AgNP
A multi-factorial design (Table S2†) was used to investigate the sulfidation kinetics of AgNP with HS− in the presence/absence of HA in oxic water. In time resolved experiments, AgNP (20, 40, 100, 200 nm) were reacted with HS− (2.5 mM, HS− in fivefold excess compared to Ag) in the presence of HA (0, 50, 250, 1000 mgHA L−1) up to 60 min and the reaction progress was monitored by determining the sulfidic and the metallic fractions of the suspension using Ag K-edge XAS. The recorded spectra and the corresponding LCF results for XANES (Fig. S3†) and EXAFS (Fig. S4†) analyses are given in the ESI† (Table S3). In all experiments, the metallic fraction steadily decreased with increasing reaction time and the decrease was faster for smaller particles (Fig. 1, Table S3†). After 15 min of reaction (1000 mgHA L−1), the 20 and 40 nm AgNP were almost completely sulfidized, whereas the 100 and 200 nm were still 20 and 30% metallic (Fig. 1A). An increasing sulfidation rate (faster decrease of the metallic fraction) was observed for increasing HA concentrations at a constant particle size (Fig. 1B). After 15 min of reaction time, the metallic fraction of the 20 nm AgNP decreased from 43% (absence of HA) over 27% (50 mgHA L−1) and 10% (250 mgHA L−1) and was completed in the presence of 1000 mgHA L−1. | ||
| Fig. 1 Average metallic Ag0 fraction from LCF (XANES and EXAFS) against time for different experiments (error bars = 1σ, derived from the average of XANES and EXAFS Ag0 fraction, Table S3†). The legend is given as inset. For better readability, the data of the 200 nm AgNP have been shifted by plus 2 min. A: different AgNP sizes at 1000 mgHA L−1, B: different HA concentrations (0, 50 and 250 mgHA L−1) for experiments with 20 and 100 nm AgNP. For better readability, the data of the 100 nm AgNP have been shifted by plus 2 min. | ||
Four different models were evaluated to describe the sulfidation kinetics of AgNP: (i) a parabolic rate model originally proposed by Jander41 (parabolic rate model, eqn (1)), which has previously been applied to describe the diffusion-limited solid state reaction of Ag0 and sulfur at elevated temperatures;42,43 (ii) a pseudo first-order rate model which has been used to describe the sulfidation kinetics of AgNP (FO model, eqn (2));21 (iii) a shrinking core model where the reaction progress is limited by diffusion through an outer, reacted (ash) layer (ash model, eqn (3));44 and (iv) a shrinking core model limited by the chemical reaction at the reactive surface of the shrinking core (chem model, eqn (4)):44
 | (1) |
| F = e−k×t | (2) |
 | (3) |
 | (4) |
The results of two selected time series (40 and 200 nm with 1000 mgHA L−1) are given for each model in Fig. 2. Qualitatively, the parabolic rate and the FO model fitted the data better than the two other models. When comparing the model fits for the sulfidation of the 200 nm AgNP, the parabolic rate model seemed to reproduce the measured data more closely than the FO model. The residual sum of squares (RSS) from each fitted data series was summed to compare the overall fit quality of the individual models. The chem and ash models had the highest values (0.70 and 0.44) followed by the FO model (0.19) and the parabolic rate model had the lowest value (0.15) indicating the best fit quality. Thus, qualitative visual inspection and the sum of the RSS both suggested that the parabolic rate model was best suited to describe the sulfidation kinetics of AgNP (results of the model with the data are given in Fig. S6 and Table S4†). The experiment conducted with 20 nm AgNP in the presence of 1000 mgHA L−1 over 5 minutes showed an unusually high metallic fraction, which was considerably higher than the metallic fraction obtained for the 40 nm AgNP for the same treatment (Fig. 1A). This high metallic fraction resulted in a substantial discrepancy between the model fit and the measured data for the 20 nm AgNP in the presence of 1000 mgHA L−1 (Fig. S6A†). As a consequence, a lower sulfidation rate was derived for the 20 nm AgNP compared to the 40 nm AgNP in the presence of 1000 mgHA L−1 (Fig. S6A†). However, this apparently lower reaction rate is most probably due to an artifact in the experiment with 20 nm AgNP in the presence of 1000 mgHA L−1 over 5 min (e.g., incomplete mixing of the AgNP with HA).
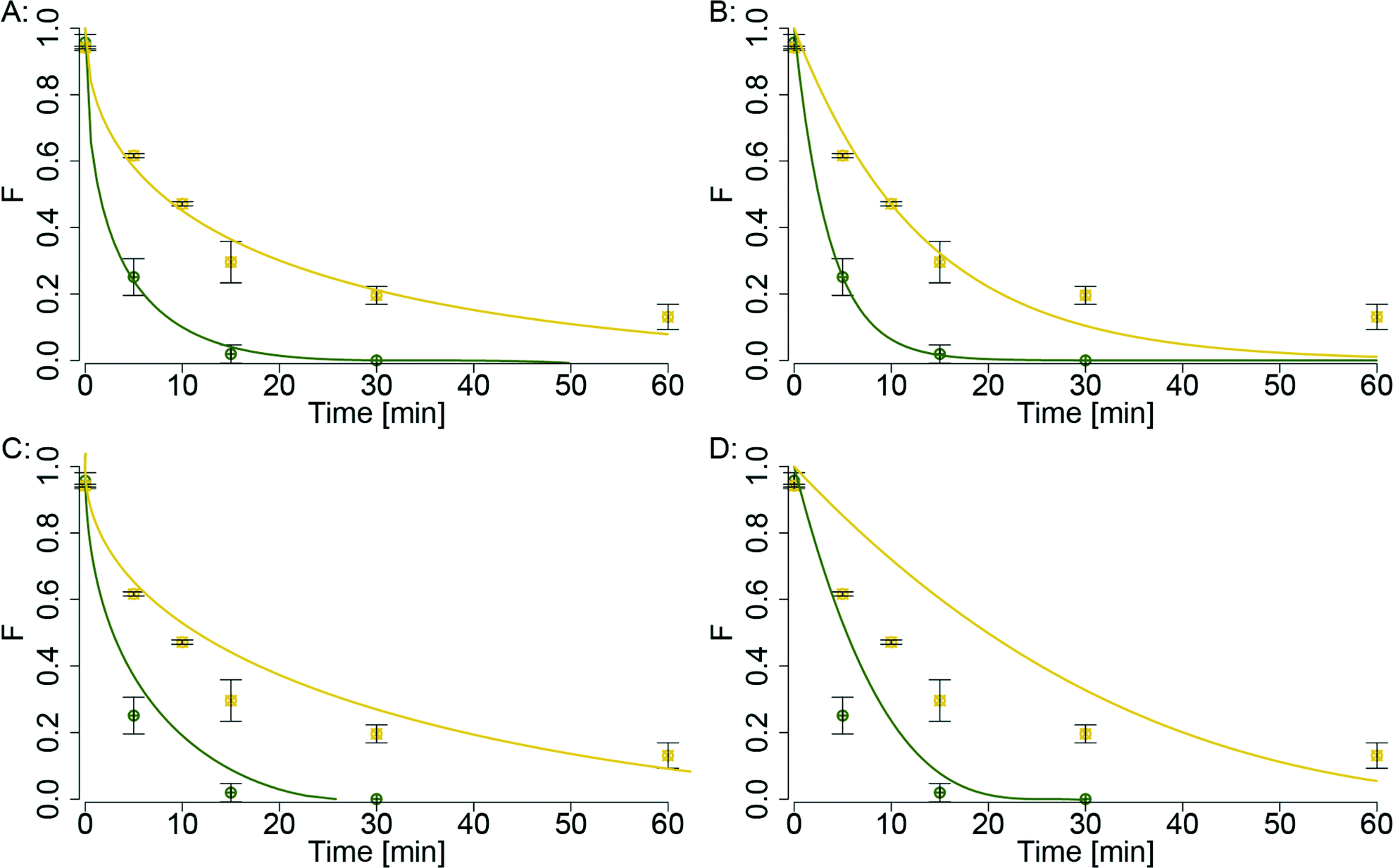 | ||
| Fig. 2 Comparison of the four different reaction models (A: parabolic rate model (eqn (1)), B: pseudo-first-order model (eqn (2)), C: ash model (eqn (3)), D: chem model (eqn (4))) for two data series (green: 40 nm AgNP, yellow: 200 nm AgNP; both with 1000 mgHA L−1). Solid lines represent model fits derived from non-linear regressions of the experimental data. | ||
The parabolic rate model implies that the reaction is diffusion-limited and dependent on the concentration gradient of the diffusing element (given that sufficient HS− for the complete sulfidation of the AgNP is available). In the following, the rate coefficients derived from the parabolic rate model are evaluated in more detail with respect to the effects of the nanoparticle size and HA concentration on the sulfidation kinetics and mechanism.
In Fig. 3, the sulfidation rate coefficients (k) derived from the parabolic rate model are plotted against the HA concentration normalized to the total AgNP surface (HAnorm, assuming ideal spherical particles). The rate coefficients increased almost linearly with increasing HAnorm, except for the rate coefficients of 100 nm AgNP reacted with 0, 50 and 250 mgHA L−1, which had significantly higher k values. The k values for 100 nm AgNP reacted in the presence of 0 and 250 mgHA L−1 were derived from only one time point (15 min) and are thus associated with a considerably higher uncertainty.
 | ||
| Fig. 3 HA concentration divided by the total AgNP surface (HAnorm) against the sulfidation rates (k) derived from the parabolic rate model (k values are given in Table S4,† error bars = 1σ). | ||
The possible explanations for the increasing rate constants with increasing HAnorm are (i) the stabilizing effect of HA preventing AgNP agglomeration and (ii) the replacement of citrate coating with HA leading to a larger AgNP surface area available for the sulfidation reaction, combined with the presence of HS− within the HA. The first hypothesis (i) was tested by measuring the hydrodynamic diameter of 20 nm AgNP (0, 50 or 250 mgHA L−1) during the reaction with HS− over ~45 min (Fig. S7†). In the absence of HA, the diameter increased from 32 to 42 nm and sedimentation was observed, indicating that agglomeration of AgNP induced by the sulfidation with HS− occurred. In the presence of HA, however, the diameter only increased by 2–4 nm regardless of the HA concentration. Thus, the stabilizing effect of HA may be responsible for the initial increase of the sulfidation rates (difference between the absence and the presence of HA), but with increasing HA concentrations no further stabilizing effect was observed. The increased reaction rates can thus not exclusively be explained by the colloidal stabilization of the AgNP by the HA.
To investigate the second hypothesis (ii) – replacement of citrate by HA bringing HS− in close contact with the AgNP surface – we studied the interaction of HA with AgNP and HA with HS−.
Interaction of HA with AgNP
The hydrodynamic diameter of the AgNP in the presence of selected amounts of HA was measured by DLS (Table 1). The diameter of the 20 nm AgNP slightly decreased with the addition of 25 mgHA L−1 HA concentration but then steadily increased with increasing HA concentration to a maximum of 79 nm. The polydispersity index (PDI) followed the same trend with an initial slight decrease from 0.29 to 0.21 followed by a steady increase up to 0.836. However, the highest values might be unreliable due to the very high PDI of up to 0.84 (Table 1). Comparable results were obtained for experiments conducted with the 40 nm and the 100 nm AgNP, where the diameter increased to 63 nm and 117.7 nm with the PDI of 0.414 and 0.218, respectively. The hydrodynamic diameter of the 200 nm AgNP did not follow any consistent trend. The relative change in the size for the 200 nm AgNP is expected to be even smaller than for the 100 nm AgNP and, therefore, may not be detectable with DLS anymore.| AgNP diametera [nm] (PDI)b | 0 mgHA L−1 | 25 mgHA L−1 | 50 mgHA L−1 | 100 mgHA L−1 | 200 mgHA L−1 | 400 mgHA L−1 | 800 mgHA L−1 |
|---|---|---|---|---|---|---|---|
| a The data refer to number weighted particle size distributions using the cumulant method. b PDI = polydispersity index. | |||||||
| 20 | 29.3 (0.29) | 27.3 (0.21) | 27.7 (0.22) | 31.1 (0.32) | 37.9 (0.40) | 55.8 (0.74) | 79.1 (0.84) |
| 40 | 39.4 (0.19) | 38.4 (0.19) | 39.8 (0.20) | 40.3 (0.21) | 43.3 (0.24) | 49.6 (0.35) | 63.3 (0.41) |
| 100 | 103.7 (0.22) | 98.6 (0.15) | 96.3 (0.12) | 98.2 (0.13) | 108.1 (0.16) | 116.2 (0.17) | 117.7 (0.22) |
| 200 | 228.8 (0.23) | 254.3 (0.25) | 233.2 (0.18) | 242.4 (0.20) | 233.9 (0.18) | 213.4 (0.17) | 235.8 (0.32) |
The initial decrease of the PDI indicates that the AgNP were stabilized by HA (and possibly disaggregated to some extent), leading to a more monodisperse suspension, which also has been previously observed by other researchers.11,46,47 The particle diameter mostly remained within 10% of the original value (determined in the absence of HA) for a polydispersity index smaller than 0.3. Higher PDI were mainly observed at high HA concentrations and resulted in larger particle sizes. However, the DLS results with PDI > 0.3 are difficult to interpret and are thus excluded from the discussion. Furthermore, additional measurements confirmed that the particle size remained constant over 60 min, indicating a high colloidal stability of the suspensions. We did not observe a decreasing particle size with time as previously reported.12 This apparent discrepancy can most likely be explained by the considerably shorter run times in our experiments (1 h) compared to the aforementioned study (24 h).
Although our DLS measurements did not allow us to assess whether citrate was displaced from the surface of the AgNP, it is generally assumed that citrate is only weakly bound to the AgNP surface. The stronger interaction of the AgNP surface with NOM, especially with sulfur and nitrogen groups in the form of thiols and amines, will lead to the displacement of citrate by NOM as suggested by Gunsolus, et al.11 Furthermore, the high molecular weight fraction extracted from a specific NOM source was shown to have stronger interactions with citrate coated AuNP compared to the lighter weight fraction.46,47 Thus, we assume that especially the heavy weight fraction of the NOM would displace the citrate from the AgNP surface, which would be in favour of hypothesis (ii). Whether the sorption of HA to the surface could increase the HS− concentration at the AgNP surface was therefore investigated in additional experiments.
Interaction of HA with HS−
Different concentrations of HS− were mixed with 250 mgHA L−1, followed by a centrifugal separation of the HA for 60 min. The HS− concentration in the supernatant was measured. The HS− associated with the HA, and thus removed by centrifugation, and divided by the total HA concentration (q) was plotted against the free HS− concentration measured in the supernatant (C, Fig. 4). A Freundlich sorption isotherm was fitted to the data (eqn (5)):| q = l × Cn | (5) |
 | ||
| Fig. 4 Adsorbed HS− on HA (q) versus HS− in solution (C) (for both values, the error bars (1σ) were derived from duplicate experiments). Sorption of HS− on HA follows a Freundlich isotherm (solid line, eqn (5)). | ||
The two constants l and n were 0.00404(2) and 0.64(6), respectively (a standard error of 1σ is given in the brackets referring to the last digit). The sorption capacity of HA for metal cations is in the range of 0.1–0.2 μmol mgHA−1,48 which is substantially lower compared to the observed sorption capacity of up to 7 μmol mgHA−1 for HS−. These high (apparent) sorption capacities may be explained by an oxidative loss of HS−, as it was recently shown that HS− can react with HA to form higher oxidized sulfur species.49 However, the methylene blue method is specific to sulfides50 and in the experiments, where the HA was not separated by centrifugation, we did not observe a significant decrease of HS− over the time period of 60 min, and thus, the oxidative loss of HS− can be excluded. Alternatively, a fraction of HS− may have reacted with the metals present in the HA and thus, may have precipitated as metal sulfides and was removed during the centrifugation. Also this can be excluded, as the ICP-OES measurement of the HA revealed only very small amounts of Fe (0.254 μmolFe mgHA−1), which could only explain a loss of 63.5 μmol of HS− or 13% of the observed loss after centrifugation. Therefore, we speculate that due to the lower pKa of HA compared to HS−, the HS− becomes protonated within the Donnan volume to form H2S which may be sorbed to non-polar groups of the HA. The replacement of the citrate with HA and the sorption of HS− to HA may thus bring the sulfide close to the AgNP surface and could explain the increased sulfidation rates observed with increasing HA concentrations.
Microscopic insights into the sulfidation mechanism
To study the mechanism of AgNP sulfidation in more detail, analytical STEM analyses were performed. Samples were collected from partially and fully sulfidized 20 and 100 nm AgNP after specific reaction times (5 and 45 min for 20 nm and 5 and 15 min or 4 h for 100 nm AgNP) in the absence and presence of HA (250 mgHA L−1 for 20 nm and 1000 mgHA L−1 for 100 nm particles, Fig. 5, Table S2†).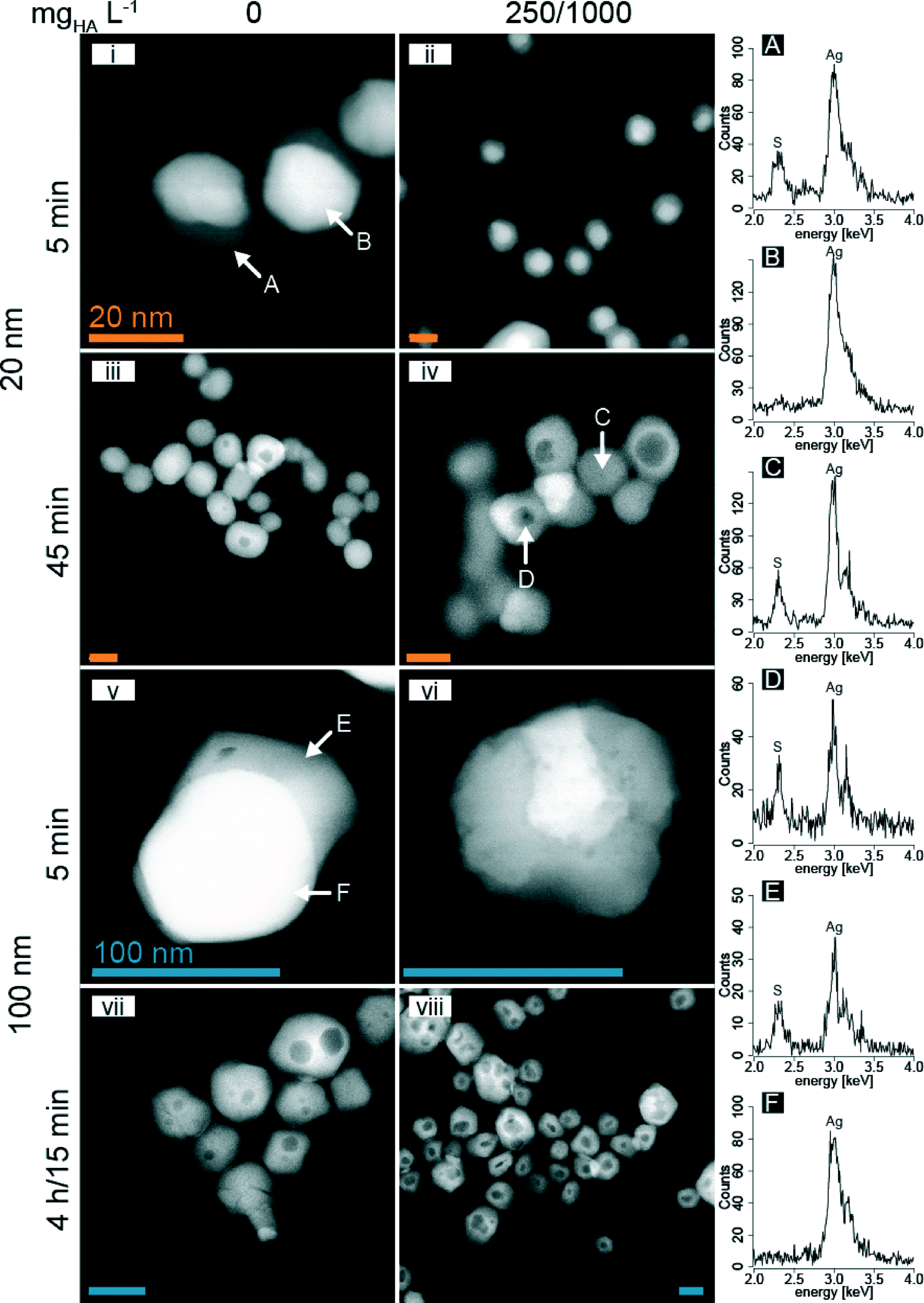 | ||
| Fig. 5 HAADF images of partially to fully sulfidized 20 and 100 nm AgNP in the absence and presence of HA (250 or 1000 mgHA L−1 for 20 and 100 nm AgNP, respectively) after 5, 15 and 45 min or 4 h (vii: 100 nm and 0 mgHA L−1) of reaction time (orange bars are 20 nm (i–iv), blue bars are 100 nm (v–viii)). EDX spectra (right side, A–F) corresponding to specific locations in i, iv, and v are indicated by arrows. | ||
During sulfidation, the 20 nm AgNP aggregated to form larger colloids, as already suggested by the DLS experiments, but the primary particles preserved their shapes and remained spherical. In the absence of HA and after 5 min, the HAADF images of the 20 nm AgNP revealed a bright, spherical area with an adjacent grey sickle-shaped part. The EDX analysis revealed that the bright part corresponded to metallic Ag and the grey areas represented Ag2S (Fig. 5i, A and B). With increasing reaction time, the Ag2S increasingly replaced the metallic Ag resulting in mostly light-grey, spherical particles (Fig. 5iii).
In the presence of 250 mgHA L−1 and after 5 min of reaction time, the metallic Ag (bright areas) was concentrically surrounded by a grey (Ag2S) shell, forming core–shell type structures (Fig. 5ii; the high resolution TEM image is shown in Fig. S8†). After 45 min of reaction time, the contrast of the core–shell structures was reversed and a dark core was often surrounded by a light-grey shell. The EDX analysis of the core and the shell revealed that both parts consisted of Ag2S (Fig. 5iv, C and D).
For the 100 nm AgNP and in the absence of HA, the sulfidation resulted in similar structures as observed for the 20 nm AgNP (Fig. 5v and vii). However, in the presence of 1000 mgHA L−1, the 100 nm AgNP developed more heterogeneous structures than without HA, with a distinct surface structure, probably representing adsorbed HA (Fig. S9†). Furthermore, we observed the reduction of freshly sulfidized AgNP to metallic Ag under the electron beam within a minute (Fig. 5vi; a time sequence over one minute revealing the formation of metallic Ag under the electron beam is provided in the ESI,† Fig. S10). Highly crystalline Ag2S particles are very stable under the electron beam as demonstrated by the lattice fringes in the high-resolution images (HR-TEM).32,34 Therefore, we speculate that the presence of large amounts of HA (1000 mg L−1) interfered with the formation of highly crystalline Ag2S nanoparticles and resulted in poorly crystalline Ag2S which was unstable under the electron beam. For the 100 nm AgNP, larger and in some cases even multiple dark spots were observed within a single particle (Fig. 5vii and viii).
The dark spots in the center of the sulfidized AgNP observed in Fig. 5 could be explained by either different elemental compositions (e.g., lighter elements concentrated in the center) or by a reduced thickness.51 Elemental analysis of the central dark spots revealed the same elemental distribution as for the rim of the particles. Therefore, the contrast observed on the HAADF image can only be explained by a thickness contrast. Additional secondary electron images (SE), probing the topography of the particles, revealed that the sulfidized particles remained spherical (Fig. S11†). Therefore, the central dark spots observed in the HAADF images can only be explained by a central cavity.
The formation of hollow nanoparticles is well known in materials science52–54 and recently has also been described in environmental systems.55 The formation of central voids can be explained by the different diffusion coefficients of two counter-diffusing species across an interfacial layer, which is compensated by the (inward) diffusion of vacancies. When the concentration of the vacancies that migrated into the center of the particle reaches a certain threshold value, the vacancies collapse into the central cavity. This phenomenon is known as the Kirkendall effect52 and has been used to synthesize hollow Ag2Se nanoparticles in organic solvents.53 We observed the formation of Kirkendall voids during the sulfidation of AgNP in oxic water, at room temperature and more prominently in the presence of HA. Furthermore, the 100 nm AgNP also had multiple voids within one particle, most likely due to the polycrystalline nature of larger AgNP. Similar core–shell structures as reported in this study may also have been observed by Impellitteri et al.27 (Fig. 3A and 4A in their publication) for AgNP that were sulfidized in real wastewater samples, indicating that Kirkendall-like core–shell structures may also form in real wastewater. The occurrence of hollow AgNP may therefore be indicative of the release of (engineered) AgNP into the wastewater.
In the Ag(0)–S(−II) system, presumably a thin layer of Ag2S initially forms and Ag diffuses faster through the Ag2S than S, which was previously also reported for bulk Ag(0) with S(0) at elevated temperatures (220 °C).43 These observations are in line with the parabolic rate model which is based on the assumption of diffusion limitation.
Furthermore, the TEM analysis revealed that in the absence of HA, the sulfidation of AgNP mostly started from one side of the particle, whereas in the presence of HA, the sulfidation resulted in more symmetrical core–shell structures. We hypothesize that citrate was preventing the even sulfidation of the AgNP and the replacement of citrate with HA increased the reaction rate as an increased area of the particle surface was available for sulfidation, resulting in the observed core–shell structures. This hypothesis is also supported by the formation of larger and central Kirkendall voids that were generally observed in the presence of HA (20 nm AgNP). The ‘asymmetrical’ sulfidation observed in the absence of HA favors the formation of voids which are either off center or completely lost at the surface of the particles.
The results from the TEM investigations are thus in line with the hypothesis that the replacement of the citrate coating with HA increases the AgNP surface area available for the sulfidation. In addition, the adsorption of HS− within HA brings the HS− close to the AgNP surface and thus facilitates the sulfidation. The combination of both effects eventually led to increased sulfidation rates with increasing HAnorm (Fig. 3). The diffusion of S and Ag along subgrain boundaries29 resulting in the formation of more heterogeneous structures observed for larger, polycrystalline AgNP additionally increases the complexity of the sulfidation pathways of AgNP.
Conclusions
The results of this study demonstrate that HA increases the sulfidation rates of AgNP. The half-life (t1/2, the time when 50% of the total Ag was transformed to Ag2S) of AgNP decreased from 12 to 1 min with an increase of the HA concentration from 0 and 1000 mgHA L−1 for the 20 nm AgNP. Based on the results from the current study, it is likely that the sulfidation of AgNP in the presence of HA is substantially accelerated and completed within one hour or less. The sulfidation of AgNP in the presence of HA initially led to the formation of core (Ag0)–shell (Ag2S) particles that transformed into hollow Ag2S particles via the Kirkendall effect.Furthermore, we found that the sulfidation kinetics is best described by a parabolic rate model, which implies that neither the overall Ag nor the HS− concentration influences the sulfidation rate. Therefore, in the presence of sufficient amounts of HS− to completely sulfidize all AgNP (which should be the case in urban wastewater systems),24,56 the half-life of AgNP can be estimated based on their size and the concentration of the HA in the respective solutions. The sulfidation of polycrystalline AgNP (100 nm) resulted in Ag2S particles that quickly degraded under the electron beam, in contrast to highly crystalline Ag2S generally formed in the absence of organics. This might indicate that the Ag2S formed by sulfidation of Ag in the presence of HA is more reactive than the Ag2S formed in the HA-free systems. Further work is required to evaluate this hypothesis and its potential implications for the assessment of the fate and impact of Ag in environmental systems.
Acknowledgements
We acknowledge the Electron Microscopy Centers at ETH Zurich (EMEZ, Zurich, Switzerland) and at Empa (Swiss Federal Institute for Materials Science and Technology, Duebendorf, Switzerland) for providing access to the microscopes. The staff at the Swiss–Norwegian Beamline at the European Synchrotron Radiation Facility (SNBL, ESRF, Grenoble, France) are acknowledged for the allocation of beamtime. We thank Dipanjan Banerjee for support at the Dutch Belgian Beamline (DUBBLE) at ESRF. This work is part of the project “Behavior of silver nanoparticles in a wastewater treatment plant” within the Swiss National Research Program NRP 64 “Opportunities and Risks of Nanomaterials”. Additionally, we would like to thank Brian Sinnet for support in the laboratories at Eawag.References
- I. Woodrow Wilson, Nanotechnology Consumer Products, Informa Healthcare, 2015 Search PubMed.
- A. D. Russell and W. B. Hugo, Prog. Med. Chem., 1994, 31, 351–370 CrossRef CAS PubMed.
- H. T. Ratte, Environ. Toxicol. Chem., 1999, 18, 89–108 CrossRef CAS.
- J. Farkas, H. Peter, P. Christian, J. A. Gallego Urrea, M. Hassellov, J. Tuoriniemi, S. Gustafsson, E. Olsson, K. Hylland and K. V. Thomas, Environ. Int., 2011, 37, 1057–1062 CrossRef CAS PubMed.
- L. Geranio, M. Heuberger and B. Nowack, Environ. Sci. Technol., 2009, 43, 8113–8118 CrossRef CAS PubMed.
- T. Benn, B. Cavanagh, K. Hristovski, J. D. Posner and P. Westerhoff, J. Environ. Qual., 2010, 39, 1875–1882 CrossRef CAS PubMed.
- F. Gottschalk, T. Sonderer, R. W. Scholz and B. Nowack, Environ. Sci. Technol., 2009, 43, 9216–9222 CrossRef CAS PubMed.
- T. Y. Sun, F. Gottschalk, K. Hungerbühler and B. Nowack, Environ. Pollut., 2014, 185, 69–76 CrossRef CAS PubMed.
- R. Kaegi, A. Voegelin, B. Sinnet, S. Zuleeg, H. Hagendorfer, M. Burkhardt and H. Siegrist, Environ. Sci. Technol., 2011, 45, 3902–3908 CrossRef CAS PubMed.
- Y. Yin, M. Shen, Z. Tan, S. Yu, J. Liu and G. Jiang, Environ. Sci. Technol., 2015, 49, 6581–6589 CrossRef CAS PubMed.
- I. L. Gunsolus, M. P. Mousavi, K. Hussein, P. Bühlmann and C. L. Haynes, Environ. Sci. Technol., 2015, 49, 8078–8086 CrossRef CAS PubMed.
- V. Manoharan, A. Ravindran and C. H. Anjali, Cell Biochem. Biophys., 2014, 68, 127–131 CrossRef CAS PubMed.
- N. Akaighe, R. I. Maccuspie, D. A. Navarro, D. S. Aga, S. Banerjee, M. Sohn and V. K. Sharma, Environ. Sci. Technol., 2011, 45, 3895–3901 CrossRef CAS PubMed.
- F. Maurer, I. Christl, M. Hoffmann and R. Kretzschmar, Environ. Sci. Technol., 2012, 46, 8808–8816 CrossRef CAS PubMed.
- T. S. Peretyazhko, Q. Zhang and V. L. Colvin, Environ. Sci. Technol., 2014, 48, 11954–11961 CrossRef CAS PubMed.
- R. Ma, C. Levard, S. M. Marinakos, Y. Cheng, J. Liu, F. M. Michel, G. E. Brown and G. V. Lowry, Environ. Sci. Technol., 2012, 46, 752–759 CrossRef CAS PubMed.
- W. Zhang, Y. Yao, N. Sullivan and Y. Chen, Environ. Sci. Technol., 2011, 45, 4422–4428 CrossRef CAS PubMed.
- J. Liu and R. H. Hurt, Environ. Sci. Technol., 2010, 44, 2169–2175 CrossRef CAS PubMed.
- C. Levard, S. Mitra, T. Yang, A. D. Jew, A. R. Badireddy, G. V. Lowry and G. E. Brown, Jr., Environ. Sci. Technol., 2013, 47, 5738–5745 CrossRef CAS PubMed.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, Fla, 95 edn, 2014 Search PubMed.
- J. Liu, K. G. Pennell and R. H. Hurt, Environ. Sci. Technol., 2011, 45, 7345–7353 CrossRef CAS PubMed.
- R. Kaegi, A. Voegelin, C. Ort, B. Sinnet, B. Thalmann, J. Krismer, H. Hagendorfer, M. Elumelu and E. Mueller, Water Res., 2013, 47, 3866–3877 CrossRef CAS PubMed.
- G. Brunetti, E. Donner, G. Laera, R. Sekine, K. G. Scheckel, M. Khaksar, K. Vasilev, G. De Mastro and E. Lombi, Water Res., 2015, 77, 72–84 CrossRef CAS PubMed.
- P. H. Nielsen, K. Raunkjær and T. Hvitved-Jacobsen, Water Sci. Technol., 1998, 37, 97–104 CrossRef CAS.
- R. Ma, C. Levard, J. D. Judy, J. M. Unrine, M. Durenkamp, B. Martin, B. Jefferson and G. V. Lowry, Environ. Sci. Technol., 2014, 48, 104–112 CrossRef CAS PubMed.
- C. Doolette, M. McLaughlin, J. Kirby, D. Batstone, H. Harris, H. Ge and G. Cornelis, Chem. Cent. J., 2013, 7, 1–18 CrossRef PubMed.
- C. A. Impellitteri, S. Harmon, R. G. Silva, B. W. Miller, K. G. Scheckel, T. P. Luxton, D. Schupp and S. Panguluri, Water Res., 2013, 47, 3878–3886 CrossRef CAS PubMed.
- G. V. Lowry, B. P. Espinasse, A. R. Badireddy, C. J. Richardson, B. C. Reinsch, L. D. Bryant, A. J. Bone, A. Deonarine, S. Chae, M. Therezien, B. P. Colman, H. Hsu-Kim, E. S. Bernhardt, C. W. Matson and M. R. Wiesner, Environ. Sci. Technol., 2012, 46, 7027–7036 CrossRef CAS PubMed.
- B. Thalmann, A. Voegelin, B. Sinnet, E. Morgenroth and R. Kaegi, Environ. Sci. Technol., 2014, 48, 4885–4892 CrossRef CAS PubMed.
- E. Navarro, F. Piccapietra, B. Wagner, F. Marconi, R. Kaegi, N. Odzak, L. Sigg and R. Behra, Environ. Sci. Technol., 2008, 42, 8959–8964 CrossRef CAS PubMed.
- B. C. Reinsch, C. Levard, Z. Li, R. Ma, A. Wise, K. B. Gregory, G. E. Brown, Jr. and G. V. Lowry, Environ. Sci. Technol., 2012, 46, 6992–7000 CrossRef CAS PubMed.
- B. Thalmann, A. Voegelin, U. von Gunten, R. Behra, E. Morgenroth and R. Kaegi, Environ. Sci. Technol., 2015, 49, 10911–10919 CrossRef CAS PubMed.
- V. Réveillé, L. Mansuy, É. Jardé and É. Garnier-Sillam, Org. Geochem., 2003, 34, 615–627 CrossRef.
- C. Levard, B. C. Reinsch, F. M. Michel, C. Oumahi, G. V. Lowry and G. E. Brown, Environ. Sci. Technol., 2011, 45, 5260–5266 CrossRef CAS PubMed.
- R. D. Kent, J. G. Oser and P. J. Vikesland, Environ. Sci. Technol., 2014, 48, 8564–8572 CrossRef CAS PubMed.
- C. Levard, E. M. Hotze, B. P. Colman, A. L. Dale, L. Truong, X. Y. Yang, A. J. Bone, G. E. Brown, Jr., R. L. Tanguay, R. T. Di Giulio, E. S. Bernhardt, J. N. Meyer, M. R. Wiesner and G. V. Lowry, Environ. Sci. Technol., 2013, 47, 13440–13448 CrossRef CAS PubMed.
- A. D. Eaton, M. A. H. Franson, A. P. H. Association, A. W. W. Association and W. E. Federation, Standard Methods for the Examination of Water & Wastewater, American Public Health Association, 2005 Search PubMed.
- J. Cenens and R. A. Schoonheydt, Clays Clay Miner., 1988, 36, 214–224 CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- D. Mavrocordatos, C.-P. Lienemann and D. Perret, Microchim. Acta, 1994, 117, 39–47 CrossRef CAS.
- W. Jander, Z. Anorg. Allg. Chem., 1927, 163, 1–30 CrossRef CAS.
- I. Bartkowicz and A. Stokłosa, Solid State Ionics, 1987, 24, 45–49 CrossRef.
- K. N. Strafford, Metall. Rev., 1969, 14, 153–174 CAS.
- O. Levenspiel, Ind. Eng. Chem. Res., 1999, 38, 4140–4143 CrossRef CAS.
- R. C. Team, R: A Language and Environment for Statistical Computing, R Foundation for Statictical Computing, 2012 Search PubMed.
- S. M. Louie, E. R. Spielman-Sun, M. J. Small, R. D. Tilton and G. V. Lowry, Environ. Sci. Technol., 2015, 49, 2188–2198 CrossRef CAS PubMed.
- S. M. Louie, R. D. Tilton and G. V. Lowry, Environ. Sci. Technol., 2013, 47, 4245–4254 CrossRef CAS PubMed.
- H. Kerndorff and M. Schnitzer, Geochim. Cosmochim. Acta, 1980, 44, 1701–1708 CrossRef CAS.
- Z.-G. Yu, S. Peiffer, J. Göttlicher and K.-H. Knorr, Environ. Sci. Technol., 2015, 49, 5441–5449 CrossRef CAS PubMed.
- J. Small and H. Hintelmann, Anal. Bioanal. Chem., 2007, 387, 2881–2886 CrossRef CAS PubMed.
- S. Utsunomiya and R. C. Ewing, Environ. Sci. Technol., 2003, 37, 786–791 CrossRef CAS PubMed.
- W. Wang, M. Dahl and Y. Yin, Chem. Mater., 2013, 25, 1179–1189 CrossRef CAS.
- Y. Tang and M. Ouyang, Nat. Mater., 2007, 6, 754–759 CrossRef CAS PubMed.
- B. D. Anderson and J. B. Tracy, Nanoscale, 2014, 6, 12195–12216 RSC.
- F.-A. Weber, A. Voegelin, R. Kaegi and R. Kretzschmar, Nat. Geosci., 2009, 2, 267–271 CrossRef CAS.
- F. Gottschalk, T. Sonderer, R. W. Scholz and B. Nowack, Environ. Toxicol. Chem., 2010, 29, 1036–1048 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional supporting information is provided for the AgNP characterization (Table S1, Fig. S1 and Fig. S2), XANES (Fig. S3) and EXAFS (Fig. S4) spectra, comparison of LCF derived Ag fractions from XANES and EXAFS (Fig. S5), fits of the parabolic rate model to the data (Fig. S6), AgNP size measurement over time (Fig. S7), HR-TEM image of partially sulfidized 20 nm AgNP (Fig. S8), SE image of 100 nm AgNP (Fig. S9), TEM time sequence of 100 nm AgNP (Fig. S10), SE and HAADF image of 100 nm AgNP (Fig. S11), run table (Table S2), LCF results (Table S3) and rate coefficients (Table S4). See DOI: 10.1039/c5en00209e |
| This journal is © The Royal Society of Chemistry 2016 |