
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ni-based bimetallic heterogeneous catalysts for energy and environmental applications
Sudipta
De
a,
Jiaguang
Zhang
a,
Rafael
Luque
b and
Ning
Yan
*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, 117585, Singapore. E-mail: ning.yan@nus.edu.sg
bDepartamento de Quimica Organica, Universidad de Cordoba, Campus de Rabanales, Edificio Marie Curie (C-3), Ctra Nnal IV, Km. 396, E-14014, Cordoba, Spain
First published on 15th September 2016
Abstract
Bimetallic catalysts have attracted extensive attention for a wide range of applications in energy production and environmental remediation due to their tunable chemical/physical properties. These properties are mainly governed by a number of parameters such as compositions of the bimetallic systems, their preparation method, and their morphostructure. In this regard, numerous efforts have been made to develop “designer” bimetallic catalysts with specific nanostructures and surface properties as a result of recent advances in the area of materials chemistry. The present review highlights a detailed overview of the development of nickel-based bimetallic catalysts for energy and environmental applications. Starting from a materials science perspective in order to obtain controlled morphologies and surface properties, with a focus on the fundamental understanding of these bimetallic systems to make a correlation with their catalytic behaviors, a detailed account is provided on the utilization of these systems in the catalytic reactions related to energy production and environmental remediation. We include the entire library of nickel-based bimetallic catalysts for both chemical and electrochemical processes such as catalytic reforming, dehydrogenation, hydrogenation, electrocatalysis and many other reactions.
 Sudipta De | Sudipta De received his BSc (2008) and MSc (2010) degrees in chemistry from the University of Calcutta. He obtained a PhD from the University of Delhi in 2015. After a short postdoctoral stay with Prof. Rafael Luque at Universidad de Cordoba, Spain, he joined Prof. Ning Yan's group at the National University of Singapore as a research fellow. His current research is focused on the preparation of single atom catalysts and their application in diverse catalytic processes. |
 Jiaguang Zhang | Jiaguang Zhang obtained his BSc and PhD degrees from Peking University in 2009 and 2014, respectively. Then he joined the National University of Singapore as a research fellow in Prof. Ning Yan's group. His research focuses on the catalytic valorization of biomass including lignin, cellulose and chitin into useful chemicals and materials. |
 Rafael Luque | Rafael Luque has extensively published in the areas of (nano)materials science, heterogeneous (nano)catalysis, microwave and flow chemistry, and biofuels and green chemical methods in synthetic organic chemistry, filed a few patent applications and edited several books as well as numerous contributions to book chapters and invited guest, keynote and plenary lectures in scientific events worldwide. Among the recent awards, Rafael has received the TR35 Spain from Technology Review and MIT as one of the top 10 young entrepreneurs in Spain (2012) and the RSC Environment, Sustainability and Energy Early Career Award (2013) from the Royal Society of Chemistry UK. |
 Ning Yan | Ning Yan obtained his bachelor and PhD degrees from Peking University in 2004 and 2009, respectively. Thereafter, he worked as a Marie-Curie research Fellow with Prof. Paul Dyson at École Polytechnique Fédérale de Lausanne, Switzerland. He joined the National University of Singapore (NUS) as an Assistant Professor and established the Lab of Green Catalysis in 2012. Very recently, he won the NUS Young Investigator Award and the G2C2 Young Researcher Award. |
Broader contextTo address the increasing energy demand while mitigating environmental concerns, numerous research efforts have been devoted to finding the most sustainable routes of energy production. Catalysis can offer attractive solutions to such processes, with the possibility of designing highly effective advanced catalytic systems as the basis of future industrial implementation. Bimetallic catalysts emerged as materials of a new category, which often show electronic and chemical properties different from their monometallic counterparts, thus offering an opportunity to design new catalysts with enhanced selectivity, activity, and stability. Since the infancy of bimetallic catalysts in the 1960's, an enormous number of catalysts have been explored, most of which were based on having noble metals as the main components. However, the industrial application of these noble metal catalysts is limited by their exceptionally high prices and low availability, which has turned the attention towards more abundant transition metal-based catalysts. Nickel is the most widely used element among the transition metal-based catalysts and has the highest ability to form bimetallic systems with other metals. As a result, the library of bimetallic Ni catalysts has been enriched very rapidly in the last decade. This review provides an overview of the recent progress in the design of bimetallic nickel-based catalysts for use in energy production and environmental remediation. Design aspects of the catalysts and the fundamental understanding of their catalytic properties are also critically discussed. |
1. Introduction
Population increase, urbanization, and rising living standards have rapidly increased global energy consumption and environmental burdens in the last half century. Energy consumption is expected to continue to increase dramatically in the coming years along with its associated environmental issues. Although there is no universal solution to solve all energy and environment-related problems,1 catalysis certainly plays a critical role in the design of efficient processes and systems able to maximize the value of starting materials while minimizing waste generation and energy requirements. Metal-based catalysts, in this regard, have been of immense importance in a wide range of diverse catalytic applications with high efficiency. However, fine-tuning of the structure and the properties of these catalysts is highly desirable to meet the stringent requirements in energy and environmental applications.Nickel is one of the most widely used elements in metal based catalysts (see Fig. 1). It is the fourth most abundant transitional metal on earth after Fe, Ti and Zr. It currently costs 3.8 USD per lb, which is only 1/5000 of the price of gold. In fact, nickel has a long history in modern catalysis – its first application as a hydrogenation catalyst dates back to the 19th century which led to the Nobel Prize in Chemistry in 1912. Afterwards Ni was found to be active in a number of processes, in particular in hydrogenation and reforming reactions. Despite the huge success and wide applications of the Ni catalyst in industry, single component Ni catalysts are not able to meet the activity, selectivity, and stability requirements in many emerging applications related to energy and environments.
 | ||
| Fig. 1 Fact sheet showing the historical background, physical properties and uses of nickel. | ||
Bimetallic catalysts, which often have electronic and chemical properties that are distinct from those of their parent metals, may show enhanced performances. Since the pioneering and systematic work by Exxon Research and Engineering in the early 1960s, the library of bimetallic catalysts has been significantly enriched, particularly in the past two decades. Among all bimetallic catalysts, platinum-group metals are the most studied catalyst components.2,3 However, high cost and low availability limit their applications in large scale processes. Therefore, attention turned towards nickel, a low-cost alternative, which has similar electronic properties and can perform many of the same elementary reactions as palladium or platinum.4–6 In fact, Ni is known for its high alloying efficiency with all noble metals as well as many transition metals in different mass ratios, which makes it easier to develop a wide range of composition-dependent bimetallic Ni systems for diversified catalytic applications. Recently, there have been many reports on the facile synthesis of bimetallic Ni systems, which have shown the potential to replace expensive noble metal catalysts in terms of catalytic activity and stability.
After a detailed survey of recent publications on bimetallic Ni, we found that the majority of them were focused on catalytic reactions relating to energy, and to a lower extent the environment, which reflects the growing concern of bimetallic heterogeneous catalysis in these domains. This contribution aims to provide a critical overview of the entire library of Ni-based heterogeneous bimetallic catalysts with their recent synthesis protocols in view of their applications in a wide range of chemical processes. A rationalized approach is provided to identify suitable metal combinations that may be able to provide improved activities in selected applications compared to conventional and/or presently reported systems. Different energy-related catalytic processes will be discussed where the influence of modification of the Ni catalysts by other metals and the relation to their catalytic properties, will be introduced. Additionally, a brief discussion on the fundamental approach using classical theories of chemistry (such as the d-band theory) to understand the origin of the improved efficiency and stability of the bimetallic systems will also be provided.
Recently, several reviews based on homogeneous Ni-based complexes were published focusing mainly on the C–C coupling reactions.7,8 Two excellent reviews based on heterogeneous Ni-based catalysts were published focusing exclusively on reforming reactions.9,10 Nevertheless, to the best of our knowledge, there is currently no review focused on bimetallic Ni-based catalysts that provides a detailed discussion on their energy and environmental applications. We hope that this contribution will be a good addition to the existing literature, and will provide useful information for future research.
2. Preparation of bimetallic Ni catalysts
A number of factors are responsible for the structure of bimetallic catalysts (see Fig. 2). At present, there are many well established methods for preparing bimetallic catalysts (either unsupported colloidal NPs or supported bimetallic NPs).2,3 The choice of method often depends on the desired surface and the bulk structure of the catalyst. For instance, the co-impregnation process is mainly used to produce supported bimetallic or alloy type materials. On the other hand, the sequential impregnation method is often adopted to obtain core–shell type materials, where a less active metal (generally a 3d transition metal) core is prepared first and then the active metal (generally a noble metal) is deposited onto it. Wet impregnation methods are widely applicable for the syntheses of bimetallic catalysts with well-controlled shape, size, and composition.11,12 Various wet-chemical schemes have been applied to synthesize bimetallic NPs. They can be classified into two categories according to the formation mechanisms: seed-mediated growth and one-pot co-reduction. The two strategies have similar fundamental principles. In the seed-mediated growth process, the metal seeds of the comparatively inactive metal are first prepared and then dispersed in a solution of active metal precursors. The controlled reduction of the second metal forms uniform layers over the seeds and a core–shell structure is obtained. In the one-pot co-reduction method, two metallic precursors are added at the same time. In this case, the reduction potentials of the metals play a key role in determining the final architecture. Two possibilities arise here: (1) metals with a similar reduction potential are reduced simultaneously and form an alloyed structure, and (2) metals with different reduction potentials are reduced in a successive manner and form a core–shell structure. However, there is no straightforward relationship between reduction kinetics and reduction potential. The reduction kinetics also depends on the synthesis conditions applied. For example, the use of surfactants, stronger reducing agents (such as NaBH4) or high temperature solvothermal processes can simultaneously reduce metals with different reduction potentials resulting in an alloyed structure.13–16 Alongside the wet impregnation processes, much attention has been paid to colloidal chemistry to synthesize unsupported NPs with controlled size, shape, and stability.17 While colloidal chemistry is highly applicable towards the syntheses of noble metal bimetallic systems, its application in 3d transition metals is less studied.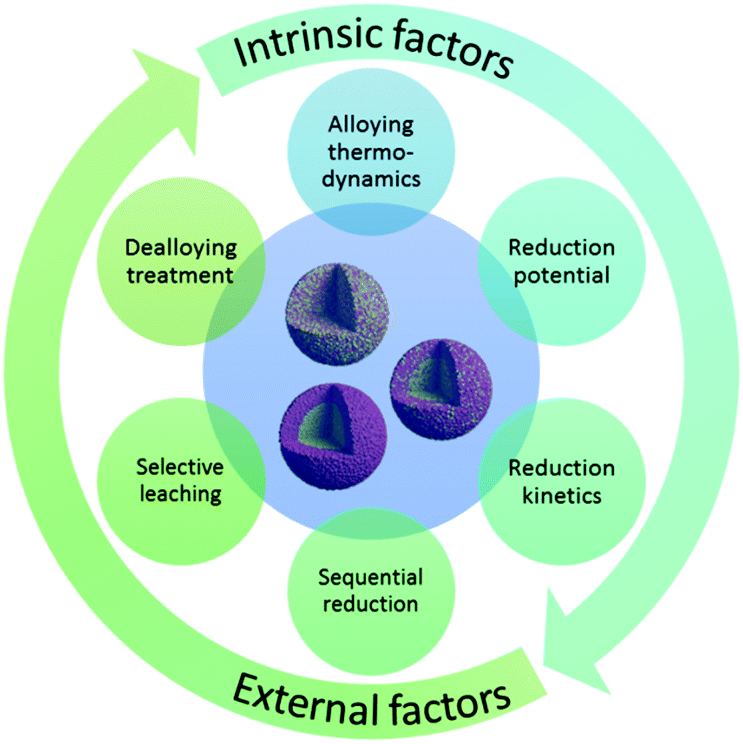 | ||
| Fig. 2 Factors responsible for the formation of different bimetallic structures. | ||
Apart from the preparation methods, another factor that determines the structure of a bimetallic system is the relative position of two metals in the periodic table. A solid solution or miscible system is formed when the two metals are very close in the periodic table. For example, nickel is miscible with Co, Cu, Rh, Pd, and Ir at any proportions as shown in Fig. 3, and therefore can easily form alloys. The metals which are further from nickel generally form intermetallic compounds. For example, Pt forms different intermetallic compounds with Ni at different specific ratios, among which Pt3Ni is the most stable compound and is very well known for its electrocatalytic applications. Although the predicted structure can be derived from the experimental data presented in the periodic table, it is not necessary that the final structure should be similar to that of the predicted one. Different variable structures can also be obtained by using a controlled preparation method. The reduction potential of the metal plays a crucial role in the formation of a definite structure since in most cases the bimetallic catalysts are prepared by reducing their salt precursors. In fact, multiple factors need to be considered when predicting the relationship between the predicted and experimentally obtained structure of a bimetallic system.
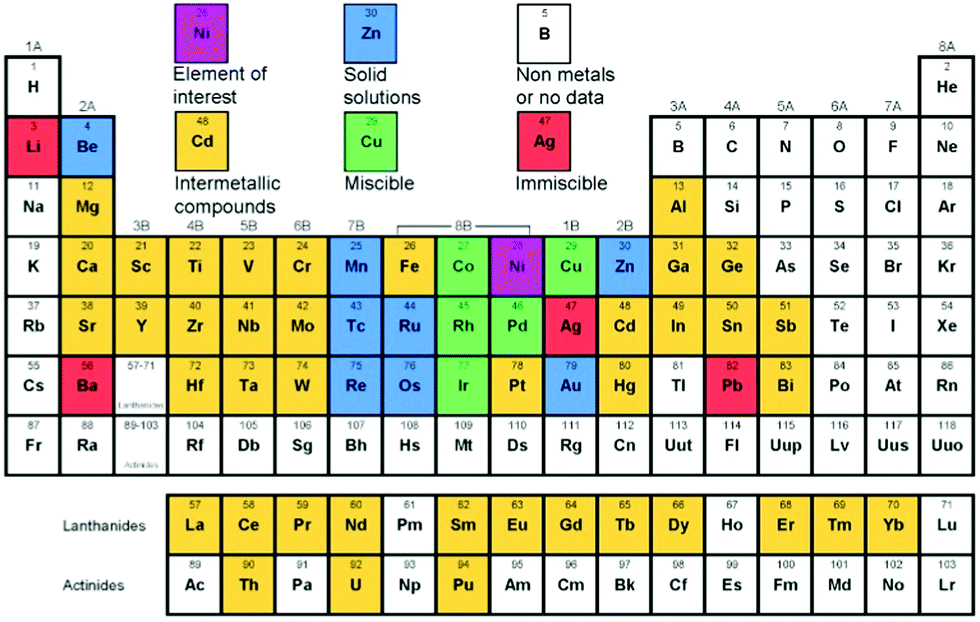 | ||
| Fig. 3 Possibility of alloy formation of Ni with other elements in the periodic table (figure generated from data in ref. 18).18 | ||
Structural modification largely enhances the capability of tuning the catalytic performance, therefore it is important to have the knowledge of the surface structure and properties of the catalyst to derive the relationship between their structural features and catalytic activity. In the past decade, significant efforts have been made to understand the surface properties of bimetallic systems using advanced characterization techniques (e.g. Auger electron spectroscopy (AES), X-ray photoelectron spectroscopy (XPS), extended X-ray absorption fine structure (EXAFS) spectroscopy, low-energy electron diffraction (LEED), etc.) as well as theoretical calculations (e.g. density functional theory (DFT)).2,3,19,20 To reveal the origin of the novel catalytic properties, bimetallic surfaces have also gained a considerable amount of interest in surface science research.21–23 Since the characterization methods for bimetallic systems have already been discussed in some excellent reviews,2,3 we would like to pay our attention to discussing how different structures of bimetallic Ni catalysts are obtained depending on their synthesis method.
2.1 Core–shell structure
In principle, bimetallic core–shell structures can be obtained either by seed-mediated growth or by one-pot co-reduction of metallic precursors. In particular, for the bimetallic Ni@noble metal core–shell structure, Ni serves as a core and the noble metal serves as a shell. This kind of structure is generally prepared using a seed-mediated growth process, where Ni cores are prepared by reducing their salt precursor prior to the deposition of the noble metal on their surface. In most cases, capping ligands are used in this step to prevent aggregation.24,25 It should be noted that the one-pot co-reduction method cannot form this structure because of the higher reduction potential of noble metals than that of Ni. In this case, noble metals will be reduced first and Ni will be deposited subsequently as a shell over the noble metal core.The reverse structure, i.e. noble metal@Ni core–shell, can be obtained in a one-pot sequential reduction. For example, the preparation of Au@Ni core–shell NPs with varying Ni shell thicknesses was reported by mixing both metal precursors in one-pot in the presence of oleylamine and 1-octadecene.26 The reduction of the Au precursor to Au NPs was achieved by using NaBH4 as the reducing agent at room temperature. At an increased temperature (210 °C), the as-formed Au seeds acted as the nucleation sites on which nickel ions were reduced. The different shell thicknesses could be controlled by using different ratios of two precursors. Because of the higher electron density, noble metal cores sometimes induce the reduction of Ni,27 which is termed as noble-metal induced reduction where no external reducing agent is required.
The core-multishell is a more complex architecture in this category, which is well known for its good optical and electronic properties.29 More precise kinetic control over the reaction is required to achieve the structure. Recently, well-shaped Pd–Ni–Pt core–sandwich–shell NPs were synthesized using cubic and octahedral Pd substrates in the aqueous phase at low temperature with a cationic surfactant as the capping agent and hydrazine as the reducing agent.28 The combination of shaped Pd substrates and mild reduction conditions directed the overgrowth of Ni and Pt in an oriented, layer-by-layer fashion. Scheme 1 demonstrates how Pd cubes function as shaped crystal substrates to catalyze and direct the oriented overgrowth of Ni. Pt ions were added after the Ni overgrowth to “trap” the metallic Ni phase and complete the layer-by-layer synthesis of the shaped ternary metal NPs.
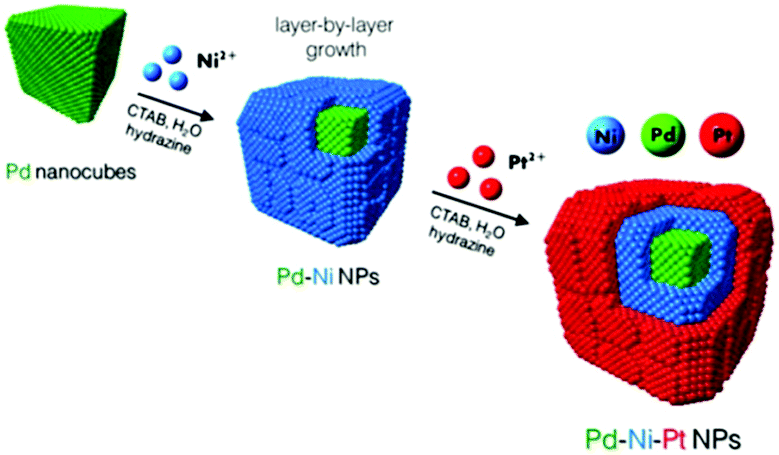 | ||
| Scheme 1 Schematic illustration for the synthesis of cubic Pd–Ni–Pt core–sandwich–shell NPs.28 (Copyright 2014 American Chemical Society.) | ||
Post-modification of an alloy precursor through dealloying and surface oxidation is another way to prepare a core–shell structure. This method was recently applied by the Strasser group to prepare IrNi@IrOx core–shell NPs from IrNix precursor alloys.30,31 Ni rich bimetallic Ir–Ni NPs were first prepared using a conventional polyol method involving 1,2-tetradecanediol as the reducing agent and oleylamine along with oleic acid as the capping ligands. The hybrid IrNi@IrOx core–shell materials were prepared in two ways (Fig. 4). In the first approach, the IrNix NP precursor alloys (PA-IrNix) were electrochemically dealloyed to form metallic core–shell NPs (“D-IrNix”) with an Ir enriched surface, and subsequently they were selectively surface oxidized to form “SO-IrNi@IrOx” metal oxide core–shell NPs. Alternatively, the dealloying and oxidation could be performed directly in one step to obtain DO-IrNi@IrOx.
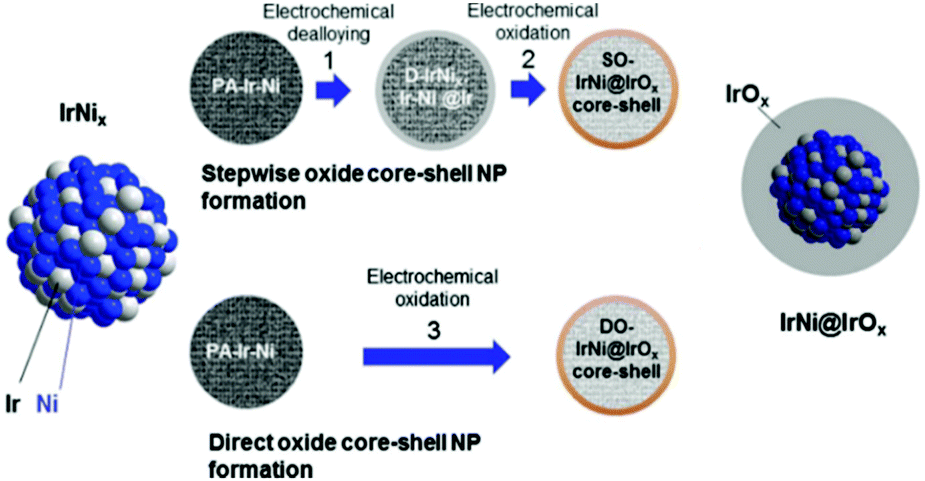 | ||
| Fig. 4 Synthetic protocol for the preparation of the SO-IrNi@IrOx and DO-IrNi@IrOx hybrid core–shell NP catalysts. Precursor IrNi alloys (“PA-IrNi”, and alloy scheme on left, blue: Ni, and grey: Ir) are stepwise (SO) or directly (DO) dealloyed and surface oxidized. “D-IrNix” denotes the dealloyed stage.30 (Copyright 2014 Royal Society of Chemistry.) | ||
2.2 Alloyed structure
Alloy systems are formed when two metal atoms have a homogeneous distribution in one particle. Some metals are thermodynamically more stable when mixing together, while for others the reaction kinetics must be rigidly controlled to produce an alloyed structure. One approach is to use strong reducing agents that reduce all metal precursors simultaneously to form a homogeneous mixture of metals. For example, bimetallic Ni–Fe alloy NPs could be formed using NaBH4 as the reducing agent to reduce Ni2+ and Fe2+ ions in an aqueous solution, containing cetyltrimethylammonium chloride (CTAB) as the surfactant, although these two metals cannot be mixed with a random ratio thermodynamically.13,32The appropriate selection of surfactants or counterions adjusts the redox potentials of metals through specific adsorption or coordination, leading to the simultaneous reduction of different metal ions.33 Surfactants can also play a key role in directing the reaction and crystal growth pathways in yielding particles with a desired geometry. For example, (111) surface-dominant Pt3Ni alloy nanocrystals – the best-known catalyst for the oxygen reduction reaction (ORR) – can be prepared by reducing the metal precursors using CO, in the presence of oleylamine (OAm) and oleic acid (OA) as the capping ligands.34,35
Well-faceted nanocrystal alloys were also reported to form without any capping agents. Carpenter et al. reported the solvothermal synthesis of ORR active Ni–Pt alloy catalysts using a mild reducing agent, N,N-dimethylformamide, which also served as the solvent.15 The effect of temperature on the reduction kinetics of the metal precursors was studied. It was observed that Ni(acac)2 was more difficult to reduce than Pt(acac)2 and did not react up to 150 °C. When the temperature was further increased to 200 °C, 90% Ni reduction was observed. Another controlled reaction with only the Ni precursor at the same temperature showed merely 42% Ni reduction, suggesting that the presence of Pt and/or Pt(acac)2 enhances the deposition of Ni. The converse also appeared to hold true because the apparent reaction onset temperature of the mixed precursor solution (115 °C) was lower than that of the Pt(acac)2-only solution (137 °C). These observations suggest that the free energy of formation of the Ni–Pt alloy NPs from the reaction mixture is more negative than the free energy of formation of platinum NPs from the same solution. Another probable reason could be that under the reaction conditions Pt proto-particles of only a few atoms were formed initially and acted as seed crystals for subsequent Ni and Pt3Ni deposition, which was also supported by the previous finding by Deivaraj et al. that the reduction of Ni ions by hydrazine at room temperature requires the presence of Pt nuclei.36 This method was recently extended to prepare trimetallic Pt–Ni–Co alloy NPs at 130 °C.37
Alongside the wet chemistry methods, gas phase synthesis has also been reported for bimetallic Ni alloys.38 As a “bottom-up” approach, the gas phase method is more complex since it starts from atomic-level precursors and needs better control over nucleation and growth of nanocrystals. The advantage of this method is that it does not require any reducing agent or surfactant. Lin and Sankaran reported a plasma-assisted scalable method for the synthesis of NiCu alloys from the vapors of organometallic precursors, bis(cyclopentadienyl)nickel [Ni(Cp)2] and copper acetylacetonate [Cu(acac)2].38 By carefully combining precursor vapors and varying the flow rates, a wide range of compositionally controlled alloyed NPs (less than 5 nm) with narrow size distributions were obtained.
2.3 Porous structure
Generally, bimetallic alloys form crystalline particles with very low surface areas, which is a limitation for applications in catalytic processes. The surface area is one of the crucial factors for a good catalytic performance. As such, porous structured alloys – which have the advantages of high surface area, high gas permeability, high mass diffusion ability and low density – are more promising in catalytic applications than their solid counterparts. In 1927, Murray Raney produced porous Ni (RANEY® Ni) by selective leaching of a block of Ni–Al alloy (NiAl3 and Ni2Al3) with concentrated sodium hydroxide;39 this has been used as a heterogeneous catalyst for more than 80 years due to its low cost and high catalytic activity. After this report, chemical dealloying became a widely accepted route to develop many porous alloys and is still used in many applications. Wang et al. developed a facile chemical dealloying process to produce porous NPs using nanocrystalline alloys as precursors.40 The non-noble metal components were selectively dissolved with an excess amount of concentrated nitric acid to obtain a nanoporous residue. This general pathway could be applied to any nanocrystalline alloys of noble and non-noble metals (such as Ni–Pt, Ni–Rh) to produce the corresponding nanoporous NPs with increased surface areas and narrow pore-size distributions.Recently, an electrochemical dealloying technique was reported using an improved and more selective process for leaching of an alloy precursor material to form porous alloys. The dealloying mechanism is based on the selective dissolution of the less-noble component from a binary or multicomponent solid solution at a potential where the remaining more-noble component is free to diffuse along the surface at which 3D porosity evolves. The process intrinsically forms a core–shell nanoporous structure where the surface is passivated by the more-noble component and the interior of the ligaments maintains a significant residual fraction of the less-noble component. Chen et al. reported a nanoporous PdNi (np-PdNi) bimetallic catalyst fabricated by electrochemically dealloying a Pd20Ni80 alloy in an acid solution at a low applied potential.41 The key advantage of this process is that the residual Ni in the nanoporous alloy could be easily controlled by tuning the dealloying potentials. The electrochemical dealloying was also applied to make nanoporous Ni–Pt alloy NPs with a hierarchical structure and a high surface area.42
Apart from the dealloying process, templating pathways can also be used to generate a porous structure. For example, porous Ni@Pt core–shell nanotubes were prepared via the electrodeposition method using ZnO nanorods as templates.43Scheme 2 describes the synthetic pathway for well-defined porous Ni@Pt core–shell nanotube arrays. ZnO nanorod arrays were first synthesized on conductive substrates utilized as templates. Then, the electrodeposition of Ni layers was carried out on the surfaces of the ZnO nanorods to prepare ZnO@Ni core–shell nanorod arrays, and Pt thin layers were further deposited on the surfaces of the Ni shells to prepare ZnO@Ni@Pt triple-layered core–shell nanorod arrays. Finally, porous Ni@Pt core–shell nanotube arrays were synthesized from the ZnO@Ni@Pt triple-layered core–shell nanorod arrays by dissolving ZnO in a weak alkali solution. This method is facile and suitable for large-scale and low-cost production under mild conditions in the absence of organic surfactants.
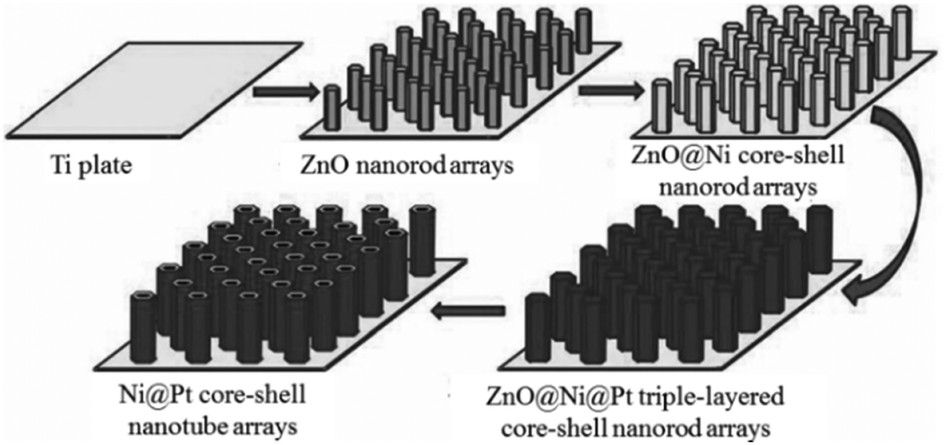 | ||
| Scheme 2 Schematic illustration of the synthesis of Ni@Pt core–shell nanotube arrays.43 (Copyright 2012 Wiley-VCH.) | ||
An analogous method was reported to prepare a Ni–Pt framework structure using a metal–organic framework as the template (Fig. 5).44 In the first step, a Ni-enriched truncated octahedral Ni–Pt alloy was prepared through a surfactant-assisted solvothermal method, which was then dispersed in N,N-dimethylformamide containing an appropriate amount of dihydroxyterephthalic acid and autoclaved at 110 °C for 12 h to form a Ni–Pt frame@Ni-MOF. Finally, the Ni–Pt frame@Ni-MOF was dispersed in dilute acetic acid to decompose the Ni-MOF, resulting in the formation of a bare Ni–Pt frame. This is an example of using combined top-down and bottom-up strategies, where organic linkers captured the abandoned Ni2+ ion during the dealloying process, to build a shell of MOFs on the surface of the Ni–Pt alloy in situ.
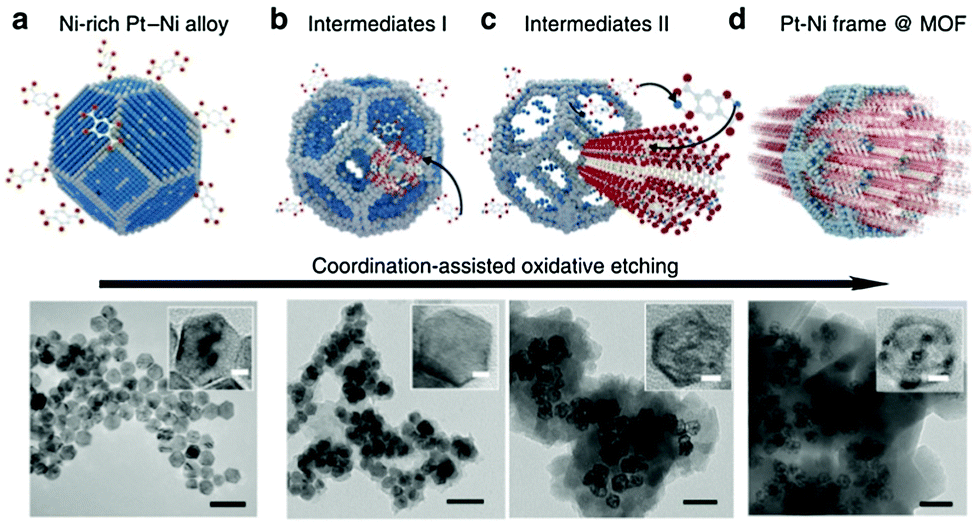 | ||
| Fig. 5 Scheme and corresponding TEM images of the coordination-assisted oxidative etching process. (a) Initial solid Ni–Pt polyhedra. (b) Ni–Pt frame@MOF intermediates I. (c) Ni–Pt frame@MOF intermediates II. (d) Final Ni–Pt frame@MOF. The scale bar is 50 nm. (The insets show the magnified TEM images. The scale bar is 5 nm).44 (Copyright 2015 Nature Publishing Group.) | ||
2.4 Others
Apart from the discussions above (i.e. core–shell, alloy and porous structures), some other structures are also reported for bimetallic Ni catalysts. For example, hollow bimetallic (Ni/Au, Ni/Ag, Ni/Pt, and Ni/Pd) spheres were synthesized via a decomposition and reduction route by using hollow nickel hydroxide spheres as precursors.45 Hollow Ni(OH)2 microspheres were first prepared through a hydrothermal method and subsequently calcined in air to produce a NiO sphere. Hollow metallic nickel spheres were then obtained by the H2 reduction of NiO hollow spheres. Different hollow bimetallic spheres were then obtained via a replacement reaction route by using hollow metallic Ni spheres and the corresponding noble metal precursors, where the Ni spheres acted as sacrificial templates. The driving force of these reactions comes from the large standard reduction potential gap between the Ni2+/Ni redox pair (−0.25 V vs. standard hydrogen electrode (SHE)) and the Mx+/M redox pair (1.00 V for AuCl4−/Au, 0.80 V for Ag+/Ag, 0.74 V for PtCl62−/Pt, and 0.83 V for Pd2+/Pd vs. (SHE), respectively).3. Modification of the surface properties of bimetallic Ni catalysts: fundamental understanding
Understanding the origins of the novel catalytic properties of bimetallic surfaces has gained considerable interest in fundamental surface science research. Two critical factors can be considered. First, the formation of heteroatom bonds changes the electronic environment of the metal surface, thereby modifying its electronic structure through the ligand effect. Second, the geometry of the bimetallic structure is typically different from that of the parent metals (e.g. the average metal–metal bond lengths change, resulting in the strain effect that can modify the electronic structure of the metal through changes in the orbital overlap).46 Theoretical approaches, such as density functional theory (DFT), have recently been extensively applied for a better understanding of the surface properties and design principles of different bimetallic systems.47 In this section, we will emphasize the fundamental roles of added guest metals in modifying the catalytic behavior of the host metal.In a bimetallic system, the catalytic reactivity depends on the combined electronic effect of two metal components. d-Band theory can be applied to understand the combined effect of two metal species. The chief principle underlying the d-band theory is that the binding energy of an adsorbate to a metal surface is largely dependent on the electronic structure of the surface itself. The metal d-band hybridizes with the bonding (σ) orbital of the adsorbate to form bonding (d–σ) and antibonding (d–σ)* states (Fig. 6a). For the metals we are concerned with, the (d–σ) state is full, but the extent of filling of the (d–σ)* state depends on the local electronic structure of the metal at the surface, i.e. the surface density of states. An increased filling of the antibonding (d–σ)* state corresponds to a destabilization of the metal–adsorbate interaction and hence weaker binding, which induces higher activity.
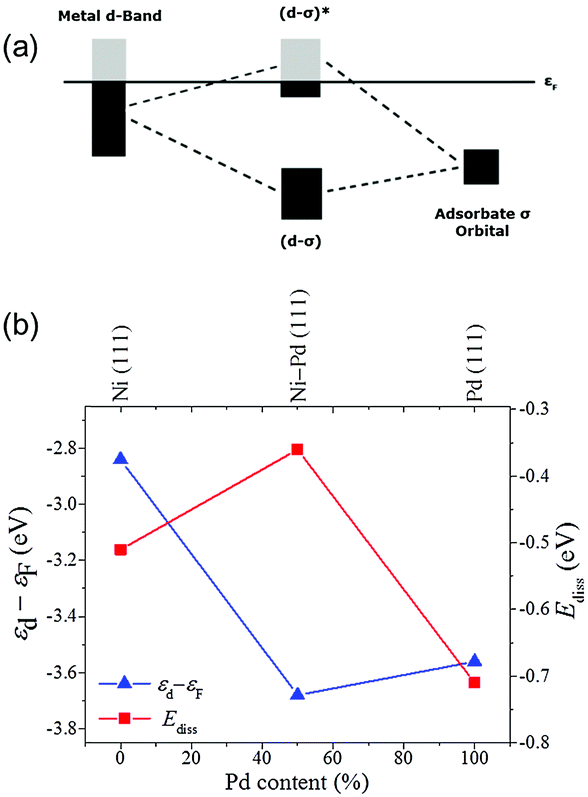 | ||
| Fig. 6 (a) Hybridization of the d-band of metal and the σ orbital of the adsorbate. (b) d-Band center with respect to the Fermi level and dissociative absorption energy as a function of Pd content (figure generated from data in ref. 48).48 | ||
Taking the Ni–Pd nanoalloy system as an example, the d-band theory can be applied to determine the effect of Pd on pure Ni in the hydrogenation reaction.48 The energy with which hydrogen is bound to the metal surface – a decisive factor in catalytic activity – strongly depends on the Ni/Pd ratio. The nanoalloys containing approximately equal amounts of Ni and Pd show a higher catalytic activity than pure particles of either metal, and the weakest binding and Gibbs free energies of hydrogen adsorption (close to zero) are calculated. This result can also be explained by the d-band model (Fig. 6b). The d-band center of the mixed Ni–Pd(111) surface is at lower energies than that of the pure Ni(111) and Pd(111) surface, which is in accordance with the higher Ediss of Ni–Pd(111) and hence higher activity. The same observation has been made for metal clusters of different sizes as well as for bulk surfaces, and therefore is of general applicability.
Another example is the well-known Ni–Pt system for the ORR process. The oxygen binding energy on the catalyst surface is a key descriptor in determining ORR activity. Due to the high oxophilic nature, Pt binds oxygen too strongly, implying that its d-band center is too high. Alloying Ni and Pt in a specific composition lowers the d-band center by both altering the electronics and inducing a degree of irregularity in the Pt lattice, which subsequently causes the binding to oxygen weaker than that on Pt.46
The geometry of Ni in the bimetallic catalysts is also a key factor governing catalytic activity. In general, two possibilities arise when Ni is alloyed with a noble metal: Ni either remains on the surface of the noble metal or it diffuses inside to form a subsurface region. To explore this fundamental structural modification, bimetallic Ni–Pt has been widely studied. Ni atoms underneath the surface Pt layer are thermodynamically stable in an ultra-high vacuum environment and under hydrogenation reactions, exhibiting good activity and stability. In other types of reactions, such as oxidation, dehydrogenation, and reforming, Ni-terminated surfaces are generally favored. The segregation energy (Eseg) for transition metal alloys is considered as the thermodynamic driving potential to move the subsurface admetal from the bulk to the surface of the host metal. Ruban et al. performed DFT calculations of the values of Eseg for admetal atoms (M) on many host substrates (H).49 If Eseg is sufficiently negative, the admetal segregates to the surface to produce a M–H–H monolayer structure. If Eseg is sufficiently positive, the surface layer is dominated by the host metal, leading to the formation of an H–M–H subsurface monolayer structure.
Besides determining the relationship between the Eseg value and the structure, it is important to study the influence of adsorbates on surface segregation using different adsorbates (such as hydrogen and oxygen), to predict the favorable structure responsible for a particular type of reaction. Taking the particular case of the bimetallic Ni–Pt system, the segregation of subsurface Ni was verified under ambient pressure using X-ray absorption near edge structure (XANES) spectroscopy under in situ conditions.51 Using a polycrystalline Pt foil as the substrate, the Ni atoms were found to undergo inward diffusion upon exposure to H2 and surface segregation after exposure to O2. This reversible behavior was also applicable to bimetallic Ni–Pt NPs.50Fig. 7 shows the XPS Ni/Pt intensity ratio, which oscillates with the oxidation–reduction cycles. The XPS results indicate that the Ni–Pt NP surface is NiO-rich in oxidizing gases and Pt-terminated in reducing gases. In turn, such structural changes modify the catalytic performance in both hydrogenation and oxidation reactions, and could be the main reason behind the observation that the subsurface Pt–Ni–Pt structure works better in hydrogenation reactions, while the surface Ni–Pt–Pt structure is more suitable for oxidation or reforming reactions.
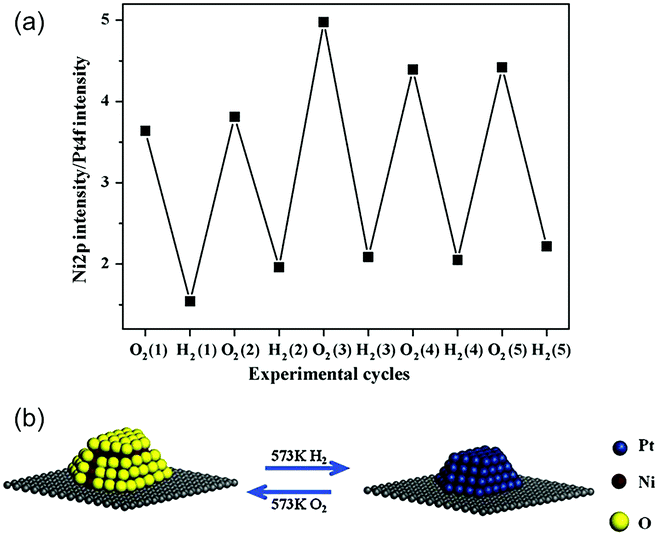 | ||
| Fig. 7 (a) Dependence of the ratio of XPS Ni 2p intensity on XPS Pt 4f intensity and (b) reversible change in the surface structure of Ni–Pt transition metal NPs with the oxidation and reduction cycles.50 (Copyright 2009 Elsevier.) | ||
4. Catalysis applications of bimetallic Ni catalysts
Since Ni has similar electronic properties as Pd and Pt, its application is widely explored in similar reactions as the other two. Here we will critically discuss recent advances in bimetallic Ni catalysts where both noble metals (Ru, Rh, Ir, Pd, Pt, Au, and Ag) and transition metals (Fe, Co, and Cu) have been used as guest metal counterparts (see Fig. 8). However, in some cases Ni also acts as a guest metal or promoter to modify the catalytic activity of reactive noble metals. One issue with Ni-based bimetallic catalysts is that Ni is often partially oxidized when exposed on the catalyst surface due to its relatively low reduction potential. However, oxidation of Ni is not necessarily detrimental since Ni in the oxidized form can enhance catalytic performance by changing the geometric and/or electronic environment of the second metal. For this reason, we will include those examples in the discussion even if both metal elements are not in the metallic state.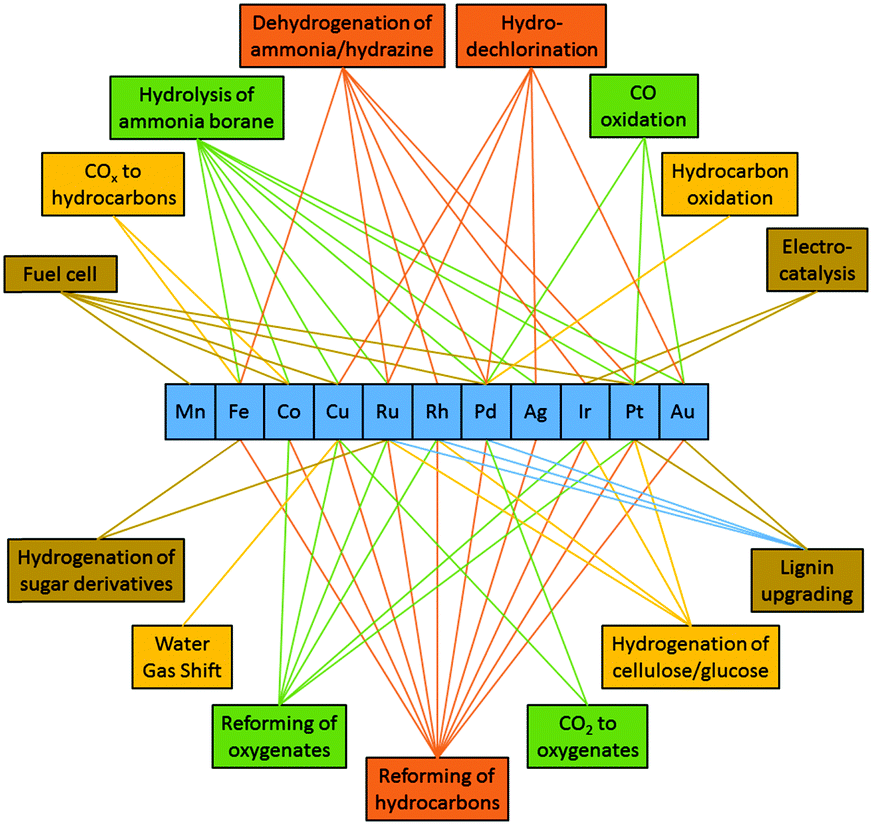 | ||
| Fig. 8 Different guest metals in bimetallic NiM catalysts and their corresponding catalytic applications. | ||
This section will be divided into two subsections focusing on energy and environmental applications, respectively. The first section is further divided by the type of energy carrier, including hydrogen, hydrocarbons, oxygenates, and electricity, whereas the second section on environmental remediation is categorized by the type of targeted pollutant. Wherever possible, we will compare the catalytic activities of bimetallic Ni catalysts with those of their monometallic counterparts, and provide reasons for the improved activities on the basis of their electronic and geometric configuration. We will also look into fundamental studies of bimetallic Ni catalysts in order to understand the catalytic mechanisms at the molecular level.
4.1 Energy production
4.1.1.1 Catalytic reforming. Catalytic reforming is a widely used technique for the production of hydrogen from different resources such as natural gas, oil, coal, and alcohols. Specifically, high-temperature steam reforming of hydrocarbons (i.e. methane at over 800 °C) accounts for a significant portion of worldwide commercial hydrogen generation (about 50%).60 Another potential application of steam reforming is the on-board hydrogen generation for fuel-cell powered vehicles.62 Noble metals are known to be the best catalysts for hydrocarbon and alcohol reforming due to their greater ability to break C–C bonds.57,63,64 However, recent attention has been slightly shifted to 3d transition metals, preferably Ni-based catalysts, which are also effective in breaking C–H and C–C bonds.10 Nevertheless, nickel-based catalysts are very sensitive to deactivation by sintering and carbon deposition.65 Unfortunately, both desired reforming reaction and undesired coke deposition are plausibly initiated by the same elementary hydrocarbon activation step,66 and under steam-reforming conditions metal surfaces are covered with various CHx intermediates. Without a fast steam gasification step to convert these intermediates to CO and H2, these adsorbed CHx species on Ni can undergo further dehydrogenation, polymerization, and rearrangement into highly stable carbon.67 It has been reported that the addition of noble metals to Ni catalysts can promote the reducibility of Ni, and stabilize it during the catalytic process.68,69 Indeed, significant efforts have been made in the formulation of bimetallic Ni catalysts during the last decade, with major research focused on how to prevent catalyst deactivation by carbon deposition. In this section, we will include some specific examples of these bimetallic systems and discuss the fundamental role of each component in overcoming this issue.
4.1.1.1.1 Reforming of hydrocarbons. Steam reforming and dry reforming of light hydrocarbons, such as methane and butane provide a promising method for hydrogen production. Although steam reforming of methane yields synthesis gas with a high H2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO ratio of about 3:1, dry (CO2) reforming of methane has certain advantages since it utilizes two abundantly available green-house gases to produce industrially important syngas and can reduce net emissions of these gases.57 However, compared to H2O reforming, CO2 reforming causes more severe coke formation because of the increased C/H molar ratio in the feedstock. Therefore, the development of a coke-resistant catalyst is the major challenge for CO2 reforming of hydrocarbons.
:CO ratio of about 3:1, dry (CO2) reforming of methane has certain advantages since it utilizes two abundantly available green-house gases to produce industrially important syngas and can reduce net emissions of these gases.57 However, compared to H2O reforming, CO2 reforming causes more severe coke formation because of the increased C/H molar ratio in the feedstock. Therefore, the development of a coke-resistant catalyst is the major challenge for CO2 reforming of hydrocarbons.
Dry reforming of methane (DRM) has been investigated over both noble (Rh, Ru, Pd and Pt) and non-noble metal (Ni, Co and Fe) based catalysts.70,71 In spite of the high cost, noble metal catalysts have drawn attention due to their superior coking resistance, higher stability, and activity especially for higher temperature applications (>750 °C). Among different noble metals, Rh has been identified as the best candidate with the following trend observed in the catalytic activity and stability of a series of alumina supported noble metal catalysts: Rh/α-Al2O3 > Ru/α-Al2O3 > Ir/α-Al2O3 > Pd/α-Al2O3 > Pt/α-Al2O3.72 The addition of a small amount of Rh to the Ni catalyst, encouragingly, further enhanced catalytic activity without any coke formation. According to Rostrup-Nielsen, the coke resistance of Ni-based catalysts can be enhanced by enhancing the adsorption of steam (in the case of the steam reforming process) or CO2, by enhancing the rate of the surface reaction, or by decreasing the rate and the degree of methane activation and dissociation.73 Various promoters such as alkali or alkaline earth metals can be used to achieve this.71 However, major research has been focused on noble metals as promoters because of their high activity and excellent coking resistance. It has been proved that carbon formation occurs less on noble metals than on Ni, mainly because the lower solubility of carbon in noble metals favors the gasification of carbon. In fact, many previous works have reported that Ni catalysts can exhibit high efficiency and better resistance against carbon deposition when modified with noble metals such as Pd,74 Pt,75–83 Ru,84,85 Rh,86–91 Ir,92 Au,93–97 and Ag.98–100 The effects of these secondary noble metals are diverse; they may function to reduce and stabilize the metal particle size of the Ni catalysts (e.g. Pt,75,77,78 Rh101), to tailor the ensemble size of the Ni catalysts (e.g. Ag,98 Au93), to block the step sites (e.g. Au93), or to modify the surface electronic properties of the Ni catalysts (e.g. Au93). The modification of Ni catalysts with noble metals was reviewed in some early reviews and it has been demonstrated as a promising approach to design catalysts with excellent performances in methane reforming.6,9,102,103 We will mainly focus on the fundamental role of noble metals in altering the catalytic and coke resistance properties of bimetallic Ni systems.
A landmark paper by Nørskov and co-workers reported that the Au/Ni surface alloy on the Ni particles (supported on MgAl2O4) was active for steam reforming and more resistant towards carbon formation than the pure Ni catalyst.93 The catalyst was designed based on the fact that when Au is added to any of the low index Ni surfaces, an alloy is formed in the first atomic layer.104 Since the Au atoms have a high electron density, the neighboring Ni atoms experience an enhanced electron density and a higher effective coordination number simultaneously. The advantage of Ni surface modification by Au can be determined by two factors: (i) the ability of the surface to activate hydrocarbon molecules and (ii) the tendency of the surface to bind C and form graphite. The abstraction of the first H atom from CH4 is considered as the rate-limiting step in the steam-reforming process over pure Ni catalysts. The addition of Au to the Ni surface could slightly increase the energy barrier of the CH4 dissociation step, as confirmed through DFT calculations and experimental methods. Therefore, Au impeded CH4 dissociation as expected. However, when the effect of Au addition was considered for the second factor, i.e. tendency to bind C, it was observed that the Au-modified Ni surface had a much lower ability to adsorb C. On the pure Ni surface, the most stable adsorption site was the threefold hexagonal close-packed site. However, the threefold sites adjacent to a gold atom were unstable, and even the threefold sites that were the next nearest neighbors to the Au atoms were substantially destabilized. The effect of Au on atomic C adsorption was thus considerably stronger than the effect on CH4 activation, and eventually led to a coke resisting catalyst without much compromise of activity.
Bimetallic Ni–Pt catalysts behave in a similar way to Ni–Au. In a recent work, supported Ni/Pt bimetallic NPs with a controlled surface composition and structure were prepared.81 The surface restructuring of the bimetallic catalysts upon thermal treatment was investigated, which demonstrated the structure evolution of the bimetallic NPs from Pt monolayer island-modified Ni NPs to core–shell bimetallic NPs composed of a Ni-rich core and a Ni/Pt alloy shell. The surface modification of the Ni-based catalysts by adding Pt atoms effectively enhanced the catalytic activities and resistance towards carbon formation in the dry reforming process. To assess the surface and bulk structure according to Pt/Ni elemental composition, a series of alumina-supported Ni/Pt bimetallic NPs with various Pt coverages were prepared and tested. DFT calculations suggested that the addition of Pt to the Ni surface might facilitate the CH oxidation pathway and inhibit the carbon oxidation pathway. This led to enhanced catalytic activity and suppressed carbon formation as the Pt coverage increased.
Modification by Ag follows a different mechanism to improve the catalytic and coke resistance properties of the Ni catalysts. Kang et al. reported a Ni/Ag/MgAl2O4 catalyst containing equimolar amounts of Ni and Ag for the steam reforming of butane.99 During butane reforming, the Ag and Ni components played a role in the oxidation of the feed gases. The main products from steam reforming over the Ni/MgAl2O4 catalyst without the Ag component were H2, CO, CO2, and CH4, with a small amount of C2∼. However, the addition of Ag reduced the degree of carbon deposition and improved the H2 product selectivity by eliminating the formation of all C2∼ products. The unmodified Ni/MgAl2O4 catalyst experienced strong Ni sintering at a high temperature. It was also proved that the catalytic performances differed according to the order in which the metal precursors were added. Fig. 9 illustrates the phase changes of different catalysts during the butane reforming process. Simultaneous addition of Ag/Ni depressed the NiAlO3 spinal structure and induced catalytic deactivation due to their strong sintering. When Ag was added between Ni and Al, deactivation was reduced significantly, while simultaneously improving the catalytic activity.
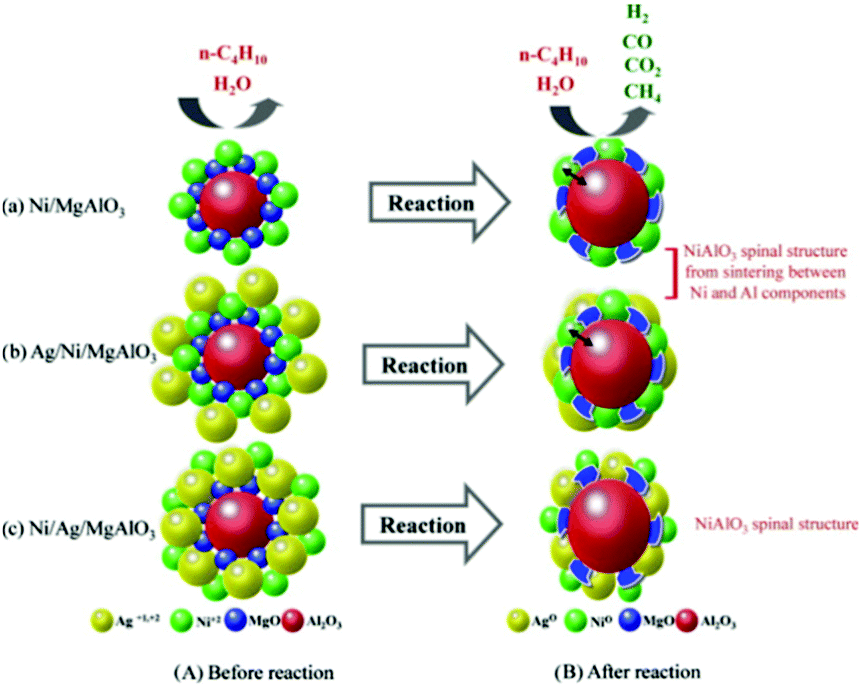 | ||
| Fig. 9 Expected phase transformation in the butane reforming mechanism before and after the butane reforming reaction.99 (Copyright 2010 Elsevier.) | ||
In addition to carbon deposition on the catalysts, sulfur poisoning is another serious issue, which is often seen during the steam reforming of hydrocarbons due to sulfur contaminants.105 The bimetallic Ni–Rh catalyst on the CeO2–Al2O3 support exhibited better sulfur tolerance than their monometallic counterparts at 550 °C, although the catalytic performance of the bimetallic catalyst was inferior compared with the pure Rh catalyst.106 Unlike the Ni catalyst, the Rh catalyst dramatically improved its sulfur tolerance upon increasing the temperature to 800 °C. The superior sulfur tolerance of the Rh catalyst at this high temperature could be associated with its better capability in sulfur oxidation than Ni, which formed different sulfur species on the metal surface. It was observed that the metal sulfide and organic sulfide were the dominant sulfur species on the Ni catalyst, while sulfonate and sulfate predominated on the Rh catalyst. The presence of sulfur induced the formation of nickel sulfide which suppressed the carbon gasification and caused severe carbon deposition on the Ni catalyst at 800 °C. This could be one of the reasons for the lower activity of the bimetallic Rh–Ni catalyst compared with pure Rh.
Along with the use of noble metal promoters, 3d transition metals, especially Fe, Co and Cu, also improved the activity and coke resistance properties in the methane reforming reaction.107–115 In a recent study, a series of bimetallic Fe–Ni/MgAl2O4 catalysts with Fe/Ni ratios between 0 and 1.5 were examined for methane dry reforming at 650–800 °C.107 In H2-TPR, Fe2O3 and NiO were reduced above 700 °C to form a Fe–Ni alloy, constituting the active phase for the methane dry reforming reaction (Fig. 10). This alloy remained stable in a flowing gas stream of CO2 during reoxidation until 627 °C, but was decomposed to metallic Ni and Fe3O4 above this temperature. The process of dry reforming on Ni–Fe could be described by the Mars–van Krevelen mechanism, where CO2 oxidizes Fe to FeOx and CH4 is activated on the Ni sites to form H2 and surface carbon. This surface carbon was oxidized by FeOx lattice oxygen to produce CO, thereby reducing the probability of carbon deposition.
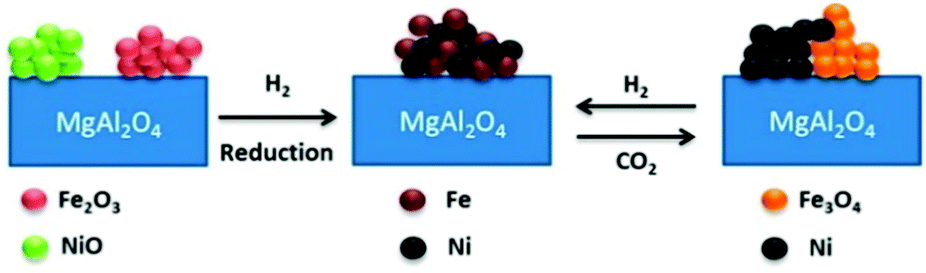 | ||
| Fig. 10 Schematic diagram of Ni–Fe alloy formation, during H2-reduction, and decomposition, during CO2 oxidation.107 (Copyright 2015 American Chemical Society). | ||
4.1.1.1.2 Reforming of oxygenates. The production of H2 from renewable resources such as ethanol and other small molecule oxygenates (i.e. acetone, glycerol, ethylene glycol, etc.) has received special attention in recent years.116–122 Among different metal catalysts investigated, late transition metals such as Ni, Co, Pt, and Ru are still the major active components due to their high activity in breaking C–H and C–C bonds.123 Although oxygenate reforming requires comparatively lower temperature than that of hydrocarbons, coke formation on the metal surface is more inclined to take place. In a recent review article, Li and Gong explained the different possible pathways of alcohol reforming on oxide and supported metal catalysts, where the metal surface is proposed to be the primary active site for the reforming process.10 In the case of Ni, carbon deposition becomes severe upon the aggregation of Ni particles.124 Therefore, metal dispersion and inhibition of metal sintering are critical in the development of an efficient nickel reforming catalyst.
Skeletal Ni is considered to be a highly active catalyst due to its considerable nickel dispersion and large exposed surface area. Gong et al. reported low-temperature steam reforming of ethanol using skeletal Ni (Ni–Al alloy powder prepared from 50 wt% Ni and 50 wt% Al)-based catalysts in combination with different assistant metals such as Pt, Cu, and Co.125 It was observed that three different assistant metals play different roles in the reaction; Pt and Cu suppress the methanation and enhance H2 production, while Co promotes the methanation and increases CH4 selectivity. It has been proved that dissociative adsorption of CO is generally suppressed from left to right and from 3d to 5d in the periodic table of transition-metal elements.126 Therefore, it can be rationalized that Cu and Pt could suppress the dissociation of CO on the Ni surface, and consequently suppress the methanation reaction, whereas Co would enhance the dissociation of CO on the Ni surface.
The incorporation of noble metals such as Pt and Rh can simultaneously improve the reforming activity and prevent the catalyst deactivation.117,127–133 Aqueous phase reforming of ethylene glycol over supported Ni–Pt/C and Ni–Pt/γ-Al2O3 catalysts revealed that the enhanced activity of the bimetallic catalysts was correlated with the changes in the catalyst structure.129 Under the reaction conditions, Ni segregated to the surface of the catalysts, resembling Ni-terminated bimetallic surfaces that were more active than Pt as identified in theoretical and experimental studies on model surfaces. Very recently, Moraes et al. investigated the effect of Pt addition on the performance of a Ni/CeO2 catalyst for low temperature steam reforming of ethanol.127,133 Based on different characterization techniques, they provided an explanation addressing why Ni/CeO2 deactivated more extensively than Ni–Pt/CeO2. In the first step, both catalysts are able to decompose the ethoxy species, the dehydrogenated species (acetaldehyde, acetyl species), and the acetate species into H2, CO, and CHx. Following that, there are two possibilities: (i) the CHx species can be hydrogenated and desorbs as methane or may react with water producing H2 and CO or, (ii) these CHx species may be further dehydrogenated to carbon.123 Therefore, when the rate of this reaction pathway is higher than the rate of desorption of CHx species such as CH4, carbon is accumulated over the surface and the catalyst deactivates. In situ X-ray absorption studies revealed the formation of a nickel carbide phase during the reforming process over the Ni/CeO2 catalyst, which was associated with amorphous carbon deposition and catalyst deactivation. The addition of Pt to the Ni/CeO2 catalyst promoted the decomposition of dehydrogenated and acetate species into hydrogen, methane, CO and carbonate species (Fig. 11). The segregated Pt effectively produced reactive hydrogen species by dissociative adsorption of hydrogen and spillover of adsorbed hydrogen atoms to the Ni surface. These highly reactive hydrogen atoms hydrogenated the adsorbed carbon precursor species, which eventually desorbs as methane. As such, the segregation of Pt on the surface of the Ni particles minimized the formation of nickel carbide and promoted the catalyst stability (Table 1).
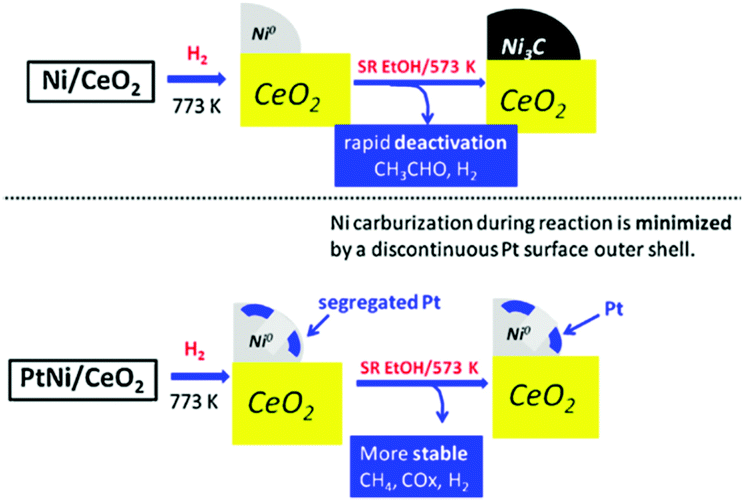 | ||
| Fig. 11 Model depicting the dynamic transformations of the Ni containing phase upon activation and steam reforming of ethanol for both Ni/CeO2 and Ni–Pt/CeO2 catalysts.127 (Copyright 2016 Elsevier.) | ||
| Catalyst | M/Ni (wt/wt) | Synthesis method | Reaction | T (°C) | Catalytic performance | Note | Ref. |
|---|---|---|---|---|---|---|---|
| IWI: incipient wet impregnation; DP: deposition–precipitation; HT: hydrothermal method; DRM: dry reforming of methanol; SRE: steam reforming of ethanol; ATR: auto-thermal reforming; OSR: oxidative steam reforming; APR: aqueous phase reforming. | |||||||
| Ni–Pt/γ-Al2O3 | 0.33 | IWI | DRM | 700 | 69% CH4 conv. | Ni increased the electron-withdrawing ability of Pt | 76 |
| Ni–Pt/γ-Al2O3 | 0.05 | IWI | DRM | 750 | 78% CH4 conv. | Carbon deposition was markedly lowered | 79 |
| Ni–Pt@Hol S-1 | 0.34 | IWI | DRM | 800 | >70% CH4 conv., GHSV 72 L g−1 h−1 | Hollow silicalite shell prevented coke deposition on encapsulated Ni | 82 |
| Ni–Pd/ZrO2–La2O3 | 0.25 | IWI | DRM | 700 | 73% CH4 conv. | — | 74 |
| Ni–Rh/Al2O3 | 0.33 | IWI | DRM | 700 | 87% CH4 conv. | Segregation of metals led to the formation of Ni-rich surface alloy | 86 |
| Ni–Rh/CeO2–Al2O3 | 0.1 | IWI | DRM | 750 | 85% CH4 conv. | Higher activity and stability due to the presence of the redox couple Ce+4/Ce+3 and Rh0/Rhδ+ | 90 |
| Ni–RhCe2Zr1.51 | 0.1 | Pseudo sol–gel | DRM | 800 | >90% CH4 conv. | Ce–Zr mixed oxide support increased the gasification of surface coke | 89 |
| Ni–Au/MgAl2O4 | 0.057 | IWI | DRM | 800 | 100% CH4 conv. | Au prevented the formation of carbon nanotubes | 97 |
| Ni–Co/CeO2–ZrO2 | 1.5 | DP, HT | DRM | 800 | >55% CH4 conv. | Defective CeO2–ZrO2 crystalline lattice increased carbon oxidation | 108 |
| Ni–Co/ZrO2 | 1.0 | IWI | DRM | 750 | 90% CH4 conv., GHSV 150 L g−1 h−1 | Co produced more H2 and maintained the reduced state of Ni | 109 |
| Ni–Co/MSN | 1.0 | In situ electrolysis | DRM | 783 | 97% CH4 conv., GHSV 38.7 L g−1 h−1 | Co induced the formation of spinel-type NiCo2O4 solid solution to result in d-electron transfer from Co to Ni | 112 |
| Ni–Fe/MgAl2O4 | 0.7 | IWI | DRM | 800 | 51% CH4 conv. | Surface carbon oxidized FeOx lattice oxygen | 107 |
| Ni–Rh/CeZrO2 | 0.017 | IWI | SR of n-butane | 680–740 | H2 yield 10.6 mol mol−1n-butane | Oxidized Ni and Rh species increased the activity | 88 |
| Ni–Ru/CeO2–Al2O3 | 0.1 | Sol–gel | ATR of n-dodecane | 750 | H2 yield 1.6 mol mol−1n-dodecane | High activity due to high dispersion, high metal area and high reducibility of Ni | 85 |
| Ni–Ag/MgAl2O4 | 0.11 | IWI | SR of n-butane | 700 | 100% butane conv., 68% H2 yield | Addition of Ag increased H2 selectivity by reducing C2∼ products | 99 |
| Ni–Pt/CeO2–Al2O3 | 0.08 | IWI | SRE | 500 | 60.8% H2 yield | Ni cleaved the C–C bond and Pt hydrogenated the coke precursors (CxHy) | 130 |
| Ni–Pt/CeO2–Al2O3 | 0.17 | IWI | APR of glycerol | 240 | 86% H2 yield | Ni modified the crystallite and electronic structure of Pt | 119 |
| Ni–Rh/Y2O3–Al2O3 | 0.13 | IWI | SRE | 402 | 44.9% H2 yield | Lewis acidic NiAl2O4 phase decreased coke formation | 117 |
| Ni–Cu/ZrO2 | 0.54 | DP | OSR of methanol | 360 | 60% H2 yield | High activity due to bimetallic and support effects | 134 |
| Ni–Cu/ZrO2 | 0.33 | Urea co-precipitation | SRE | 600 | 84% H2 yield | Cu enhanced the WGS reaction and favored acetaldehyde decomposition and reforming over the ethanol dehydrogenation | 135 |
| Ni–Cu/MWNT | 0.08 | Reflux | APR of glycerol | 240 | 72% H2 yield | Cu suppressed undesirable methanation reaction | 136 |
Among the noble metal-free catalysts, bimetallic Ni–Cu catalysts are one of the most studied bimetallic systems for ethanol steam reforming.116,119,134,135,137–142 It has been shown that the addition of Cu to Ni catalysts highly promotes the WGS reaction to instantly convert the adsorbed CO into CO2.137 Furthermore, Cu addition induces the decomposition of CH3CHO, which is one of the intermediates in the coke formation process.139
Apart from the dry reforming and steam reforming processes, another promising alternative way to produce syngas is the partial oxidation of methane (POM) and other hydrocarbons. POM is an exothermic catalytic process in which methane is converted to form H2 and CO in the presence of a limited amount of oxygen (or air). Compared to the early success of methane steam reforming, catalytic partial oxidation remained almost unexplored until 1990. Since the first reports by Huszar et al. and Gavalas et al., a significant amount of research has been carried out on Ni/Al2O3.143 However, deactivation occurred in all cases due to the formation of NiAl2O4. Furthermore, carbon deposition was another major issue on nickel catalysts, which reduced the number of active sites of the catalysts. It was observed that both the support and the promoter had a considerable effect on the activity and the stability of the catalysts.144 Al2O3 could be replaced by other supports such as La2O3, MgO, SiO2, CeO2, ZrO2, and TiO2, which act as promoters and reduce the sintering of the active Ni phase into the support. Different bimetallic Ni catalysts consisting of noble metals such as Pt,145 Ru,146–149 Rh,150 and Ir151 as well as non-noble metals such as Co,149,152,153 Cu,154 lanthanides (Pr, Gd, Lu),155 and actinides (Th, U)156 have been studied. In all cases, the added second metals exhibited increased reducibility and higher coke resistance of the catalysts as observed in the bimetallic Ni catalysts for reforming reactions.
4.1.1.2 Production of H2 through dehydrogenation and hydrolysis. Developing hydrogen storage materials is essential for a viable hydrogen economy. An ideal hydrogen storage material should satisfy several technical requirements. These include: sufficiently high volumetric and gravimetric capacities, facile release of hydrogen at a reasonably low temperature, and efficient regeneration at a practical temperature.59,157 Hydrides of boron and nitrogen have drawn significant interest because they are light atoms with high gravimetric hydrogen capacities. In addition, hydridic B–H and protic N–H bonds can thermally or catalytically dissociate to yield hydrogen. Many chemical storage materials, such as ammonia, ammonia borane, hydrazine, hydrazine borane, and formic acid, are reported, where two main processes, dehydrogenation and hydrolysis, are generally involved in the production of H2.
4.1.1.2.1 Dehydrogenation of ammonia. Decomposition of ammonia has recently gained increased attention due to the potential of ammonia to be used as a hydrogen storage medium. Ammonia can be liquefied easily at a pressure of 8 atm at 20 °C, leading to high energy densities. As a result of its high hydrogen storage capacity, ammonia can serve as a fuel to provide COx free hydrogen through catalytic decomposition.158 It has been shown that the heat of nitrogen chemisorption is a good descriptor for ammonia synthesis and decomposition.159,160 The binding energy of the nitrogen atom to the surface must be strong enough for dehydrogenation of the NHx species to occur, but sufficiently weak that the nitrogen recombines to desorb from the surface to complete the catalytic cycle. Initial studies on single-metal catalysts showed that Ru is the most active decomposition catalyst,161 but it is expensive and therefore not employable for large scale applications. Later on, studies on bimetallic catalysts showed that Co–Mo is even more active than Ru, which triggered more exploration on a series of transition metal-based bimetallic systems that can potentially replace the precious single noble metals.159,160
Vlachos et al. reported an interesting, rational approach to designing bimetallic Ni catalysts with a comparable ammonia decomposition activity to Ru.162–164 Using microkinetic modeling combined with DFT studies, the Ni–Pt–Pt(111) surface was predicted to be a catalytically active surface. According to the calculation, the Ni–Pt–Pt(111) bimetallic surface has a nitrogen binding energy of 130.7 kcal mol−1, slightly lower than Ru (141.6 kcal mol−1), and is a potentially active catalyst. While on the other hand, the subsurface configuration, Pt–Ni–Pt(111), and the parent metals, Pt(111) and Ni(111), were expected to have lower activities because of their weaker nitrogen binding energies (87.5, 102.1 and 113.8 kcal mol−1 respectively). This was verified using temperature-programmed desorption and high-resolution electron energy loss spectroscopy experiments. 3 L (where L indicates Langmuir, 1 L = 1 × 10−6 Torr s) of ammonia was dosed at 350 K, then the temperature was ramped at a heating rate of 3 K s−1 and desorption of nitrogen was monitored using a mass spectrometer. The Ni–Pt–Pt surface was the only one that showed activity towards ammonia decomposition under these conditions, as indicated by the peak at 626 K (Fig. 12). This confirmed the model predictions of the Ni–Pt–Pt surface being active towards ammonia decomposition based on an optimal nitrogen binding energy and the other three surfaces being inactive due to a nitrogen binding energy that was too low. The activity of the Ni–Pt–Pt surface was also compared with that of the Ru(0001) surface. In a previous study of ammonia decomposition on Ru(0001), an exposure of 3500 L of ammonia was dosed at 500 K to achieve a nitrogen saturation coverage.165 In comparison, saturation coverage was achieved with 3 L at 375 K on the Ni–Pt–Pt surface. The significantly lower dosing temperature and ammonia exposure clearly indicated that the overall dehydrogenation barrier was much lower than that for the bimetallic surface. Along with the success of the Ni–Pt catalyst, noble metal-free Ni-based bimetallic catalysts (such as Ni–Fe alloy NPs, and core–shell Ce–NiO@SiO2) were also explored for the decomposition of ammonia.166,167 However, the temperature applied in these cases was very high (773–1073 K) to achieve a high conversion.
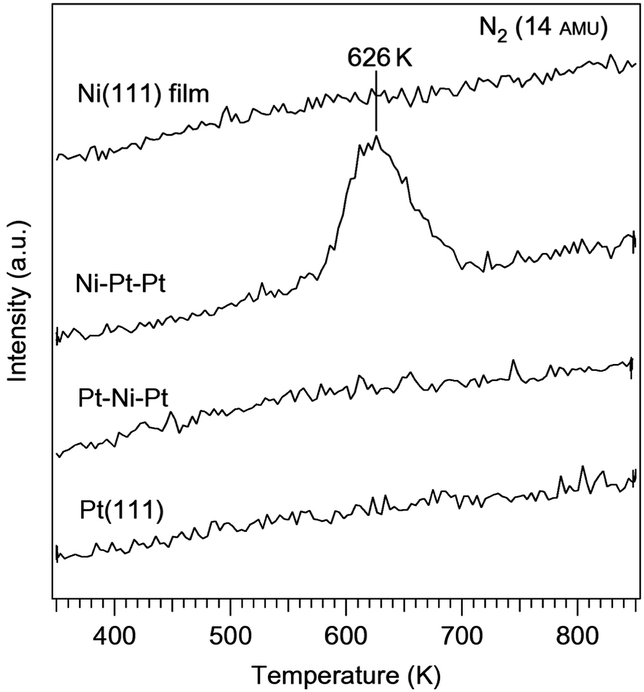 | ||
| Fig. 12 Ammonia decomposition on different Ni–Pt surfaces. TPD results of nitrogen desorption from the decomposition of ammonia on Pt(111), Pt–Ni–Pt, Ni–Pt–Pt and a Ni(111) film.162 (Copyright 2010 Nature Publishing Group.) | ||
4.1.1.2.2 Hydrolysis of ammonia borane. Although ammonia is easily available and can be stored safely, for on-board applications it is desirable that the storage materials are able to release hydrogen at a moderate temperature. Consequently, an air stable compound ammonia borane (NH3BH3, AB), which has a hydrogen capacity of 19.6 wt%, which is well above the US Department of Energy targets (2015) of a gravimetric density (9 wt%),59 and can be catalytically hydrolyzed at room temperature, received considerable attention.
There are many reports on Ni–noble metal catalytic systems for the hydrolysis of AB. Noble metal containing hollow bimetallic (Ni/Au, Ni/Ag, Ni/Pt, and Ni/Pd) catalysts were prepared via a decomposition and reduction route and tested for hydrogen generation from AB.45 The Ni/Pt catalyst was most active among all the combinations. In another study, the same results were found using a combination of Pt and transition metals such as Fe, Co and Ni.168 Ni–Pt NPs with a ratio of 1:4 exhibited the best catalytic activity. The Ni oxidation state in the Ni–Pt NPs seems to be responsible for the corresponding catalytic activity in AB decomposition. As confirmed by a XANES study, Ni remains in a metallic state in the Ni–Pt (1:4) NPs but a higher oxidation state of Ni is found in the Ni–Pt (1:1) NPs and Ni–Pt (4:1) NPs leading to a reduced catalytic performance (Fig. 13).
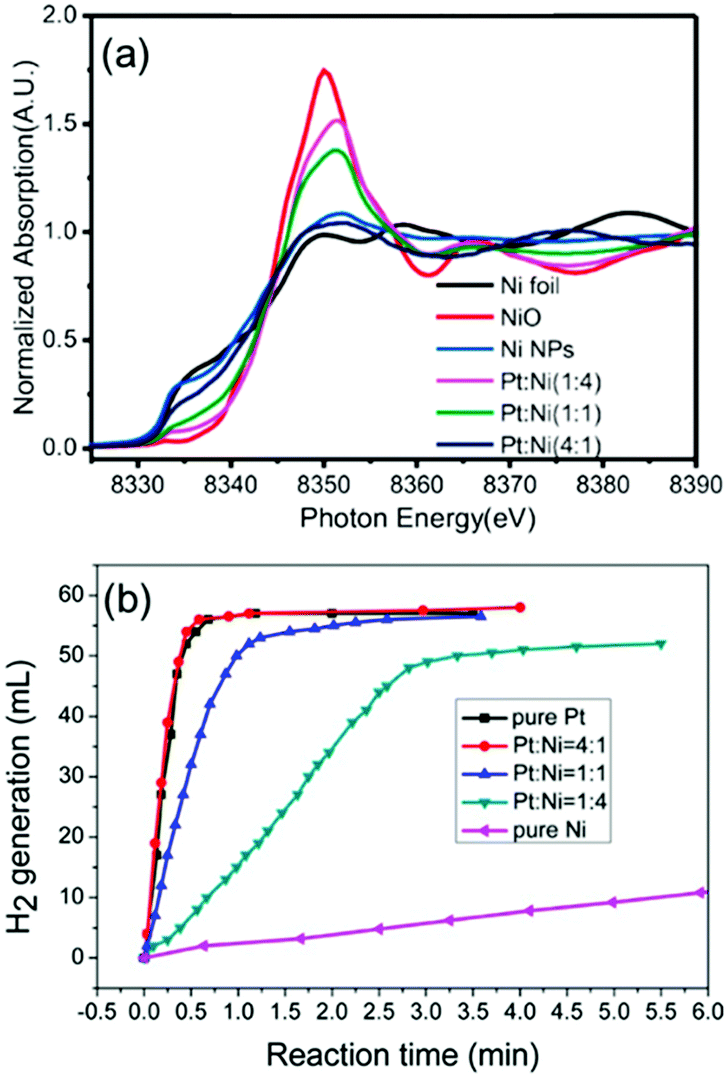 | ||
| Fig. 13 (a) Normalized absorption spectra at the Ni K-edge for the corresponding Pt–Ni catalysts with three different ratios, pure Ni NPs, and references of Ni foil, Ni NPs, and NiO. (b) Hydrogen evolution of hydrolysis of AB aqueous solution (0.5 wt%, 5 mL) catalyzed by pure Pt NPs, pure Ni NPs, and Pt–Ni NPs with different ratios under an ambient atmosphere.168 (Copyright 2014 American Chemical Society.) | ||
Chen et al. reported ultra-small magnetic Ni@Ru core–shell NPs, which showed enhanced activity in the hydrolysis of AB.24 A TOF value of 114 min−1 was achieved for the Ni@Ru NPs which was higher than that of monometallic Ru NPs (31.5 min−1). The same group reported NixRu1−x (x = 0.56–0.74) alloy NPs with different Ni/Ru ratios, among which the Ni0.74Ru0.26 sample performed as the best catalyst for AB hydrolysis.169 The hydrolysis activation energy for the Ni0.74Ru0.26 alloy catalyst was approximately 37 kJ mol−1, which is considerably lower than the values measured for monometallic Ni (≈70 kJ mol−1) and Ru NPs (≈49 kJ mol−1). This value is also lower than that previously reported for Ni@Ru (≈44 kJ mol−1) and most of the noble metal-containing bimetallic NPs reported in the literature.170 Ru@Ni core–shell NPs supported on graphene were synthesized via one-step in situ co-reduction, affording a TOF value of 340 min−1.171 The number is much higher than the reversed Ni@Ru NPs, which could be due to the presence of a more reactive Ru in the shell.
Ni–Au NPs of 3–4 nm diameter embedded in silica nanospheres were prepared via in situ reduction in an aqueous solution of NaBH4/NH3BH3. Compared to monometallic Au@SiO2 and Ni@SiO2, the as-synthesized Ni–Au@SiO2 catalyst showed a higher catalytic activity and better durability in the hydrolysis of ammonia borane, generating a nearly stoichiometric amount of hydrogen at 18 °C.172 Triple-layered Ag@Co@Ni core–shell NPs containing a silver core, a cobalt inner shell, and a nickel outer shell were prepared via an in situ chemical reduction method.173 Compared with its bimetallic core–shell counterparts, this catalyst showed a higher catalytic activity for the hydrolysis of AB. Ni–Co double shells surrounding the silver core in the special triple-layered core–shell structure provided increasing amounts of active sites on the surface to facilitate the catalytic reaction.
3d transition metal-based catalysts comprised of Ni were also explored for the hydrolysis of AB with good activity, where Ni–Fe and Ni–Cu combinations were found to be the most active.174–177 Xu and coworkers reported magnetically recyclable Fe1−xNix nano-alloy catalysts which exhibited Pt-like high catalytic activity.175 Kim et al. prepared bimetallic NiCu nanorods (NRs) incorporated into carbon nanofibers (NFs) that showed superior catalytic activity toward H2 release from AB as well as excellent recyclability and chemical stability.177 The activation energy (∼28.9 kJ mol−1) for the reaction was very low over the catalyst. Recently, CeO2 supported bimetallic Ni–Fe NPs were reported as efficient noble-metal-free catalysts for AB hydrolysis.174 The catalyst is comprised of highly disperse and partially oxidized amorphous Ni–Fe NPs stabilized by strong interactions with the CeO2 support via Ni–O–Ce and Fe–O–Ce bonding. The influence of Fe/Ni ratio on catalytic activity revealed a volcano-shaped relationship with a maximum at Fe/Ni = 1:1 (Fig. 14). The volcano-shaped activity order clearly suggested that the formation of a uniform FeNi alloy structure (at Fe/Ni = 1:1) on the surface of CeO2 and the synergistic effect originated from the integration of Fe with Ni.
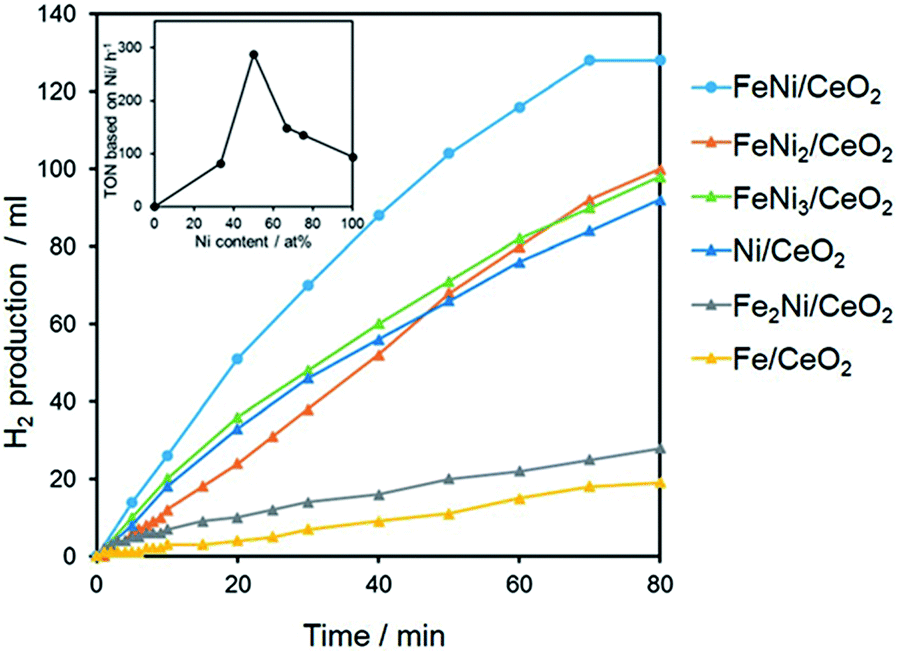 | ||
| Fig. 14 Effect of the Fe/Ni molar ratio in the hydrogen production from AB. The inset shows the TON versus Ni content.174 (Copyright 2015 Wiley-VCH). | ||
4.1.1.2.3 Dehydrogenation of hydrazine and hydrazine borane. Anhydrous hydrazine (N2H4) is another promising hydrogen storage material because of its high H2 content (12.5 wt%) and its liquid state at room temperature. However, the direct use of anhydrous hydrazine is restricted because it is extremely toxic, highly reactive, and potentially explosive. Therefore, hydrous hydrazine (N2H4·H2O) is often used due to safety concerns. Complete decomposition of hydrazine yields only hydrogen and nitrogen according to the following equation:
| N2H4 → N2(g) + 2H2(g) [ΔH = −95.4 kJ mol−1] | (1) |
| 3N2H4 → 4NH3(g) + N2(g) [ΔH = −157 kJ mol−1] | (2) |
From the perspective of hydrogen storage, reaction (1) must be selectively promoted and reaction (2) restrained. The dissociation pathways depend significantly on the catalyst and reaction conditions. Initial research findings showed that noble metals such as Ir and Rh were highly effective for the decomposition of hydrazine.178,179 However, hydrogen selectivity is highly dependent on the reaction conditions (mainly temperature) and the nature of the catalysts. Earlier studies achieved maximum hydrogen selectivity up to only 43.8%, which was due to these two competing reactions during the decomposition. Later on, non-noble metals such as Ni were incorporated to reduce the material cost and Ni-based bimetallic catalysts such as Ni–Ir,180,181 Ni–Rh,13,182–187 Ni–Pt,14,188–197 and Ni–Pd,198,199 and exhibited a superior catalytic performance compared to their monometallic counterparts.
Xu and coworkers reported that the combination of Rh, Pt, and Ir with Ni could catalyze the complete decomposition of hydrous hydrazine at room temperature with 100% H2 selectivity.13,180,188 A surfactant-assisted co-reduction process was applied to synthesize bimetallic Rh–Ni catalysts with different Rh/Ni ratios. Despite nickel itself being inactive to the reaction, the presence of nickel drastically enhanced H2 selectivity to a maximum of 100% at Rh/Ni = 4:1 (Fig. 15).13 Physically mixed Rh NPs and Ni NPs with the same Rh/Ni ratio did not show any activity enhancement over Rh, confirming the role of the bimetallic synergistic effect. Alloying of Rh and Ni led to a modification of the catalyst surface and tuned the interactions of Rh with the N–N and N–H bonds as well as the stability of the reaction intermediates on the catalyst surface. Although the catalytic activity of this Rh–Ni catalyst is very impressive, the use of a high amount of Rh (80 mol% in Rh4Ni) is not appealing from an economical point of view. Therefore, the same group synthesized Ni–Pt and Ni–Ir catalysts with much lower noble metal content (7 mol% Pt and 5 mol% Ir, respectively) and achieved similar results to the Ni–Rh catalysts.180,188 The surfactant-assisted process enhanced the activity by suppressing the agglomeration of the NPs, without affecting the bimetallic compositions of the NPs.
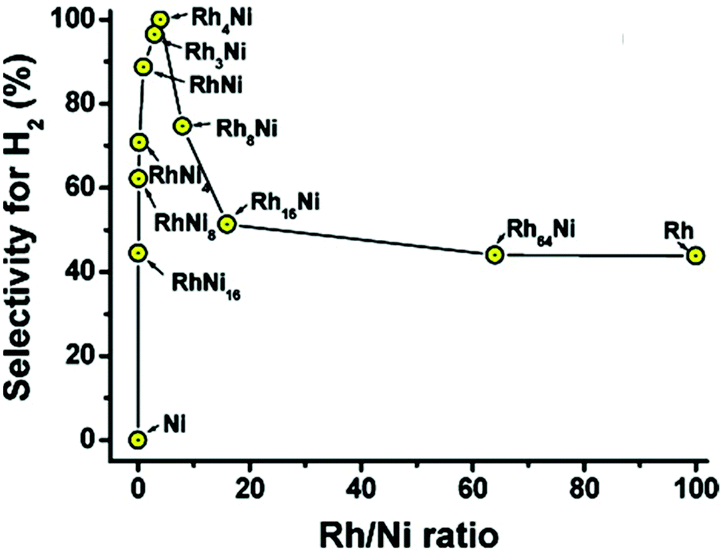 | ||
| Fig. 15 Selectivity for hydrogen generation from hydrous hydrazine (0.5 M) catalyzed by RhxNiy (x = 0–64; y = 0–16) with Rh/N2H4 = 1:10 at room temperature.13 (Copyright 2009 American Chemical Society). | ||
In spite of the quantitative selectivity, sluggish reaction kinetics is a major issue in hydrazine decomposition. In the above discussed work by Xu et al., the addition of surfactants during the preparation of the NPs makes it difficult to separate the catalysts from the reactants and also significantly reduces the reaction rate.13,180,188 Wang et al. reported a surfactant-free method to prepare a supported catalyst by depositing Ni–Rh NPs on graphene oxide, where the support played a key role in obtaining highly dispersed Ni–Rh NPs.183 Ni–Rh catalysts prepared this way exhibited 100% H2 selectivity and remarkably high activity to complete the decomposition reaction of hydrous hydrazine within only 49 min in the presence of NaOH at room temperature, which was more than three times faster than that of the previously reported single metallic catalysts.179,200,201
Luo et al. proposed a facile liquid impregnation approach for the immobilization of ultrafine bimetallic Ni–Pt NPs inside the pores of MIL-101 (Fig. 16).194,195 Highly dispersed bimetallic Ni–Pt NPs with different compositions were obtained that showed remarkable activity, selectivity, and durability towards hydrogen generation from aqueous alkaline solution of hydrazine. The catalysts showed composition dependent catalytic activity, among which Ni88Pt12@MIL-101 exhibited the highest catalytic activity, with a turnover frequency (TOF) value of 375.1 h−1 at 50 °C and 65.2 h−1 at room temperature. The same group reported Ni–Pt NPs dispersed on MIL-96, which exhibited a TOF of 114.3 h−1 and 100% hydrogen selectivity at room temperature with a composition of Ni64Pt36/MIL-96.193 In both cases, excellent catalytic performances were due to the synergistic effect of the metal organic framework (MOF) support and Ni–Pt NPs, since Ni–Pt NPs supported on other conventional supports – such as SiO2, carbon black, γ-Al2O3, poly(N-vinyl-2-pyrrolidone) (PVP), and the physical mixture of Ni–Pt and MIL-96 – exhibited inferior catalytic activities. Unfortunately, the exact nature of the interaction between the metal NPs and MOF was not clear. When comparing the activities of the two catalysts, Ni88Pt12@MIL-101 and Ni64Pt36/MIL-96, MIL-96-supported catalysts showed better activity at room temperature. This could be understood by the structures of these two MOF materials. The pore size of MIL-96 is about 1.2 nm, which is much smaller than that of MIL-101 (2.9 nm). As a result, most of the metal NPs are expected to remain on the surface of MIL-96 instead of going inside the pores. Therefore, it is expected that the Ni–Pt NPs on the surface of MIL-96 are more exposed to the substrate molecules. However, at an increased temperature (50 °C), the Ni88Pt12@MIL-101 catalyst showed increased activity, which could likely be due to the higher diffusion of the substrate through the pores and better contact with the Ni–Pt NPs located inside the pores. In a recent work, Luo and Cheng reported ultrafine monodisperse bimetallic Ni–Pt NPs on graphene by the co-reduction of nickel acetylacetonate and platinum acetylacetonate with borane-tert-butylamine in oleylamine.191 The catalyst with a composition of Ni84Pt16/graphene exhibited the highest TOF of 415 h−1 with 100% hydrogen selectivity at 50 °C.
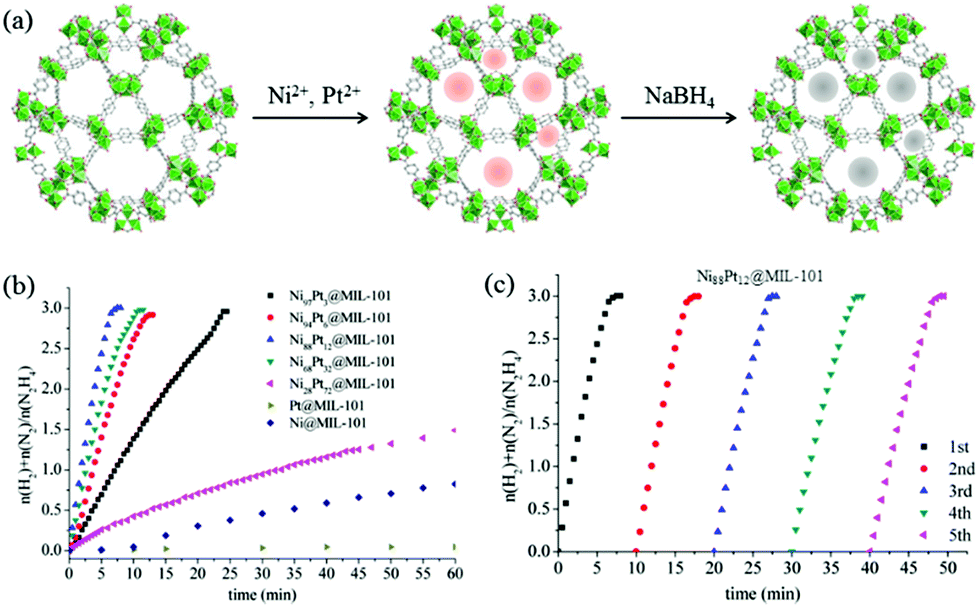 | ||
| Fig. 16 (a) Synthesis of NiPt@MIL-101 nanocatalysts. (b) Time course plots and (c) durability test for the decomposition of aqueous solution of hydrazine over NiPt@MIL-101 with NaOH (0.5 M) at 50 °C (catalyst = 0.1 g; N2H4·H2O = 0.1 mL).194 (Copyright 2014 American Chemical Society.) | ||
The development of a noble-metal-free catalyst is of economic advantage and is crucial for promoting the potential application of hydrazine as a hydrogen storage material. Many groups reported the Ni–Fe combination to be an effective catalyst for the dehydrogenation of hydrous hydrazine.32,202–204 Xu et al. reported high-performance bimetallic Ni–Fe alloy NPs for the complete and selective decomposition of hydrous hydrazine under moderate conditions.32 The H2 selectivity was 80% at 50 °C and 100% at 70 °C in the presence of 0.5 M NaOH, but the catalyst was inactive at room temperature. Tang et al. prepared monodispersed Ni3Fe single-crystalline nanospheres on carbon with a specific surface area of 182.3 m2 g−1 which resulted in 100% H2 selectivity and exceedingly high activity (TOF = 528 h−1) at room temperature.202 Wei and co-workers synthesized bifunctional Ni–Fe-alloy/MgO catalysts containing both an active center and a solid base center.203 The catalyst showed a comparable activity to most reported noble metal catalysts, exhibiting 100% conversion and 99% H2 selectivity at room temperature. The strongly basic MgO support excluded the requirement of an externally added base (such as NaOH), which was used in many other studies to increase activity.
Hydrazine borane (N2H4BH3, HB) is another H2 storage material which has a gravimetric hydrogen storage capacity of 15.4 wt% and can be easily prepared by mixing sodium borohydride and hydrazine hemisulfate at room temperature. The aqueous solution of HB is stable against spontaneous hydrolysis. Similar to hydrazine, efforts were made in the dehydrogenation of HB using bimetallic Ni catalysts.205–207 A series of Ni-based bimetallic systems were investigated with Pt, Ru, Rh, or Ir as the second metal. The results showed that most of the Ni1−xMx nanocatalysts outperformed the monometallic Ni, Pt, Ru, Rh, and Ir catalysts.206 The best performance achieved was 5.1 ± 0.05 mol (H2 + N2) per mol (HB) with Ni0.89Rh0.11 and Ni0.89Ir0.11. Using Ni0.89Pt0.11 NPs, 5.79 ± 0.05 equiv. (H2 + N2) per HB could be released, corresponding to a H2 selectivity as high as 93 ± 1%.205 The Ni1−xPtx NPs were capable of hydrolyzing the BH3 group of HB and then decomposing the N2H4 group at 50 °C, whereas the monometallic Ni and Pt were inactive for the second reaction. Xu and co-workers reported a sodium-hydroxide-assisted reduction approach to synthesize ultrafine surfactant-free bimetallic Ni–Pt NPs supported on nanoporous carbon, Maxsorb MSC-30.207 The catalyst exhibited remarkable catalytic activity towards the complete dehydrogenation of hydrazine borane with 100% H2 selectivity at room temperature. It has been found that catalytic activity and H2 selectivity are strongly dependent on the Ni/Pt ratio (Fig. 17). Both monometallic Ni and Pt nanocatalysts showed activity for hydrogen release by hydrolysis of the BH3 group in HB only, whereas the Ni1−xPtx/MSC-30 nanocatalysts with platinum contents in the range of 15–70 mol% exhibited high catalytic activities and 100% hydrogen selectivity with 5.95 ± 0.05 equiv. (H2 + N2) per HB released. NaOH served as an efficient dispersing agent to control the particle size during the formation of NiPt NPs and also played an important role as a catalyst promoter (Table 2).
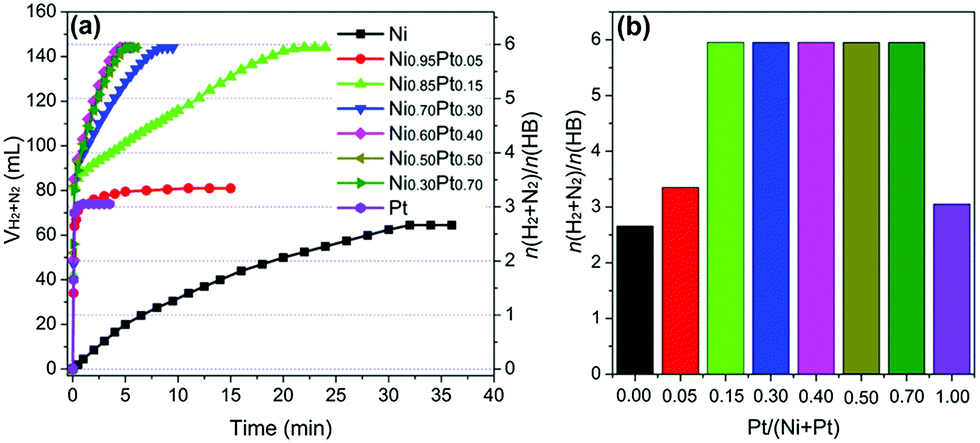 | ||
| Fig. 17 (a) Volume of the generated gas (H2 + N2) versus time and (b) Pt-content dependence of n(H2 + N2)/n(HB) for the dehydrogenation of HB over Ni–Pt/MSC-30 with different Ni/Pt molar ratios prepared with NaOH (nmetal/nHB = 0.1, 30 °C).207 (Copyright 2014 American Chemical Society.) | ||
| Catalyst | M/Ni (wt/wt) | Synthesis method | H2 storage material | T (°C) | Conv. (%)/H2 sel. (%) | TOF | Note | Ref. |
|---|---|---|---|---|---|---|---|---|
| DP: deposition–precipitation. EISA: evaporation-induced self-assembly. | ||||||||
| Ni@Ru NPs | 1.73 | Seeded growth | NH3–BH3 | RT | — | 114 min−1 | Ni seeds resulted in heterogeneous nucleation of Ru NPs to form a smaller size | 24 |
| Ru@Ni/graphene | 0.23 | Co-reduction | NH3BH3 | RT | — | 340 min−1 | Catalyst was magnetically recyclable | 171 |
| RuCuNi/CNTs | 1:1:1 |
Co-reduction | NH3BH3 | RT | — | 311 min−1 | Trimetallic synergistic effects | 208 |
| Pd10Ni6@MIL-101 | 3.3 | Co-reduction | NH3BH3 | RT | — | 83 min−1 | Bifunctional effects between Ni–Pd alloy NPs and the host of MIL-101 | 209 |
| Ni–Rh NPs | 7.01 | Co-reduction | N2H4·H2O | RT | 100/100 | — | Alloying resulted in high reducibility of each metal | 13 |
| Ni–Rh/graphene | 7.57 | Co-reduction | N2H4·H2O | RT | 100/100 | — | High activity due to the synergistic effect of the graphene support and Ni–Rh NPs and the promotion effect of NaOH | 183 |
| Ni–Rh/graphene | 0.19 | Co-reduction | N2H4·H2O | 50 | 100/100 | — | High activity even at very low noble-metal content | 182 |
| Rh/Ni@SiO2 NPs | 0.21 | Thermal hydrolysis | N2H4·H2O | RT | 100/99 | — | Catalyst was magnetically recyclable | 184 |
| Ni66Rh34@ZIF-8 | 0.9 | Liquid impregnation | N2H4·H2O | 50 | 100/100 | 140 h−1 | High activity due to the synergistic molecular-scale alloying effect and the promotion effect of ZIF-8 | 185 |
| Rh55Ni45/Ce(OH)CO3 | 2.23 | Co-reduction | N2H4·H2O | 50 | 100/100 | 395 h−1 | High activity due to strain and ligand effects between Rh and Ni | 187 |
| Ni0.95Ir0.05–B | 0.17 | Co-reduction | N2H4·H2O | RT | 100/100 | — | High activity at very low Ir content | 180 |
| NiIr0.059/Al2O3 | 0.2 | DP | N2H4·H2O | 30 | 100/99 | — | Ni–Ir alloy might tune the interaction strength between N2H4 and the catalyst | 181 |
| Ni60Pd40 NPs | 1.21 | Co-reduction | N2H4·H2O | RT | 100/100 | — | NaOH acted as a promoter for complete N2H4 decomposition | 199 |
| NiPt0.057/Al2O3 | 0.19 | DP | N2H4·H2O | 30 | 100/98 | — | Enhanced reaction rate due to weak interaction between surface Ni atoms and adspecies produced | 189 |
| Ni3Pt7/graphene | 7.76 | Co-reduction | N2H4·H2O | RT | 100/100 | 68 h−1 | Strong interaction of Ni–Pt alloy with graphene | 197 |
| Ni88Pt12@MIL-101 | 0.44 | Liquid impregnation | N2H4·H2O | RT | 100/100 | 65 h−1 | Activity was highly composition dependent | 194 |
| Ni84Pt16/graphene | 0.66 | Co-reduction | N2H4·H2O | RT | 100/100 | 133 h−1 | High activity due to the ultrafine size, narrow size distribution and synergistic effect between Ni and Pt | 191 |
| Ni60Pt40/CeO2 | 2.22 | One-pot EISA | N2H4·H2O | 30 | 100/100 | 293 h−1 | Enrichment of strongly basic sites and high resistance to alkaline solution of the CeO2 | 192 |
| Ni64Pt36/MIL-96 | 1.83 | Liquid impregnation | N2H4·H2O | RT | 100/100 | 114 h−1 | Excellent activity due to the synergistic effect of the MIL-96 support and Ni–Pt NPs | 193 |
| Ni–Fe NPs | 0.95 | Co-reduction | N2H4·H2O | 70 | 100/100 | — | Catalyst was inactive at room temperature but active at higher temperature in the presence of NaOH | 32 |
| Ni3Fe/C | 0.32 | Co-reduction | N2H4·H2O | RT | 100/100 | 9.26 min−1 | Catalyst started to deactivate because of the oxidation of Ni and Fe | 202 |
| Ni–Fe/MgO | 0.63 | Calcination–reduction | N2H4·H2O | RT | 100/99 | — | High activity due to the Ni–Fe synergistic effect and the strong basicity of MgO | 203 |
4.1.1.3 Production of H2 through the WGS reaction. Water gas shift reaction (WGS) is a reversible exothermic reaction in which carbon monoxide reacts with water (steam) to form carbon dioxide and hydrogen (eqn (3)). WGS is an important reaction typically used during and after reforming to increase the yield of H2. CO is oxidized by H2O to produce an additional mole of H2 with CO2 as a waste gas. Generally, a two-step WGS reactor is employed in large-scale industrial plants: high-temperature shift (with a Fe2O3/Cr2O4 catalyst) and low-temperature shift (with a Cu/ZnO/Al2O3 catalyst).210 For the low-temperature copper-based WGS catalysts, the first step (H abstraction from H2O) is rate-limiting for the entire process. Therefore, much importance has been given to develop new WGS catalysts with high activity toward H2O dissociation at low temperatures.211
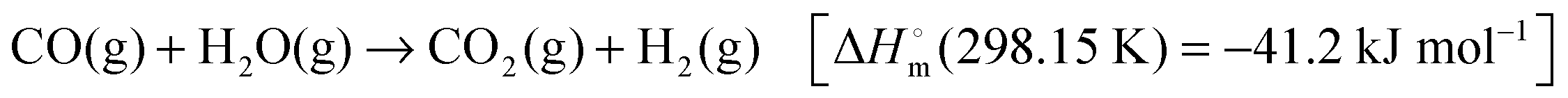 | (3) |
One of the most important detrimental factors in the WGS process is the dissociation of CO which leads to the formation of CH4. In addition to the high H2O dissociation activity of Ni, it also has a high activity towards CO activation. Therefore, an optimum concentration of Ni is required to achieve high WGS selectivity by minimizing the undesired methanation reaction. Zhao et al. performed detailed DFT calculations to study H2O and CO dissociations on a set of Ni–Cu bimetallic surfaces aiming at exploring the optimal Ni ensemble on Cu(111) for an efficient WGS process.215Fig. 18 illustrates the favorable process on different bimetallic Ni–Cu catalysts with different compositions. It was found that Ni additives in the Cu(111) surface layer including a Ni monomer remarkably enhance water splitting. As for CO dissociation on the three selected CuNi surfaces (Ni monomer, dimer, and trimer-α), the reaction barrier on the Ni monomer was as high as the value on pure Cu(111), implying that C–O breaking was unfavorable on the Ni monomer. In contrast, CO dissociation on the Ni dimer and trimer-α was highly promoted, and the calculated barriers were very close to that on pure Ni(111). The conclusion from these studies is that increasing Ni loading can induce the promotional effects on C–O bond breaking to facilitate the undesired methane yield. Therefore, bimetallic Ni–Cu catalysts with highly dispersed Ni ensembles containing a lower Ni concentration are desirable for better activity and selectivity toward WGS.
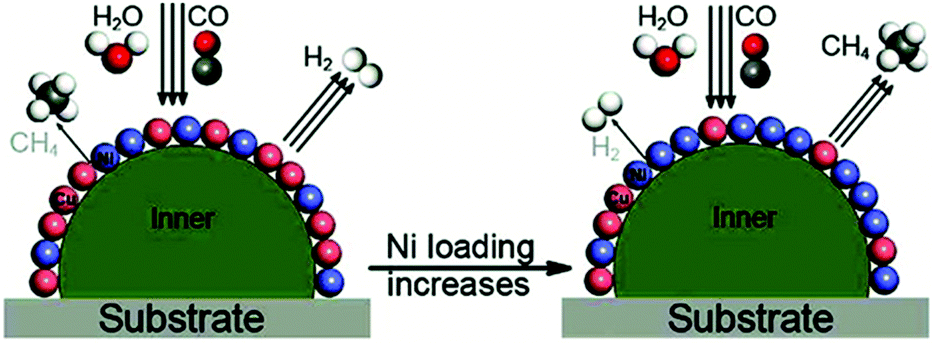 | ||
| Fig. 18 Different dissociation modes on the bimetallic Ni–Cu surface with different Ni ensembles in the WGS process.215 (Copyright 2012 American Chemical Society.) | ||
In the last few years, the WSG reaction has been extensively studied over a bimetallic Ni–Cu system where the main concern was to suppress the methanation reaction by increasing the CO adsorption.216–223 In a recent study, experimental analyses such as CO-TPR-MS, CO-TPD-MS and in situ DRIFTS have proved that the CO adsorption can be enhanced on a Ni–Cu alloy at a high temperature.220 The Ni–Cu/CeO2 catalyst with a Ni/Cu ratio of one exhibited a high reaction rate with the least methane formation due to the formation of a Ni–Cu alloy phase. Strong CO adsorption strength at a high adsorption temperature implied that the formation of a Ni–Cu alloy prevents the dissociation of CO. At the same time, it can prevent the formation of the carbon species “formate”, which could block the reaction active site and produce methane as an undesired side product. It is noteworthy that the surface lattice oxygen mobility of ceria could be activated with the formation of a Ni–Cu alloy particularly at high reaction temperatures.
4.1.1.4 Production of H2 through electrocatalysis. Electrocatalytic splitting of water is expected to play a key role in the future sustainable production of hydrogen from electricity. This process can be divided into two half-redox reactions, hydrogen evolution reaction (HER) on the cathode and oxygen evolution reaction (OER) on the anode. The key challenge in electrocatalytic water splitting is the anodic OER because of its high overpotential and sluggish surface kinetics in virtually all known materials. Currently, typical catalysts used in water splitting reactions are mostly based on noble metals such as Pt, Ru, Ir, and their alloys/compounds, among which Ir oxide is one of the most appropriated polymer electrolyte membrane electrolyzer OER catalysts providing excellent activity and stability.224–228 However, Ir is an extremely rare element with an abundance 10 times lower than Pt, and therefore the amount of Ir required must be reduced to a minimum to make its application feasible on a large scale.
Recently, Strasser's group derived dealloyed metal–oxide hybrid IrNi@IrOx core–shell NPs, which provide substantial advances towards more efficient and less expensive electrolytic water splitting.30 The IrNi@IrOx NPs were synthesized from IrNix precursor alloys through selective surface Ni dealloying and controlled surface oxidation of Ir. The final materials contained a nanometer scale, an almost pure IrOx surface, while the inner core region became increasingly metallic and enriched with Ni. The catalysts showed 3-fold activity enhancement in the electrochemical OER process over the IrO2 and RuO2 benchmark catalysts on a noble metal mass basis. In the next report published by this group, the catalysts were modified by introducing a support, where core–shell IrNix@IrOx NPs supported on high-surface-area mesoporous antimony-doped tin oxide (IrNiOx/Meso-ATO) were synthesized from bimetallic IrNix precursor alloys (PA-IrNix/Meso-ATO) using electrochemical Ni leaching and concomitant Ir oxidation.31 This work was designed based on the structural hypothesis regarding the active catalyst/support couple, with the OER proceeding at thin IrOx shells on Ir-low/Ir-free cores, thereby reducing the required Ir amount significantly (Fig. 19). The materials were annealed at different temperatures (T = 180, 250, 300, 400, 500 °C) and the effects of thermal treatment on the atomic structure of the PA-IrNix/Meso-ATO-T and on the OER activity of the obtained core–shell catalysts were investigated (Fig. 19). The IrNiOx/Meso-ATO-T catalysts with T ≤ 300 °C were significantly more OER active on both the geometric surfaces and on an Ir mass basis compared to the IrOx/C and IrOx/com.-ATO benchmarks. In fact, the lower annealing temperature maintained the desired IrNi metallic alloy phase and the Ni content in the particle core remained high. It is assumed that, as a result of this, electronic and/or strain effects modified the chemisorption and reactivity of intermediates at the surface. In contrast, the catalysts annealed at 400 °C and 500 °C showed significantly lower OER activities due to the phase segregation into a mixture of NiO and Ir-rich nanophases.
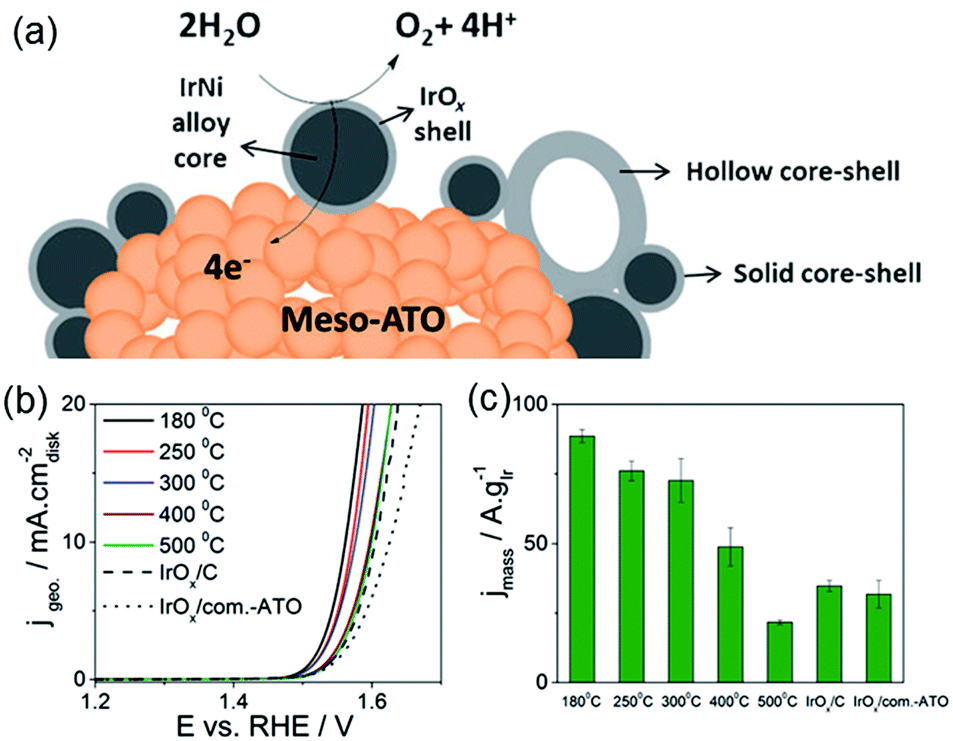 | ||
| Fig. 19 (a) Scheme of the oxygen evolution reaction on the IrOx shell of IrNiOx core–shell NPs supported on Meso-ATO. (b) Electrocatalytic oxygen evolution reaction (OER) activities of IrNiOx core–shell NPs supported on mesoporous ATO (IrNiOx/Meso-ATO-T), pure Ir NPs supported on carbon (IrOx/C), and on commercial ATO (IrOx/com.-ATO) measured using linear sweep voltammetry. (c) Ir-mass-based activity at η = 280 mV overpotential of IrOx/C, IrOx/com.-ATO, and IrNiOx/Meso-ATO-T.31 (Copyright 2015 Wiley-VCH.) | ||
The same group reported Ni–Ir mixed oxide thin film catalysts for the OER with an unprecedented 20-fold improvement in Ir mass-based activity over pure Ir oxide.229 The initial variation of the Ir to Ni ratio resulted in a volcano type OER activity curve with a maximum at high Ni contents (67–79 at%) (Fig. 20a). The intuitive model for the formation of the active state of the catalytic surface suggested that the coverage of the reactive surface hydroxyls serves as a useful descriptor for OER activity (Fig. 20c). During the electrocatalytic OER protocol, Ni is leached from these oxides, yielding Ir-rich oxides. Consequently, oxygen atoms lose binding partners and take up protons from the electrolyte to form surface hydroxyl groups. Upon Ni leaching the surface OH fraction increased significantly up to 67 at% initial Ni content (Fig. 20b). Interestingly, the surface-specific OER activity revealed a rather similar trend to the OH fraction, i.e. both increased with increasing Ni content and reached saturation at 67 at% Ni. These results demonstrated that the ratio of weakly bonded surface hydroxyls is directly related to the surface specific catalytic OER activity of the Ir oxides.
 | ||
| Fig. 20 (a) Electrocatalytic measurements of OER activity and stability of Ni–Ir mixed oxide films with different Ir to Ni ratios. (b) Hydroxyl group (OH) fraction to the total oxide related oxygen (hydroxyl groups and both lattice oxygen species) as determined by XPS. (c) Model of Ni leaching from the surface of Ni–Ir mixed oxides.229 (Copyright 2015 American Chemical Society.) | ||
Apart from Ir-based bimetallic systems, recent studies also focused on cheap metal based catalysts for OER.230–233 In a recent work, 3d transition metal layered double hydroxide (LDH) nanosheets based on Ni and Fe were synthesized and supported on conductive graphene oxide.231 The catalytic activity of the Ni–Fe LDH catalysts increased with the increase of Fe content, and reached the highest value for Ni2/3Fe1/3–rGO composition. The synergistic effect originated from the face-to-face interfacial hybridization of redoxable LDH nanosheets and conductive graphene at a molecular scale in the superlattice structure.
| CO2 + 4H2 → CH4 + 2H2O [ΔH298K = −165 kJ mol−1] | (4) |
| CO + 3H2 → CH4 + H2O [ΔH298K = −206 kJ mol−1] | (5) |
| (2n + 1)H2 + nCO → CnH(2n+2) + nH2O | (6) |
According to the thermodynamics, methanation of CO2 and CO is the most advantageous reaction as it is considerably faster than other reactions which form hydrocarbons or alcohols.239 However, the process needs effective catalysts to occur. In fact, a huge number of studies have been performed on the design of effective methanation catalysts during the past few years.240–242 Since the earliest work by Sabatier and Senderens in 1902, nickel-based catalysts have been considered as the most active catalysts for the methanation process.241 In 1975, M. A. Vannice compared the specific activity and product distributions of group VIII metals dispersed on Al2O3 in the production of hydrocarbons from H2–CO mixtures and found that CO methanation can occur readily over these metals.243,244 The specific activity followed the order of Ru ≫ Fe > Ni > Co > Rh > Pd > Pt > Ir, and the reaction rate of CO methanation was observed to be closely related to CO dissociation. Among all these catalysts, Ni supported on Al2O3 is one of the most widely studied catalysts in methanation reactions due to its high performance-cost ratio. However, the performance of Ni catalysts toward methanation is dependent on various parameters such as the effect of support, Ni loading, the presence of a second metal and the preparation method. In this section, we will focus our discussion on the influence of the second metals on the activity of various Ni-based catalysts for the methanation of CO2 and CO.
In the earlier section on the dry reforming process, we discussed that conventional Ni-based catalysts suffer from severe catalyst deactivation due to carbon deposition as a result of the CO2 methanation reaction. The same strategy is also applied here to overcome this problem, where the addition of second metal such as Fe, Zr, Co, Cu, Mn, La, Y, and Mg has been attempted to enhance the stability and the catalytic activity of the nickel-based catalysts.245–255 Hwang et al. reported the effect of a second metal (M = Fe, Zr, Ni, Y, and Mg) in mesoporous Ni (35 wt%)–M (5 wt%)–alumina xerogel (denoted as 35Ni5MAX) catalysts for methane production from CO2 and H2.247 In the CO2 methanation reaction, the yield of CH4 decreased in the order 35Ni5FeAX > 35Ni5ZrAX > 35Ni5NiAX > 35Ni5YAX > 35Ni5MgAX. This indicated that the catalytic performance was greatly influenced by the identity of the second metal in the CO2 methanation reaction. The CO dissociation energy and metal–support interaction of the catalyst played key roles in determining the catalytic performance of the 35Ni5MAX catalysts in the reaction, in which the 35Ni5FeAX catalyst retained the most optimal CO dissociation energy and the weakest metal–support interaction, and exhibited the best catalytic performance in terms of conversion of CO2 and yield of CH4.
Iron has been widely used as the second metal in Ni catalysts to improve CO2 conversion and CH4 yield.248–251 The performance of the catalysts was highly dependent on the metal loading, the nature of the support and the preparation methods. For example, supported Ni–Fe catalysts with a total metal loading containing 75 wt% Ni and 25 wt% Fe showed a significantly better CO2 conversion and higher CH4 yield compared to the Ni and Fe supported catalysts.250 Kang et al. studied the effect of Fe metal on NiAl2O3 for CO2 methanation and their results showed that the Ni0.7Fe0.3/Al2O3 catalyst achieved the maximum carbon conversion and CH4 selectivity.248 The increase in Fe content led to the enhancement of the water gas shift reaction and hydrocarbon production. Therefore, achieving the optimum ratio of the two metals is important in order to obtain a maximum CH4 yield. Pandey and Deo studied CO2 methanation over different oxide supports (such as Al2O3, ZrO2, TiO2, SiO2 and Nb2O5) containing a bimetallic Ni–Fe catalyst in the ratio of 3:1.251 It was observed that the relative enhancement in yield for the most active catalyst of each series was support dependent, and that the maximum enhancement was achieved over Al2O3 supported catalysts. The authors proposed that the maximum enhancement for the Al2O3 supported catalysts was due to the ability of the support to adsorb CO2. However, the effect of CO2 adsorption on the supported catalysts was not studied to support the statement.
Among the noble metals, Ru shows the highest activity for CO2 methanation and therefore it has been used to promote the catalytic efficiency of Ni catalysts.256,257 The effect of Ru content in the mesoporous Ni (35 wt%)–Fe (5 wt%)–Ru (x wt%)–alumina xerogel (denoted as 35Ni5Fex–RuAX) catalysts was investigated.256 Both the conversion of CO2 and the yield of CH4 showed volcano-shaped trends with respect to Ru content, which indicates that optimal Ru content was required for the maximum production of CH4 (Fig. 21). The metal surface area and the amount of desorbed CO2 of the catalysts also showed the same trends with respect to Ru content, which explained the good correlation between the catalytic performance and their physicochemical properties. Zhen et al. reported CO2 methanation over γ-Al2O3-supported bimetallic Ni–Ru NPs, which were prepared by co-impregnation and sequential impregnation methods.257 The catalytic activities were found to be highly dependent on the preparation sequence. Catalysts prepared by the co-impregnation method showed higher catalytic activities, selectivities, and excellent stabilities for CO2 methanation. It was also found that the segregation phenomenon of Ru occurred on the catalyst surface in the co-impregnation method, by which more active Ni and Ru species (metallic Ru) could be provided on the surface of the catalyst. Based on these characterizations, a possible reaction mechanism was proposed, where the individual role of each metal site was explained (Fig. 22). In the first step, CO2 was dissociated and activated to form carbon species (COads) on the Ru species surface. At the same time, H2 was dissociated into H on the metallic Ni surface. In the second step, carbon species (COads) was dissociated into C and O on the catalyst surface. Finally, the C species could react with H to produce CH4 on metallic Ru, and H and O atoms formed H2O.
 | ||
| Fig. 21 Conversion of CO2 and yield of CH4 in the methanation of CO2, plotted as a function of Ru content of 35Ni5FexRuAX catalysts.256 (Copyright 2013 Elsevier.) | ||
 | ||
| Fig. 22 The proposed reaction mechanism of CO2 methanation over the 10Ni–1.0Ru catalyst.257 (Copyright 2014 Royal Society of Chemistry.) | ||
Product selectivity is one of the major concerns in the FT process. In the FT process, the product composition primarily includes paraffins, olefins, and different classes of oxygenates (alcohols in major fraction). Therefore, one of the primary interests in this research is to generate a higher olefin fraction, which can then be further oligomerized to generate the target transportation fuel or chemicals and reduce/avoid CH4 and CO2 formation from the syngas hydrogenation. In a recent work by Ramasamy et al., a dual bed configuration has been introduced to convert oxygenates generated in the CO hydrogenation process to produce targeted hydrocarbon compounds.258 In the first reactor of the multi-step process, the Ni–Co bimetallic catalyst containing a total metal loading less than 10 wt% was introduced, which produced 40% of short-chain olefinic compounds (C2–C7) along with 10% oxygenates. An acidic alumina containing reactor was added followed by the Ni–Co containing reactor for the deoxygenation of oxygenates, which could effectively reduce the oxygenate products from 10% to 1.3%.

4.1.3.1 Hydrogenation of cellulose and glucose. Cellulose and glucose can be directly converted into various polyols (e.g. ethylene glycol (EG), propylene glycol, sorbitol, and mannitol) through hydrogenation or hydrogenolysis over metal catalysts.267 Gallezot et al. studied the effect of adding promoters such as Sn, Mo, Cr and Fe to RANEY® Ni and found that the addition of Cr increased the rate of glucose hydrogenation by up to 5 times.268 Cr(III) acted as a Lewis acid to adsorb glucose and favor the nucleophilic attack on the carbon atom by H2 dissociated on Ni. Shrotri et al. reported an aqueous phase hydrolysis–hydrogenation process to convert cellulose into sorbitol using a bimetallic Ni–Pt catalyst.269 Monometallic Ni catalysts showed little activity for the reaction, but the addition of a small amount of Pt to Ni (Ni
:Pt = 22:1 atom ratio) greatly enhanced the catalytic activity. The bimetallic Ni–Pt catalysts supported on mesoporous alumina gave a hexitol (sorbitol + mannitol) yield of 32.4% compared to 5% with the pure Ni catalyst. The presence of Pt promoted the protonation of water and hydrogen, which could spill over to Ni sites creating in situ acid sites to catalyze the hydrolysis of cellulose. In another report, hydrogenation of cellulose was performed over bimetallic Ni catalysts supported on mesoporous carbon (MC).270 By efficiently coupling the hydrolysis reaction and “in situ” hydrogenation of oligosaccharides, the hexitol yield reached 59.8%. The monometallic Ni catalyst (20%Ni/MC) produced a 42.1% hexitol yield at a cellulose conversion of 84.5%, while the addition of Rh and Ir showed increasing activity, producing 59.8% and 57.5% hexitols, respectively. The stability of the catalyst also increased upon addition of promoters. The monometallic Ni catalyst deactivated rapidly, and a 20% yield of hexitols was achieved after 5 cycles, while 4%Ir/4%Ni/MC retained its activity for 4 cycles with yields around 60%.
A highly active bimetallic Ni–Pt/ZSM-5 was reported for the hydrolytic hydrogenation of cellulose in hot-compressed water.271 Ni–Pt NPs were formed with a Pt-enriched alloy surface in the catalyst. A remarkable yield of hexitols (76.9%) was obtained on Ni–Pt/ZSM-5 which was higher than the 52.7% obtained on Ni/ZSM-5 at similar conversion levels, indicating that the presence of a Pt-enriched alloy surface significantly enhances the selectivity to hexitols. The Ni–Pt/ZSM-5 presented much higher hydrogenation activity compared to Ni/ZSM-5. The H2-TPD of Ni–Pt/ZSM-5 showed that more H species migrate from the Pt-like sites to the Ni-like ones, which means that more activated hydrogen species can participate in glucose hydrogenation.
The product distribution of the cellulose hydrogenation/hydrogenolysis process depends on the nature of the second metal used. For example, noble metals such as Pt and Ru show improved hydrogenation properties along with Ni to produce mainly hexitols as observed in previous cases. In contrast, tungsten species are very effective for cellulose degradation due to their high activity in breaking the C–C bond and as a result, lower polyols (such as ethylene glycol) are obtained through the hydrogenolysis process.272,273 However, the yield of polyols with pure tungsten catalysts is low because of the poor hydrogenation properties. Ni, in principle, can act here as a promoter to increase the hydrogenation ability to obtain ethylene glycol with a higher yield.
4.1.3.2 Hydrogenation of furfurals and levulinic acid. Biomass-derived platform molecules such as furfural, 5-hydroxymethylfurfural (HMF) and levulinic acid (LA) are of immense importance because these can be upgraded into other useful chemicals and fuels by oxidation, hydrogenation, aldol condensation, etherification and many other reactions.264,274 Here we will focus on hydrogenation and hydrogenolysis, as these are the reactions where bimetallic Ni catalysts can be used.
2,5-Dimethylfuran (DMF) has received increasing attention as a promising liquid transportation fuel lately. DMF can be obtained via selective hydrogenation of HMF over metal-based catalysts.275 However, obtaining a high selectivity of DMF is challenging due to the highly reactive nature of HMF. HMF reductive chemistries include C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond reduction, hydrogenation of the furan ring as well as C–O hydrogenolysis. Therefore, the catalysts must be carefully designed to perform the desired reaction to achieve the desired product. Huang et al. reported a highly efficient non-noble bimetallic catalyst based on nickel–tungsten carbide for the hydrogenolysis of HMF to DMF with excellent yields.276 Using different catalysts, metal ratios and reaction conditions, a maximum DMF yield of 96% was obtained. To understand the role of Ni and W2C components in the hydrogenolysis reaction, Ni/AC and W2C were prepared and individually tested. The results suggested two different roles of metals: Ni particles mainly contributed to hydrogenation activity while W2C offers an additional deoxygenation activity. The W2C catalyst is well known for its bifunctional nature, which contains both acidic and metallic sites, and can therefore catalyze both deoxygenation and hydrogenation reactions. The addition of Ni was proved to be essential for increasing the active hydrogen concentration to improve the hydrogenation rates. Higher Ni loadings were also shown to result in a remarkable improvement in the hydrogenolysis reaction upon increasing the hydrogenation ability. Despite the in-depth studies on the individual roles of the two components, the interactions between Ni and W2C, and the structures and properties of the interface remain elusive.
O bond reduction, hydrogenation of the furan ring as well as C–O hydrogenolysis. Therefore, the catalysts must be carefully designed to perform the desired reaction to achieve the desired product. Huang et al. reported a highly efficient non-noble bimetallic catalyst based on nickel–tungsten carbide for the hydrogenolysis of HMF to DMF with excellent yields.276 Using different catalysts, metal ratios and reaction conditions, a maximum DMF yield of 96% was obtained. To understand the role of Ni and W2C components in the hydrogenolysis reaction, Ni/AC and W2C were prepared and individually tested. The results suggested two different roles of metals: Ni particles mainly contributed to hydrogenation activity while W2C offers an additional deoxygenation activity. The W2C catalyst is well known for its bifunctional nature, which contains both acidic and metallic sites, and can therefore catalyze both deoxygenation and hydrogenation reactions. The addition of Ni was proved to be essential for increasing the active hydrogen concentration to improve the hydrogenation rates. Higher Ni loadings were also shown to result in a remarkable improvement in the hydrogenolysis reaction upon increasing the hydrogenation ability. Despite the in-depth studies on the individual roles of the two components, the interactions between Ni and W2C, and the structures and properties of the interface remain elusive.
Recently, a carbon nanotube-supported bimetallic Ni–Fe (Ni–Fe/CNT) catalyst was investigated for the selective hydrogenation and hydrogenolysis of HMF with H2 as the hydrogen donor.277 Two different metal components presented different behaviors when used individually. The Fe/CNT catalyst showed low catalytic activity at both low and high temperatures, which confirmed the poor hydrogenation properties of Fe. Meanwhile, the monometallic Ni/CNT catalyst showed high conversion but poor selectivity under the same conditions. Scheme 3 represents the pathway of HMF conversion over different catalysts, which shows that different byproducts including the decarbonylation product [5-methylfurfural (MF)], the ring hydrogenation product [2,5-dimethyltetrahydrofuran (DMTHF)], the ring-opening product [1,2-hexanediol (HD)], and the etherification product were detected over Ni/CNTs. The combination of Ni and Fe in an appropriate atomic ratio of Ni/Fe (2.0) significantly increased the selectivity to 2,5-furandimethanol or 2,5-dimethylfuran depending on the reaction temperature. The selectivities to 2,5-furandimethanol and 2,5-dimethylfuran were as high as 96.1% at 383 K and 91.3% at 473 K, respectively. The improved selectivity could be attributed to the formation of Ni–Fe alloy species, which was beneficial for the selective cleavage of the C–O bond.
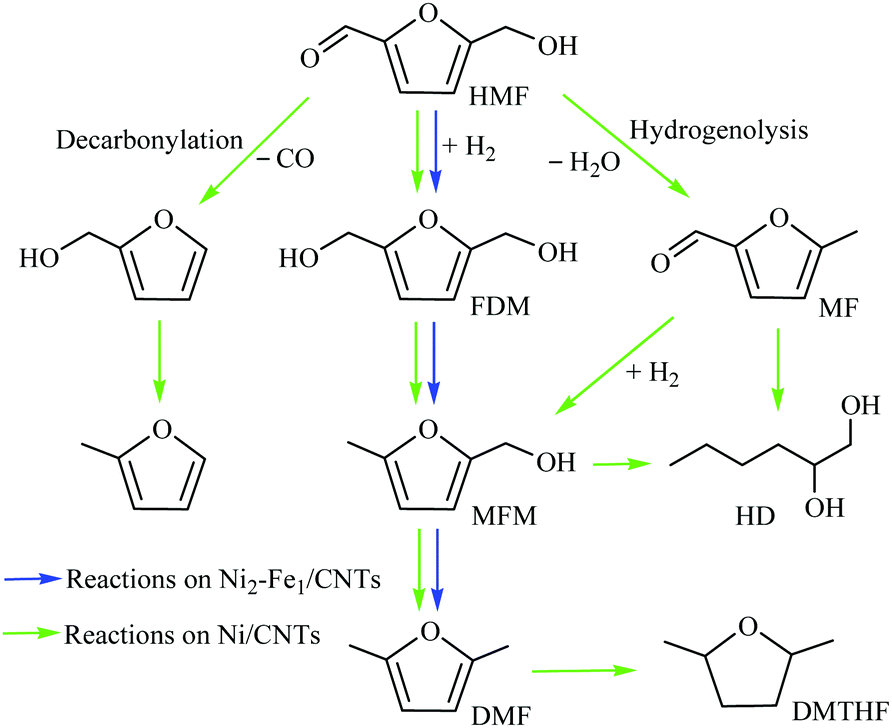 | ||
| Scheme 3 Possible reaction pathway of HMF hydrogenation over different catalysts.277 (Copyright 2015 Wiley-VCH). | ||
Sitthisa et al. investigated SiO2-supported Ni and Ni–Fe bimetallic catalysts for the conversion of furfural.278 Furfuryl alcohol and furan were primary products over monometallic Ni/SiO2, resulting from hydrogenation and decarbonylation of furfural. Comparatively, Fe–Ni bimetallic catalysts predominantly produced 2-methylfuran (2-MF) with reduced yields of furan and C4 products. The results proved that the addition of Fe suppressed the decarbonylation activity of Ni and at the same time promoted CO hydrogenation (at low temperatures) and C–O hydrogenolysis (at high temperatures). The strong interaction between O (from the carbonyl group) and the oxyphilic Fe atoms resulted in a preferential hydrogenolysis reaction on the bimetallic alloy. On the other hand, the pure Ni surface promoted the conversion of η2-(C,O) species into a surface acyl species, which was subsequently decomposed into furan and CO. In another report, furfural was converted into cyclopentanone (CPO) over Ni–Cu bimetallic catalysts in an aqueous medium under a H2 atmosphere.279 Furfuryl alcohol, 4-hydroxy-2-cyclopentenone and 2-cyclopentenone were identified as three key intermediates during the transformation. The rearrangement of the furan ring was independent of catalytic hydrogenation, starting from furfuryl alcohol rather than furfural. The opening and closure of the furan ring were closely related to the attack of the H2O molecule in the 5-position of furfuryl alcohol. The Ni/SBA-15 catalyst produced 39% CPO selectivity at a furfural conversion of 46%, while on the Ni–Cu-50/SBA-15 catalyst (Cu:Ni = 50% in atomic ratio), nearly complete conversion of furfural was achieved with 62% selectivity of CPO. Chen et al. recently reported liquid-phase hydrogenation of furfural using bimetallic Ni–Pd catalysts supported over TiO2–ZrO2 binary oxides.280 The addition of a small proportion of Pd directly transferred the catalytic performance of supported Ni from the partial hydrogenation catalyst to the total hydrogenation catalyst. The yield of the total hydrogenation product, tetrahydrofurfuryl alcohol (THFA), was 93.4% using the catalyst with a Ni–Pd molar ratio of 5:1.
Levulinic acid is another important building block that can be converted into various fuel units including ethyl levulinate (EL), γ-valerolactone (GVL), and 2-methyltetrahydrofuran (2-MeTHF).281 Hydrogenation of levulinic acid to GVL has attracted a significant amount of interest recently, as GVL can be used as a fuel blend, as a solvent, or be converted into other liquid fuels.282 Hydrogenation of LA was performed over a composition-tuned bimetallic Ni–Ru catalyst supported on ordered mesoporous carbons.283 Ru0.9Ni0.1–OMC catalysts demonstrated unprecedented catalytic activity (TOF > 2000 h−1), producing a 97% yield of GVL. The high activity of the catalyst could be ascribed to the homogeneous distribution and strong metal–support interaction. The non-noble metal-based bimetallic Ni–Cu/Al2O3 catalyst exhibited synergetic effects allowing higher activity and improved selectivity compared to the monometallic catalysts in the hydrogenation of LA to 2-MeTHF.284 The activity of the Ni-based catalytic system was highly dependent on the solvent. Water resulted in high GVL yields but inhibited MTHF formation. In contrast, hydrogen donating solvents (such as 2-PrOH) facilitated the transformation of the highly stable GVL intermediate into MTHF.
4.1.3.3 Lignin upgrading. Lignin has shown significant potential as a source for the sustainable production of fuels and bulk chemicals. Hydrogenolysis of the C–O linkages in lignin is regarded as an effective way to transform lignin into depolymerized aromatic platform compounds.259 Different bimetallic systems including noble metals combined with a transition metal (e.g. Fe, Ni, Cu, Zn or Sn) have also been found to be highly selective for the removal of oxygen even under mild HDO conditions.285,286 Among the non-noble metal-based catalysts, Ni catalysts have shown promising activity for the selective C–O cleavage in lignin model compounds under mild reaction conditions.287,288 However, pure Ni-based catalysts are unsatisfactory for β-O–4C–O bond hydrogenolysis in real lignin, due to both their limited activity (TOFs of 5–30 h−1) and low dispersions.
In our recent work, we developed a series of core–shell bimetallic catalysts, NiM (M = Ru, Rh, Pd, and Au), to achieve a better performance in the hydrogenolysis of lignin model compounds and organosolv lignin.289,290 Due to the difference in the reduction kinetics, the bimetallic catalysts have a core–shell structure and Ni was enriched in the shell. XPS and XANES studies confirmed the charge transfer from noble metals to Ni, which made the Ni surface electron-enriched. In addition, the core–shell bimetallic catalysts were formed in ultrasmall size, typically much smaller than the pure Ni catalyst (∼2 nm for Ni85Ru15 and Ni85Rh15, and ∼4 nm for Ni7Au3), which afforded an enhanced fraction of surface atoms. The as-prepared bimetallic Ni catalyst showed an excellent synergistic effect in the hydrogenolysis of a lignin model compound, 2-phenoxy-1-phenylethanol, plausibly due to an electron transfer from noble metals to Ni. While the pure Ni catalyst achieved a high selectivity to monomer products (desired hydrogenolysis products) with a low conversion, and pure Ru and Rh catalysts exhibited a high catalytic activity with low selectivity towards the desired monomer products, all the bimetallic core–shell catalysts demonstrated a high conversion as well as a high selectivity towards monomers. Our synthesized catalysts were also applied to the hydrogenolysis of organosolv lignin and it was found that the bimetallic catalysts exhibited three to ten times higher activity than pure Ni, Ru, Rh, and Pd NPs.
Sometimes it is observed that the high activity of noble metals results in undesired aromatic ring hydrogenation, which is a severe problem in lignin hydrogenolysis. One possible way to address the issue is to modify the catalyst through partially blocking of the active sites that can hinder the coordination of the aromatic rings on the catalyst surface. Recently, we developed a few effective strategies to modify the bimetallic catalyst surface, thereby making it selective for C–O bond hydrogenolysis over benzene ring hydrogenation. In the approach, the surface of highly active Rh nanoparticles was blocked by inactive NiOx, which segregates the surface terrace zones into smaller segments, thereby preventing the easy access to the Rh surface for benzene rings (Fig. 23).291 The introduction of NiOx did not exhibit a pronounced electronic interaction with Rh but significantly modified its geometric properties, inhibiting benzene ring hydrogenation without compromising the hydrogenolysis activity in the aryl ethers. The second approach was to use an inert metal such as Ag to decorate the active Ni catalyst surface, thus inhibiting the coordination and hydrogenation of the aromatic rings.292 These are examples of bimetallic systems where one component shows a detrimental effect on the reactivity of the other components to achieve improved selectivity (Table 3).
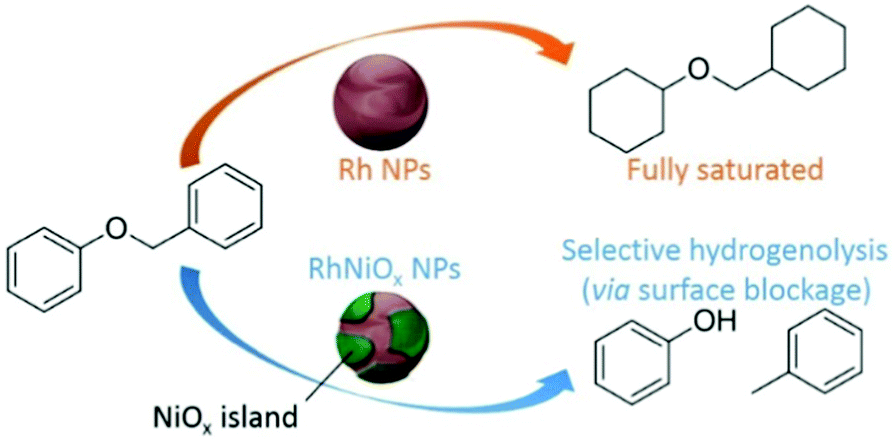 | ||
| Fig. 23 Catalytic activity of NiOx/Rh NPs in the hydrogenolysis of the C–O bond in the lignin model compound.291 (Copyright 2016 Elsevier.) | ||
| Catalyst | M/Ni (wt/wt) | Reactant | Reaction conditions | Key product(s) | Conv. (%)/yield (%) | Note | Ref. |
|---|---|---|---|---|---|---|---|
| BHTF: 2,5-bis(hydroxymethyl)tetrahydrofuran. CPO: cyclopentanone. THFA: tetrahydrofurfuryl alcohol. | |||||||
| Ni5–W25/SBA-15 | 5 | Cellulose | 245 °C, 60 bar H2, 30 min | Ethylene glycol | 100/75.4 | W performed C–C cracking and Ni performed hydrogenation | 272 |
| 1%Rh–5%Ni/MC | 0.33 | Cellulose | 245 °C, 60 bar H2, 30 min | Hexitols (sorbitol + mannitol) | 100/59.8 | Noble metal enhanced hydrogenation and decreased dehydration of sugars to increase hexitol yield | 270 |
| Ni–Pt/Beta_75 | 0.17 | Cellulose | 200 °C, 50 bar H2, 6 h | Hexitols (sorbitol + mannitol) | 51.3/36.6 | Pt created in situ acid sites by protonation and H2 spillover to catalyze the hydrolysis of cellulose | 269 |
| Ni–W/AC | 4 | Cellulose | 215 °C, 65 bar H2, 3 h | Ethylene glycol | 88.4/43.7 | W performed C–C cracking and Ni performed hydrogenation | 273 |
| Ni–Pt/ZSM-5 | 0.06 | Cellulose | 240 °C, 40 bar H2, 4 h | Hexitols | 100/76.9 | Pt showed remarkable hydrogen spillover and inhibited the oxidation of Ni | 271 |
| Ni–Pd/SiO2 | 0.26 | HMF | 40 °C, 80 bar H2, 2 h | BHTF | 99/96 | Catalyst was more active than commercial RANEY® Ni and more selective than Pd/C | 293 |
| 7Ni–30W2C/AC | 4 | HMF | 180 °C, 40 bar H2, 3 h | DMF | 100/96 | Ni showed good hydrogenation ability and W2C showed good HDO activity | 276 |
| Ni2–Fe1/CNTs | 0.48 | HMF | 200 °C, 30 bar H2, 3 h | DMF | 100/91.3 | Ni–Fe alloy selectively cleaved the C–O bond | 277 |
| NiCu-50/SBA-15 | 0.54 | Furfural | 160 °C, 40 bar H2, 4 h | CPO | 99/62 | High selectivity of CPO was ascribed to the presence of 2-cyclopentenone | 279 |
| CuNi@C | 2.17 | Furfural | 130 °C, 50 bar H2, 5 h | CPO | 99.3/96.9 | Porous carbon matrix acted as a supporter and prevented the accumulation of metal particles | 294 |
| Ni–Pd/TiO2–ZrO2 | 0.36 | Furfural | 130 °C, 50 bar H2, 8 h | THFA | 99/93.4 | Addition of Pd transferred the reaction selectivity from partial hydrogenation to total hydrogenation | 280 |
| Ru0.9Ni0.1–OMC | 11.71 | LA | 150 °C, 45 bar H2, 2 h | GVL | 99/97 | High TOF (>2000 h−1), catalyst was recyclable up to 15 times | 283 |
| 23Ni–12Cu/Al2O3 | 0.52 | LA | 250 °C, 70 bar H2, 5 h | 2-MeTHF | 100/56 | Bimetallic catalyst showed improved activity and selectivity for 2-MeTHF | 284 |
4.1.3.4 Conversion of CO2 into oxygenates. Small molecule oxygenates such as methanol, dimethyl ether, and formic acid are considered to be potential energy careers, which can be produced from CO2 through catalytic hydrogenation.234,238 Several catalytic systems have been studied for CO2 hydrogenation to methanol; among them Cu-based catalysts have long been recognized as the active catalyst component.295 There are many reports where different promoters have been introduced to improve the efficiency of Cu catalysts. Early studies have shown that Ni has a very good promotional effect on Cu catalysts for methanol synthesis, where the deposition of Ni leads to a dramatic increase in the rate of methanol formation from CO, CO2, and H2.296 It was also observed that the CuNi/SiO2 catalyst had the same level of turnover frequency and a slightly higher selectivity to methanol than the best known industrially used Cu/ZnO/Al2O3 catalyst.297 Recently, bimetallic CuxNiy/γ-Al2O3 alloy catalysts were studied for methanol synthesis from CO/CO2 hydrogenation.298 The activity was shown to be highly composition dependent where the Cu3Ni7/γ-Al2O3 catalyst exhibited the highest methanol formation rate of 5.86 mmol g−1 h−1, much higher than the commercial Cu/ZnO/Al2O3 catalyst under the same reaction conditions. The highest activity of the Cu3Ni7/γ-Al2O3 catalyst could be attributed to the larger specific surface area, the smallest alloy particle size and the lower reduction temperature.
A catalyst based on a Ni–Pd alloy on a carbon nanotube–graphene support was used for the production of pure formic acid via CO2 hydrogenation.299 The thermodynamics of the pure FA formation from CO2 hydrogenation is not favorable  even at a high temperature and pressure, and therefore organic/inorganic bases are often added to the reaction mixture to increase the conversions.300,301 The authors claimed this to be the first ever report where the heterogeneously catalyzed reaction was carried out under milder conditions (40 °C and 50 bar) in water without any base additive. Nevertheless, the highest formic acid yield obtained was very low (1.92 mmol) with a turnover number of 6.4 and a turnover frequency of 1.2 × 10−4 s−1. Based on previous results, a reaction mechanism for the formation of formic acid over a Ni–Pd alloy was proposed (Scheme 4). Noble metals (such as Pt, Ru, Rh, and Pd) are active for splitting H2 into H atoms,302 while the transition metal (such as Ni) has a high reactivity in CO2 reduction.303,304 DFT calculations also proved that CO2 is adsorbed as a formate intermediate on the Ni surface and consecutively reacts with subsurface H in Ni, producing formic acid as the final product.304 The proposed mechanism was derived based on the XPS results, which confirmed the electron transfer from Ni to Pd. The role of the Ni/Pd atomic ratio of the Ni–Pd bimetallic system could also be explained from the mechanism. The experimentally optimized composition of Pd (Ni/Pd = 2.33 for Pd3Ni7) was close to the ratio (Ni/Pd = 2) in Scheme 4, which could explain why Pd3Ni7 composition exhibited a better performance than others.
even at a high temperature and pressure, and therefore organic/inorganic bases are often added to the reaction mixture to increase the conversions.300,301 The authors claimed this to be the first ever report where the heterogeneously catalyzed reaction was carried out under milder conditions (40 °C and 50 bar) in water without any base additive. Nevertheless, the highest formic acid yield obtained was very low (1.92 mmol) with a turnover number of 6.4 and a turnover frequency of 1.2 × 10−4 s−1. Based on previous results, a reaction mechanism for the formation of formic acid over a Ni–Pd alloy was proposed (Scheme 4). Noble metals (such as Pt, Ru, Rh, and Pd) are active for splitting H2 into H atoms,302 while the transition metal (such as Ni) has a high reactivity in CO2 reduction.303,304 DFT calculations also proved that CO2 is adsorbed as a formate intermediate on the Ni surface and consecutively reacts with subsurface H in Ni, producing formic acid as the final product.304 The proposed mechanism was derived based on the XPS results, which confirmed the electron transfer from Ni to Pd. The role of the Ni/Pd atomic ratio of the Ni–Pd bimetallic system could also be explained from the mechanism. The experimentally optimized composition of Pd (Ni/Pd = 2.33 for Pd3Ni7) was close to the ratio (Ni/Pd = 2) in Scheme 4, which could explain why Pd3Ni7 composition exhibited a better performance than others.
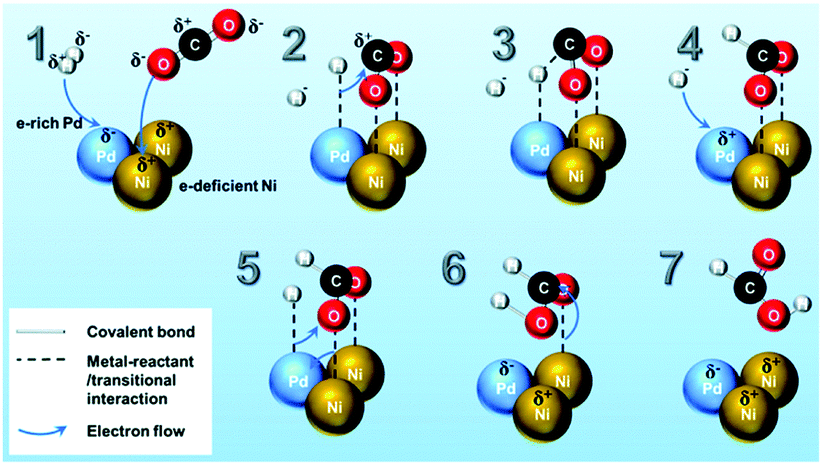 | ||
| Scheme 4 Proposed reaction mechanism of the selective formation of HCOOH from CO2 hydrogenation over the Ni–Pd bimetallic surface.299 (Copyright 2015 Royal Society of Chemistry.) | ||
Because of the high price of Pt-based catalysts, less expensive and more widely available Pd has recently been exploited as a substitute for Pt in ORR.41,309,310 Chen et al. reported a nanoporous Ni–Pd (np-Ni–Pd) bimetallic catalyst using the dealloying method where a Pd20Ni80 alloy was electrochemically dealloyed in an acid solution.41 With ∼9 at% Ni, the np-Ni–Pd bimetallic catalyst exhibited superior electrocatalytic performances in oxygen reduction in comparison with commercial Pd/C and nanoporous Pd (np-Pd). The excellent electrocatalytic properties of the dealloyed np-Ni–Pd appeared to arise from the combined effect of unique bicontinuous nanoporosity and bimetallic synergistic action. Wang et al. synthesized Ni–Pd hollow NPs via a modified galvanic replacement method using Ni NPs as sacrificial templates (Fig. 24).309 Compared with the commercially available Pt/C or Pd/C catalysts, the synthesized Ni–Pd/C exhibited a superior electrocatalytic performance towards the ORR process, which could be due to the unique hollow porous structure and changes in the electronic structures when a second metal (Ni) was introduced. Recently, a noble metal free catalyst based on bimetallic Ni–Fe layer double hydroxide (NiFe-LDH) has been reported for fuel cell applications.311 The two-phase bifunctional oxygen reduction and evolution (ORR and OER) electrocatalyst (physical mixture of NiFe-LDH and Fe–N–C) exhibited the lowest combined OER/ORR overpotential ever recorded in 0.1 M KOH.
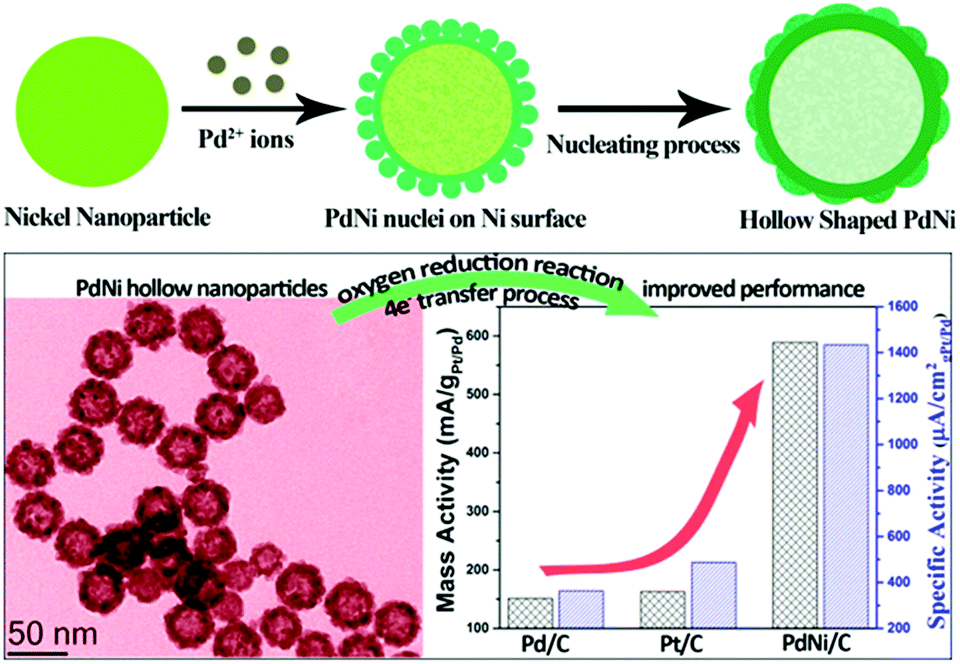 | ||
| Fig. 24 Schematic illustration of the formation of hollow Ni–Pd NPs and their ORR activity.309 (Copyright 2013 American Chemical Society.) | ||
Direct methanol fuel cells are a class of PEMFCs where methanol is used as the fuel component to supply the protons. The main reactions occurring within the methanol fuel cell are the cathodic oxygen reduction reaction (ORR) and the anodic methanol oxidation reaction (MOR). Bimetallic 3d transition metal catalysts such as Ni–Mn,312 Ni–Co,313–315 and Ni–Cu316,317 were explored for MOR with good electrochemical activities. Very recently, Cui et al. investigated the role of cobalt in bimetallic NimCon electrocatalysts for MOR.315 In their study, the composition, the surface morphology, and the crystal phase structure of the bimetallic NimCon electrocatalysts significantly changed with an increased content of cobalt. The mechanism study based on electrochemical experiments and DFT calculations indicated that doping of Co into NimCon can significantly improve the surface coverage of the redox species, weaken the CO adsorption, as well as adjust the CH3OH adsorption. Furthermore, the adsorption energies of CH3OH adsorbed on the Ni atom are higher than those of CH3OH adsorbed on the Co atom (Fig. 25a), suggesting that CH3OH prefers to bind to Ni than Co for Ni3Co and Ni2Co2. This confirms a fast kinetic rate reaction of CH3OH molecules on the surface of Ni3Co and Ni2Co2. The adsorption energies of CO adsorbed on Co are slightly higher than those of CO adsorbed on Ni, indicating that CO prefers to bind to Co than Ni. As shown in Fig. 25b, compared with other cases, Ni2Co2 has a relatively low adsorption energy, resulting in less CO poisoning of catalysts.
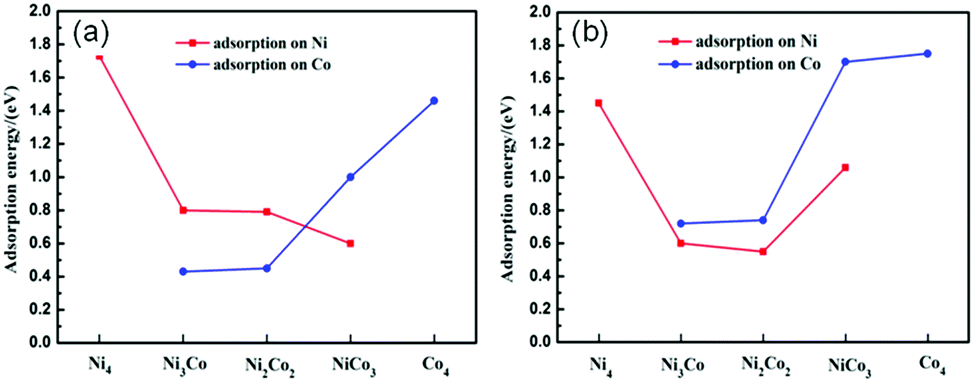 | ||
| Fig. 25 (a) Adsorption energy of CH3OH adsorbed on NimCon (m + n = 4) clusters. (b) Adsorption energy of CO adsorbed on NimCon (m + n = 4) clusters.315 (Copyright 2015 American Chemical Society.) | ||
4.2 Environmental applications
Rapid developments in industrialization, population expansion, and urbanization have largely contributed to severe air pollution. Carbon monoxide, short hydrocarbons (CxHy, especially methane), different halogenated species, and nitrogen oxides (NOx) are the main pollutants produced by industrial processes and vehicles running on petroleum fuels. Improving air quality has been a hot research topic for the past few decades and many different paths have been taken to try and formulate a solution. Catalytic oxidation is one of the most powerful ways for the remediation of air pollution, which can effectively convert the toxic compounds into less harmful and more ecofriendly compounds. For example, a three way catalyst (TWC) is highly effective for this purpose as it simultaneously removes CO, CxHy, and NOx by both oxidation and reduction pathways.| Oxidation: 2CO + O2 → 2CO2 |
| Oxidation: CxH2x+2 + [(3x + 1)/2]O2 → xCO2 + (x + 1)H2O |
| Reduction: 2NOx → xO2 + N2 |
Bimetallic Ni–Pd catalysts have been explored in CO oxidation and have a higher activity than monometallic Pd catalysts.327–329 Ni–Pd nanoalloy catalysts exhibit remarkable tunability in terms of phase state, bimetallic composition, and atomic-scale structure, which are responsible for the origin of the structural synergy for the CO oxidation reaction. Al2O3-supported bimetallic Ni–Pd catalysts were recently tested in CO oxidation to examine the effect of nickel addition on the activity and stability of Pd based catalysts.328 The improved activity could be rationalized by an interaction between Pd and Ni, which improved the reducibility of both Ni and Pd metals.
Apart from Pd, some excellent bimetallic catalysts based on Pt and Au were also reported for CO oxidation.330–333 Mu et al. reported supported sandwich type bimetallic Ni–Pt catalysts, which demonstrated high activity in CO oxidation with 100% CO conversion near room temperature.330 The Ni–Pt(111) surface consisted of both surface 2D NiO1−x nanoislands and subsurface Ni atoms. The surface Ni oxide monolayer nanoislands contained coordinatively unsaturated cations at the island edges, which provided active sites for O2 dissociative adsorption. On the other hand, the subsurface Ni atoms enhanced the elementary reaction of CO oxidation, with atomic O species produced at the edges of the surface oxide islands. In another report on the bimetallic Ni–Au catalyst, Au was the predominant species on the surface, while no surface Ni0 or NiO was observed.331 Based on the reduction potential values of Ni2+/Ni and AuCl4−/Au (−0.25 V vs. 1.00 V), Au is expected to be reduced first to form a core–shell structure with Au as the core. However, the synthesis protocol in this study employed a dendrimer stabilization approach combined with decanethiol extraction, which induced the system toward the formation of Ni@Au core–shell NPs. Au is thermodynamically more stable on the particle surface due to its lower work function than Ni. In this case, the difference was further enhanced by introducing a thiol containing extracting agent via the formation of strong Au–S bonds, which provided an additional driving force to bring Au to the NP surface. DFT calculations indicated that the incorporation of Ni into the Au slabs resulted in the stronger adsorption of O and CO on the Au surfaces. Kinetics studies also revealed that the apparent activation energies decreased by more than 50% and that the O2 reaction orders increased from 0.2 to 0.9 after Ni incorporation. Despite the promotional effect, the addition of Ni reduced the relative number of active sites on the catalyst.
Bimetallic core–shell Au–Ni NPs supported on a CeO2 support were studied for CO oxidation and compared with their monometallic counterparts.333 As expected, the Au/CeO2 catalysts exhibited a better catalytic performance than the Ni/CeO2 sample. However, Au–Ni/CeO2 was surprisingly more active than the Au/CeO2 catalyst, even if the characterization data proved that only nickel atoms are exposed on the surface during the reaction. It was believed that the Au atoms in the core of the Au@Ni NPs induced an electronic effect on the local density of the Ni d states via the presence of core Au atoms (Fig. 26), similar to that proposed for the Au–Ni surface alloys earlier.334
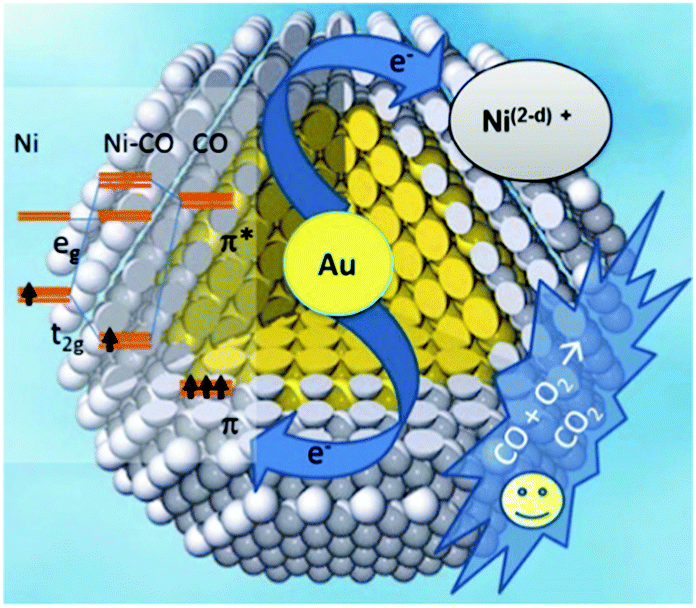 | ||
| Fig. 26 Modification of the electronic d state of Ni by Au atoms in the Au@Ni NPs.333 (Copyright 2013 American Chemical Society.) | ||
Iron–nickel hydroxide–platinum nanoparticles were reported by Chen et al. for CO oxidation at room temperature with high efficiency.335 It was shown that Ni2+ played a key role in stabilizing the interface against dehydration. In general, the stability of the Pt/Fe(OH)x catalyst was highly dependent on the humidity of air. A decline in CO oxidation activity from 100% to 27% in 70 min occurred when the reaction was switched from humid air to dry air. The proposed reason behind the decrease in activity was the instability of the interfacial Fe3+–OH–Pt sites with respect to dehydration, considering the fact that CO oxidation is an exothermic process. Since Ni2+ can form a stable layered structure of Ni(OH)2 with nearly perfect octahedral coordination, it was incorporated into the Fe(OH)x sub-monolayer to prevent the dehydration-induced loss of Fe3+–OH–Pt sites. The TiO2-supported Pt/FeNi(OH)x catalyst was stable in the reaction stream for more than 28 h without any decrease in activity at room temperature and achieved 100% CO conversion (Fig. 27). The OH:O ratio in the Pt/FeNi(OH)x catalyst was maintained at 5.3 even after a 2 hour heat treatment at 453 K under the reaction atmosphere. DFT and isotope-labeling experiments revealed that the OH groups at the Fe3+–OH–Pt interfaces readily reacted with CO adsorbed nearby to directly convert the CO into CO2 and simultaneously produce coordinatively unsaturated Fe sites for O2 activation.
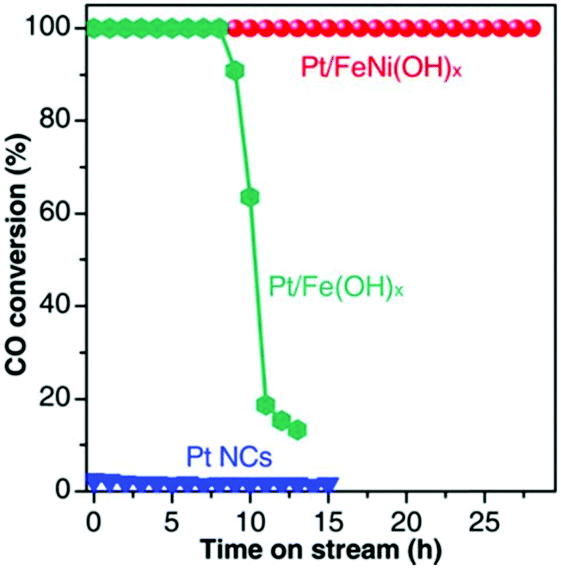 | ||
| Fig. 27 Catalytic performances of TiO2-supported Pt/FeNi(OH)x NPs, Pt NCs, and Pt/Fe(OH)x NPs as a function of time-on-stream. Reaction conditions: 1% CO; 16% O2; N2 balance; T = 303 K; SV = 400 L g−1 Pt h−1; relative humidity = 50%; pressure = 0.1 MPa.335 (Copyright 2014 American Association for the Advancement of Science.) | ||
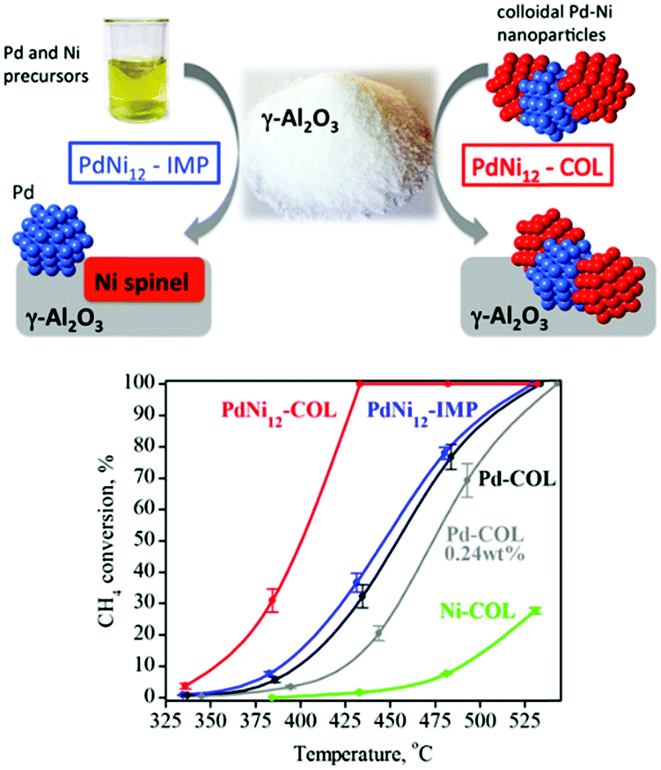 | ||
| Fig. 28 Schematic diagram for the preparation of bimetallic Ni–Pd catalysts and their catalytic performances in the methane combustion. Reaction conditions: 4100 ppm methane, 5 mol% water, a pressure of 1.1 bar, 1.2 mg of Pd, and 7.6 mg of Ni (0.029 wt% Pd and 0.190 wt% Ni loading in relevant catalysts, except for the 0.24 wt% Pd catalyst).329 (Copyright 2015 American Chemical Society.) | ||
Nickel is a good candidate to promote hydrodechlorination. Indeed, the catalytic properties of Ni have been investigated in the transformation of chlorinated aromatic compounds such as chlorobenzene.339,340 In the case of vicinal chloroalkanes (such as 1,2-dichloropropane), employing Ni catalysts involved more severe reaction conditions, i.e. high temperature and high hydrogen pressure, than those used for Pd and Pt catalysts.337 In this regard, Ni catalysts modified by noble metals such as Pd, Ru, Ag and Au are advantageous,341–346 where the noble metal shows high activity and Ni shows high selectivity. Simagina et al. reported liquid phase HDC of hexachlorobenzene over carbon supported Ni–Pd bimetallic catalysts, where the catalyst was less active than the pure Pd catalysts for the complete conversion of hexachlorobenzene into benzene, but presented a reasonable activity to obtain the compounds of interest, mono- and di-chlorobenzene.341 The degree of dechlorination was proportional to the surface Pd concentration. Interestingly, isolated Pd atoms located at the surface of Ni rich bimetallic particles were more active than those lying on larger ensembles.
Recently, bimetallic Ni–Cu catalysts were also tested for the HDC process to fully replace the use of noble metals.347–351 While hydrodechlorination of 1,2-dichloroethane over pure Ni mainly produces ethane, increasing the Cu content in the bimetallic catalysts results in an increase in ethylene selectivity. The specific consumption rate of 1,2-dichloroethane decreases when Cu loading increases, however, the turnover frequency seems to be independent of the surface composition of the alloy particles.
5. Conclusions and perspectives
The present contribution provides an overview on the design principles of different bimetallic nickel catalysts and their use in different types of catalysis processes related to energy production and environmental applications. In most cases, the role of the bimetallic surface and the role of each metal counterpart in the reaction have been critically discussed. Some reactions were favored and proceed on specific surfaces of the bimetallic catalysts. Consequently, continued efforts on the rational design of surface specific bimetallic catalysts, and the understanding of structure–activity relationships are critical to achieve superior catalytic performances.In addition to an appropriate catalyst design based on fundamental understanding, another major issue relates to the stability of bimetallic catalysts under the reaction conditions. It is often seen that at high reaction temperatures (such as under reforming conditions), the high diffusion rate of Ni over noble metals causes the destruction of the original structure of the bimetallic catalysts and the loss of its initial activity. Diffusion of Ni can be prevented by introducing a diffusion barrier layer into the surface Ni metal atoms.355,356 For instance, replacing bulk Pt with WC in Ni–Pt catalysts can retain the unique catalytic properties of the bimetallic surfaces, i.e. the subsurface Pt–Ni-WC structure for hydrogenation and the surface Ni-WC configuration for reforming reactions. We expect more of such systems to be developed in the future.
Coke and sulfur deposition are serious problems in hydrocarbon reforming processes. Although noble metals can significantly improve the coke or sulfur resistant properties of Ni-based catalysts, their high cost limits their application in industrial processes. Some efforts have been made with 3d transition metals, but their stability under high temperature conditions was not sufficient.107,110 One possible way to reduce catalyst costs without compromising their activity/stability could be the introduction of non-metal elements such as boron and phosphorus,357 since nickel-borides and phosphides have shown high activity and poison resistant properties towards different hydrotreating reactions.357,358 Alternatively, “noble-metal-like” compounds can be combined with Ni. For example, tungsten carbide (WC) could be a replacement for Pt since it exhibits “Pt-like” behavior and possesses high chemically stability, resistance to poisoning, and high electronic conductivity.359,360 We believe that the introduction of these non-metal additives can further improve the catalytic properties of simple bimetallic Ni–M systems, which is certainly an interesting area with room for further developments.
Acknowledgements
We thank the Young Investigator Award from the National University of Singapore (WBS: R-279-000-464-133) for the financial support.Notes and references
- B. Everett, G. Boyle, S. Peake and J. Ramage, Energy Systems and Sustainability: Power for a Sustainable Future, Oxford University Press, 2nd edn, 2012 Search PubMed.
- F. Liao, T. W. B. Lo and S. C. E. Tsang, ChemCatChem, 2015, 7, 1998–2014 CrossRef CAS.
- W. Yu, M. D. Porosoff and J. G. Chen, Chem. Rev., 2012, 112, 5780–5817 CrossRef CAS PubMed.
- D. Li, I. Atake, T. Shishido, Y. Oumi, T. Sano and K. Takehira, J. Catal., 2007, 250, 299–312 CrossRef CAS.
- K. Nagaoka, A. Jentys and J. A. Lercher, J. Catal., 2005, 229, 185–196 CrossRef CAS.
- D. Li, Y. Nakagawa and K. Tomishige, Appl. Catal., A, 2011, 408, 1–24 CrossRef CAS.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- H. Mu, L. Pan, D. Song and Y. Li, Chem. Rev., 2015, 115, 12091–12137 CrossRef CAS PubMed.
- C.-j. Liu, J. Ye, J. Jiang and Y. Pan, ChemCatChem, 2011, 3, 529–541 CrossRef CAS.
- S. Li and J. Gong, Chem. Soc. Rev., 2014, 43, 7245–7256 RSC.
- H. Zhang, M. Jin and Y. Xia, Chem. Soc. Rev., 2012, 41, 8035–8049 RSC.
- H.-l. Liu, F. Nosheen and X. Wang, Chem. Soc. Rev., 2015, 44, 3056–3078 RSC.
- S. K. Singh and Q. Xu, J. Am. Chem. Soc., 2009, 131, 18032–18033 CrossRef CAS PubMed.
- H.-L. Wang, J.-M. Yan, Z.-L. Wang, S.-I. O and Q. Jiang, J. Mater. Chem. A, 2013, 1, 14957–14962 CAS.
- M. K. Carpenter, T. E. Moylan, R. S. Kukreja, M. H. Atwan and M. M. Tessema, J. Am. Chem. Soc., 2012, 134, 8535–8542 CrossRef CAS PubMed.
- Y. Wu, S. Cai, D. Wang, W. He and Y. Li, J. Am. Chem. Soc., 2012, 134, 8975–8981 CrossRef CAS PubMed.
- J. Gu, Y.-W. Zhang and F. Tao, Chem. Soc. Rev., 2012, 41, 8050–8065 RSC.
- P. Nash, Phase diagrams of binary nickel alloys, ASM International, 1991 Search PubMed.
- A. I. Frenkel, Chem. Soc. Rev., 2012, 41, 8163–8178 RSC.
- J. C. Yang, M. W. Small, R. V. Grieshaber and R. G. Nuzzo, Chem. Soc. Rev., 2012, 41, 8179–8194 RSC.
- C. T. Campbell, Annu. Rev. Phys. Chem., 1990, 41, 775–837 CrossRef CAS.
- J. Rodriguez, Surf. Sci. Rep., 1996, 24, 223–287 CrossRef CAS.
- J. G. Chen, C. A. Menning and M. B. Zellner, Surf. Sci. Rep., 2008, 63, 201–254 CrossRef CAS.
- G. Chen, S. Desinan, R. Nechache, R. Rosei, F. Rosei and D. Ma, Chem. Commun., 2011, 47, 6308–6310 RSC.
- S. Carenco, C.-H. Wu, A. Shavorskiy, S. Alayoglu, G. A. Somorjai, H. Bluhm and M. Salmeron, Small, 2015, 11, 3045–3053 CrossRef CAS PubMed.
- A. B. Vysakh, C. L. Babu and C. P. Vinod, J. Phys. Chem. C, 2015, 119, 8138–8146 CAS.
- J. Xiang, P. Li, H. Chong, L. Feng, F. Fu, Z. Wang, S. Zhang and M. Zhu, Nano Res., 2014, 7, 1337–1343 CrossRef CAS.
- B. T. Sneed, A. P. Young, D. Jalalpoor, M. C. Golden, S. Mao, Y. Jiang, Y. Wang and C.-K. Tsung, ACS Nano, 2014, 8, 7239–7250 CrossRef CAS PubMed.
- M. B. Gawande, A. Goswami, T. Asefa, H. Guo, A. V. Biradar, D.-L. Peng, R. Zboril and R. S. Varma, Chem. Soc. Rev., 2015, 44, 7540–7590 RSC.
- H. N. Nong, L. Gan, E. Willinger, D. Teschner and P. Strasser, Chem. Sci., 2014, 5, 2955–2963 RSC.
- H. N. Nong, H.-S. Oh, T. Reier, E. Willinger, M.-G. Willinger, V. Petkov, D. Teschner and P. Strasser, Angew. Chem., Int. Ed., 2015, 54, 2975–2979 CrossRef CAS PubMed.
- S. K. Singh, A. K. Singh, K. Aranishi and Q. Xu, J. Am. Chem. Soc., 2011, 133, 19638–19641 CrossRef CAS PubMed.
- Y. W. Lee, M. Kim, S. W. Kang and S. W. Han, Angew. Chem., Int. Ed., 2011, 50, 3466–3470 CrossRef CAS PubMed.
- J. Zhang, H. Yang, J. Fang and S. Zou, Nano Lett., 2010, 10, 638–644 CrossRef CAS PubMed.
- J. Wu, L. Qi, H. You, A. Gross, J. Li and H. Yang, J. Am. Chem. Soc., 2012, 134, 11880–11883 CrossRef CAS PubMed.
- T. C. Deivaraj, W. Chen and J. Y. Lee, J. Mater. Chem., 2003, 13, 2555–2560 RSC.
- R. M. Arán-Ais, F. Dionigi, T. Merzdorf, M. Gocyla, M. Heggen, R. E. Dunin-Borkowski, M. Gliech, J. Solla-Gullón, E. Herrero, J. M. Feliu and P. Strasser, Nano Lett., 2015, 15, 7473–7480 CrossRef PubMed.
- P. A. Lin and R. M. Sankaran, Angew. Chem., Int. Ed., 2011, 50, 10953–10956 CrossRef CAS PubMed.
- M. Raney, US Pat., 1628190, 1927 Search PubMed.
- D. Wang, P. Zhao and Y. Li, Sci. Rep., 2011, 1, 37 Search PubMed.
- L. Chen, H. Guo, T. Fujita, A. Hirata, W. Zhang, A. Inoue and M. Chen, Adv. Funct. Mater., 2011, 21, 4364–4370 CrossRef CAS.
- J. Snyder, I. McCue, K. Livi and J. Erlebacher, J. Am. Chem. Soc., 2012, 134, 8633–8645 CrossRef CAS PubMed.
- L.-X. Ding, G.-R. Li, Z.-L. Wang, Z.-Q. Liu, H. Liu and Y.-X. Tong, Chem. – Eur. J., 2012, 18, 8386–8391 CrossRef CAS PubMed.
- Z. Li, R. Yu, J. Huang, Y. Shi, D. Zhang, X. Zhong, D. Wang, Y. Wu and Y. Li, Nat. Commun., 2015, 6, 8248 CrossRef CAS PubMed.
- R. Yi, R. Shi, G. Gao, N. Zhang, X. Cui, Y. He and X. Liu, J. Phys. Chem. C, 2009, 113, 1222–1226 CAS.
- J. R. Kitchin, J. K. Nørskov, M. A. Barteau and J. G. Chen, Phys. Rev. Lett., 2004, 93, 156801 CrossRef CAS PubMed.
- J. K. Norskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef CAS PubMed.
- L. Leppert, R. Kempe and S. Kummel, Phys. Chem. Chem. Phys., 2015, 17, 26140–26148 RSC.
- A. V. Ruban, H. L. Skriver and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 15990–16000 CrossRef.
- R. Mu, Q. Fu, H. Liu, D. Tan, R. Zhai and X. Bao, Appl. Surf. Sci., 2009, 255, 7296–7301 CrossRef CAS.
- C. A. Menning and J. G. Chen, J. Power Sources, 2010, 195, 3140–3144 CrossRef CAS.
- K. Mazloomi and C. Gomes, Renewable Sustainable Energy Rev., 2012, 16, 3024–3033 CrossRef CAS.
- A. Züttel, A. Remhof, A. Borgschulte and O. Friedrichs, Philos. Trans. R. Soc. London, Ser. A, 2010, 368, 3329–3342 CrossRef PubMed.
- U. Eberle, M. Felderhoff and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef CAS PubMed.
- S.-i. Orimo, Y. Nakamori, J. R. Eliseo, A. Züttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111–4132 CrossRef CAS PubMed.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS PubMed.
- D. Pakhare and J. Spivey, Chem. Soc. Rev., 2014, 43, 7813–7837 RSC.
- P. Makowski, A. Thomas, P. Kuhn and F. Goettmann, Energy Environ. Sci., 2009, 2, 480–490 CAS.
- M. Yadav and Q. Xu, Energy Environ. Sci., 2012, 5, 9698–9725 CAS.
- Z. Wei, J. Sun, Y. Li, A. K. Datye and Y. Wang, Chem. Soc. Rev., 2012, 41, 7994–8008 RSC.
- V. Dal Santo, A. Gallo, A. Naldoni, M. Guidotti and R. Psaro, Catal. Today, 2012, 197, 190–205 CrossRef CAS.
- C. Song, Catal. Today, 2002, 77, 17–49 CrossRef CAS.
- J. H. Sinfelt and D. J. C. Yates, J. Catal., 1967, 8, 82–90 CrossRef CAS.
- N. Bion, D. Duprez and F. Epron, ChemSusChem, 2012, 5, 76–84 CrossRef CAS PubMed.
- J. Sehested, Catal. Today, 2006, 111, 103–110 CrossRef CAS.
- H. S. Bengaard, J. K. Nørskov, J. Sehested, B. S. Clausen, L. P. Nielsen, A. M. Molenbroek and J. R. Rostrup-Nielsen, J. Catal., 2002, 209, 365–384 CrossRef CAS.
- D. L. Trimm, Catal. Today, 1997, 37, 233–238 CrossRef CAS.
- Y.-g. Chen, K. Tomishige, K. Yokoyama and K. Fujimoto, Appl. Catal., A, 1997, 165, 335–347 CrossRef CAS.
- K. Tomishige, S. Kanazawa, M. Sato, K. Ikushima and K. Kunimori, Catal. Lett., 2002, 84, 69–74 CrossRef CAS.
- S. Wang, G. Q. Lu and G. J. Millar, Energy Fuels, 1996, 10, 896–904 CrossRef CAS.
- Y. H. Hu and E. Ruckenstein, Advances in Catalysis, Academic Press, 2004, vol. 48, pp. 297–345 Search PubMed.
- Z. Hou, P. Chen, H. Fang, X. Zheng and T. Yashima, Int. J. Hydrogen Energy, 2006, 31, 555–561 CrossRef CAS.
- J. R. Rostrup-Nielsen, Catal. Today, 2000, 63, 159–164 CrossRef CAS.
- B. Steinhauer, M. R. Kasireddy, J. Radnik and A. Martin, Appl. Catal., A, 2009, 366, 333–341 CrossRef CAS.
- N. V. Parizotto, D. Zanchet, K. O. Rocha, C. M. P. Marques and J. M. C. Bueno, Appl. Catal., A, 2009, 366, 122–129 CrossRef CAS.
- M. García-Diéguez, E. Finocchio, M. Á. Larrubia, L. J. Alemany and G. Busca, J. Catal., 2010, 274, 11–20 CrossRef.
- M. García-Diéguez, I. S. Pieta, M. C. Herrera, M. A. Larrubia and L. J. Alemany, J. Catal., 2010, 270, 136–145 CrossRef.
- M. García-Diéguez, I. S. Pieta, M. C. Herrera, M. A. Larrubia and L. J. Alemany, Appl. Catal., A, 2010, 377, 191–199 CrossRef.
- S. R. de Miguel, I. M. J. Vilella, S. P. Maina, D. San José-Alonso, M. C. Román-Martínez and M. J. Illán-Gómez, Appl. Catal., A, 2012, 435–436, 10–18 CrossRef CAS.
- X. Yu, F. Zhang, N. Wang, S. Hao and W. Chu, Catal. Lett., 2013, 144, 293–300 CrossRef.
- L. Li, L. Zhou, S. Ould-Chikh, D. H. Anjum, M. B. Kanoun, J. Scaranto, M. N. Hedhili, S. Khalid, P. V. Laveille, L. D'Souza, A. Clo and J.-M. Basset, ChemCatChem, 2015, 7, 819–829 CrossRef CAS.
- C. Dai, S. Zhang, A. Zhang, C. Song, C. Shi and X. Guo, J. Mater. Chem. A, 2015, 3, 16461–16468 CAS.
- N. H. Elsayed, N. R. M. Roberts, B. Joseph and J. N. Kuhn, Appl. Catal., B, 2015, 179, 213–219 CrossRef CAS.
- V. S. Guggilla, J. Akyurtlu, A. Akyurtlu and I. Blankson, Ind. Eng. Chem. Res., 2010, 49, 8164–8173 CrossRef CAS.
- V. S. Guggilla, V. P. S. Mangalampalli, J. F. Akyurtlu and A. Akyurtlu, Ind. Eng. Chem. Res., 2013, 52, 338–345 CAS.
- M. Nowosielska, W. K. Jozwiak and J. Rynkowski, Catal. Lett., 2008, 128, 83–93 CrossRef.
- H. Arbag, S. Yasyerli, N. Yasyerli and G. Dogu, Int. J. Hydrogen Energy, 2010, 35, 2296–2304 CrossRef CAS.
- M. Ferrandon, A. J. Kropf and T. Krause, Appl. Catal., A, 2010, 379, 121–128 CrossRef CAS.
- A. Horváth, G. Stefler, O. Geszti, A. Kienneman, A. Pietraszek and L. Guczi, Catal. Today, 2011, 169, 102–111 CrossRef.
- M. Ocsachoque, F. Pompeo and G. Gonzalez, Catal. Today, 2011, 172, 226–231 CrossRef CAS.
- Z. Rui, D. Feng, H. Chen and H. Ji, Int. J. Hydrogen Energy, 2014, 39, 16252–16261 CrossRef CAS.
- V. L. Dagle, R. Dagle, L. Kovarik, A. Genc, Y.-G. Wang, M. Bowden, H. Wan, M. Flake, V.-A. Glezakou, D. L. King and R. Rousseau, Appl. Catal., B, 2016, 184, 142–152 CrossRef CAS.
- F. Besenbacher, I. Chorkendorff, B. S. Clausen, B. Hammer, A. M. Molenbroek, J. K. Nørskov and I. Stensgaard, Science, 1998, 279, 1913–1915 CrossRef CAS PubMed.
- A. M. Molenbroek, J. K. Nørskov and B. S. Clausen, J. Phys. Chem. B, 2001, 105, 5450–5458 CrossRef CAS.
- Y.-H. Chin, D. L. King, H.-S. Roh, Y. Wang and S. M. Heald, J. Catal., 2006, 244, 153–162 CrossRef CAS.
- I. Gavrielatos, V. Drakopoulos and S. G. Neophytides, J. Catal., 2008, 259, 75–84 CrossRef CAS.
- L. Guczi, G. Stefler, O. Geszti, I. Sajó, Z. Pászti, A. Tompos and Z. Schay, Appl. Catal., A, 2010, 375, 236–246 CrossRef CAS.
- N. V. Parizotto, K. O. Rocha, S. Damyanova, F. B. Passos, D. Zanchet, C. M. P. Marques and J. M. C. Bueno, Appl. Catal., A, 2007, 330, 12–22 CrossRef CAS.
- H. Jeong and M. Kang, Appl. Catal., B, 2010, 95, 446–455 CrossRef CAS.
- Y. Xu, C. Fan, Y.-A. Zhu, P. Li, X.-G. Zhou, D. Chen and W.-K. Yuan, Catal. Today, 2012, 186, 54–62 CrossRef CAS.
- D. Li, T. Shishido, Y. Oumi, T. Sano and K. Takehira, Appl. Catal., A, 2007, 332, 98–109 CrossRef CAS.
- K. Tomishige, J. Jpn. Pet. Inst., 2007, 50, 287–298 CrossRef CAS.
- H. Wu, V. La Parola, G. Pantaleo, F. Puleo, A. Venezia and L. Liotta, Catalysts, 2013, 3, 563 CrossRef CAS.
- L. Pleth Nielsen, F. Besenbacher, I. Stensgaard, E. Laegsgaard, C. Engdahl, P. Stoltze, K. W. Jacobsen and J. K. Nørskov, Phys. Rev. Lett., 1993, 71, 754–757 CrossRef PubMed.
- M. Rangan, M. M. Yung and J. W. Medlin, J. Catal., 2011, 282, 249–257 CrossRef CAS.
- C. Xie, Y. Chen, Y. Li, X. Wang and C. Song, Appl. Catal., A, 2010, 390, 210–218 CrossRef CAS.
- S. A. Theofanidis, V. V. Galvita, H. Poelman and G. B. Marin, ACS Catal., 2015, 5, 3028–3039 CrossRef CAS.
- P. Djinović, I. G. Osojnik Črnivec, B. Erjavec and A. Pintar, Appl. Catal., B, 2012, 125, 259–270 CrossRef.
- V. M. Gonzalez-delaCruz, R. Pereñiguez, F. Ternero, J. P. Holgado and A. Caballero, J. Phys. Chem. C, 2012, 116, 2919–2926 CAS.
- H. Ay and D. Üner, Appl. Catal., B, 2015, 179, 128–138 CrossRef CAS.
- W. An, X. C. Zeng and C. H. Turner, J. Chem. Phys., 2009, 131, 174702 CrossRef PubMed.
- S. M. Sidik, S. Triwahyono, A. A. Jalil, Z. A. Majid, N. Salamun, N. B. Talib and T. A. T. Abdullah, Chem. Eng. J., 2016, 295, 1–10 CrossRef CAS.
- H. Arbag, S. Yasyerli, N. Yasyerli, G. Dogu and T. Dogu, Appl. Catal., B, 2016, 198, 254–265 CrossRef CAS.
- X. Zhang, C. Yang, Y. Zhang, Y. Xu, S. Shang and Y. Yin, Int. J. Hydrogen Energy, 2015, 40, 16115–16126 CrossRef CAS.
- M. Abdollahifar, M. Haghighi, A. A. Babaluo and S. K. Talkhoncheh, Ultrason. Sonochem., 2016, 31, 173–183 CrossRef CAS PubMed.
- L. De Rogatis, T. Montini, B. Lorenzut and P. Fornasiero, Energy Environ. Sci., 2008, 1, 501–509 CAS.
- A. Le Valant, N. Bion, F. Can, D. Duprez and F. Epron, Appl. Catal., B, 2010, 97, 72–81 CrossRef CAS.
- G. Zhou, L. Barrio, S. Agnoli, S. D. Senanayake, J. Evans, A. Kubacka, M. Estrella, J. C. Hanson, A. Martínez-Arias, M. Fernández-García and J. A. Rodriguez, Angew. Chem., Int. Ed., 2010, 49, 9680–9684 CrossRef CAS PubMed.
- M. M. Rahman, T. L. Church, M. F. Variava, A. T. Harris and A. I. Minett, RSC Adv., 2014, 4, 18951–18960 RSC.
- R. M. Navarro, R. Guil-Lopez, A. A. Ismail, S. A. Al-Sayari and J. L. G. Fierro, Catal. Today, 2015, 242(Part A), 60–70 CrossRef CAS.
- D. Zanchet, J. B. O. Santos, S. Damyanova, J. M. R. Gallo and J. M. C. Bueno, ACS Catal., 2015, 5, 3841–3863 CrossRef CAS.
- P. Mierczynski, K. Vasilev, A. Mierczynska, W. Maniukiewicz, M. I. Szynkowska and T. P. Maniecki, Appl. Catal., B, 2016, 185, 281–294 CrossRef CAS.
- L. V. Mattos, G. Jacobs, B. H. Davis and F. B. Noronha, Chem. Rev., 2012, 112, 4094–4123 CrossRef CAS PubMed.
- K. O. Christensen, D. Chen, R. Lødeng and A. Holmen, Appl. Catal., A, 2006, 314, 9–22 CrossRef CAS.
- C. Zhang, P. Zhang, S. Li, G. Wu, X. Ma and J. Gong, Phys. Chem. Chem. Phys., 2012, 14, 3295–3298 RSC.
- R. I. Masel, Principles of Adsorption and Reaction on Solid Surfaces, Wiley-IEEE, New York, 1996 Search PubMed.
- T. S. Moraes, R. C. Rabelo Neto, M. C. Ribeiro, L. V. Mattos, M. Kourtelesis, S. Ladas, X. Verykios and F. B. Noronha, Appl. Catal., B, 2016, 181, 754–768 CrossRef CAS.
- M. C. Sanchez-Sanchez, R. M. Navarro Yerga, D. I. Kondarides, X. E. Verykios and J. L. G. Fierro, J. Phys. Chem. A, 2010, 114, 3873–3882 CrossRef PubMed.
- S. A. Tupy, A. M. Karim, C. Bagia, W. Deng, Y. Huang, D. G. Vlachos and J. G. Chen, ACS Catal., 2012, 2, 2290–2296 CrossRef CAS.
- M. C. Sanchez-Sanchez, R. M. Navarro, I. Espartero, A. A. Ismail, S. A. Al-Sayari and J. L. G. Fierro, Top. Catal., 2013, 56, 1672–1685 CrossRef CAS.
- R. M. Navarro, R. Guil-Lopez, J. M. Gonzalez-Carballo, A. Cubero, A. A. Ismail, S. A. Al-Sayari and J. L. G. Fierro, Appl. Catal., A, 2014, 474, 168–177 CrossRef CAS.
- C. He, J. Zheng, K. Wang, H. Lin, J.-Y. Wang and Y. Yang, Appl. Catal., B, 2015, 162, 401–411 CrossRef CAS.
- T. S. Moraes, R. C. R. Neto, M. C. Ribeiro, L. V. Mattos, M. Kourtelesis, S. Ladas, X. Verykios and F. Bellot Noronha, Catal. Today, 2015, 242(Part A), 35–49 CrossRef CAS.
- R. Pérez-Hernández, G. Mondragón Galicia, D. Mendoza Anaya, J. Palacios, C. Angeles-Chavez and J. Arenas-Alatorre, Int. J. Hydrogen Energy, 2008, 33, 4569–4576 CrossRef.
- P. K. Sharma, N. Saxena, A. Bhatt, C. Rajagopal and P. K. Roy, Catal. Sci. Technol., 2013, 3, 1017–1026 CAS.
- M. M. Rahman, Int. J. Hydrogen Energy, 2015, 40, 14833–14844 CrossRef CAS.
- A. J. Vizcaíno, A. Carrero and J. A. Calles, Int. J. Hydrogen Energy, 2007, 32, 1450–1461 CrossRef.
- A. Carrero, J. A. Calles and A. J. Vizcaíno, Appl. Catal., A, 2007, 327, 82–94 CrossRef CAS.
- P. Biswas and D. Kunzru, Catal. Lett., 2007, 118, 36–49 CrossRef CAS.
- J. A. Calles, A. Carrero and A. J. Vizcaíno, Microporous Mesoporous Mater., 2009, 119, 200–207 CrossRef CAS.
- F. Wang, Y. Li, W. Cai, E. Zhan, X. Mu and W. Shen, Catal. Today, 2009, 146, 31–36 CrossRef CAS.
- L.-C. Chen and S. D. Lin, Appl. Catal., B, 2011, 106, 639–649 CrossRef CAS.
- B. Christian Enger, R. Lødeng and A. Holmen, Appl. Catal., A, 2008, 346, 1–27 CrossRef.
- S. A. Al-Sayari, Open Catal. J., 2013, 6, 17–28 CrossRef CAS.
- P. Corbo and F. Migliardini, Int. J. Hydrogen Energy, 2007, 32, 55–66 CrossRef CAS.
- M. Shiraga, D. Li, I. Atake, T. Shishido, Y. Oumi, T. Sano and K. Takehira, Appl. Catal., A, 2007, 318, 143–154 CrossRef CAS.
- D. Li, M. Shiraga, I. Atake, T. Shishido, Y. Oumi, T. Sano and K. Takehira, Appl. Catal., A, 2007, 321, 155–164 CrossRef CAS.
- J. A. Velasco, C. Fernandez, L. Lopez, S. Cabrera, M. Boutonnet and S. Järås, Fuel, 2015, 153, 192–201 CrossRef CAS.
- H. E. Figen and S. Z. Baykara, Int. J. Hydrogen Energy, 2015, 40, 7439–7451 CrossRef CAS.
- P. Benito, V. Dal Santo, V. De Grandi, M. Marelli, G. Fornasari, R. Psaro and A. Vaccari, Appl. Catal., B, 2015, 179, 150–159 CrossRef CAS.
- K. Nakagawa, N. Ikenaga, Y. Teng, T. Kobayashi and T. Suzuki, Appl. Catal., A, 1999, 180, 183–193 CrossRef CAS.
- A. C. W. Koh, L. Chen, W. Kee Leong, B. F. G. Johnson, T. Khimyak and J. Lin, Int. J. Hydrogen Energy, 2007, 32, 725–730 CrossRef CAS.
- L. Li, P. Lu, Y. Yao and W. Ji, Catal. Commun., 2012, 26, 72–77 CrossRef CAS.
- L. De Rogatis, T. Montini, A. Cognigni, L. Olivi and P. Fornasiero, Catal. Today, 2009, 145, 176–185 CrossRef CAS.
- A. C. Ferreira, A. M. Ferraria, A. M. B. do Rego, A. P. Gonçalves, M. R. Correia, T. A. Gasche and J. B. Branco, J. Alloys Compd., 2010, 489, 316–323 CrossRef CAS.
- A. C. Ferreira, A. P. Gonçalves, T. A. Gasche, A. M. Ferraria, A. M. B. d. Rego, M. R. Correia, A. M. Bola and J. B. Branco, J. Alloys Compd., 2010, 497, 249–258 CrossRef CAS.
- S. K. Singh and Q. Xu, Catal. Sci. Technol., 2013, 3, 1889–1900 CAS.
- A. Klerke, C. H. Christensen, J. K. Norskov and T. Vegge, J. Mater. Chem., 2008, 18, 2304–2310 RSC.
- C. J. H. Jacobsen, S. Dahl, B. S. Clausen, S. Bahn, A. Logadottir and J. K. Nørskov, J. Am. Chem. Soc., 2001, 123, 8404–8405 CrossRef CAS PubMed.
- A. Boisen, S. Dahl, J. K. Nørskov and C. H. Christensen, J. Catal., 2005, 230, 309–312 CrossRef CAS.
- S.-F. Yin, Q.-H. Zhang, B.-Q. Xu, W.-X. Zhu, C.-F. Ng and C.-T. Au, J. Catal., 2004, 224, 384–396 CrossRef CAS.
- D. A. Hansgen, D. G. Vlachos and J. G. Chen, Nat. Chem., 2010, 2, 484–489 CrossRef CAS PubMed.
- W. Guo, M. Stamatakis and D. G. Vlachos, ACS Catal., 2013, 3, 2248–2255 CrossRef CAS.
- W. Guo and D. G. Vlachos, Nat. Commun., 2015, 6, 8619 CrossRef CAS PubMed.
- H. Dietrich, K. Jacobi and G. Ertl, J. Chem. Phys., 1996, 105, 8944–8950 CrossRef CAS.
- S. B. Simonsen, D. Chakraborty, I. Chorkendorff and S. Dahl, Appl. Catal., A, 2012, 447–448, 22–31 CrossRef CAS.
- L.-F. Zhang, M. Li, T.-Z. Ren, X. Liu and Z.-Y. Yuan, Int. J. Hydrogen Energy, 2015, 40, 2648–2656 CrossRef CAS.
- S. Wang, D. Zhang, Y. Ma, H. Zhang, J. Gao, Y. Nie and X. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 12429–12435 CAS.
- G. Chen, S. Desinan, R. Rosei, F. Rosei and D. Ma, Chem. – Eur. J., 2012, 18, 7925–7930 CrossRef CAS PubMed.
- G. P. Rachiero, U. B. Demirci and P. Miele, Int. J. Hydrogen Energy, 2011, 36, 7051–7065 CrossRef CAS.
- N. Cao, J. Su, W. Luo and G. Cheng, Int. J. Hydrogen Energy, 2014, 39, 426–435 CrossRef CAS.
- H.-L. Jiang, T. Umegaki, T. Akita, X.-B. Zhang, M. Haruta and Q. Xu, Chem. – Eur. J., 2010, 16, 3132–3137 CrossRef CAS PubMed.
- F. Qiu, G. Liu, L. Li, Y. Wang, C. Xu, C. An, C. Chen, Y. Xu, Y. Huang, Y. Wang, L. Jiao and H. Yuan, Chem. – Eur. J., 2014, 20, 505–509 CrossRef CAS PubMed.
- K. Mori, T. Taga and H. Yamashita, ChemCatChem, 2015, 7, 1285–1291 CrossRef CAS.
- J.-M. Yan, X.-B. Zhang, S. Han, H. Shioyama and Q. Xu, J. Power Sources, 2009, 194, 478–481 CrossRef CAS.
- H.-L. Jiang, T. Akita and Q. Xu, Chem. Commun., 2011, 47, 10999–11001 RSC.
- A. Yousef, N. A. M. Barakat, M. El-Newehy and H. Y. Kim, Int. J. Hydrogen Energy, 2012, 37, 17715–17723 CrossRef CAS.
- S. Cho, J. Lee, Y. Lee and D. Kim, Catal. Lett., 2006, 109, 181–186 CrossRef CAS.
- S. K. Singh, X.-B. Zhang and Q. Xu, J. Am. Chem. Soc., 2009, 131, 9894–9895 CrossRef CAS PubMed.
- S. K. Singh and Q. Xu, Chem. Commun., 2010, 46, 6545–6547 RSC.
- L. He, Y. Huang, X. Y. Liu, L. Li, A. Wang, X. Wang, C.-Y. Mou and T. Zhang, Appl. Catal., B, 2014, 147, 779–788 CrossRef CAS.
- A. K. Singh, M. Yadav, K. Aranishi and Q. Xu, Int. J. Hydrogen Energy, 2012, 37, 18915–18919 CrossRef CAS.
- J. Wang, X.-B. Zhang, Z.-L. Wang, L.-M. Wang and Y. Zhang, Energy Environ. Sci., 2012, 5, 6885–6888 CAS.
- J. B. Yoo, H. S. Kim, S. H. Kang, B. Lee and N. H. Hur, J. Mater. Chem. A, 2014, 2, 18929–18937 CAS.
- B. Xia, N. Cao, H. Dai, J. Su, X. Wu, W. Luo and G. Cheng, ChemCatChem, 2014, 6, 2549–2552 CrossRef CAS.
- J. Wang, W. Li, Y. Wen, L. Gu and Y. Zhang, Adv. Energy Mater., 2015, 5, 1401879 CrossRef.
- J. Chen, Q. Yao, J. Zhu, X. Chen and Z.-H. Lu, Int. J. Hydrogen Energy, 2016, 41, 3946–3954 CrossRef CAS.
- S. K. Singh and Q. Xu, Inorg. Chem., 2010, 49, 6148–6152 CrossRef CAS PubMed.
- L. He, Y. Huang, A. Wang, Y. Liu, X. Liu, X. Chen, J. J. Delgado, X. Wang and T. Zhang, J. Catal., 2013, 298, 1–9 CrossRef CAS.
- Y. Jiang, Q. Kang, J. Zhang, H.-B. Dai and P. Wang, J. Power Sources, 2015, 273, 554–560 CrossRef CAS.
- Y. Du, J. Su, W. Luo and G. Cheng, ACS Appl. Mater. Interfaces, 2015, 7, 1031–1034 CAS.
- Y.-Y. Jiang, H.-B. Dai, Y.-J. Zhong, D.-M. Chen and P. Wang, Chem. – Eur. J., 2015, 21, 15439–15445 CrossRef CAS PubMed.
- L. Wen, X. Du, J. Su, W. Luo, P. Cai and G. Cheng, Dalton Trans., 2015, 44, 6212–6218 RSC.
- N. Cao, L. Yang, H. Dai, T. Liu, J. Su, X. Wu, W. Luo and G. Cheng, Inorg. Chem., 2014, 53, 10122–10128 CrossRef CAS PubMed.
- N. Cao, J. Su, W. Luo and G. Cheng, Int. J. Hydrogen Energy, 2014, 39, 9726–9734 CrossRef CAS.
- A. K. Singh and Q. Xu, Int. J. Hydrogen Energy, 2014, 39, 9128–9134 CrossRef CAS.
- N. Cao, L. Yang, C. Du, J. Su, W. Luo and G. Cheng, J. Mater. Chem. A, 2014, 2, 14344–14347 CAS.
- S. K. Singh, Y. Iizuka and Q. Xu, Int. J. Hydrogen Energy, 2011, 36, 11794–11801 CrossRef CAS.
- D. Bhattacharjee, K. Mandal and S. Dasgupta, J. Power Sources, 2015, 287, 96–99 CrossRef CAS.
- S.-H. Wu and D.-H. Chen, J. Colloid Interface Sci., 2003, 259, 282–286 CrossRef CAS PubMed.
- D. G. Tong, X. L. Zeng, W. Chu, D. Wang and P. Wu, Mater. Res. Bull., 2010, 45, 442–447 CrossRef CAS.
- D. G. Tong, D. M. Tang, W. Chu, G. F. Gu and P. Wu, J. Mater. Chem. A, 2013, 1, 6425–6432 CAS.
- W. Gao, C. Li, H. Chen, M. Wu, S. He, M. Wei, D. G. Evans and X. Duan, Green Chem., 2014, 16, 1560–1568 RSC.
- K. V. Manukyan, A. Cross, S. Rouvimov, J. Miller, A. S. Mukasyan and E. E. Wolf, Appl. Catal., A, 2014, 476, 47–53 CrossRef CAS.
- J. Hannauer, O. Akdim, U. B. Demirci, C. Geantet, J.-M. Herrmann, P. Miele and Q. Xu, Energy Environ. Sci., 2011, 4, 3355–3358 CAS.
- Ç. Çakanyıldırım, U. B. Demirci, T. Şener, Q. Xu and P. Miele, Int. J. Hydrogen Energy, 2012, 37, 9722–9729 CrossRef.
- Q.-L. Zhu, D.-C. Zhong, U. B. Demirci and Q. Xu, ACS Catal., 2014, 4, 4261–4268 CrossRef CAS.
- X. Xiong, L. Zhou, G. Yu, K. Yang, M. Ye and Q. Xia, Int. J. Hydrogen Energy, 2015, 40, 15521–15528 CrossRef CAS.
- L. Zhang, L. Zhou, K. Yang, D. Gao, C. Huang, Y. Chen, F. Zhang, X. Xiong, L. Li and Q. Xia, J. Alloys Compd., 2016, 677, 87–95 CrossRef CAS.
- C. V. Ovesen, B. S. Clausen, B. S. Hammershøi, G. Steffensen, T. Askgaard, I. Chorkendorff, J. K. Nørskov, P. B. Rasmussen, P. Stoltze and P. Taylor, J. Catal., 1996, 158, 170–180 CrossRef CAS.
- A. A. Gokhale, J. A. Dumesic and M. Mavrikakis, J. Am. Chem. Soc., 2008, 130, 1402–1414 CrossRef CAS PubMed.
- N. Schumacher, A. Boisen, S. Dahl, A. A. Gokhale, S. Kandoi, L. C. Grabow, J. A. Dumesic, M. Mavrikakis and I. Chorkendorff, J. Catal., 2005, 229, 265–275 CrossRef CAS.
- C. A. Callaghan, S. A. Vilekar, I. Fishtik and R. Datta, Appl. Catal., A, 2008, 345, 213–232 CrossRef CAS.
- S.-C. Huang, C.-H. Lin and J. H. Wang, J. Phys. Chem. C, 2010, 114, 9826–9834 CAS.
- L.-Y. Gan, R.-Y. Tian, X.-B. Yang, H.-D. Lu and Y.-J. Zhao, J. Phys. Chem. C, 2012, 116, 745–752 CAS.
- J.-H. Lin and V. V. Guliants, Appl. Catal., A, 2012, 445–446, 187–194 CrossRef CAS.
- V. M. Shinde and G. Madras, Appl. Catal., B, 2012, 123–124, 367–378 CrossRef CAS.
- J.-H. Lin and V. V. Guliants, ChemCatChem, 2012, 4, 1611–1621 CrossRef CAS.
- A. R. S. Rad, M. B. Khoshgouei, S. Rezvani and A. R. Rezvani, Fuel Process. Technol., 2012, 96, 9–15 CrossRef CAS.
- E. T. Saw, U. Oemar, X. R. Tan, Y. Du, A. Borgna, K. Hidajat and S. Kawi, J. Catal., 2014, 314, 32–46 CrossRef CAS.
- Y. C. Huang, T. Zhou, H. Liu, C. Ling, S. Wang and J. Y. Du, ChemPhysChem, 2014, 15, 2490–2496 CrossRef CAS PubMed.
- E. T. Saw, U. Oemar, M. L. Ang, K. Hidajat and S. Kawi, ChemCatChem, 2015, 7, 3358–3367 CrossRef CAS.
- O. Arbeláez, T. R. Reina, S. Ivanova, F. Bustamante, A. L. Villa, M. A. Centeno and J. A. Odriozola, Appl. Catal., A, 2015, 497, 1–9 CrossRef.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- J. R. Greer, Science, 2014, 343, 1319–1320 CrossRef CAS PubMed.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, V. van der, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300–306 CrossRef PubMed.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- E. Ortel, T. Reier, P. Strasser and R. Kraehnert, Chem. Mater., 2011, 23, 3201–3209 CrossRef CAS.
- T. Reier, Z. Pawolek, S. Cherevko, M. Bruns, T. Jones, D. Teschner, S. Selve, A. Bergmann, H. N. Nong, R. Schlögl, K. J. J. Mayrhofer and P. Strasser, J. Am. Chem. Soc., 2015, 137, 13031–13040 CrossRef CAS PubMed.
- W.-F. Chen, K. Sasaki, C. Ma, A. I. Frenkel, N. Marinkovic, J. T. Muckerman, Y. Zhu and R. R. Adzic, Angew. Chem., Int. Ed., 2012, 51, 6131–6135 CrossRef CAS PubMed.
- W. Ma, R. Ma, C. Wang, J. Liang, X. Liu, K. Zhou and T. Sasaki, ACS Nano, 2015, 9, 1977–1984 CrossRef CAS PubMed.
- H. Chen, X. Huang, L.-J. Zhou, G.-D. Li, M. Fan and X. Zou, ChemCatChem, 2016, 8, 992–1000 CrossRef CAS.
- B. M. Hunter, W. Hieringer, J. R. Winkler, H. B. Gray and A. M. Muller, Energy Environ. Sci., 2016, 9, 1734–1743 CAS.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 CAS.
- C. van der Giesen, R. Kleijn and G. J. Kramer, Environ. Sci. Technol., 2014, 48, 7111–7121 CrossRef CAS PubMed.
- Y. Xuecheng, G. Han, Y. Dongjiang, Q. Shilun and Y. Xiangdong, Curr. Org. Chem., 2014, 18, 1335–1345 CrossRef.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 CAS.
- T. Inui and T. Takeguchi, Catal. Today, 1991, 10, 95–106 CrossRef CAS.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono and A. Ahmad, Green Chem., 2015, 17, 2647–2663 RSC.
- J. Gao, Q. Liu, F. Gu, B. Liu, Z. Zhong and F. Su, RSC Adv., 2015, 5, 22759–22776 RSC.
- S. Tada and R. Kikuchi, Catal. Sci. Technol., 2015, 5, 3061–3070 CAS.
- M. A. Vannice, J. Catal., 1975, 37, 449–461 CrossRef CAS.
- M. A. Vannice, J. Catal., 1975, 37, 462–473 CrossRef CAS.
- G. Zhi, X. Guo, Y. Wang, G. Jin and X. Guo, Catal. Commun., 2011, 16, 56–59 CrossRef CAS.
- H. Zhu, R. Razzaq, C. Li, Y. Muhmmad and S. Zhang, AIChE J., 2013, 59, 2567–2576 CrossRef CAS.
- S. Hwang, U. G. Hong, J. Lee, J. H. Baik, D. J. Koh, H. Lim and I. K. Song, Catal. Lett., 2012, 142, 860–868 CrossRef CAS.
- S.-H. Kang, J.-H. Ryu, J.-H. Kim, S.-J. Seo, Y.-D. Yoo, P. S. Sai Prasad, H.-J. Lim and C.-D. Byun, Korean J. Chem. Eng., 2011, 28, 2282–2286 CrossRef CAS.
- S. Hwang, U. G. Hong, J. Lee, J. G. Seo, J. H. Baik, D. J. Koh, H. Lim and I. K. Song, J. Ind. Eng. Chem., 2013, 19, 2016–2021 CrossRef CAS.
- D. Pandey and G. Deo, J. Mol. Catal. A: Chem., 2014, 382, 23–30 CrossRef CAS.
- D. Pandey and G. Deo, J. Ind. Eng. Chem., 2016, 33, 99–107 CrossRef CAS.
- D. Pandey and G. Deo, Chem. Eng. Commun., 2016, 203, 372–380 CrossRef CAS.
- J. Ren, X. Qin, J.-Z. Yang, Z.-F. Qin, H.-L. Guo, J.-Y. Lin and Z. Li, Fuel Process. Technol., 2015, 137, 204–211 CrossRef CAS.
- K. Zhao, Z. Li and L. Bian, Front. Chem. Sci. Eng., 2016, 1–8, DOI:10.1007/s11705-016-1563-5.
- F. Ocampo, B. Louis, L. Kiwi-Minsker and A.-C. Roger, Appl. Catal., A, 2011, 392, 36–44 CrossRef CAS.
- S. Hwang, J. Lee, U. G. Hong, J. H. Baik, D. J. Koh, H. Lim and I. K. Song, J. Ind. Eng. Chem., 2013, 19, 698–703 CrossRef CAS.
- W. Zhen, B. Li, G. Lu and J. Ma, RSC Adv., 2014, 4, 16472–16479 RSC.
- K. K. Ramasamy, M. Gray, H. Job and Y. Wang, Chem. Eng. Sci., 2015, 135, 266–273 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC.
- S. De, B. Saha and R. Luque, Bioresour. Technol., 2015, 178, 108–118 CrossRef CAS PubMed.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- P. Gallezot, P. J. Cerino, B. Blanc, G. Flèche and P. Fuertes, J. Catal., 1994, 146, 93–102 CrossRef CAS.
- A. Shrotri, A. Tanksale, J. N. Beltramini, H. Gurav and S. V. Chilukuri, Catal. Sci. Technol., 2012, 2, 1852–1858 CAS.
- J. Pang, A. Wang, M. Zheng, Y. Zhang, Y. Huang, X. Chen and T. Zhang, Green Chem., 2012, 14, 614–617 RSC.
- G. Liang, L. He, M. Arai and F. Zhao, ChemSusChem, 2014, 7, 1415–1421 CrossRef CAS PubMed.
- M.-Y. Zheng, A.-Q. Wang, N. Ji, J.-F. Pang, X.-D. Wang and T. Zhang, ChemSusChem, 2010, 3, 63–66 CrossRef CAS PubMed.
- K. Fabicovicova, O. Malter, M. Lucas and P. Claus, Green Chem., 2014, 16, 3580–3588 RSC.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- L. Hu, L. Lin and S. Liu, Ind. Eng. Chem. Res., 2014, 53, 9969–9978 CrossRef CAS.
- Y.-B. Huang, M.-Y. Chen, L. Yan, Q.-X. Guo and Y. Fu, ChemSusChem, 2014, 7, 1068–1072 CrossRef CAS PubMed.
- L. Yu, L. He, J. Chen, J. Zheng, L. Ye, H. Lin and Y. Yuan, ChemCatChem, 2015, 7, 1701–1707 CrossRef CAS.
- S. Sitthisa, W. An and D. E. Resasco, J. Catal., 2011, 284, 90–101 CrossRef CAS.
- Y. Yang, Z. Du, Y. Huang, F. Lu, F. Wang, J. Gao and J. Xu, Green Chem., 2013, 15, 1932–1940 RSC.
- B. Chen, F. Li, Z. Huang and G. Yuan, Appl. Catal., A, 2015, 500, 23–29 CrossRef CAS.
- F. M. A. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2010, 49, 5510–5514 CrossRef CAS PubMed.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS PubMed.
- Y. Yang, G. Gao, X. Zhang and F. Li, ACS Catal., 2014, 4, 1419–1425 CrossRef CAS.
- I. Obregón, I. Gandarias, N. Miletić, A. Ocio and P. L. Arias, ChemSusChem, 2015, 8, 3483–3488 CrossRef PubMed.
- M. Saidi, F. Samimi, D. Karimipourfard, T. Nimmanwudipong, B. C. Gates and M. R. Rahimpour, Energy Environ. Sci., 2014, 7, 103–129 CAS.
- S. Dutta, K. C. W. Wu and B. Saha, Catal. Sci. Technol., 2014, 4, 3785–3799 CAS.
- J. He, C. Zhao and J. A. Lercher, J. Am. Chem. Soc., 2012, 134, 20768–20775 CrossRef CAS PubMed.
- Q. Song, F. Wang and J. Xu, Chem. Commun., 2012, 48, 7019–7021 RSC.
- J. Zhang, H. Asakura, J. van Rijn, J. Yang, P. Duchesne, B. Zhang, X. Chen, P. Zhang, M. Saeys and N. Yan, Green Chem., 2014, 16, 2432–2437 RSC.
- J. Zhang, J. Teo, X. Chen, H. Asakura, T. Tanaka, K. Teramura and N. Yan, ACS Catal., 2014, 4, 1574–1583 CrossRef CAS.
- J. Zhang, M. Ibrahim, V. Collière, H. Asakura, T. Tanaka, K. Teramura, K. Philippot and N. Yan, J. Mol. Catal. A: Chem., 2016, 422, 188–197 CrossRef CAS.
- J. Zhang and N. Yan, Part. Part. Syst. Charact., 2016, 33, 610–619 CrossRef CAS.
- Y. Nakagawa and K. Tomishige, Catal. Commun., 2010, 12, 154–156 CrossRef CAS.
- Y. Wang, S. Sang, W. Zhu, L. Gao and G. Xiao, Chem. Eng. J., 2016, 299, 104–111 CrossRef CAS.
- S. G. Jadhav, P. D. Vaidya, B. M. Bhanage and J. B. Joshi, Chem. Eng. Res. Des., 2014, 92, 2557–2567 CrossRef CAS.
- J. Nerlov and I. Chorkendorff, J. Catal., 1999, 181, 271–279 CrossRef CAS.
- F. Studt, F. Abild-Pedersen, Q. Wu, A. D. Jensen, B. Temel, J.-D. Grunwaldt and J. K. Nørskov, J. Catal., 2012, 293, 51–60 CrossRef CAS.
- F. Zhao, M. Gong, Y. Zhang and J. Li, J. Porous Mater., 2016, 1–8, DOI:10.1007/s10934-016-0128-9.
- L. T. M. Nguyen, H. Park, M. Banu, J. Y. Kim, D. H. Youn, G. Magesh, W. Y. Kim and J. S. Lee, RSC Adv., 2015, 5, 105560 RSC.
- D. Preti, C. Resta, S. Squarcialupi and G. Fachinetti, Angew. Chem., Int. Ed., 2011, 50, 12551–12554 CrossRef CAS PubMed.
- S. Wesselbaum, U. Hintermair and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 8585–8588 CrossRef CAS PubMed.
- L. Wang and R. T. Yang, Energy Environ. Sci., 2008, 1, 268–279 CAS.
- G. Peng, S. J. Sibener, G. C. Schatz, S. T. Ceyer and M. Mavrikakis, J. Phys. Chem. C, 2012, 116, 3001–3006 CAS.
- G. Peng, S. J. Sibener, G. C. Schatz and M. Mavrikakis, Surf. Sci., 2012, 606, 1050–1055 CrossRef CAS.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- J. W. Hong, S. W. Kang, B.-S. Choi, D. Kim, S. B. Lee and S. W. Han, ACS Nano, 2012, 6, 2410–2419 CrossRef CAS PubMed.
- Y. Nie, L. Li and Z. Wei, Chem. Soc. Rev., 2015, 44, 2168–2201 RSC.
- M. Wang, W. Zhang, J. Wang, D. Wexler, S. D. Poynton, R. C. T. Slade, H. Liu, B. Winther-Jensen, R. Kerr, D. Shi and J. Chen, ACS Appl. Mater. Interfaces, 2013, 5, 12708–12715 CAS.
- J. Wu, S. Shan, V. Petkov, B. Prasai, H. Cronk, P. Joseph, J. Luo and C.-J. Zhong, ACS Catal., 2015, 5, 5317–5327 CrossRef CAS.
- S. Dresp, F. Luo, R. Schmack, S. Kuhl, M. Gliech and P. Strasser, Energy Environ. Sci., 2016, 9, 2020–2024 CAS.
- I. Danaee, M. Jafarian, A. Mirzapoor, F. Gobal and M. G. Mahjani, Electrochim. Acta, 2010, 55, 2093–2100 CrossRef CAS.
- N. A. M. Barakat and M. Motlak, Appl. Catal., B, 2014, 154–155, 221–231 CrossRef CAS.
- N. A. M. Barakat, M. Motlak, B.-S. Kim, A. G. El-Deen, S. S. Al-Deyab and A. M. Hamza, J. Mol. Catal. A: Chem., 2014, 394, 177–187 CrossRef CAS.
- X. Cui, W. Guo, M. Zhou, Y. Yang, Y. Li, P. Xiao, Y. Zhang and X. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 493–503 CAS.
- I. Danaee, M. Jafarian, F. Forouzandeh, F. Gobal and M. G. Mahjani, Int. J. Hydrogen Energy, 2008, 33, 4367–4376 CrossRef CAS.
- R. Ding, J. Liu, J. Jiang, F. Wu, J. Zhu and X. Huang, Catal. Sci. Technol., 2011, 1, 1406–1411 CAS.
- F. Gao, Y. Wang, Y. Cai and D. W. Goodman, J. Phys. Chem. C, 2009, 113, 174–181 CAS.
- L. Liu, F. Zhou, L. Wang, X. Qi, F. Shi and Y. Deng, J. Catal., 2010, 274, 1–10 CrossRef CAS.
- K. Liu, A. Wang and T. Zhang, ACS Catal., 2012, 2, 1165–1178 CrossRef CAS.
- K. Persson, A. Ersson, K. Jansson, N. Iverlund and S. Järås, J. Catal., 2005, 231, 139–150 CrossRef CAS.
- A. Iglesias-Juez, A. B. Hungría, A. Martínez-Arias, J. A. Anderson and M. Fernández-García, Catal. Today, 2009, 143, 195–202 CrossRef CAS.
- A. B. Hungría, N. D. Browning, R. P. Erni, M. Fernández-García, J. C. Conesa, J. A. Pérez-Omil and A. Martínez-Arias, J. Catal., 2005, 235, 251–261 CrossRef.
- A. B. Hungría, M. Fernández-García, J. A. Anderson and A. Martínez-Arias, J. Catal., 2005, 235, 262–271 CrossRef.
- A. B. Hungría, J. J. Calvino, J. A. Anderson and A. Martínez-Arias, Appl. Catal., B, 2006, 62, 359–368 CrossRef.
- S. E. Golunski and S. A. Roth, Catal. Today, 1991, 9, 105–112 CrossRef CAS.
- S. Shan, V. Petkov, L. Yang, J. Luo, P. Joseph, D. Mayzel, B. Prasai, L. Wang, M. Engelhard and C.-J. Zhong, J. Am. Chem. Soc., 2014, 136, 7140–7151 CrossRef CAS PubMed.
- Y. Hilli, N. M. Kinnunen, M. Suvanto, A. Savimäki, K. Kallinen and T. A. Pakkanen, Appl. Catal., A, 2015, 497, 85–95 CrossRef CAS.
- J. Shen, R. E. Hayes, X. Wu and N. Semagina, ACS Catal., 2015, 5, 2916–2920 CrossRef CAS.
- R. Mu, Q. Fu, H. Xu, H. Zhang, Y. Huang, Z. Jiang, S. Zhang, D. Tan and X. Bao, J. Am. Chem. Soc., 2011, 133, 1978–1986 CrossRef CAS PubMed.
- B. D. Chandler, C. G. Long, J. D. Gilbertson, C. J. Pursell, G. Vijayaraghavan and K. J. Stevenson, J. Phys. Chem. C, 2010, 114, 11498–11508 CAS.
- D. Wang and Y. Li, J. Am. Chem. Soc., 2010, 132, 6280–6281 CrossRef CAS PubMed.
- J. P. Holgado, F. Ternero, V. M. Gonzalez-delaCruz and A. Caballero, ACS Catal., 2013, 3, 2169–2180 CrossRef CAS.
- P. Kratzer, B. Hammer and J. K. Norskov, J. Chem. Phys., 1996, 105, 5595–5604 CrossRef CAS.
- G. Chen, Y. Zhao, G. Fu, P. N. Duchesne, L. Gu, Y. Zheng, X. Weng, M. Chen, P. Zhang, C.-W. Pao, J.-F. Lee and N. Zheng, Science, 2014, 344, 495–499 CrossRef CAS PubMed.
- M. A. Keane, ChemCatChem, 2011, 3, 800–821 CrossRef CAS.
- V. I. Kovalchuk and J. L. d’Itri, Appl. Catal., A, 2004, 271, 13–25 CrossRef CAS.
- M. Martin-Martinez, L. M. Gómez-Sainero, M. A. Alvarez-Montero, J. Bedia and J. J. Rodriguez, Appl. Catal., B, 2013, 132–133, 256–265 CrossRef CAS.
- N. Lingaiah, M. A. Uddin, A. Muto, T. Iwamoto, Y. Sakata and Y. Kusano, J. Mol. Catal. A: Chem., 2000, 161, 157–162 CrossRef CAS.
- K. V. Murthy, P. M. Patterson, G. Jacobs, B. H. Davis and M. A. Keane, J. Catal., 2004, 223, 74–85 CrossRef CAS.
- V. Simagina, V. Likholobov, G. Bergeret, M. T. Gimenez and A. Renouprez, Appl. Catal., B, 2003, 40, 293–304 CrossRef CAS.
- A. Śrbowata, W. Juszczyk, Z. Kaszkur and Z. Karpiński, Catal. Today, 2007, 124, 28–35 CrossRef.
- A. Śrbowata, M. Sadowska, W. Juszczyk, Z. Kaszkur, Z. Kowalczyk, M. Nowosielska and Z. Karpiński, Catal. Commun., 2007, 8, 11–15 CrossRef.
- G. Yuan, C. Louis, L. Delannoy and M. A. Keane, J. Catal., 2007, 247, 256–268 CrossRef CAS.
- Z. Wu, Z. Zhao and M. Zhang, ChemCatChem, 2010, 2, 1606–1614 CrossRef CAS.
- A. Śrbowata, I. Zielińska, R. Baran, G. Słowik and S. Dzwigaj, Catal. Commun., 2015, 69, 154–160 CrossRef.
- Y. H. Choi and W. Y. Lee, J. Mol. Catal. A: Chem., 2001, 174, 193–204 CrossRef CAS.
- S. Mallick, S. Rana and K. Parida, Ind. Eng. Chem. Res., 2011, 50, 12439–12448 CrossRef CAS.
- A. Śrbowata, R. Baran, S. Casale, I. I. Kamińska, D. Łomot, D. Lisovytskiy and S. Dzwigaj, Appl. Catal., B, 2014, 152–153, 317–327 CrossRef.
- R. Baran, A. Srebowata, S. Casale, D. Łomot and S. Dzwigaj, Catal. Today, 2014, 226, 134–140 CrossRef CAS.
- S. L. Pirard, J. G. Mahy, J.-P. Pirard, B. Heinrichs, L. Raskinet and S. D. Lambert, Microporous Mesoporous Mater., 2015, 209, 197–207 CrossRef CAS.
- J.-C. Chen, M.-Y. Wey, C.-L. Yeh and Y.-S. Liang, Appl. Catal., B, 2004, 48, 25–35 CrossRef CAS.
- F. Rahmaninejad, V. S. Gavaskar and J. Abbasian, Appl. Catal., B, 2012, 119–120, 297–303 CrossRef CAS.
- R. Jin, Y. Liu, Y. Wang, W. Cen, Z. Wu, H. Wang and X. Weng, Appl. Catal., B, 2014, 148–149, 582–588 CrossRef CAS.
- P. Gouy-Pailler and Y. Pauleau, J. Vac. Sci. Technol., A, 1993, 11, 96–102 CAS.
- H. H. Hwu and J. G. Chen, Chem. Rev., 2005, 105, 185–212 CrossRef CAS PubMed.
- S. Carenco, D. Portehault, C. Boissière, N. Mézailles and C. Sanchez, Chem. Rev., 2013, 113, 7981–8065 CrossRef CAS PubMed.
- R. Prins and M. E. Bussell, Catal. Lett., 2012, 142, 1413–1436 CrossRef CAS.
- S. T. Hunt, M. Milina, A. C. Alba-Rubio, C. H. Hendon, J. A. Dumesic and Y. Román-Leshkov, Science, 2016, 352, 974–978 CrossRef CAS PubMed.
- S. T. Hunt, T. Nimmanwudipong and Y. Román-Leshkov, Angew. Chem., Int. Ed., 2014, 53, 5131–5136 CAS.
| This journal is © The Royal Society of Chemistry 2016 |