
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron based catalysts from novel low-cost organic precursors for enhanced oxygen reduction reaction in neutral media microbial fuel cells†
Carlo
Santoro‡
a,
Alexey
Serov‡
a,
Lydia
Stariha
a,
Mounika
Kodali
a,
Jonathan
Gordon
a,
Sofia
Babanova
b,
Orianna
Bretschger
b,
Kateryna
Artyushkova
a and
Plamen
Atanassov
*a
aDepartment of Chemical and Biological Engineering, Center for Micro-Engineered Materials (CMEM) University of New Mexico, Albuquerque, NM 87131, USA. E-mail: plamen@unm.edu
bJ. Craig Venter Institute, 4120 Capricorn Lane, La Jolla, CA 92037, USA
First published on 13th May 2016
Abstract
Two iron-based platinum group metal-free catalysts for the oxygen reduction reaction (ORR) were synthesized from novel and low cost organic precursors named niclosamide and ricobendazole. These catalysts have been characterized, incorporated in a gas diffusional electrode and tested in “clean” conditions as well as in operating microbial fuel cell (MFC) for 32 days. Both catalysts demonstrated unprecedented performance yielding a power density 25% higher than that of platinum (Pt) and roughly 100% higher than activated carbon (AC) used as a control. Durability tests were performed and showed that Pt-based cathodes lost their activity within the first week of operation, reaching the level of the supporting AC-based electrode. Fe–ricobendazole, however, demonstrated the highest performance during the long-term study with a power density of 195 ± 7 μW cm−2 (day 2) that slightly decreased to 186 ± 9 μW cm−2 at day 29. Fe–niclosamide also outperformed Pt and AC but the power density roughly decreased with 20% for the 32 days of the study. Accelerated poisoning test using S2− as pollutant showed high losses in activity for Pt. Fe–niclosamide suffered higher losses compared to Fe–ricobendazole. Importantly, Fe–ricobendazole represents a 55-fold cost reduction compared to platinum.
Broader contextThe oxygen reduction reaction (ORR) is critical for fuel cells, metal air batteries and electrolysis technologies that use oxygen depolarization. Cost-effective non-platinum group metal (Non-PGM) catalysts have become of paramount interest as fuel cell technologies advance into hydrogen fuel cell-powered electric vehicles and other industrial applications. Microbial fuel cells and related microbial bio-reactors for energy harvesting and fuel generation are viewed as potentially disruptive technologies in wastewater treatment and other waste-to-energy applications. However, microbial fuel cells operate at a neutral pH and with solutions having many pollutants that cause conventional oxygen reduction catalysts to have low activity and durability. To address these issues and increase the overall power output from microbial fuel cells, we have developed innovative platinum-free iron based catalysts synthesized using low cost novel organic precursors. The evaluation of these materials resulted in power densities of about 200 μW cm−2, which are 20–25% higher than platinum-based catalysts and over 90% higher than previously reported activated carbon-based catalysts. Those catalysts resulted to be much more durable than platinum in operating MFCs. A cost-model analysis was performed for these novel catalysts and reveal their utility for practical large scale devices with a 55-fold cost reduction relative to Pt-based catalysts. |
Introduction
Bioelectrochemical systems have captured the attention of scientists in the past decade due to the possibility of transforming organic waste into electricity through biological, biochemical, and electrochemical reactions. In particular, microbial fuel cells (MFCs) utilize electroactive bacteria capable of degrading organic compounds and releasing electrons on the conductive anode electrode directly or through mediators or nanowires.1 Electrons move through an external circuit from the anode to the cathode where oxygen is reduced, generating electricity. Presently, high overpotentials and slow reaction kinetics of the oxygen reduction reaction (ORR) are one of the most limiting factors for practical implementation of MFCs.2 In fact, in a neutral environment, inorganic or carbonaceous catalysts suffer from tremendous activation overpotentials resulting in low catalytic activity. On the contrary, enzymes such as laccase or bilirubin oxidase have shown the lowest activation overpotentials at pH = 7,3,4 but the elevated cost and low endurance makes these enzymes unsuitable for MFC applications. Carbonaceous or metal-based catalysts have thus far been the most widely utilized catalysts for ORR in MFCs.5,6 The carbonaceous catalysts display interesting properties that make them well suited for MFC applications. The most important characteristic of these catalysts for enhancing ORR is undoubtedly their high surface area. Additionally, carbonaceous materials possess levels of mechanical strength, electrical conductivity, stability, low cost and durability in polluted environments all of which are advantageous for utilization in MFCs. Activated carbon (AC),7,8 carbon nanotubes,9 carbon nanofibers,10 and others11 have previously been reported as MFC cathode materials. Despite all these positive features, the activation overpotential of such catalysts remains high. These losses must be overcome to enhance MFC performance. Metal-based catalysts represent another avenue of exploration followed by the MFC field. Platinum (Pt) catalysts were initially used at the cathode of MFCs, but the extremely high cost12 compared to the electricity produced, and the presence of contaminants in wastewater that easily deactivate Pt active sites10,13,14 have steered efforts in the field toward the utilization of cheaper and more durable catalysts.5,6 The best catalyst substitutes have revealed themselves to be the so-called non-platinum group metal (non-PGM) M–N–C systems, where M is a transitional metal such as iron (Fe), manganese (Mn), cobalt (Co) or nickel (Ni) and N and C are nitrogen and carbon respectively.These transition metals are abundant, inexpensive and suitable for MFC applications.5,6 Several successful applications of the above materials as cathode catalysts in MFCs have been previously demonstrated.15–23 In our previous studies, we demonstrated the high performances of iron-aminoantipyrine (Fe–AAPyr) in double chamber MFCs,24 in ceramic separator MFC,25 and in single chamber MFCs.26 In this manuscript, we have synthesized two new catalysts based on iron and novel low cost organic precursors using the previously presented sacrificial support method (SSM).27,28 These novel catalysts have been integrated into an air breathing gas diffusion electrode and studied in “clean” conditions and integrated in working MFCs. Electrocatalytic activities were compared those of AC and Pt catalysts in an air-breathing MFC gas diffusion electrode, which were both used as controls. Additionally, durability studies were conducted, in which MFCs were run continuously for 32 days with periodic evaluation of the cathode performances.
Materials
The specifics of the SSM dictate that the precursors used for the synthesis should contain high amounts of carbon and nitrogen atoms. Full details on the selection can be found in our previous works.27,28 The structural formula of selected novel organic molecules is shown in Fig. 1a and b. | ||
| Fig. 1 The structural formula of organic precursors ricobendazole (a) and niclosamide (b). SEM images of Fe–ricobendazole derived catalyst at 15 K (c) and 30 K (d) magnification. High resolution N 1s XPS spectrum of Fe–ricobendazole catalyst (e) and Fe–niclosamide catalyst (f). | ||
A wet impregnation method was used for preparing the different Fe–N–C catalysts. In general, a calculated amount of iron precursor Fe(NO3)3·9H2O (Sigma Aldrich) was dispersed in a minimal amount of water and deposited onto the surface of fumed silica (Cab-O-Sil LM150, surface area ∼150 m2 g−1). The obtained mixture was stirred for 120 minutes resulting in a homogeneous colloidal solution. A solution of the organic precursor in water was added to Fe(NO3)3–SiO2 mixture and stirred for 20 minutes. Then the final mixture was dried overnight in an oven at T = 85 °C, solid material was ground in a mortar and treated at high temperature. The heat treatment (HT) step involved heating to 975 °C with a fast ramp rate of 10 °C min−1 under a nitrogen atmosphere generated by UHP nitrogen with flow rate 100 mL min−1. The pyrolysis time was selected as 45 minutes. After pyrolysis, the silica was removed by etching in 20 wt% solution of hydrofluoric acid (HF). The powder obtained was carefully washed with DI water and dried at T = 80 °C. The catalysts tested in this manuscript were named as a function of the precursor used: Fe–ricobendazole and Fe–niclosamide.
The cathodes were prepared by pressing a pellet based on activated carbon (AC, Norit SX Ultra, Sigma Aldrich), carbon black (CB, Alfa Aesar), PTFE (60 wt% solution, Sigma Aldrich) and our novel Fe–N–C catalysts. AC, CB, and PTFE were in weight percentages of 70%, 10% and 20% as reported previously.29 A mixture with 40 ± 2 mg cm−2 of the above materials was inserted into a metallic pellet die and compacted under a mechanical press at 3 mT for 5 minutes. The obtained cathode based on activated carbon (named as AC) was used as a control. In the case of catalyst utilization, the respective material was added to the above mixture, mixed vigorously and then pressed as previously described. The cathodes obtained had a loading of 2 ± 0.1 mg cm−2 Fe–N–C. Platinum on carbon (Pt/C, 50 wt% Alpha Aesar) was also mixed with AC + CB + PTFE to make additional cathodes (named as Pt) with the same catalyst loading of 2 ± 0.1 mg cm−2 for comparison with our Fe–N–C catalysts. The mixtures were pressed on a stainless steel (SS, McMaster) mesh that was used as a current collector. Cathodes were mounted on the on the flange of a glass bottle with geometric area exposed to the solution of 2.8 cm2. Preliminary cathode polarization curves were recorded in a “clean” abiotic environment: a solution of 0.1 M potassium phosphate buffer and 0.1 M KCl (K-PBS). After cathode polarization curves, cathodes were then inserted in running MFCs containing a solution of 50% by volume of K-PBS (0.1 M), and 50% by volume of activated sludge with the addition of 2 g L−1 of sodium acetate added from a stock solution. The anode was based on a high surface area carbon brush (Millirose, USA) with diameter and height of 3 cm (projected geometrical surface area of 42.4 cm2) occupying a theoretical volume of 21.2 cm3. The anode was chosen to be much larger than the cathode in order to make the cathode, which is actually the electrode of interest, the limiting factor of the MFC. In fact, the projected surface area of the anode was more than 3 times higher than the geometric area of the cathode (2.8 cm2). The anodes were previously colonized and running for 6 months (using acetate as a carbon source) prior to introducing the new cathodes. Consequently the anode was colonized by well-formed electroactive biofilm.
Method
The set-up used as an electrochemical cell and the three-electrodes configuration utilized for the electrochemical measurement have been reported previously.29 A potentiostat was used for the experiments with (i) cathode as the working electrode, (ii) titanium wire as a counter electrode; (iii) Ag/AgCl (3 M KCl) as a reference electrode. The cathodes were exposed to the solution (based on K-PB 0.1 M and 0.1 M KCl) for at least 16 hours before the experiments were carried out to allow a stable open circuit potential (OCP) to be reached. Linear sweep voltammetry (LSV) in the range between OCP and −0.35 V vs. Ag/AgCl at a scan rate of 0.2 mV s−1 was carried out for cathode characterization. An adjusted Luggin capillary was used to reduce the ohmic resistance of the solution during LSV measurements. After transferring the cathode into a working MFC and soaking it for least 6 hours to fully stabilize, overall MFC polarization curves were instead examined in the range between open circuit voltage (OCV) and 0 mV with a scan rate of 0.2 mV s−1. In this case, the anode was made to be the working electrode (WE), the cathode to be the counter electrode (CE), and Ag/AgCl (3 M KCl) was the reference electrode (RE). While running the overall polarization curves, profiles of the anode and cathode electrodes were recorded against a reference electrode. Durability tests were performed separately in MFCs for 32 days. These tests were started with new electrodes, which were connected to an external resistance (100 Ω) and the voltage collected through a data log system (Personal DAQ/56) every 20 minutes. The electrolyte was fully replaced every time the MFC voltage dropped below 50 mV indicating that the acetate was depleted. After a day of each solution replacement, at day 2, 10, 20 and 29, the MFCs were disconnected for at least 3 hours and polarization curves were measured between open circuit voltage (OCV) and 0 mV (scan rate of 0.2 mV s−1). Also in this case, the anode and cathode polarizations were run separately in order to understand the influence of each separate electrode. Current densities and power densities (P = V × I) were expressed as functions of the geometric area of the cathode. Abiotic LSVs were run in independent triplicates as well as were the initial (uninoculated) MFC baseline operation. Independent triplicates were run for the durability tests (32 days). The experiments were conducted in Albuquerque (NM, USA), which is located at 1600 meters above sea level with an atmospheric pressure roughly 20% lower than at the sea level, which resulted in a lower partial pressure of oxygen.Accelerated poisoning tests were conducted via chronoamperometry in a three electrodes configuration using Ti-wire as counter electrode, the cathode as the working electrode and Ag/AgCl (3 M KCl) as the reference electrode. Cathode potential was kept constant at −0.1 V (vs. 3 M Ag/AgCl). The solution was based on 0.1 M potassium phosphate buffer and 0.1 M KCl (K-PBS). After 1 hour of stable current production, 0.5 mM, 1 mM, 2 mM, 3 mM, 4 mM and 5 mM of Na2S were added in the solution every 30 minutes. The current was then normalized and current losses (μA) over time as a function of S2− concentration were plotted.
The bulk and individual particle morphology of the synthesized catalysts were characterized using scanning electron microscopy (SEM). XPS measurements were performed on powders, as well as fresh and tested electrodes using a Kratos Axis Ultra DLD X-ray photoelectron spectrometer using a monochromatic Al Ka source operating at 225 W. Survey and high-resolution spectra were acquired at pass energies of 80 and 20 eV, respectively. Three areas per sample were analysed. No charge compensation was necessary. Data analysis and quantification were performed using the CASAXPS software. A linear background was used for C 1s, F 1s, N 1s, S 2p and O 1s spectra. Quantification utilized sensitivity factors that were provided by the manufacturer. A 70% Gaussian/30% Lorentzian [GL(30)] line shape was used for the curve fits.
Results and discussion
Morphology and surface chemistry of the novel catalysts
Morphology of Fe–ricobendazole (Fig. 1c and d) and Fe–niclosamide (not shown) were found to be similar with previously synthesized SSM materials.27,28 XPS analysis detected about 3% of atomic nitrogenwith a distribution of different types of nitrogen species typical for M–N–Cs obtained by SSM.27,28 Pyridinic nitrogen and nitrogen coordinated to metal, which have been shown to be important species for ORR27,28 were detected in significant amounts as shown in Fig. 1e and f. Importantly, the Fe–riconbendazole catalyst displayed a larger pyridinic to pyrrolic ratio than Fe–niclosamide which is an important metric of ORR activity in acidic media30 as well as in neutral media.21Initial electrochemical performance
Initial electrochemical performance measurements were done before connecting the MFCs to the external resistance. For simplicity the results from these measurements will be named day 0. Despite the fact that cathodic OCPs of approximately 300 mV vs. Ag/AgCl are among the highest reported for MFCs, the theoretical value for the ORR at pH 7.5 is approximately 586 mV (vs. Ag/AgCl), indicating that the overpotential still remains considerably high. Cathode polarization curves conducted in “clean” abiotic conditions are shown in Fig. 2a. Our measurements show that Fe–ricobendazole and Fe–niclosamide display higher catalytic activity than Pt despite identical loadings investigated and with the added benefit of a much lower cost. The differences between the data obtained from two Fe-based cathodes were negligible indicating excellent reproducibility within three replicates. In general, at day 0, the MFCs had a similar OCV around 719 ± 18 mV, with the exception of the MFCs equipped with AC-based cathodes, which exhibited slightly lower OCV (661 ± 9 mV) (Fig. 2b). Power curves confirmed the previously identified trend of the cathode polarization curves in clean conditions (Fig. 2a). At day 0, Fe–ricobendazole containing MFCs (204 ± 10 μW cm−2) had the highest power density recorded followed by Fe–niclosamide (197 ± 8 μW cm−2), Pt (167 ± 5 μW cm−2) and AC (108 ± 6 μW cm−2) (Fig. 2c). It should be noted that the difference in power generation was due only to cathode performances since the anodes performed nearly identically (Fig. 2d). | ||
| Fig. 2 Cathode polarization curves in K-PBS (a), overall MFC polarization curves (b), power curves (c) and anode and cathode polarization trend (d) at day 0. | ||
Long-term operation
Durability test were carried out for 32 days to track the stability of the cathodes over time under operational conditions. The durability tests were performed utilizing MFCs with pre-colonized anodes and an electrolyte consisting of a mixture of K-PBS (0.1 M) and activated sludge with sodium acetate (2 g L−1) as a carbon source. The electrolyte composition was designed to simulate a real wastewater with “harsh” and polluted conditions. The latter contains a large amount of bacteria, various pollutants and organic compounds. Bacteria and pollutants are well known to decrease the cathode performance.31 Bacteria can quickly colonize the cathode surface and dramatically hinder the proton mass transfer from the electrolyte to the catalyst.32 In addition the pollutants present in wastewater (e.g. sulfur) can permanently bind to the catalyst surface (e.g. platinum) deactivating the catalyst centers.33 Each MFC type was tested in triplicate. The utilization of pre-colonized anodes, allowed immediate MFC operation instead of the 2–3 weeks start up time necessary for electroactive biofilm formation and establishment. The voltage generated by the MFCs with the different cathode catalysts (Fig. 3) showed a slight initial decrease most likely due to a combination of several factors: (i) initial adaptation of the cathode to the solution and progressive loss of hydrophobicity; (ii) consumption of oxygen adsorbed on the substrate surface; (iii) adsorption and binding of ions on the catalytic sites; (iv) initial precipitation of compounds on the cathode surface; and (v) bacteria colonization of the cathode.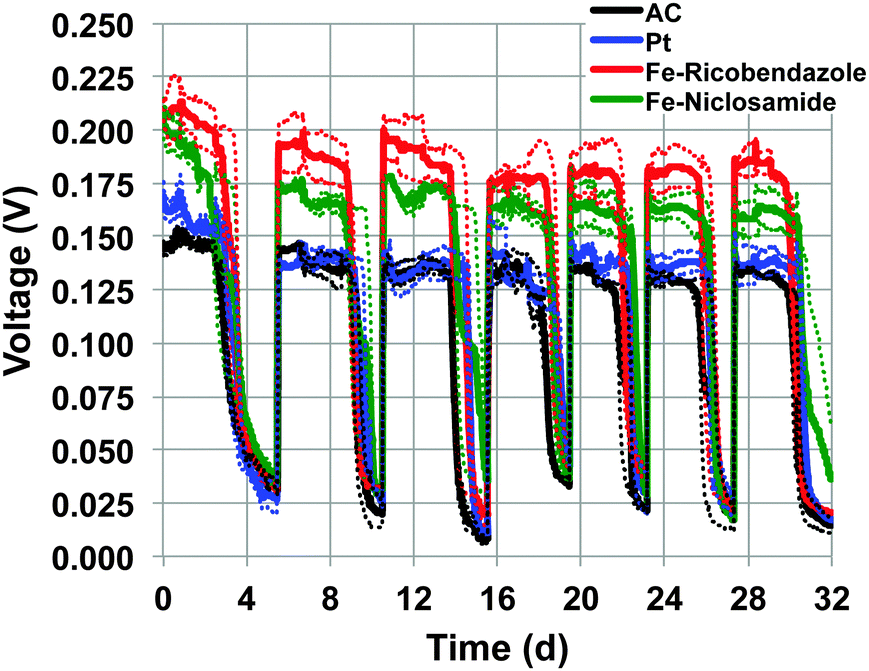 | ||
| Fig. 3 Voltage trend recorded for 32 days for MFCs equipped with brush anodes and four different catalysts at the cathodes as specified in the figure legend. The voltage was recorded across a 100 Ω resistor. | ||
As it can be seen in Fig. 3, Fe–ricobendazole had the highest recorded voltage within the four weeks of operation. The initial voltage was 212 ± 13 mV (first day) and stabilized to a slightly lower value of 201 ± 12 mV during the second week and 183 ± 16 mV for the remainder of the study. The decrease of the voltage during the 32 days of operation was within 15%. Fe–niclosamide had the second highest voltage recorded. The initial voltage for Fe–niclosamide was 205 ± 7 mV (first day) and dropped more significantly compared to Fe–ricobendazole (180 ± 5 mV second cycle) and stabilized during the additional cycles to 163 ± 11 mV. The decrease of the Fe–niclosamide voltage during the 32 days of operation was roughly 20%. Initially, Pt had higher voltage output (170 ± 8 mV, first day) compared to AC (152 ± 11 mV, first day), however, after several days, Pt and AC displayed similar voltage output during the remainder of the experiment, which stabilized at 135 ± 15 mV. The decreased performance of Pt was likely due to the deactivation of its active centers caused by anion poisoning. Once Pt lost its activity, the voltage trend followed that of the AC-based MFC because AC was used as the support material for the Pt-cathode as well. The relatively stable voltage trend for the AC-based cathode is in agreement with previously reported data in which AC showed a 25% decrease in activity during 1 year of operation.31 On the other hand, the results in our study are in disagreement with previous findings that report AC catalysts performing better than Pt during long-term experiments. However, it has been reported that the Pt catalyst decreased in activity significantly and yielded less than half of the AC activity after 3.5 months experiments.34 This disagreement can be attributed to the fact that the Pt cathode in the Zhang's study was built in combination with carbon black (CB), not AC, and once Pt was poisoned, only the CB substrate was acting as catalyst for ORR. The supremacy of AC compared to CB and ORR in neutral media has been shown previously.35
Power trend during long-term operation
Polarization curves of the different MFCs were carried out periodically to monitor the maximum power trend during the course of the experiment. The maximum power generated by the MFCs strictly followed the voltage trend, as shown in Fig. 4. Power and polarization curves of the MFCs at day 2, 10, 20 and 29 are reported separately in the ESI† (Fig. S1). Fe–ricobendazole had the highest recorded maximum power (195 ± 7 μW cm−2) after day two, followed by Fe–niclosamide (184 ± 8 μW cm−2). Fe-based MFCs had higher performance than Pt (145 ± 3 μW cm−2) and AC (120 ± 1 μW cm−2) during the first cycle. Fe–ricobendazole decreased by 6.5% at day 10 (183 ± 7 μW cm−2) but the power output stabilized by day 20 (182 ± 11 μW cm−2) and day 29 (186 ± 9 μW cm−2). Fe–ricobendazole was shown to be the most efficient catalyst losing no more than 10% of its power density during the 32 days of operation. The power density of Fe–niclosamide decreased by 11% at day 10 (167 ± 6 μW cm−2) and continuously decreased to 159 ± 9 μW cm−2 at day 20 and 156 ± 7 μW cm−2 at day 29. The overall power density of Fe–niclosamide decreased by 18% during the course of the investigation. During the first cycle, Pt outperformed AC but Pt lost its activity over time (especially after the first cycle) and Pt and AC had comparable performances after just a few days of operation (Fig. 4). Pt and AC had power densities of 121 ± 13 μW cm−2 and 114 ± 11 μW cm−2, respectively. The decrease in the Pt-based MFC output was 23%.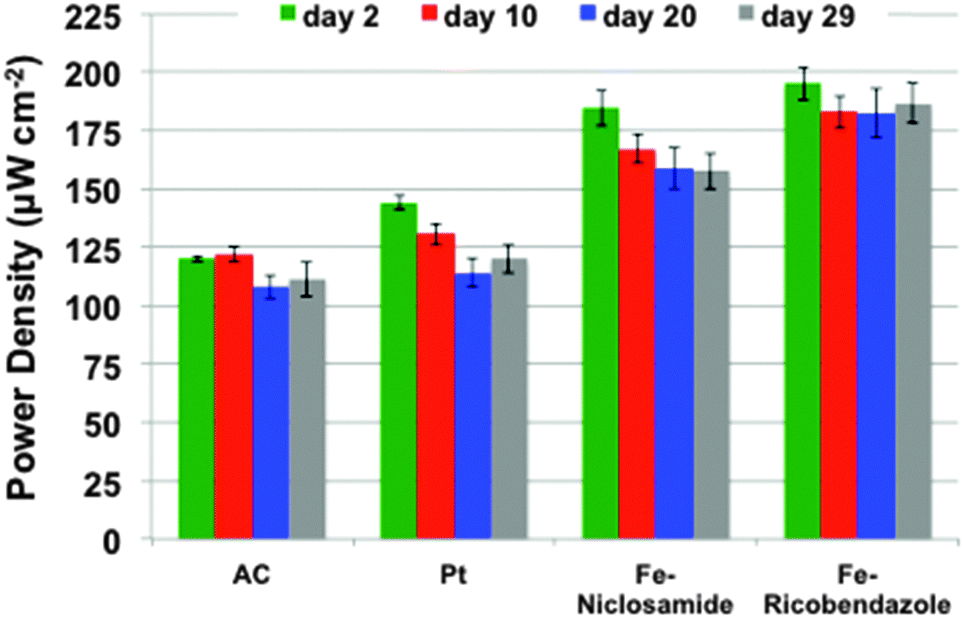 | ||
| Fig. 4 Power density of the MFCs measured after 2, 10, 20 and 29 days of continuous operation where AC, Pt, Fe–ricobendazole, and Fe–niclosamide represent MFCs loaded with each respective catalyst. | ||
Accelerated poisoning tests
Different pollutants are naturally and commonly present in wastewater or activated sludge. Several of these mainly based on sulphur, are well known to attack the platinum catalytic active centers and significantly diminishing their activity in a relatively short amount of time.33It is known from the literature that S2− poisons Pt and it is the main reason for Pt degradation in MFC systems. In this work, accelerated poisoning tests of Fe–ricobendazole, Fe–niclosamide and AC catalyst were performed and compared to Pt (Fig. S2, ESI†). Current losses expressed as a function of the concentration of S2− are presented in Fig. 5. It must be noted that AC and Fe–ricobendazole have a current density loss of roughly 100 μA cm−2 at 5 mM S2− concentration. Higher current losses were detected for Fe–niclosamide with losses of 160 μA cm−2 at 5 mM S2− concentration. As expected, the current losses of Pt were the most pronounced among the samples investigated. Pt showed a loss of current density of 400 μA cm−2 at 5 mM S2− concentration, which was 4 times more current loss than AC and Fe–ricobendazole and 2.5 times more current loss than Fe–niclosamide. These results support the measurements obtained from the durability tests (Fig. 3) in which Pt lost its activity in a very short amount of time and Fe–niclosamide had lower performances compared to Fe–ricobendazole, which is likely due to the low tolerance for S2−. XPS was performed on the cathodes of the non-platinum based catalyst, before and after the accelerated poisoning test. The results of those experiments are shown in Table 1. The initial composition of M–N–C-containing electrodes had high amounts of fluorine from PTFE and carbon from the catalyst, AC, CB and PTFE. PTFE showed significant oxidation possibly due to interaction with the other constituents of the mixture. In the high resolution F 1s spectra of the electrodes, a peak at 698.5 eV was observed due to the main PTFE CF2 chain. A peak at 692.5 eV was also observed, which was due to the oxidized CF2 chains that were present. In the C 1s spectrum, graphitic carbon peaks (at 284.4 eV) and aliphatic carbon peaks (at 285 eV) were detected in significant amounts along with surface oxides (peaks between 286 and 288 eV). PTFE contributes to a higher binding energy range with peaks due to the main chain around 290 eV and peaks due to oxidized PTFE between 291 eV and 295 eV.
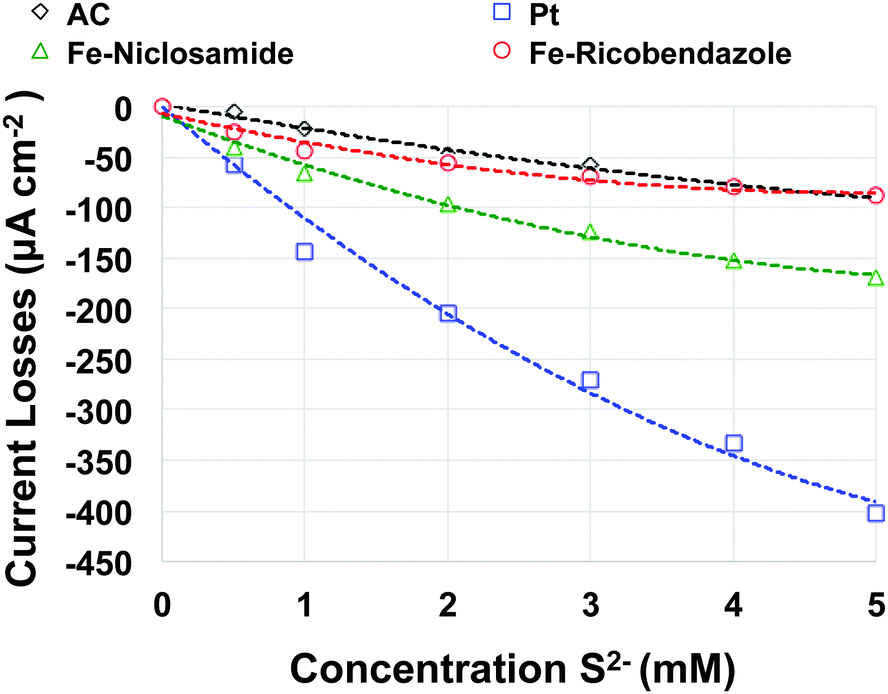 | ||
| Fig. 5 Current losses as a function of the concentration of S2− present in the electrolyte for the catalysts investigated. | ||
| Sample | Elemental composition | Carbon speciation | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| C 1s | F 1s | N 1s | O 1s | S 2p | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C C |
C–C | CxOy | PTFE | PTFE oxidized | |
| Fe–NICLO fresh | 51.8 | 45.2 | 0.3 | 2.7 | 34.0 | 20.7 | 23.4 | 8.4 | 13.5 | |
| Fe–NICLO after test | 78.7 | 14.2 | 2.1 | 4.9 | 0.16 | 37.9 | 20.6 | 26.5 | 8.3 | 6.7 |
| Fe–RICO fresh | 44.5 | 52.5 | 0.2 | 2.8 | 41.1 | 21.6 | 21.5 | 7.0 | 8.8 | |
| Fe–RICO after test | 65.7 | 29.9 | 1.1 | 3.3 | 0.05 | 31.4 | 23.9 | 21.5 | 14.3 | 8.9 |
| Sample | Flourine speciation | Nitrogen speciation | ||||||
|---|---|---|---|---|---|---|---|---|
| CF2 | CFxOy | N imine | N pyrid | N–Me | N pyrrolic | N gr | NOx | |
| Fe–NICLO fresh | 35.4 | 64.6 | 7.3 | 13.3 | 14.0 | 36.0 | 16.4 | 12.9 |
| Fe–NICLO after test | 51.3 | 48.7 | 6.3 | 18.5 | 17.0 | 27.5 | 13.7 | 16.9 |
| Fe–RICO fresh | 40.4 | 59.6 | 5.1 | 8.7 | 11.5 | 36.7 | 19.2 | 18.8 |
| Fe–RICO after test | 61.7 | 38.3 | 5.0 | 18.6 | 19.9 | 27.0 | 13.6 | 15.7 |
Nitrogen is naturally present from the M–N–C electrocatalysts themselves as a result of the precursors. Smaller than expected atomic % N was detected in the uninoculated electrodes, which can be explained by the segregation of PTFE to the top surface. Both M–N–C electrodes have similar initial speciation of nitrogen compounds, as shown in Fig. 1. The XPS analysis of tested electrodes was also performed after the accelerated poisoning test, and a significant change in composition can be noticed. This is likely due to the loss of PTFE and the rearrangement of the catalyst and PTFE within the top 8–10 nm of the analysis sampling depth. The decrease in F detected after the test indicates a partial loss of PTFE. The loss is significantly larger for Fe–niclosamide compared to Fe–ricobendazole. In the case of Fe–niclosamide, a decrease of both the main chain of PTFE and oxidized PTFE is observed to a large extent, while most of the main chains of PTFE stay intact, while the oxidized part of PTFE decreases significantly in Fe–ricobendazole. Carbon speciation after testing confirms the higher amount of PTFE preserved in the Fe–ricobendazole-based catalyst. Electrodes tested after exposure to S2− exhibit larger atomic % of nitrogen at amounts usually present in M–N–C based electrocatalysts, due to better mixing of the PTFE and the partial loss of its oxidized portions. After the accelerated poisoning test, smaller amounts of graphitic and pyrrolic types of nitrogen are observed, and higher amounts of pyridinic and nitrogen coordinated to Fe are detected. This final structure of the catalyst is characteristic for a material with the optimal composition with high amounts of active centers such as pyridinic and Nx–Fe nitrogen moieties. After the poisoning test, 0.16% of S was detected in Fe–niclosamide, while only 0.05% was detected in Fe–ricobendazole. Larger amounts of S along with the more significant loss of oxidized PTFE for the Fe–niclosamide compared to Fe–ricobendazole, explains the higher stability of the Fe–ricobendazole-based material.
Comparison with existing literature
The results obtained in this work are among the highest ever shown in the literature during 32 days of operate using Fe–N–C catalysts. Previous reports have demonstrated 270 μW cm−2 using carbonate buffer solution, Pt catalyst, in a 26 mL volume MFC.36 In this case, no durability tests were reported.29 To the best of our knowledge, there are few reports of non-PGM catalysts mixed with AC in air-breathing gas diffusion electrode configurations. In the few that exist, Fe, Co and Mn were used as the non precious metal (M) in the M–C–N catalyst. Particularly, concerning Fe-based catalysts, Fe–EDTA was used as a catalyst mixed with AC and pressed on a stainless steel current collector.22 Results achieved showed a power density of 158 μW cm−2, which was 10% higher relative to the plain AC control.22 In another case, Fe3O4 was incorporated with AC into an air-breathing cathode which produced a maximum power density of 143 μW cm−2. Few others manuscripts illustrate the utility of Co-based catalysts incorporated within an AC matrix for enhancing ORR compared to plain AC alone.37 The MFCs with Co3O4/AC reached a maximum power density of 142.1 ± 5.4 μW cm−2.38 Similar results have been demonstrated using the same CO3O4/AC catalyst for cathode ORR by Ge et al.39 Also, MnO2 mixed with AC has been utilized as a cathode catalyst, yielding a power density of 155 μW cm−2.40 The majority of the works report on initial cathode performances, in relatively “clean” conditions and not in long-term experiments. Only a few studies address the durability of cathodes for periods of time longer than 30 days. However, long-term experiments have been conducted on AC for a period of 12 months.31 The results of these experiments showed that AC cathodes had relatively high and mostly stable performance with a loss in power density of roughly 25% in 12 months due to biofouling. In another study, the same research group, tested plain AC, AC mixed with carbon black (CB), heat treated AC and AC mixed with Fe–EDTA.34 The authors found a decrease of 10–16% in the performance during the first month of operation which is in agreement with the current data presented despite the fact that the authors utilized a simple phosphate buffer solution with no addition of bacteria or calcium/magnesium that could precipitate on the cathode. Moreover, after 5.5 months, all the materials lost roughly 30% of their initial performance. This was attributed to the biofilm growth and biofouling of the cathode. In a previous study using micro-tomography, it was shown that the main contribution of cathode losses was carbonate precipitation.32 At the end of the experiments (after 32 days), the MFC systems were disassembled and photographs were taken of the hydrated and fully dried cathodes (Fig. S3, ESI†). The images suggest that a thick biofilm had developed on the cathodes and fouled the surfaces (Fig. S3, ESI†).Outlook
In this study, we showed that cathodes containing Fe–ricobenzadole, and Fe–niclosamide containing cathodes outperformed those containing Pt and AC-based cathodes during initial evaluation in clean conditions and in MFC operation in bottle reactors. The highest power generation reported is 270 μW m−2 using carbonate buffer solution and a Pt catalyst, in a 26 mL volume MFC of a different design.36 To the best of our knowledge, the results presented here reflect the highest power density ever reported for single bottle MFCs with a volume larger than 0.1 L with phosphate buffer and activated sludge solution and acetate as the fuel source. The initial power densities achieved in this study were 20–30% higher than the previously reported performance of Fe–AAPyr catalysts26 evaluate in the same bottle systems, indicating that the utilization of novel organic precursors is one way to further improve the operation of Pt-free catalysts for MFC applications. In this work, we confirm that Pt quickly lost its catalytic activity due to the presence of pollutants in the solution in a very short amount of operational time; however, Fe–ricobendazole and Fe–niclosamide showed high stability along the 32 days operations. Additionally, accelerated poisoning tests confirm that Fe–ricobendazole is more tolerant to S2− compared to Fe–niclosamide. In our previous studies, we have determined that the cost of the Fe–N–C catalyst is around 3.3–3.5 US$ g−1, which is ≈46 times cheaper than Pt (≈150 US$ g−1).24–26 This means that the cost of Fe-based catalysts is roughly 31–33 US$ per W produced, leading to an approximately 55-fold cost reduction compared to Pt (1770 US$ per W). Because of its high cost and low durability Pt is not a promising catalyst for MFC applications. Fe–N–C cathodes, on the other hand, outperform Pt and showed almost double the performance of AC-based cathodes, while at the same time providing a significant reduction in cost.Conclusion
Iron-based catalysts for ORR have been synthesized using novel low cost novel organic precursors (ricobendazole and niclosamide). Cathodes based of these materials have been investigated in neutral media and in operating MFCs. The electrocatalytic activity of Fe–ricobendazole, and Fe–niclosamide was found to be substantially higher than that of Pt during cathode polarization tests and generate more power in working MFCs. The overall performances for the Fe–ricobendazole, and Fe–niclosamide based MFCs were 20–25% higher than Pt and roughly 100% higher than AC. The power density decreased only slightly for Fe–ricobendazole and Fe–niclosamide and AC during when exposed to high concentrations of sulphur. Pt lost its activity after 1 week of operation. The higher performance and durability of Fe–ricobendazole and Fe–niclosamide make these novel low cost catalysts suitable for large-scale MFC cathodes. In the future, additional improvements to the performance and durability of MFC cathodes could be achieved by utilizing other novel organic precursors.Acknowledgements
This project was funded in part by: (i) a grant from the Bill & Melinda Gates Foundation: “Efficient Microbial Bio-electrochemical Systems” (OPP1139954); (ii) a contract with J. Craig Venter Institute and UNM Center for Micro-Engineered Materials.References
- O. Schaetzle, F. Barrière and U. Schröder, Energy Environ. Sci., 2009, 2, 96–99 CAS.
- H. Rismani-Yazdi, S. M. Carver, A. D. Christy and O. H. Tuovinen, J. Power Sources, 2008, 180(2), 683–694 CrossRef CAS.
- C. Santoro, S. Babanova, P. Atanassov, B. Li, I. Ieropoulos and P. Cristiani, J. Electrochem. Soc., 2013, 160(10), H720–H726 CrossRef CAS.
- C. Agnès, M. Holzinger, A. Le Goff, B. Reuillard, K. Elouarzaki, S. Tingry and S. Cosnier, Energy Environ. Sci., 2014, 7, 1884–1888 Search PubMed.
- K. B. Liew, W. R. W. Daud, M. Ghasemi, J. X. Leong, S. S. Lim and M. Ismail, Int. J. Hydrogen Energy, 2014, 39, 4870–4883 CrossRef.
- E. Antolini, Biosens. Bioelectron., 2015, 69, 54–70 CrossRef CAS PubMed.
- I. Gajda, J. Greenman, C. Melhuish and I. Ieropoulos, Bioelectrochemistry, 2015, 104, 58–64 CrossRef CAS PubMed.
- J. Wei, P. Liang and X. Huang, Bioresour. Technol., 2011, 102, 9335–9344 CrossRef CAS PubMed.
- Y. Jiang, Y. Xu, Q. Yang, Y. Chen, S. Zhu and S. Shen, Int. J. Energy Res., 2014, 38(11), 1416–1423 CrossRef CAS.
- C. Santoro, A. Stadlhofer, V. Hacker, G. Squadrito, U. Schröder and B. Li, J. Power Sources, 2013, 243, 499–507 CrossRef CAS.
- X.-W. Liu, W.-W. Li and H.-Q. Yu, Chem. Soc. Rev., 2014, 43, 7718–7745 RSC.
- Z. Wang, C. Cao, Y. Zheng, S. Chen and F. Zhao, ChemElectroChem, 2014, 1, 1813–1821 CrossRef CAS.
- D. E. Grove, Platinum Met. Rev., 2003, 47(1), 44 Search PubMed.
- V. A. Sethuraman and J. W. Weidner, Electrochim. Acta, 2010, 55, 5683–5694 CrossRef CAS.
- B. Li, X. Zhou, X. Wang, B. Liu and B. Li, J. Power Sources, 2014, 272, 320–327 CrossRef CAS.
- F. Zhao, F. Harnisch, U. Schröder, F. Scholz, P. Bogdanoff and I. Herrmann, Electrochem. Commun., 2005, 7, 1405–1410 CrossRef CAS.
- F. Zhao, F. Harnisch, U. Schröder, F. Scholz, P. Bogdanoff and I. Herrmann, Environ. Sci. Technol., 2006, 40, 5191–5199 Search PubMed.
- E. H. Yu, S. Cheng, B. E. Logan and K. Scott, Electrochemical reduction of oxygen with iron phthalocyanine in neutral media, J. Appl. Electrochem., 2009, 39, 705–711 CrossRef CAS.
- B. Liu, C. Brückner, Y. Lei, Y. Cheng, C. Santoro and B. Li, J. Power Sources, 2014, 257, 246–253 CrossRef CAS.
- X. Li, B. Hu, S. Suib, Y. Lei and B. Li, J. Power Sources, 2010, 195(9), 2586–2591 CrossRef CAS.
- G. Lu, Y. Zhu, L. Lu, K. Xu, H. Wang, Y. Jin, Z. J. Ren, Z. Liu and W. Zhang, J. Power Sources, 2016, 315, 302–307 CrossRef CAS.
- X. Xia, F. Zhang, X. Zhang, P. Liang, X. Huang and B. E. Logan, ACS Appl. Mater. Interfaces, 2013, 5(16), 7862–7866 CAS.
- P. Zhang, K. Li and X. Liu, J. Power Sources, 2014, 264, 248–253 CrossRef CAS.
- C. Santoro, A. Serov, C. W. Narvaez Villarrubia, S. Stariha, S. Babanova, A. J. Schuler, K. Artyushkova and P. Atanassov, ChemSusChem, 2015, 8(5), 828–834 CrossRef CAS PubMed.
- C. Santoro, K. Artyushkova, I. Gajda, S. Babanova, A. Serov, P. Atanassov, J. Greenman, A. Colombo, S. Trasatti, I. Ieropoulos and P. Cristiani, Int. J. Hydrogen Energy, 2015, 40, 14706–14715 CrossRef CAS.
- C. Santoro, A. Serov, C. W. Narvaez Villarrubia, S. Stariha, S. Babanova, K. Artyushkova, A. J. Schuler and P. Atanassov, Sci. Rep., 2015, 5, 16596 CrossRef PubMed.
- A. Serov, K. Artyushkova and P. Atanassov, Adv. Energy Mater., 2014, 4, 1301735, DOI:10.1002/aenm.201301735.
- A. Serov, K. Artyushkova, N. I. Andersen, S. Stariha and P. Atanassov, Electrochim. Acta, 2015, 179, 154–160 CrossRef CAS.
- C. Santoro, K. Artyushkova, S. Babanova, P. Atanassov, I. Ieropoulos, M. Grattieri, P. Cristiani, S. Trasatti, B. Li and A. J. Schuler, Bioresour. Technol., 2014, 163, 54–63 CrossRef CAS PubMed.
- K. Artyushkova, A. Serov, S. Rojas-Carbonell and P. Atanassov, J. Phys. Chem. C, 2015, 119, 25917–25928 CAS.
- F. Zhang, D. Pant and B. E. Logan, Biosens. Bioelectron., 2011, 30(1), 49–55 CAS.
- M. Santini, M. Guilizzoni, M. Lorenzi, P. Atanassov, E. Marsili, S. Fest-Santini, P. Cristiani and C. Santoro, Biointerphases, 2015, 10, 031009 Search PubMed.
- K. M. Minachev, N. I. Shuikin and I. D. Rozhdestivenskaya, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1952, 1(4), 567–575 CrossRef.
- X. Zhang, D. Pant, F. Zhang, J. Liu and B. E. Logan, ChemElectroChem, 2014, 1(11), 1859–1866 CrossRef CAS.
- V. J. Watson, C. N. Delgado and B. E. Logan, J. Power Sources, 2013, 242, 756–761 CrossRef CAS.
- Y. Fan, H. Hu and H. Liu, Environ. Sci. Technol., 2007, 41(23), 8154–8158 CrossRef CAS PubMed.
- Z. Fu, L. Yan, K. Li, B. Ge, L. Pu and X. Zhang, Biosens. Bioelectron., 2015, 74, 989–995 CrossRef CAS PubMed.
- Z. Liu, B. Ge, K. Li, X. Zhang and K. Huang, Fuel, 2016, 176, 173–180 CrossRef CAS.
- B. Ge, K. Li, Z. Fu, L. Pu and X. Zhang, Bioresour. Technol., 2015, 195, 180–187 CrossRef CAS PubMed.
- P. Zhang, K. Li and X. Liu, J. Power Sources, 2014, 264, 248–253 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ee01145d |
| ‡ The two authors have contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2016 |