
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlasmonic photothermic directed broadband sunlight harnessing for seawater catalysis and desalination†
Minmin
Gao
a,
Peh Kang Nuo
Connor
a and
Ghim Wei
Ho
*ab
aDepartment of Electrical and Computer Engineering, National University of Singapore, Singapore 117576, Singapore. E-mail: elehgw@nus.edu.sg; Fax: +65 67754710; Tel: +65 65168121
bEngineering Science Programme, National University of Singapore, 4 Engineering Drive 3, 117576, Singapore
First published on 6th June 2016
Abstract
Using readily available renewable resources, i.e. solar energy and seawater, to secure sustainable fuel and freshwater for humanity is an impactful quest. Here, we have designed solar thermal collector nanocomposites (SiO2/Ag@TiO2 core–shell) that possess efficient photothermic properties for highly targeted interfacial phase transition reactions that are synergistically favorable for both seawater catalysis and desalination reactions. The photothermic effect arising from plasmonic metal nanoparticles causes localized interfacial heating which directly triggers surface-dominated catalysis and steam generation processes, with minimal heat losses, reduced thermal masses and optics implementation. The solar thermal collector nanocomposites are seawater/photo stable for practical solar conversion of seawater to simultaneously produce clean energy and water. Finally, a proof-of-concept all-in-one compact solar hydrogen and distillate production prototype demonstrates the viability of sustainable photothermic driven catalysis and desalination of seawater under natural sunlight. Importantly, this approach holds great promise for enhancing energy and water productivity without considerable capital, infrastructure and environmental ramifications.
Broader contextOur work demonstrates the proposition of parallel production of energy and water resources that are freely powered by renewable energy and seawater. We have developed a broadband solar thermal collector nanocomposite that is endowed with integrated photocatalytic, plasmonic and photothermic attributes. Such a multifunctional nanocomposite offers synergistic capabilities of distillate production and clean fuel generation. The solution processable nanocomposite made up of several functionalities incorporated within its structure forms the basis of a compact, lightweight and enhanced performance material. Having such solar thermal nanocomposites directly dispersed in the reactant fluid can lead to efficient and instantaneous initiation of chemical reactions and steam generation based on the plasmonic photothermic effect in their immediate surrounding. We have shown seawater seeded with solar thermal collector nanocomposites to efficiently produce steam and distillates at temperatures well below its boiling point. This work addresses the intricate nexus between water, energy and environmental impacts. |
Introduction
One of the most pressing issues of mankind is the lack of clean fuel and freshwater supplies which are the two inseparable commodities that govern the lives of humanity.1–3 Though water is one of the most abundant resources on earth covering three-quarters of the earth's surface, 97% of it is seawater.4–6 Therefore, finding means of producing fuel and freshwater from seawater is invaluable. Besides being environmentally attractive, catalysis and desalination of seawater based on renewable solar energy will also become sustainable and economically attractive as fossil fuel prices and greenhouse gas emissions continue to exacerbate.Heat is imperative for catalysis and desalination as these reactions are endothermic in nature, requiring heat to conceive vapor phases from liquid reactants and products. The main problems associated with direct sunlight to heat conversion in a liquid medium are the relatively low photothermal efficiency, poor productivity rate, and considerable use of land.7,8 Another challenge faced is that these processes typically occur in large systems with a continuous stream of reactants that are converted to desired products via bulk heating.9 It is apparent that the large thermal mass of these bulky systems limits the efficiency of the reaction. Moreover, an overlooked fact is that catalysis and desalination processes are very much surface-dominated interfacial transition processes, confined to water/catalyst and water/air interfaces.10 Furthermore, catalytic hydrogen production from water suffers from not being able to utilize the full spectrum of sunlight due to the stringent bandgap requirements of semiconductor catalysts.11 Most of the literature focuses on band structure engineering,12 incorporating visible active materials13 or upconversion nanoparticles14 for increased absorption of the solar spectrum for catalytic hydrogen production. Utilization of visible to near infrared (NIR) solar spectrum for localized heat generation at the catalyst surface has largely been ignored and underestimated. Hence, it is important to recognize the strategies for minimizing the thermal mass of the system and concentrating broadband solar irradiance for targeted interfacial processes towards consequential and efficient catalysis and desalination of seawater.
Here, for the first time we have purposefully designed solar thermal collector nanocomposites (SiO2/Ag@TiO2 core–shell) to attain low thermal mass, reduced heat losses and highly targeted interfacial reactions that are synergistically beneficial for both catalysis and desalination reactions. Moreover, the nanocomposite encompasses the rational design of Ag nanoparticles embedded in a transparent dielectric SiO2 matrix for enhanced light manipulation coupled with passivation against undesirable oxidation, aggregation and migration issues. Essentially, the visible to near infrared (NIR) radiation accounts for a significant portion of the solar spectrum, however it is acknowledged that direct absorption of these broad wavelengths by conventional semiconductors is challenging. Hence, the solar thermal collector core–shell nanocomposite is devised to achieve broadband absorption where higher energy photons are taken in by the TiO2 shell for photocatalytic electron–hole pair generation, while lower energy photons are captured by the SiO2/Ag core for heat generation that is concurrently useful for the catalysis and desalination processes. Having such solar thermal nanocomposites directly dispersed in the reactant fluid can lead to instantaneous initiation of chemical reactions and steam generation in their immediate surrounding. Metal nanoparticles have been shown to allow for nonequilibrium steam production well below the system's boiling point because of reduced thermal conductivity at the metal–liquid interface.15 Such localized heating allows fine control of the site, magnitude, and span of heat generation in contrast to bulk heating which is uncontrollable and energy intensive. The use of solar thermal nanocomposites would translate into minimal implementation of focusing optics, low heat losses, reduced thermal mass and highly efficient reactions. This is especially useful for regions that have rapidly changing solar insolation due to cloud cover etc. which benefits from quick, localized heating to expedite reactions.
Subsequently, the solar thermal nanocomposites are demonstrated for practical solar conversion of seawater to simultaneously produce clean energy and water. The solar thermal nanocomposites are proven to be seawater/photo stable and can address major challenges faced in conventional catalysis and desalination of seawater. The all-in-one compact footprint solar hydrogen and distillate production proof-of-concept prototype demonstrates the viability of photothermic enhanced catalysis and desalination. To this end, a sustainable photocatalysis coupled desalination concept is designed to capture the benefits of photothermic driven catalysis and desalination of seawater, all of which hold great promise for effective increase in energy and water productivity without further stressing the environment. This is a pragmatic approach for providing fundamental energy and water needs for small communities in remote regions with limited freshwater and inaccessibility to the electricity grid.
Experimental section
Preparation of SiO2 spheres
10 ml of deionized (DI) water, 1 ml of ammonia, 20 ml of ethanol and 0.5 ml of TEOS were mixed and stirred for 2 h at room temperature to obtain silica nanospheres with an average diameter of 200 nm. The mixture was washed three times with ethanol, and then dried at 60 °C.Preparation of SiO2/Ag spheres
Silver nanoparticles were pre-synthesized using the typical procedure as follows. 10 ml of 1.0 mM silver nitrate solution was added drop by drop into 30 ml of 2.0 mM NaBH4 solution chilled in an ice bath while stirring. Upon finishing, 10 mg of PVP (Mw ∼ 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was added into the mixed solution to prevent aggregation. The resulting yellowish-brown color Ag nanoparticle solution was ready for further use. Then SiO2/Ag spheres were prepared through a facile hydrolytic process at room temperature. Firstly, 10 ml of Ag nanoparticle solution and 20 ml of ethanol were added to a 50 ml glass bottle under vigorous stirring, followed by adding 1 ml of 28–30% ammonia solution to the mixture. The hydrolytic process was carried out by dropwise adding 0.5 ml of TEOS to the solution with continuous magnetic stirring. After aging overnight, the SiO2/Ag spheres with a diameter of about 200 nm were separated from the solution and washed three times with ethanol, then dried at 60 °C.
000) was added into the mixed solution to prevent aggregation. The resulting yellowish-brown color Ag nanoparticle solution was ready for further use. Then SiO2/Ag spheres were prepared through a facile hydrolytic process at room temperature. Firstly, 10 ml of Ag nanoparticle solution and 20 ml of ethanol were added to a 50 ml glass bottle under vigorous stirring, followed by adding 1 ml of 28–30% ammonia solution to the mixture. The hydrolytic process was carried out by dropwise adding 0.5 ml of TEOS to the solution with continuous magnetic stirring. After aging overnight, the SiO2/Ag spheres with a diameter of about 200 nm were separated from the solution and washed three times with ethanol, then dried at 60 °C.
Preparation of SiO2@TiO2 and SiO2/Ag@TiO2 core–shell nanocomposites
0.1 g of as-prepared SiO2 or SiO2/Ag spheres was dispersed in 2 ml of pure ethanol. 0.1 ml of 97% titanium butoxide (TBT) was added to the above solution and stirred for 20 min. After that, a mixture of 0.3 g of polyvinylpyrrolidone (PVP), 1 ml of DI water and 20 ml of pure ethanol was added to the solution and stirred for 1 h. Then the precipitate was washed twice with ethanol and dried in an oven at 60 °C. The obtained sample was then calcined at 450 °C in an inert environment for 2 h, with a ramping rate of 2 °C per min.Photocatalysis measurements
2 ml of the glycerol sacrificial reagent was dissolved in 8 ml of DI water, to which 5 mg of SiO2/Ag@TiO2 core–shell composites were added. To prepare simulated seawater, 0.35 g of sodium chloride was added to a solution of 2 ml of glycerol and 8 ml of DI water. All experiments were carried out in 25 ml quartz vials and illuminated with a UV light source (365 nm) with a light intensity of 35.3 mW cm−2 for hydrogen generation. A 300 W Xe lamp (Excelitas, PE300BFM) with a light intensity of 100 mW cm−2 equipped with a 400 nm long-pass filter was used as a visible-NIR light source. The reaction mixture was purged with Ar gas for 10 min prior to measurements. The headspace in the reaction mixture was syringe drawn (100 μl) to sample the gas composition using a gas chromatographer (Shimadzu, GC-2014AT).For other sacrificial reagents used, 1 ml of methanol, 2.5 ml of ethylene glycol and 0.5 g of glucose were used separately to form 10 ml of sacrificial reagent–water solution. For experiments performed at elevated temperatures, the vials were purged while immersed in a hot bath of desired temperature, so as to ensure that the vials remain close to 1 atm pressure during the experiment. The headspace in the reaction mixture was syringe drawn (100 μl) to sample the gas composition using a gas chromatographer with 20 min time intervals between readings (Shimadzu, GC-2014AT).
As for the photocatalytic experiment under the real sun, the experiment was carried out on 3rd November 2015 from 10:00 to 11:00 am at an ambient temperature of 30 °C. 300 ml of simulated glycerol–seawater solution (20% v/v glycerol with 10.5 g of sodium chloride) was prepared with 150 mg of SiO2/Ag@TiO2 photocatalysts used for hydrogen generation. The mixture was placed in a quartz tube covered by an insulation layer. A 62.5 cm2 window at the bottom of the tube allows sunlight to enter. The tube was purged with Ar gas for 20 min prior to measurements. The reaction mixture was syringe drawn (100 μl) to sample the gas composition using a gas chromatographer (Shimadzu, GC-2014AT) with 10 min time interval. The intensity of sun was measured by using a solar meter, while the temperature of the system was measured using a thermocouple. The condensate was collected and measured throughout the period. Assuming 5 hours of full sunlight a day, the condensate collected was reported in liters per irradiated area per day.
Characterization
The morphology of the samples was characterized using a SEM (JEOL FEG JSM 7001F) operated at 15 kV. The crystalline structures and elemental compositions of the BHS were analyzed using XRD (D5005 Bruker X-ray diffractometer equipped with graphite monochromated Cu Kα radiation at λ = 1.541 Å), TEM, and STEM (JEOL JEM 2010F). Extinction spectra and reflectance spectra of the samples were measured using a UV-vis-NIR spectrophotometer with an integrating sphere (UV-vis, Shimadzu UV-3600). Infrared images and temperatures were taken using a FLIR i60 infrared camera. Chlorine concentration was measured using a chloride ion selective electrode (ISE) with a benchtop pH/ISE meter (Thermo Scientific Orion Star A214) during the photocatalytic hydrogen generation reaction.For localized photothermic characterization through thermal curing of poly-(dimethylsiloxane) (PDMS) (Sylgard 184 silicone elastomer), GaN substrates were first cleaned by immersion in an ultrasonic water bath in acetone, DI water and iso-propanol respectively for 5 min each. The SiO2/Ag spheres were then deposited on the cleaned GaN substrate by drop-casting its solution and drying at room temperature. After that, a layer of freshly prepared optically transparent PDMS precursor mixed with the thermosensitive curing agent in the ratio of 10:1 was spin-coated onto the substrate. The prepared substrate was illuminated under a 300 W Xe lamp with a cutoff filter (≥400 nm) for 60 s, at a distance of 15 cm away from the lamp. After irradiation, the substrate was rinsed twice with hexane to remove any non-cross-linked polymer residuals and then analyzed by SEM. Another substrate was prepared in a similar manner, with the PDMS rinsed off with hexane without illumination. All prepared samples were processed immediately after spin-coating.
Results and discussion
Ag nanoparticle embedded SiO2 spheres were synthesized through a facile hydrolytic process at room temperature based on the Stöber method. Fig. 1a shows the SEM image of Ag nanoparticle embedded SiO2 spheres with a smooth spherical morphology of an average diameter ∼200 nm. The TEM image (Fig. 1b) shows the core–shell structure of SiO2/Ag particles with Ag nanoparticles dispersed within the silica spheres. Digital photos have been taken to show the differences in the optical transparency of both plain (Fig. 1c top) and SiO2/Ag sphere coated (Fig. 1c bottom) glass substrates. The SiO2/Ag coated glass substrate has high optical transparency but not as transparent as the plain glass substrate. This highly transparent matrix allows the light to enter into SiO2 spheres and Ag nanoparticles act as the scattering center inside the spheres, thus improving the overall light absorption.16,17 The SiO2/Ag spheres were coated by the TiO2 shell using the sol–gel method, followed by annealing the synthesized SiO2/Ag@TiO2 core–shell nanospheres at 450 °C for 2 h in an inert environment. The high magnification image of the TiO2 shell (Fig. 1d) shows the adjacent lattice spacing of 0.35 nm, attributed to the (101) lattice planes of anatase TiO2. Fig. 1e shows the SEM image of monodispersed TiO2 coated SiO2/Ag core–shell nanocomposites with a fairly uniform diameter of ∼220 nm. A detailed chemical analysis was carried out using TEM EDX mapping to study the elemental composition and distribution throughout the SiO2/Ag@TiO2 core–shell nanocomposites. The images in Fig. 1f correspond to the obtained bright field image, and elemental mapping images with Si K-edge, Ti K-edge and Ag M-edge respectively. It can be seen that the Si signal is located in the center part of the nanospheres with the Ag signal confined within it and the Ti signal around it. The size distribution of Ag nanoparticles is about 10 nm to 20 nm. Moreover, it is observed that a good interface and continuity exist between the SiO2/Ag core and the TiO2 shell.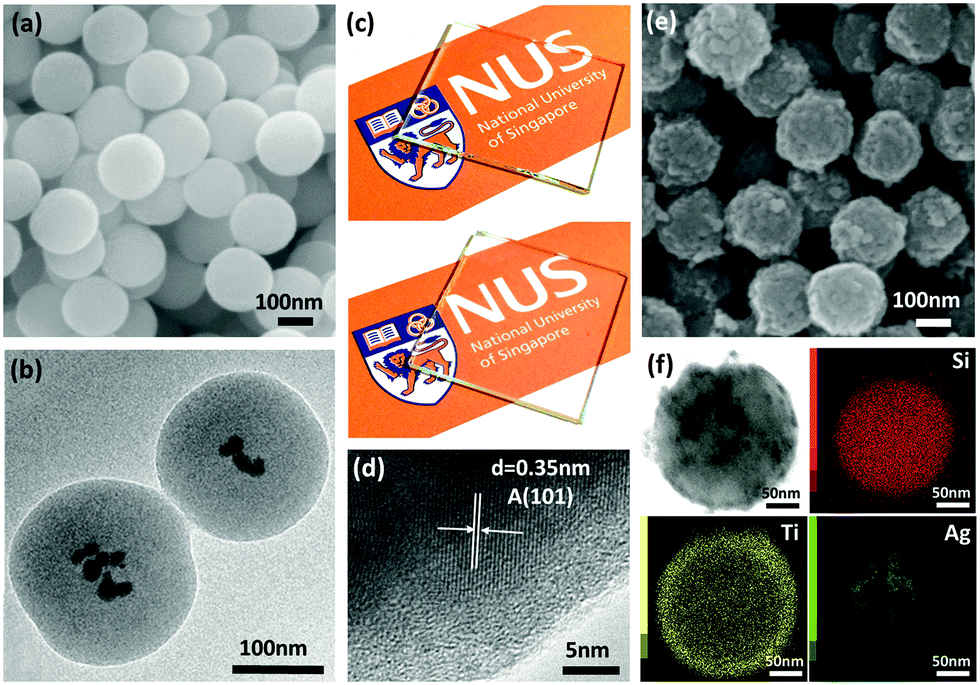 | ||
| Fig. 1 SEM image (a) and TEM image (b) of SiO2/Ag spheres. (c) Glass substrate (top) and SiO2/Ag coated glass substrate (bottom). (d) High resolution TEM image of the TiO2 coating on SiO2/Ag spheres. (e) SEM image of SiO2/Ag@TiO2 core–shell composites. (f) TEM image of SiO2/Ag@TiO2 core–shell composites and their respective STEM EDX elemental mapping images. | ||
It is well-known that localized surface plasmon resonance induced by plasmonic metal plays an important role in enhancing the rate of photocatalytic reactions. The enhancement is mostly associated with either rapid charge carrier transfer from the plasmonic metal to the semiconductor when they are in direct contact, or near-field electromagnetic where the plasmonic energy was transferred from the metal to the semiconductor to enhance the generation of electron–hole pairs in the semiconductor.18–21 On the contrary, the photothermic effect on catalytic performance is often being ignored and discounted. In essence, the photothermic effect is driven by plasmonic metal nanoparticles such as Cu, Pt, Ag, Au and Ni, where heat is effectively generated under optical excitation.22 In the presence of electromagnetic radiation, the electric field strongly drives free electrons inside the metal nanocrystals, and the energy gained by these carriers turn into heat leading to an increase in the local temperature of the system. In order to decouple the heat generation from direct charge injection and near-field electromagnetic mechanisms, SiO2/Ag@TiO2 core–shell structure was adopted for the photothermic catalysis study. Previously, it has been reported that Ag nanoparticles generate ten times higher heat than Au nanoparticles studied under resonant plasmon interaction.23,24 Accordingly, the judicious choice of Ag nanoparticles enclosed within UV-visible transparent SiO2 spheres was made and as such the Ag nanoparticles are explicitly isolated from TiO2 by a SiO2 dielectric spacer. In addition to preventing the Ag nanoparticles from making direct contact with TiO2, the thermally and chemically stable SiO2 spheres serve to prevent the Ag nanoparticles from undesirable oxidation, aggregation and migration.25 TiO2 is deposited onto the Ag/SiO2 sphere as a shell for direct light absorption without compromising its photocatalytic activity.
The XRD spectrum (Fig. 2a) of SiO2 spheres suggests that they are amorphous as no SiO2 peaks were observed, while SiO2/Ag spheres show diffraction peaks at approximately 38.1°, 44.4°, 64.3° and 77.4°, which are attributed to (111), (200), (220) and (311) planes of Ag (JCPDS no. 04-0783). XRD analysis of both SiO2@TiO2 and SiO2/Ag@TiO2 shows that they possess highly crystalline anatase phase TiO2. The TiO2 anatase diffraction peaks (101), (004), (200), (105), (211) and (204) are observed at 25.3°, 37.8°, 48.1°, 53.9°, 55.1° and 62.8° (JCPDS no. 21-1272) respectively. Due to the main Ag(111) peak located nearly at the same diffraction angles as the anatase TiO2(004) peak, it is very difficult to discriminate between these two peaks. However, the SiO2/Ag@TiO2 spectrum exhibits an additional diffraction peak at 44.4°, which can be assigned to the (200) crystal plane of Ag (JCPDS 04-0783).
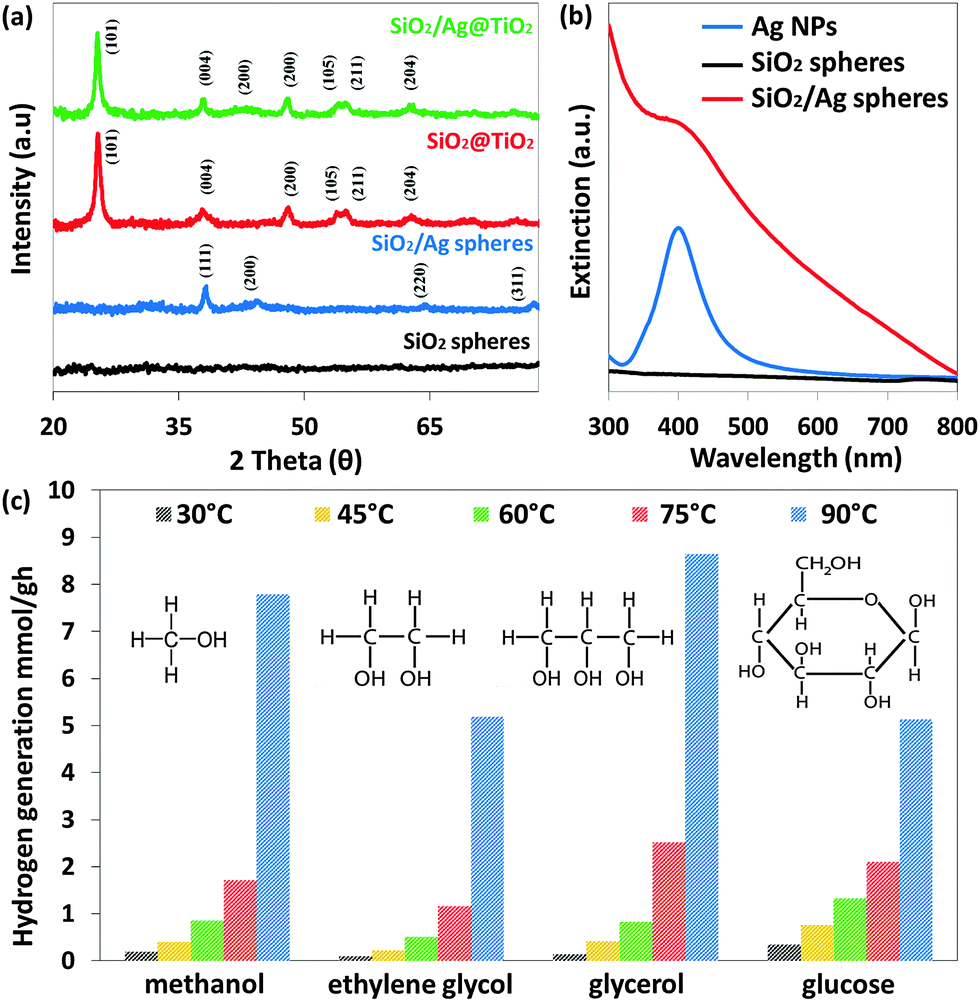 | ||
| Fig. 2 (a) XRD spectra of SiO2 spheres, SiO2/Ag nanoparticles, SiO2@TiO2 and SiO2/Ag@TiO2 core–shell nanocomposites. (b) Extinction spectra of Ag nanoparticles, SiO2 spheres and SiO2/Ag spheres. (c) Hydrogen generation by TiO2 spheres in methanol (10% v/v), ethylene glycol (25% v/v), glycerol (20% v/v) and glucose (0.5 g) solution at different temperatures. | ||
The optical properties of Ag nanoparticles, SiO2 spheres and SiO2/Ag sphere suspensions have been studied using UV-vis spectroscopy (Fig. 2b). It can be seen that the Ag nanoparticles exhibit localized surface plasmon resonance, and the plasmon band is around 402 nm. After incorporating them into the SiO2 spheres, the plasmon band of SiO2/Ag spheres shows a slight red-shifted broad absorbance band of higher intensity.26 For comparison, the absorption spectrum of SiO2 spheres was also measured with no obvious peak across the UV-visible range observed, indicating that SiO2 spheres are transparent to both UV and visible light.
Besides, a few organic compounds are combined with water as sacrificial reagents for hydrogen generation under solar radiation. Temperature elevation has also been shown to increase the efficiency of photocatalytic hydrogen production, and has been applied for the studies below.27Fig. 2c shows an increase in the hydrogen generation rate at different temperatures for all four sacrificial reagents. Among them, glycerol gives the highest improvement in hydrogen production, which is 56.6%, when temperature increased from 30 to 90 °C, followed by ethylene glycol (50.3%), methanol (40.5%) and glucose (13.9%). Glycerol is the by-product of biodiesel derived from biomass. At present, glycerol is one of the most pertinent biomass derivatives since it is a renewable commodity.28–31 It was postulated that the improvement is related to the hole-scavenging activity of sacrificial reagents from the surface of the photocatalyst which explicitly prolonged the lifetime of the photogenerated carriers.27
After establishing that temperature by means of external heating favorably affects the hydrogen generation, an inherent temperature gain from the conversion of the incoming radiation using the solar thermal collector nanocomposite was sought. This photothermic effect on the catalytic hydrogen generation of SiO2/Ag@TiO2 core–shell nanocomposites was investigated using two separate light sources; UV light (365 nm) and visible-NIR light (≥400 nm). For comparison, TiO2 spheres (Fig. S1a, ESI†) and SiO2@TiO2 (Fig. S1b, ESI†) core–shell structures were synthesized using the same sol–gel method as the reference samples for solar hydrogen generation studies. Fig. 3a shows hydrogen generation as a function of time for intrinsic TiO2 spheres, SiO2@TiO2 and SiO2/Ag@TiO2 core–shell nanocomposites. For the first 2.25 hours under UV light irradiation, TiO2 was activated for photocatalytic hydrogen generation with minimum heating effect due to low intensity single wavelength light source restriction. The hydrogen generation rates were determined to be 150 μmol g−1 h−1 for TiO2 spheres, 264 μmol g−1 h−1 for SiO2@TiO2 and 786 μmol g−1 h−1 for SiO2/Ag@TiO2 core–shell nanocomposites (Fig. S2, ESI†). As all the three samples were observed at similar temperatures of ∼30 °C (Fig. S3, ESI†), the photothermic effect plays a subsidiary role in this case. The differences in hydrogen generation rates between the samples were accredited to the structural composition of the samples. Evidently, the core–shell nanostructures (SiO2@TiO2 and SiO2/Ag@TiO2) show higher hydrogen generation rates than the pristine TiO2. It has been recognized that core–shell nanostructures composed of disparate refractive indexes possess light trapping configuration that enhances optical absorption.16 Based on Mie scattering theory, significant improvement in the light scattering efficiency can be expected for core/shell structure with the refractive index of the shell material (nTiO2 = 2.49) considerably different than that of core material (nSiO2 = 1.47).11,16,25 Unlike the TiO2 spheres, which allow most of the light to pass through, the core–shell structural design especially the metal-dielectric Ag/SiO2 configuration acquires a large absorption cross section that undergoes high internal light scattering path modulation.32,33 This greatly enhances light harnessing capability beneficial for photocatalytic reactivity.
 | ||
| Fig. 3 (a) Hydrogen generation by TiO2 spheres, SiO2@TiO2 and SiO2/Ag@TiO2 core–shell nanocomposites in glycerol–water solution under both UV light and full spectrum irradiation. (b) Calculated absorption spectra of SiO2/Ag, TiO2, SiO2@TiO2, SiO2/Ag@TiO2 spheres, and spectral solar irradiance (AM 1.5). (c) Digital and infrared images of the hydrogen generation vial reactor. (d) Infrared images of the vial reactor with SiO2@TiO2 (top) and SiO2/Ag@TiO2 (bottom) nanocomposites taken under both UV light and full spectrum irradiation as a function of time. Top-view (e) and side-view (f) SEM images of SiO2/Ag spheres on the GaN substrate. SEM images of the PDMS structures formed by thermal curing of PDMS through the localized photothermic effect, with images taken from a 30° angle (g), side view (h) and top view with its respective SEM EDX elemental mapping images (i). | ||
When the full spectrum was illuminated onto the samples, especially the SiO2/Ag@TiO2 sample showed a notable improvement of ∼95% increment (1536 μmol g−1 h−1) in the hydrogen generation rate (Fig. 3a and Fig. S2, ESI†). Similarly, TiO2 spheres and SiO2@TiO2 core–shell nanocomposites showed an improvement of ∼50–56% increment in the hydrogen evolution rate, which is nevertheless lower (Fig. 3a and Fig. S2, ESI†). It is noted that no photocatalytic hydrogen was detected for all samples when visible-NIR light was solely used for irradiation. This is apparent since the plasmonic Ag nanoparticles are isolated by SiO2 dielectric spheres and the excitation energy is inadequate to activate TiO2, hence explaining the catalytic inactivity. The significant enhancement in the photocatalytic activity, in particular, of the SiO2/Ag@TiO2 nanocomposite can be attributed to the photothermic effect. To further probe the photothermic capability of the SiO2/Ag@TiO2 sample, spectral reflectance has been measured using a UV-vis spectrophotometer equipped with an integrating sphere, and the calculated absorption spectra are shown in Fig. 3b. Total solar absorptance was obtained by weighting the spectral reflectance with the spectral solar irradiance (AM 1.5) and integrating over 300 to 2500 nm wavelengths, in which solar radiation reaches the absorber surface. The detailed calculations are shown in the ESI.† Accordingly, SiO2/Ag@TiO2 demonstrated a high solar absorptance (α) of 0.81 which is excellent for solar thermal conversion, compared to intrinsic TiO2 (α = 0.10) and SiO2@TiO2 (α = 0.40) spheres. Moreover, the solar absorptance of SiO2/Ag@TiO2 is slightly lower than SiO2/Ag spheres (α = 0.89), due to the relatively high reflectance of the TiO2 shell in the visible-NIR region. Subsequently, the temperature differences between SiO2@TiO2 and SiO2/Ag@TiO2 nanocomposites were examined using an infrared camera under both UV light and full spectrum irradiation. During the hydrogen generation process, the infrared images of the vial reactor (Fig. 3c) were taken with a time interval of 15 min. Fig. 3d shows that under UV irradiation, the final temperature difference of the two samples was less than 1 °C, while under full spectrum irradiation the temperature difference increased to 2.6 °C after 2.5 h. Notably, the plasmon mediated heat generation in this case is highly localized and spatially confined to the immediate surrounding since Ag nanoparticles are embedded in SiO2 spheres without direct physical interface with the bulk liquid. Hence, the increase in bulk liquid temperature is small by virtue of isolated Ag nanoparticles in the SiO2 matrix and the use of moderate light flux illumination unlike the reported high laser excitation and concentrated light demonstrations.34–37 Alexander et al. have also investigated the photothermic effects of colloidal metal nanoparticles, and demonstrated that the presence of concentrated colloidal metal nanoparticles has collectively led to an increase of temperature by a few degree Celsius.23 Albeit the temperature increment of the bulk solution is a few degree Celsius, the site specific or localized heat around the SiO2/Ag@TiO2 composite has sufficiently facilitated efficient hole-scavenging activity resulting in enhanced photocatalytic hydrogen production.27 Another experiment was done to isolate the photothermic effect on photocatalytic activity in which the localized heating effect due to the vis-NIR light was isolated from the bulk temperature heating using a water bath to control the temperature. This is discussed in the ESI† (Fig. S4). It can be seen that for the SiO2/Ag@TiO2 core shell structure, the concurrent illumination of UV and visible-NIR light sources resulted in enhanced H2 generation consistently across all temperatures from 0–90 °C (Fig. S5a, ESI†). In contrast, the TiO2 nanospheres show little to no increase in the rate of H2 evolution under the same measurement conditions (Fig. S5b, ESI†). This further validates the contribution of the photothermic effect leading to the enhanced photocatalytic activity of SiO2/Ag@TiO2 nanocomposites.
The localized and spatially confined heating was validated by heat-induced polymerization demonstration at the nanoscale level.38,39 PDMS was used as its polymerization rate is controlled by temperature, without involving any photo-induced reactions.40 At room temperature, thermal curing of PDMS takes about 1 day, but the curing time can be significantly reduced at higher temperatures.41 The prepared substrate was illuminated, rinsed with hexane to remove any non-cross-linked polymer residues and then analyzed using SEM. Without illumination, the particles were well-dispersed with some physical aggregations which are shown in Fig. 3e (top-view) and Fig. 3f (side-view); after illumination, localized polymerization was evidenced by the thin layer of the polymer coating around the SiO2/Ag spheres (Fig. 3g). Polymerization was caused by a localized increase in temperature in the immediate surrounding of the SiO2/Ag spheres due to the plasmonic photothermic effect. The heat was dissipated radially to the polymer which forms the hemispherical shape of the polymer shell (Fig. 3h) confined around the SiO2/Ag spheres. When the light is illuminated on the areas of the substrate without the presence of SiO2/Ag spheres, it was observed under EDX elemental mapping (Fig. 3i) that the Si signal was only detected around the SiO2/Ag spheres, while the Ga signal from the substrate was detected on those areas without SiO2/Ag spheres. It is inferred that there was no formation of PDMS on the areas without SiO2/Ag spheres due to the absence of the Si signal. Therefore, the curing of PDMS was solely caused by localized plasmonic heating due to the presence of the SiO2/Ag spheres which functioned as a solar thermal collector. In addition, the SiO2/Ag@TiO2 spheres were also tested with similar effects observed (Fig. S6, ESI†). The localized heating of the SiO2/Ag@TiO2 spheres also leads to polymerization in the immediate surrounding owing to the plasmonic photothermic effect. The heat radially spreads out to the polymer to form the hemispherical shape of the polymer shell in proximity to the SiO2/Ag@TiO2 sphere.
By and large, most studies on photocatalytic hydrogen generation have been demonstrated using precious pure water. Here, we sought to produce hydrogen from seawater, given that it is virtually unlimited in supply and does not compete appreciably with pure water for consumption or agriculture. Fig. 4a shows a similar photocatalytic performance trend in simulated seawater as observed previously in pure water (Fig. 3a) where the photothermic effect comes into play essentially under full spectrum irradiation. Likewise the SiO2/Ag@TiO2 nanocomposite showed the highest hydrogen generation rate of 816 μmol g−1 h−1 among all the samples under full spectrum irradiation, which is an increase of 97% (Fig. S2, ESI†). Both SiO2@TiO2 and TiO2 spheres also showed an increase from 156 μmol g−1 h−1 to 192 μmol g−1 h−1 and from 138 μmol g−1 h−1 to 156 μmol g−1 h−1 respectively, however to a lower extent (Fig. S2, ESI†). Hydrogen generation rates for simulated seawater were observed to be consistently lower than pure water for all three samples. This is likely to be attributed to the formation of Cl radicals by hole oxidation (eqn (1)), and the subsequent reduction of the radicals by photoelectrons (eqn (2) and (3)),10,42 thus creating an electron pathway that competes with photocatalytic hydrogen generation reaction (eqn (4)). The concentration of Cl− ions remains constant throughout the photocatalytic reaction, which is in agreement with eqn (3), due to the back-reaction with electrons.43 Another possibility is that the competitive adsorption of Cl− ions on active sites inhibits either H+ or glycerol adsorption.44
| h+ + Cl− → Cl˙ | (1) |
| Cl˙ + Cl− → Cl2˙− | (2) |
| Cl2˙− + e− → 2Cl− | (3) |
| 2e− + 2H+ → H2 | (4) |
 | ||
| Fig. 4 (a) Hydrogen generation of TiO2 spheres, SiO2@TiO2 and SiO2/Ag@TiO2 core–shell nanocomposites in glycerol-simulated seawater solution under both UV light and full spectrum irradiation as a function of time. (b) Hydrogen generation and chlorine concentration of SiO2/Ag@TiO2 core–shell nanocomposites in glycerol-simulated seawater solution under both UV light and full spectrum irradiation as a function of time. (c) Photocatalytic hydrogen generation cycles of SiO2/Ag@TiO2 core–shell nanocomposites. (d) Schematic diagram of the proposed mechanism of photothermic catalysis. | ||
The schematic of the proposed mechanism of the photothermic effect is shown in Fig. 4d. Upon solar irradiation, the TiO2 porous shell absorbs the UV portion of the solar spectrum, while the SiO2/Ag solar thermal collector core discriminately absorbed the visible and NIR light. It is recognized that visible to NIR radiation accounts for a significant portion of the solar spectrum, however direct absorption by conventional semiconductor photocatalysts proved to be difficult. Hence the proposed SiO2/Ag@TiO2 core–shell structure scheme is intended to achieve broad spectrum utilization for photocatalytic hydrogen generation, where higher energy photons are used for photocatalytic electron–hole pair generation, while lower energy photons are used for a favorable photothermic effect. In essence, the SiO2/Ag metal-dielectric composites are present as an inhomogeneous medium, where silver metal particles embedded in the SiO2 dielectric medium host scatter the propagating light to increase the light absorption capability.46 Furthermore, when Ag nanoparticles are excited with resonant photons, oscillation of electron gas results in the excitation of electrons from occupied states to unoccupied states forming hot electrons and leading to an athermal charge distribution.47 In this athermal distribution, the hot electrons decay either through re-emission of photons or through carrier multiplication via electron–electron interactions.48 The hot carriers generated from plasmon decay will redistribute their energy by electron–electron scattering processes which result in rapid increase in the localized surface temperature of the metal.49–51 After thermalization, the lattice cools via phonon–phonon coupling resulting in heat dissipation into the surrounding medium.52 The heat generated is used to increase the local temperature of the system, enhancing the hole-scavenging effect of glycerol and thus improving the photocatalytic hydrogen generation rate around the photocatalyst. This solar thermal collector enables full spectrum utilization for photocatalytic hydrogen generation, where higher energy photons are used for photocatalytic electron–hole pair generation, while lower energy photons are used for heating.
Given that the most pervasive humanitarian issue is associated with the lack of clean fuel and freshwater supplies, development of an inexpensive, effective and renewable approach to produce clean energy and water is highly desirable. Additionally, owing to the diffuse nature of solar energy, the main problems affiliated to conventional solar to thermal energy conversion infrastructure are the relatively low thermal efficiency, poor productivity rate, and considerably large footprint. On the other hand, a combination of solar thermal collector nanocomposites and a specially designed reactor for photocatalysis coupled desalination of seawater offers an effective and compact strategy towards synergistic clean energy and water solutions.
Here, we have designed a prototype reactor (Fig. 5) that is attractive for non-capital and non-energy intensive photocatalysis and desalination of seawater based on a simple construction and a largely self-operating process. The reactor design comprises of a parabolic trough that focuses sunlight onto the bottom part of a quartz tube window. The quartz tube is designed to transmit light through the window while the rest of the tube was insulated to prevent excessive heat loss. A thermocouple was built in to measure the reaction temperature in real time. A water-vapor filter was used to separate hydrogen from steam at the hydrogen outlet, while the steam was condensed into water through the condenser.
 | ||
| Fig. 5 Schematic of the prototype reactor with measurement of the time dependent graph indicating (a) solar intensity and temperature, (b) hydrogen generation, (c) volume of the condensate (top) and chlorine concentration (bottom). | ||
Fig. 5a and b show outdoor time-dependent solar intensity and its corresponding reaction temperature, while hydrogen produced by the prototype reactor was recorded accordingly. The maximum hydrogen generation rate reached as high as 13.3 mmol g−1 h−1, hence demonstrating a proof-of-concept for broadband spectrum operation exploiting photocatalytic and photothermic mechanisms under natural sunlight. Moreover, it is observed that when the reaction temperature approaches the boiling point, water can be collected from the steam passing through the condenser (Fig. 5c (top)). It has been reported that nonequilibrium steam generation can occur below the boiling point in the presence of nanoparticles which reduced thermal conductivity at the nanoparticle–liquid interface, while also functioning as nucleation sites for steam generation. This leads to enhanced steam production as compared to a system without nanoparticles.15 Importantly, heat energy harnessed for direct solar to thermal distillation of seawater with and without solar thermal collector nanocomposite suspension can produce a condensate of approximately 2.2 L m−2 day−1 as compared to 1.5 L m−2 day−1. The solar thermal collector nanocomposites contribute to efficient steam generation through the localized heating effect, as well as the conventional steam generation through bulk heating. The combination of effects leads to higher condensate production compared to a system without the solar thermal collectors.15 Steam generation solely, decoupled from condensation and distillate collection, was investigated experimentally (ESI†). It was seen that the addition of these solar thermal collector nanocomposites in water resulted in 70.8% more steam produced under solar irradiation. The localized heating effect of the nanocomposites contributed to a steam generation rate of 5.68 L m−2 h−1, which will lead to a substantial increase in distillate collected through an optimized design.
The chlorine ion concentration of the condensate was measured (Fig. 5c (bottom)) to show its absence during the whole period, indicating the production of freshwater. Moreover, the proposed hybrid photocatalysis and desalination scheme can offer further possibility of decontaminating seawater since the decomposition of pollutant constituents present in seawater has been reported in the presence of the photocatalyst under simulated light illumination.53 To the best of our knowledge, this is the first work to demonstrate a hybrid clean energy and water production prototype that has advantageously made use of photothermic enhanced catalysis and desalination. This in turn enhances clean energy and freshwater production, beneficial for small communities in remote locations with access to saline lakes or seawater and where connection to the electricity grid is either not cost effective or not available.
Conclusion
The utilization of renewable solar energy for catalysis and desalination of seawater is seen to have the potential to offer a sustainable route for increasing clean energy and water supplies. The seawater/photo stable solar thermal collector nanocomposites are able to address major challenges such as low light to heat conversion efficiency, poor productivity rate, and considerable use of land faced in conventional catalysis and desalination of seawater. Our results reveal that the photothermic effect arising from the specially designed solar thermal collector (SiO2/Ag@TiO2 core–shell) nanocomposite can result in appreciable localized thermal energy conversion intended for interfacial heating to facilitate surface-dominated catalysis and the desalination process. The hybrid seawater catalysis and desalination prototype demonstrates photothermic enhanced hydrogen production by 5.2-fold compared with intrinsic TiO2 while distillate production increased by 46.7% with the exploitation of the solar thermal collector nanocomposites.Acknowledgements
This work is supported by Ministry of Education (MOE) R-263-000-B38-112/R-263-000-B63-112 and National Research Foundation Singapore, Energy Innovation Research Programme, R-263-000-B82-279, managed on behalf by Building and Construction Authority (BCA).References
- D. Griggs, M. Stafford-Smith, O. Gaffney, J. Rockstrom, M. C. Ohman, P. Shyamsundar, W. Steffen, G. Glaser, N. Kanie and I. Noble, Nature, 2013, 495, 305–307 CrossRef CAS PubMed.
- V. G. Gude and N. Nirmalakhandan, Energy Convers. Manage., 2010, 51, 2245–2251 CrossRef CAS.
- M. He, F. Qiu and Z. Lin, Energy Environ. Sci., 2013, 6, 1352–1361 Search PubMed.
- G. N. Tiwari, H. N. Singh and R. Tripathi, Sol. Energy, 2003, 75, 367–373 CrossRef CAS.
- A. D. Khawaji, I. K. Kutubkhanah and J.-M. Wie, Desalination, 2008, 221, 47–69 CrossRef CAS.
- J. Rozema and T. Flowers, Science, 2008, 322, 1478–1480 CrossRef CAS PubMed.
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Marinas and A. M. Mayes, Nature, 2008, 452, 301–310 CrossRef CAS PubMed.
- Q. Schiermeier, Nature, 2008, 452, 260–261 CrossRef CAS PubMed.
- H. T. El-Dessouky, H. M. Ettouney and F. Mandani, Appl. Therm. Eng., 2000, 20, 1679–1706 CrossRef CAS.
- S. Iguchi, K. Teramura, S. Hosokawa and T. Tanaka, Phys. Chem. Chem. Phys., 2015, 17, 17995–18003 RSC.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- N. Serpone, J. Phys. Chem. B, 2006, 110, 24287–24293 CrossRef CAS PubMed.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- M. He, X. Pang, X. Liu, B. Jiang, Y. He, H. Snaith and Z. Lin, Angew. Chem., Int. Ed., 2016, 55, 4280–4284 CrossRef CAS PubMed.
- O. Neumann, C. Feronti, A. D. Neumann, A. Dong, K. Schell, B. Lu, E. Kim, M. Quinn, S. Thompson, N. Grady, P. Nordlander, M. Oden and N. J. Halas, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11677–11681 CrossRef CAS PubMed.
- S. Son, S. H. Hwang, C. Kim, J. Y. Yun and J. Jang, ACS Appl. Mater. Interfaces, 2013, 5, 4815–4820 CAS.
- F. Cao, K. McEnaney, G. Chen and Z. Ren, Energy Environ. Sci., 2014, 7, 1615–1627 CAS.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- D. Zheng, X. Pang, M. Wang, Y. He, C. Lin and Z. Lin, Chem. Mater., 2015, 27, 5271–5278 CrossRef CAS.
- D. Yang, X. Pang, Y. He, Y. Wang, G. Chen, W. Wang and Z. Lin, Angew. Chem., 2015, 127, 12259–12264 CrossRef.
- Y. H. Jang, Y. J. Jang, S. T. Kochuveedu, M. Byun, Z. Lin and D. H. Kim, Nanoscale, 2014, 6, 1823–1832 RSC.
- P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Nano Today, 2007, 2, 18–29 CrossRef.
- A. O. Govorov and H. H. Richardson, Nano Today, 2007, 2, 30–38 CrossRef.
- K.-S. Lee and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 19220–19225 CrossRef CAS PubMed.
- M. Gao, L. Zhu, W. L. Ong, J. Wang and G. W. Ho, Catal. Sci. Technol., 2015, 5, 4703–4726 CAS.
- D. Radziuk, R. Schuetz, A. Masic and H. Moehwald, Phys. Chem. Chem. Phys., 2014, 16, 24621–24634 RSC.
- C. K. N. Peh, M. Gao and G. W. Ho, J. Mater. Chem. A, 2015, 3, 19360–19367 CAS.
- H. Garcia, R. Ferreira, M. Petkovic, J. L. Ferguson, M. C. Leitao, H. Q. N. Gunaratne, K. R. Seddon, L. P. N. Rebelo and C. Silva Pereira, Green Chem., 2010, 12, 367–369 RSC.
- F. Yang, M. A. Hanna and R. Sun, Biotechnol. Biofuels, 2012, 5, 1–10 CrossRef PubMed.
- P. Anand and R. K. Saxena, New Biotechnol., 2012, 29, 199–205 CrossRef CAS PubMed.
- M. Pagliaro and M. Rossi, The Future of Glycerol, The Royal Society of Chemistry, 2010 Search PubMed.
- H. A. Atwater and A. Polman, Nat. Mater., 2010, 9, 205–213 CrossRef CAS PubMed.
- J. A. Schuller, E. S. Barnard, W. Cai, Y. C. Jun, J. S. White and M. L. Brongersma, Nat. Mater., 2010, 9, 193–204 CrossRef CAS PubMed.
- A. R. Mazzotti, M. G. Campbell, P. Tang, J. M. Murphy and T. Ritter, J. Am. Chem. Soc., 2013, 135, 14012–14015 CrossRef CAS PubMed.
- O. Neumann, A. S. Urban, J. Day, S. Lal, P. Nordlander and N. J. Halas, ACS Nano, 2013, 7, 42–49 CrossRef CAS PubMed.
- C. Fasciani, C. J. B. Alejo, M. Grenier, J. C. Netto-Ferreira and J. C. Scaiano, Org. Lett., 2011, 13, 204–207 CrossRef CAS PubMed.
- D. A. Boyd, L. Greengard, M. Brongersma, M. Y. El-Naggar and D. G. Goodwin, Nano Lett., 2006, 6, 2592–2597 CrossRef CAS PubMed.
- M. Fedoruk, M. Meixner, S. Carretero-Palacios, T. Lohmüller and J. Feldmann, ACS Nano, 2013, 7, 7648–7653 CrossRef CAS PubMed.
- J. M. Walker, L. Gou, S. Bhattacharyya, S. E. Lindahl and J. M. Zaleski, Chem. Mater., 2011, 23, 5275–5281 CrossRef CAS.
- C. Hubert, A. Rumyantseva, G. Lerondel, J. Grand, S. Kostcheev, L. Billot, A. Vial, R. Bachelot, P. Royer, S.-h. Chang, S. K. Gray, G. P. Wiederrecht and G. C. Schatz, Nano Lett., 2005, 5, 615–619 CrossRef CAS PubMed.
- A. A. S. Bhagat, P. Jothimuthu and I. Papautsky, Lab Chip, 2007, 7, 1192–1197 RSC.
- J. Moser and M. Grätzel, Helv. Chim. Acta, 1982, 65, 1436–1444 CrossRef CAS.
- H.-i. Kim, G.-h. Moon, D. Monllor-Satoca, Y. Park and W. Choi, J. Phys. Chem. C, 2012, 116, 1535–1543 CAS.
- M. Krivec, R. Dillert, D. W. Bahnemann, A. Mehle, J. Strancar and G. Drazic, Phys. Chem. Chem. Phys., 2014, 16, 14867–14873 RSC.
- O. Enea and A. J. Bard, pH Effects on the energetics of irradiated TiO2 suspensions in aqueous D-glucose, Centre national de la recherche scientifique, Paris, FRANCE, 1985 Search PubMed.
- K.-Y. Pan, C.-H. Chien, Y.-C. Pu, C.-M. Liu, Y.-J. Hsu, J.-W. Yeh and H. Shih, Nanoscale Res. Lett., 2014, 9, 307 CrossRef PubMed.
- S. Linic, U. Aslam, C. Boerigter and M. Morabito, Nat. Mater., 2015, 14, 567–576 CrossRef CAS PubMed.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed.
- E. Ringe, M. R. Langille, K. Sohn, J. Zhang, J. Huang, C. A. Mirkin, R. P. Van Duyne and L. D. Marks, J. Phys. Chem. Lett., 2012, 3, 1479–1483 CrossRef CAS PubMed.
- S. Link, C. Burda, M. B. Mohamed, B. Nikoobakht and M. A. El-Sayed, J. Phys. Chem. A, 1999, 103, 1165–1170 CrossRef CAS.
- J. A. Webb and R. Bardhan, Nanoscale, 2014, 6, 2502–2530 RSC.
- G. Baffou and R. Quidant, Chem. Soc. Rev., 2014, 43, 3898–3907 RSC.
- R. L. Ziolli and W. F. Jardim, J. Photochem. Photobiol., A, 2002, 147, 205–212 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ee00971a |
| This journal is © The Royal Society of Chemistry 2016 |