
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A CO2 adsorption-enhanced semiconductor/metal-complex hybrid photoelectrocatalytic interface for efficient formate production†
Xiaofeng
Huang
a,
Qi
Shen
a,
Jibo
Liu
a,
Nianjun
Yang
*b and
Guohua
Zhao
*a
aSchool of Chemical Science and Engineering, Tongji University, 1239 Siping Road, 200092, Shanghai, China. E-mail: g.zhao@tongji.edu.cn
bInstitute of Materials Engineering, University of Siegen, Paul-Bonatz Str. 9-11, Siegen 57076, Germany. E-mail: nianjun.yang@uni-siegen.de
First published on 25th July 2016
Abstract
In photoelectrochemical CO2 conversion, the concentration of fixed CO2 on the photocathode surface is of primary concern. Herein, a CO2 adsorption-enhanced semiconductor/metal-complex hybrid photoelectrocatalytic interface was established by utilizing a carbon aerogel as the CO2 fixation substrate. In CO2 reduction photoelectrocatalysis, Co3O4 was employed as the light harvester, and Ru(bpy)2dppz was utilized as the electron transfer mediator and CO2 activator. The CO2 surface concentration exhibited a 380-fold increase on this hybrid interface than that on Co3O4/FTO. The CO2 conversion to formate occurred at an onset potential of −0.45 V (vs. normal hydrogen electrode, NHE) under photoelectrochemical conditions, 160 mV more positive than its thermodynamic redox potential. At an applied potential of −0.60 V (vs. NHE), the selectivity of the formate yield reached 99.95%, with a production rate of approximately 110 μmol cm−2 h−1 and a Faradaic efficiency of 86%. Such a conversion has an electron transfer rate of 2.94 × 10−3 cm s−1. The CO2 conversion to formate was confirmed to be an instantaneous proton-coupled electron transfer process, originating from the rapid photoelectrochemical activation of bpy and dppz in Ru(bpy)2dppz as well as the synergic effect of the promoted CO2 adsorption and the applied molecular catalysis.
Broader contextIn natural photosynthesis, CO2 is fixed by ribulose-1,5-biphosphate carboxylase/oxygenase (RuBisCo) and then reduced by a regulated proton-coupled electron transfer process. Thus, there are three aspects to achieve a highly effective process of CO2 conversion to fuels by mimicking natural photosynthesis: enhanced CO2 adsorption, rapid electron transfer, and decreased energy input. For a high CO2 surface concentration, molecular catalysts, such as Ru(II) bipyridyl complexes, are promising candidates due to their unique properties in CO2 binding affinity. The application of photoelectrochemical methods that integrate photocatalysis with electrocatalysis provides a promising strategy for CO2 reduction at a low overpotential. This study demonstrates the use of highly porous and adsorptive carbon aerogels (CA) to promote CO2 fixation as well as the employment of a robust visible-light harvester Co3O4 photoelectrocatalyst and Ru(bpy)2dppz as a molecular catalyst to accelerate electron transfer for CO2 reduction. This interface enables high CO2 adsorption, a rapid electron transfer rate and a high yield of formate, an important industrial C1 stock. This reduction occurred at a relatively low onset potential under photoelectrochemical conditions and with a moderate Faradaic efficiency, originating from the multiple synergies among the enhanced CO2 adsorption from CA, the distinguished photoelectrocatalytic activity of Co3O4 and the rapid electrochemical kinetics of Ru(bpy)2dppz. |
Introduction
Natural photosynthesis converts CO2 into a series of intermediate products (e.g., malate and pyruvate) and finally to glucose with the assistance of solar light.1 In such a carbon recycling process, CO2 is fixed by ribulose-1,5-biphosphate carboxylase/oxygenase (RuBisCo). Solar light is harvested, and the electrons are generated by cytochrome. A proton-coupled electron transfer is then regulated and mediated by nicotinamide adenine dinucleotide phosphate hydride, leading to the photochemical reduction of CO2.1 Unfortunately, such a process has a low conversion efficiency. Processes mimicking natural photosynthesis, so-called artificial photosynthesis, have been developed to increase this conversion efficiency and provide more pathways to produce valuable chemicals using CO2 as the carbon stock.2–8CO2 adsorption is of the greatest concern during the CO2 reduction process because the reduction kinetics are highly correlated with the CO2 concentration. However, the free CO2 concentration in aqueous solution is only approximately 0.034 M, which severely deteriorates the aqueous heterogeneous reduction. Although various CO2 absorptive materials have been proposed,9–11 their CO2 adsorption performance and mechanism during catalytic processes have not been investigated in-depth. Moreover, the obtained efficiency for the CO2 reduction still remains to be further increased.
Semiconductor-based photoelectrochemical methods are currently popular and have been successful for CO2 reduction in that the synergetic conjunction of photoelectrocatalysis (namely, electrocatalysis and photocatalysis) facilitates the separation of photo-induced electrons and holes under applied electric fields.6,7 Light irradiation-induced band bending compensates the required overpotential for CO2 reduction, causing the CO2 conversion to occur at relatively positive potentials. Surprisingly, the majority of semiconductors exhibited non-specific CO2 surficial binding. In other words, the CO2 concentration on the surface of the semiconductors was rather low. Therefore, only slow reduction kinetics were reported.6,7 In addition, the majority of semiconductors have poor conductivities, which deteriorated the electron transfer rate as well. Thus, photoelectrochemical CO2 reduction still occurred at relatively negative potentials in these systems, for example, at −1.0 V for MoS2/TiO2 and InP/TiO2 (vs. normal hydrogen electrode, NHE, the same as below),12,13 at −1.1 V for ZnTe,5 at −1.2 V for p-GaP and GaAs,14 and at −0.9 V for Mg–CuFeO2.3 Furthermore, inevitable hydrogen evolution was observed, leading to relatively low Faradaic efficiency.
As an alternative to those semiconductors, molecular catalysts, such as Ru(II) or Re(II) bipyridyl complexes, have been employed because they present unique properties in CO2 binding affinity, and high CO2 surface concentrations are expected.15,16 Furthermore, the formation of a chemical bond between CO2 and the metal centre will lead to a high turnover rate and high selectivity of CO2 conversion during a catalytic cycle.15,16 The main drawback of these catalysts is that additional pathways are required to activate these molecular catalysts. In most cases, an extremely negative potential is required. For example, potentials of −1.52, −1.6, and −1.73 were applied for Ru(bpy)(tpy)(NCCH3),17 Re(tBu-bpy)(CO)3Cl,18 and Re(pbn)(CO)3Cl,19 respectively, to achieve the catalytic reduction of CO2. These molecular catalysts encountered destabilization (e.g., dimerization or dissolution) during a long-term electrolysis process, resulting in rapidly reduced photoelectrochemical activity for CO2 conversion.20,21
The complementary combination of a semiconductor and a molecular catalyst was then developed for photoelectrochemical CO2 conversion.14,22–24 These combinations accelerated electron transfer generation and lowered the activation barriers for both molecular catalysts and CO2 binding. The highly efficient and highly selective generation of hydrocarbon fuels, such as methanol and formic acid, has been reported on these systems.14,22–24 However, these systems have shown significant flaws, such as high overpotentials (e.g., −1.0 V on H–Si–Re(tBu-bpy)(CO)3Cl),14 poor selectivity of reduction products (e.g., the simultaneous formation of HCOOH, H2 and CO on Ag/TaON–Ru(bpy)3–(CH2)2–Ru(bpy)(CO)2Cl2, while HCOOH occupies only 56.5%),23 and low Faradaic efficiency (e.g., only 62.3% on [Ru(dcbpy)2(CO)2]n–p-InP–Zn).24
Thus, the aim of this work is to construct an adsorptive photoelectrochemical interface for CO2 reduction with better performance. The constructed interface is schematically shown in Scheme 1. In our concept, CO2 fixation is promoted by utilizing an adsorptive substrate, a conductive, micropore-dominated three-dimensional carbon aerogel (CA) with a high surface area (up to 1815 m2 g−1); solar light is efficiently harvested by Co3O4 micro-flowers, a visible-light driven and robust photoelectrocatalyst. These highly index-faceted Co(III)-enriched {12![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) } structures are grown epitaxially inside CA networks. An enzyme-mimicking molecular catalyst, Ru(bpy)2dppz, is immobilized on Co3O4/CA to accelerate and regulate the electron transfer for CO2 reduction. Using such an interface, the CO2 conversion to formate has been achieved at an onset reduction potential under photoelectrochemical conditions as low as −0.45 V, with a yield of approximately 110 μmol cm−2 h−1, a selectivity of 99.95%, and a Faradaic efficiency of 86% at −0.60 V.
} structures are grown epitaxially inside CA networks. An enzyme-mimicking molecular catalyst, Ru(bpy)2dppz, is immobilized on Co3O4/CA to accelerate and regulate the electron transfer for CO2 reduction. Using such an interface, the CO2 conversion to formate has been achieved at an onset reduction potential under photoelectrochemical conditions as low as −0.45 V, with a yield of approximately 110 μmol cm−2 h−1, a selectivity of 99.95%, and a Faradaic efficiency of 86% at −0.60 V.
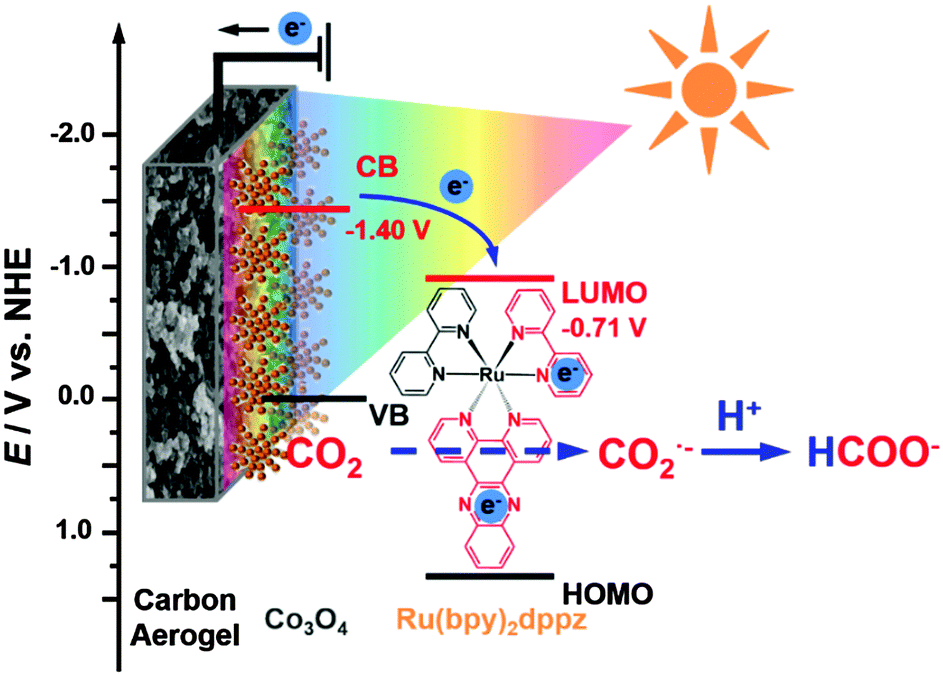 | ||
| Scheme 1 Schematic plots of the CO2 adsorption-enhanced Ru(bpy)2dppz-Co3O4/CA interface together with its energy level diagram and the possible reaction pathways for CO2 conversion on this photocathode. Such a photoelectrocatalytic interface is composed of CA as the CO2-adsorption substrate, Ru(bpy)2dppz as the molecular catalyst, and Co3O4 as the photoelectrocatalyst. | ||
Experimental section
Fabrication of the Ru(bpy)2dppz-Co3O4/CA photocathode
Monolith bulky CA was synthesized via an ambient pressure resorcinol-formaldehyde (RF) drying method. The detailed synthesis process of CA is shown as Scheme S1 in the ESI.† A solvothermal reaction was utilized to synthesize Co3O4/CA. In a 50 mL acetone–water mixture (Vacetone![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Vwater = 5:45), 1.4552 g of Co(NO3)2·6H2O, 0.2593 g of NH4F, and 1.4019 g of hexamethylenetetramine were added. Magnetic stirring of this mixture for 10 min led to the formation of a pink transparent solution. The solution was then transferred to a Teflon-lined stainless steel autoclave, where one slide of CA (4 × 1.5 cm2) was located. The solvothermal reaction was conducted at 95 °C for 24 h. After cooling down to room temperature, deposition (pink to violet colour) occurred on the CA. The as-prepared sample was carefully rinsed with acetone and then dried under vacuum at 60 °C for 2 h, followed by a calcination process in a N2 atmosphere at 450 °C for 2 h. The ramping rate was 10 °C min−1. After such a calcination process, the Co3O4/CA sample was synthesized. The amount of Co3O4/CA loading, averaged over the geometric area of the CA, was typically 8 mg cm−2. A similar procedure was applied for the preparation of the control sample (Co3O4/FTO). The loading density of Co3O4 on FTO was 5 mg cm−2. In this case, a piece of fluorine-doped tin oxide glass electrode (FTO, 4 × 1 cm2) was put into the autoclave instead of CA.
:Vwater = 5:45), 1.4552 g of Co(NO3)2·6H2O, 0.2593 g of NH4F, and 1.4019 g of hexamethylenetetramine were added. Magnetic stirring of this mixture for 10 min led to the formation of a pink transparent solution. The solution was then transferred to a Teflon-lined stainless steel autoclave, where one slide of CA (4 × 1.5 cm2) was located. The solvothermal reaction was conducted at 95 °C for 24 h. After cooling down to room temperature, deposition (pink to violet colour) occurred on the CA. The as-prepared sample was carefully rinsed with acetone and then dried under vacuum at 60 °C for 2 h, followed by a calcination process in a N2 atmosphere at 450 °C for 2 h. The ramping rate was 10 °C min−1. After such a calcination process, the Co3O4/CA sample was synthesized. The amount of Co3O4/CA loading, averaged over the geometric area of the CA, was typically 8 mg cm−2. A similar procedure was applied for the preparation of the control sample (Co3O4/FTO). The loading density of Co3O4 on FTO was 5 mg cm−2. In this case, a piece of fluorine-doped tin oxide glass electrode (FTO, 4 × 1 cm2) was put into the autoclave instead of CA.
Chemical polymerization was used to decorate the Co3O4/CA with Ru(bpy)2dppz (the detailed synthesis procedure for Ru(bpy)2dppz is provided in the ESI†). Briefly, 0.0040 g of Ru(bpy)2dppz (5.75 μmol) was dissolved in 1 mL of acetonitrile solution, denoted as solution A. Next, 0.0650 g of Fe(NO3)3·9H2O and 8 μL of pyrrole were dissolved in 1 mL of ethanol, and the mixture was denoted as solution B. The stock solution for the electrode coating was obtained by mixing solutions A and B (with equal volumes) and shaking. One slide of Co3O4/CA was then coated with 200 μL of the stock solution in a dropwise manner, and the composite was dried at 60 °C for 5 min. After repeating the coating procedure 10 times, Ru(bpy)2dppz-Co3O4/CA was obtained.
Electrochemical quartz crystal microbalance
To calculate the amount of CO2 adsorbed onto the photocathodes, electrochemical quartz crystal microbalance (EQCM) experiments were conducted on a CHI440A (CH Instruments Inc., USA) with a Au-coated AT-cut quartz crystal (a fundamental frequency of 8 MHz) as the working electrode.Approximately 10 mg of ground CA powder was ultrasonically dispersed in 1 mL of H2O along with 20 μL of 2% Nafion-117 film solution (Alfa Aesar). After intense ultrasonication for 5 min, 10 μL of the dispersed solution was dip-coated onto the Au electrode and dried in air. The CA-modified electrode was further used in the EQCM studies.
In situ IR spectroelectrochemical experiments
For the in situ IR spectroelectrochemical experiments, the mercury–cadmium–telluride detector was cooled down to 77 K by liquid nitrogen. A self-made four-bottleneck cell with a CaF2 window (2 mm thickness, 25 mm in diameter) was used as the electrochemical cell. A Pt disk electrode (5 mm in diameter), a Ag/AgCl (filled with saturated KCl) electrode, and a Pt foil (1 × 1.5 cm2) were used as the working, reference and counter electrodes, respectively. A mixture of 0.010 g mL−1 Ru(bpy)2dppz and 2% Nafion-117 was stirred in a sonication bath for 1 min. This mixture (10 μL) was dip-coated onto the Pt disk electrode and dried under ambient conditions. One hundred IR spectra were collected with a spectrum resolution of 8 cm−1 and subsequently averaged at each potential. For these experiments, the solution was purged with CO2 for at least 30 min to completely remove the dissolved oxygen and saturate the solution with CO2.Photoelectrochemical characterization and reduction of CO2
To evaluate the performance of the Ru(bpy)2dppz-Co3O4/CA catalyst, constant potential photoelectrolysis of CO2 was conducted in order to evaluate the performance of Ru(bpy)2dppz-Co3O4/CA, which was conducted in a home-made H-type cell with a maximum volume of 100 mL. The as-prepared Ru(bpy)2dppz-Co3O4/CA working electrode and the Ag/AgCl (filled with saturated KCl) reference electrode were placed in the cathodic chamber, while the counter electrode, a graphite plate (4 × 1 cm2), was placed in the anodic chamber. The two chambers were connected with 0.1 M NaHCO3 but separated with a Nafion-117 proton exchange membrane (Dupont). Prior to the experiments, the electrolyte in the cathodic chamber was purged with high-purity CO2 (99.99%) gas for more than 30 min at a flow rate of 20 mL min−1. Negative potentials (0.0, −0.2, −0.4, −0.6, −0.8, −1.0 V) were applied to the photocathode through the electrochemical workstation. An APLS-SXE300 xenon lamp with a UV cutoff (λ > 420 nm, light intensity at 9 mW cm−2) was used as the light source and illuminated on the Ru(bpy)2dppz-Co3O4/CA photocathode upon the addition of negative potential.After such constant potential photoelectrolysis for 8 h, the reduction products were collected and quantitatively determined by HPLC and GC using the same procedure as described previously.25 For the products in the aqueous phase, 0.2 mL of the liquid sample was collected and transferred into a 10 mL test-tube. The pH of the sample was adjusted to neutral by adding 0.2 mL of pH 7.6 phosphate buffer solution. Subsequently, 2,3,4,5,6-pentafluorobenzyl bromide (20 g L−1, 1.0 mL) was added. The mixture was shaken for 1 min and then kept at 60 °C for another 1 h. The esterification product was extracted with 2.0 mL of n-hexane and centrifuged at 3000 rpm for 5 min. The upper layer was the organic phase, which was filtered through a 0.45 μm membrane. A C18 column was used with a mobile phase consisting of 65% methanol and 35% H2O at a flow rate of 1.0 mL min−1. The detection wavelength was 225 nm. For gaseous products, 1.0 mL of the gas sample was collected through a syringe. The detection conditions were an injection inlet temperature of 130 °C, an oven temperature of 80 °C, a detector temperature of 150 °C, N2 carrier gas, and a gas flow rate of 0.2 L min−1.
Prior to an isotopic 13C experiment, 13CO2 (13C enrichment 98%) was purged into 0.1 M NaH13CO3 (13C enrichment 98%) electrolyte solution for at least 30 min in order to fully expel oxygen and other impurity gases. The photoelectrochemical reduction of 13CO2 saturated NaH13CO3 (0.1 M) was identical to the procedure described in the Experimental section, holding the constant potential at −1.2 V vs. Ag/AgCl. Blank experiments using nitrogen purged Na2SO4 (0.1 M) were also conducted using an identical procedure. After reduction, 0.5 mL of catholyte solution was mixed with 0.1 mL of D2O (Sigma Aldrich) containing 0.5 μL of DMSO as the internal standard. A one-dimensional 1H nuclear magnetic resonance (NMR) spectrum was recorded with water suppression using a pre-saturation method.
Results and discussion
Enhanced CO2 adsorption
For photoelectrochemical CO2 conversion, the surface concentration of CO2 on the photocathode, namely, CO2 fixation, is a chief concern. A high surface concentration of CO2 on the photocathode accelerates the photoelectrochemical kinetics. Herein, the surface concentration of CO2 (Γads), the normalized amount of adsorbed CO2 with the electrochemical active surface area (SEASA), was adopted as the parameter to evaluate the efficiency of CO2 fixation on the photocathodes.The results in Table S1 (ESI†) demonstrate the advantage of using a CA substrate for CO2 fixation. This statement is further supported by the EQCM results of CA in a CO2-saturated electrolyte under negative potentials, as shown in Fig. S1 (ESI†). The mass addition is 6–10 times heavier on activated CA than that on an Au electrode. For example, at −0.4 V, a mass addition of 174 ng cm−2 was obtained on a CA substrate, whereas it was barely observed on an Au quartz substrate. At −0.6 V, the mass addition on the CA substrate was 8.2-fold larger than that on the Au quartz substrate. At −0.9 V, the mass addition reached 143.5 ng cm−2 on the CA substrate. When the applied potentials were more negative than −0.9 V, the mass addition on the CA substrate was even larger than that on the Au quartz substrate. CA is not capable of reducing CO2 electrochemically; thus, the mass addition on the CA substrate is mainly ascribed to the promoted electro-sorption of carbonaceous species (e.g., CO2 and HCO3−) from the electrolyte. The carbonaceous species are significantly promoted because the micropores contributed a high Brunauer–Emmett–Teller surface area (SBET) of up to 1815 m2 g−1 (Fig. S2, ESI†).
This high SBET offers numerous sites for CO2 adsorption, as indicated by Fujishima et al. and Yaghi et al.,21,26 as CO2 molecules tend to be adsorbed in the micropores of a material. This value is also higher than that of common porous carbon materials, such as ordered mesoporous carbon (812.3 m2 g−1), commercially available Vulcan (237.9 m2 g−1) and carbon nanocoil (233 m2 g−1).27 The superior performance of CA supports the notion that CO2 adsorption on other high-surface-area carbon materials (e.g., activated carbon, carbon nanotubes) might result from the following aspects. First, the microporous feature of CA (Fig. S2, ESI†) offers numerous sites for CO2 adsorption. Second, the activated CA possesses a relatively high surface area (1815 m2 g−1), which provides many sites for Co3O4 loading. According to our measurements, the Co3O4 loading amount was increased to 8 mg cm−2 compared to 5 mg cm−2 on FTO. The SBET of Co3O4/CA of 713 m2 g−1 (Fig. S2a, ESI†) is considerably higher than that of commonly designed porous inorganic semiconductor electrodes.28,29 This result is further supported by the well-maintained micropore domination of the electrode (Fig. S2b, ESI†). Based on the voltammetry (Fig. S3, ESI†) and chronocoulometry (Fig. S4, ESI†) for Co3O4/CA, Γads exhibits a 20-fold increment (0.25 pmol cm−2) compared to that (0.01 pmol cm−2) of Co3O4/FTO. The electrochemically active surface area (SEASA) was determined to be 8015 cm2 (Table S1, calculated from Fig. S5, ESI†) with respect to that of Co3O4/FTO (316 cm2). Experimental tests on the effects on CO2 fixation of other porous carbon-based substrates are currently in progress.
As shown in the scanning electron microscopic (SEM) images of Co3O4/CA (Fig. S6, ESI†), the CA backbone is clearly visible even after the solvothermal growth of Co3O4 micro-flowers. The XRD patterns of both CA and Co3O4 in Co3O4/CA can also be clearly observed (Fig. S7, ESI†). In other words, such an epitaxial growth pattern fully exposes the adsorption sites to CO2, which results in an increase in the Γads for CO2. Moreover, the 3D structure of CA allows for the penetration of electrolytes into the pores of the electrode, leading to an increase in the interfacial area between the electrode and the electrolyte. Finally, CA possesses a high conductivity (electrical resistivity < 40 mΩ), similar to those of carbon nanotubes and graphene.
Γ ads further increases to 3.79 pmol cm−2 when Co3O4/CA is coated with a molecular catalyst, Ru(bpy)2dppz, which is 15.2 and 300 times larger compared to those on Co3O4/CA and Co3O4/FTO, respectively. However, the SEASA of Co3O4/CA is approximately 4 times larger than that of Ru(bpy)2dppz-Co3O4/CA, likely due to the blockage of Co(III) active sites by immobilized Ru(bpy)2dppz during the chemical polymerization. Nonetheless, the active area of this electrode is maintained at 1.64 × 10−2 cm2 (Table S1 and Fig. S3, ESI†). The existence of the Ru(bpy)2dppz molecular catalyst promotes CO2 fixation on the photocathode with an enhancement factor of 300. Γads was also normalized by the catalyst weight (Table S1, ESI†) and exhibited the same trends as that normalized by SEASA. Hence, the enhancement of CO2 adsorption and photoelectrochemical density is in fact the synergic effect of multiple factors: the electrosorption from the CA substrate, the Co(III) sites from the high index faceted Co3O4 on CA and the electrochemical activity of Ru(bpy)2dppz. These results confirm that CA, Co3O4 and Ru(bpy)2dppz co-promote CO2 fixation and lead to a high CO2 surface concentration on such an interface. Thus, accelerated kinetics are expected for photoelectrochemical CO2 conversion.
Photoelectrochemical CO2 conversion
The reactivity of Ru(bpy)2dppz as a homogenous catalyst toward electrochemical CO2 conversion was first studied in CO2-saturated NaHCO3 solution. As shown in Fig. S8 (ESI†), the CO2 reduction potential remains at −0.40 V in the current aqueous solution, which is at least 950 mV more positive than those reported in organic solvents.30 This is the first time that a molecular catalyst, polybipyridyl Ru(II), was observed to convert/reduce CO2 at such a low potential, indicating the high electron transfer ability of Ru(bpy)2dppz toward CO2 conversion.CO2 conversion on Ru(bpy)2dppz-Co3O4/CA was investigated in detail using a potentiodynamic mode. As seen in Fig. 1a, CO2 conversion/reduction occurs at an onset potential of −0.45 V under photoelectrochemical conditions (red line). This potential is an “underpotential” of approximately 160 mV with respect to the thermodynamic redox potential for CO2 to formic acid.31 The peak potential is approximately −0.61 V, considerably lower than other reported values, e.g., −1.33 V on p-Cu2O immobilized by Re(tBu-bipy)(CO)3Cl32 and −0.6 V on p-InP–Zn decorated by [Ru(dcbpy)2(CO)2]n24 On Co3O4/CA, the reduction potential is approximately −0.62 V with an onset potential of −0.52 V under photoelectrochemical conditions (dashed lines). Moreover, a second peak appears at −0.73 V. On Co3O4/FTO, the onset potential is −0.67 V (dotted lines), indicating an entirely different photoelectrochemical reduction pathway. The positive shift of the CO2 reduction potential on Ru(bpy)2dppz-Co3O4/CA is likely due to the increased surface concentration of CO2 and the catalytic role of Ru(bpy)2dppz. Based on these peak potentials, the heterogeneous electron transfer rate constant (ks) for CO2 reduction was calculated using a method provided in the literature.33ks is 2.94 × 10−3 cm s−1 for Ru(bpy)2dppz-Co3O4/CA, which is 26% larger than that (2.19 × 10−3 cm s−1) for Co3O4/CA.
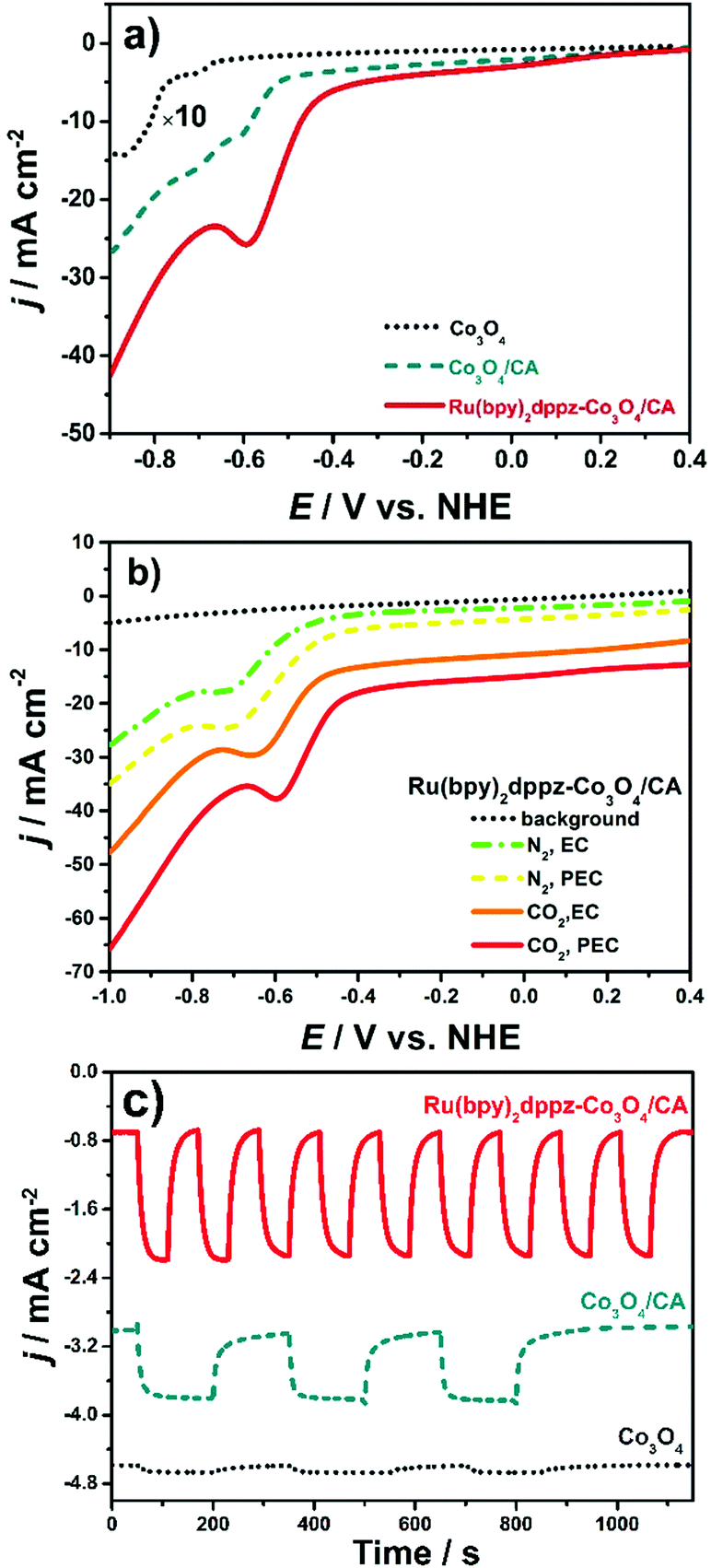 | ||
| Fig. 1 (a) Linear sweep voltammograms (LSVs) of Co3O4/FTO (dotted line, with a magnification factor of 10), Co3O4/CA (dashed line) and Ru(bpy)2dppz-Co3O4/CA (solid line) in CO2-saturated 0.1 M NaHCO3 at a scan rate of 50 mV s−1 under illumination conditions; (b) LSVs of Ru(bpy)2dppz-Co3O4/CA in N2-purged 0.1 M Na2SO4 (dotted line), N2-purged 0.1 M NaHCO3 with (dashed line)/without (dash-dotted line) light irradiation, and CO2-saturated 0.1 M NaHCO3 without (solid line) light irradiation at a scan rate of 50 mV s−1; (c) amperometric i–t curves on Co3O4/FTO (dotted line), Co3O4/CA (dashed line) and Ru(bpy)2dppz-Co3O4/CA (solid line) in CO2-saturated 0.1 M Na2SO4 at a potential of −0.40 V. The lines were shifted in the Y-axis direction for comparison. | ||
This higher ks value suggests an accelerated and enhanced electron transfer on Ru(bpy)2dppz-Co3O4/CA. Such a method to evaluate the efficiency of CO2 conversion is entirely new. According to our knowledge, this method has not been presented in the literature to date. Considering the CO2 reduction potential and ks on both electrodes, the catalytically active centre for CO2 reduction is Ru(bpy)2dppz-Co3O4/CA, not Co(III). This statement is further demonstrated from the Tafel plots described below.
The current density reaches approximately 8.1 mA cm−2 on Ru(bpy)2dppz-Co3O4/CA, which is considerably more intense than sole Ru(bpy)2dppz and those reported using similar molecular catalyst–semiconductor composites. For instance, on Ru(dcbpy)2(CO)2/p-InP and CO-dehydrogenase/p-NiO, the photocurrent density reached only the level of μA cm−2 at similar potentials. A photocurrent density larger than −2.0 mA cm−2 was obtained on a Re(tBu-bpy)(CO)3Cl/Cu2O photocathode, but a potential of −2.0 V was applied.32 So, photoelectrochemical CO2 conversion on Ru(bpy)2dppz-Co3O4/CA occurs at a low overpotential but with a high photocurrent density.
The current densities shown in this paper were obtained using the geometric areas of the electrodes. Due to their porous structures, CA-based photoelectrodes will have a higher SEASA value. Their SEASA values were determined using surface-sensitive redox probes of Fe(CN)63−/4− (Table S1 and Fig. S3, ESI†). A 2.7-fold enhancement was observed after loading the same amount of Co3O4 onto FTO. The current densities were then re-calculated using their SEASA values. The magnitude of the current densities followed the same trend, namely, in the order of Co3O4/FTO < Co3O4/CA < Ru(bpy)2dppz-Co3O4/CA, the same as that obtained from the current densities normalized by the geometric area. To compare our results with those obtained using other porous materials presented in the literature, the current densities shown throughout the paper were then calculated using the geometric areas of the photoelectrodes, the most frequently applied approach for electrochemical and photoelectrochemical CO2 reduction.
Fig. 1b shows the LSVs on Ru(bpy)2dppz-Co3O4/CA in 0.1 M NaHCO3 solution purged with N2 or CO2. The pH value of 0.1 M NaHCO3 is in the range of 8.3–8.5 after being purged with N2. Once it was saturated with CO2, the pH value decreased to the range of 6.5–7.0.34 Such a weak acidic environment provides a favourable protic environment for CO2 reduction using a Ru(bpy)2dppz catalyst.2–5 Notably, the peak potential for CO2 reduction is 50 mV more positive than that of its N2-purged counterpart, with the peak current density doubled. These results clearly confirm the involvement of protons in CO2 reduction. Fig. 1b shows the effect of light irradiation on CO2 conversion as well. On Ru(bpy)2dppz-Co3O4/CA without light irradiation, the peak current density of the CO2 reduction decreases by a ratio of 35% along with a 50 mV negative shift in the peak potential compared to the case when light irradiation is applied. Similar tendencies were observed on Co3O4/CA and Co3O4/FTO (Fig. S9, ESI†). This phenomenon could be due to the photoelectrocatalytic properties of the Ru(bpy)2dppz-Co3O4/CA electrode.
An amperometric photocurrent response was further investigated at a fixed potential of −0.40 V. As shown in Fig. 1c, the steady photocurrent density reaches approximately 1.5 mA cm−2 on Ru(bpy)2dppz-Co3O4/CA, with a stabilizing time of only 40 s, whereas on Co3O4/CA, the peak photocurrent intensity decreases by approximately 7 times compared to Ru(bpy)2dppz-Co3O4/CA. However, the peak photocurrent intensity is 8–10 times higher than that on Co3O4/FTO. The stabilization times for Co3O4/CA and Co3O4/FTO are 150 and 200 s, respectively. The potentiodynamic voltammograms in 0.1 M Na2SO4 also reflected that Ru(bpy)2dppz-Co3O4/CA showed a distinguished photoelectrochemical activity. The difference between the photocurrent (jPEC) and dark current (jEC), denoted as jPEC − jEC, is summarized in Fig. S10 (ESI†). jPEC − jEC is approximately 10 times higher on Ru(bpy)2dppz-Co3O4/CA than on FTO. This result could be explained by several reasons. First, the {12} crystal facet of Co3O4 (with an inter-planar spacing of 0.286 nm) being finely exposed (Fig. S11, ESI†), which enriches triply uncoordinated Co(III) sites. Such a high-index-facet Co3O4 facilitates the photoelectrochemical reduction of CO2, as our previous work has indicated.4 Second, the epitaxial growth manner of high-index-facet Co3O4 on CA provided a direct electron transfer channel. Photo-induced holes could rapidly transfer to the highly conductive CA network. Electrons and holes are efficiently separated. Finally, the ingenious merging of photocatalysis and electrocatalysis on a single surface has been found to efficiently separate the photo-induced carriers, resulting in a reduction of the overpotential for CO2 conversion.2,4 The applied negative potential creates a more upward bending for Co3O4, a p-type semiconductor. Then, the driving force for photoelectrons to cross the semiconductor–electrolyte junction is enlarged, resulting in an enhanced photocurrent density. In the meantime, the light-induced upward band-bending of Co3O4/CA lifts the Fermi level, which compensates part of the required applied negative potential under dark conditions, yielding a reduced overpotential for CO2 reduction. All of these effects reveal the essential role of Co3O4 as the photoelectrocatalyst in a photoelectrochemical strategy for CO2 conversion.
Comparing the cyclic voltammograms of different photoelectrodes, the lowest overpotential and the highest photocurrent density for CO2 conversion were obtained on Ru(bpy)2dppz-Co3O4/CA. Apart from the respective contributions from the enhanced adsorption on CA and distinguished photoelectrochemical properties of Co3O4/CA, the molecular catalyst Ru(bpy)2dppz also exhibits excellent electrochemical reductive activity toward CO2 reduction, as suggested by the above-mentioned ks. The synergistic effect among those effects exerted on Ru(bpy)2dppz-Co3O4/CA has superiority in magnifying the photocurrent density, reducing the overpotential, and accelerating and regulating the electron transfer pathway for the photoelectrochemical CO2 reduction process.
The photoelectrochemical CO2 conversion on Ru(bpy)2dppz-Co3O4/CA was further studied in CO2-saturated 0.1 M NaHCO3 by varying the applied potential. Its variation with the potentials applied is summarized in Fig. 2a. The amplitude of j(CO2) − j(N2) increases when the potential is more negative, following the order of Co3O4/FTO < Co3O4/CA < Ru(bpy)2dppz-Co3O4/CA. A sharp increase in the amplitude of j(CO2) − j(N2) occurs at −0.4 V. At a reduction potential of −0.6 V, the net photocurrent reaches −0.35 mA cm−2. This value is twice as high as that on Co3O4/CA and nearly 10 times higher than that on Co3O4/FTO.
 | ||
| Fig. 2 (a) Variation of j(CO2) − j(N2), on Co3O4/FTO (grey), Co3O4/CA (cyan) and Ru(bpy)2dppz-Co3O4/CA (red) with the applied potential; (b) variation of the yield rates of formate on the photocathode of Co3O4/FTO (gray triangle), Ru(bpy)2dppz/CA (green rhombus), Co3O4/CA (cyan square) and Ru(bpy)2dppz-Co3O4/CA (red circle) with the applied potentials. The dashed lines are for guiding the eyes. (c) 1H NMR spectra of the reduction product in N2-purged Na2SO4(I), CO2-saturated NaHCO3(II) and 13CO2-saturated NaH13CO3(III). The concentrations of these solutions were 0.1 M. | ||
Under identical photoelectrolysis conditions, the yield rate of formate on these four photocathodes is in the order of Ru(bpy)2dppz-Co3O4/CA > Co3O4/CA > Ru(bpy)2dppz/CA > Co3O4/FTO at the same applied potentials, as shown in Fig. 2b. For example, 869.8 μmol formate was produced at a potential of −0.6 V on Ru(bpy)2dppz-Co3O4/CA. This value is considerably higher than that presented previously (384 μmol) on hierarchical Co3O4, even at −0.7 V.5 If the electrode area is considered, the predicted yield rate of formate on Ru(bpy)2dppz-Co3O4/CA is approximately 450 μmol cm−2 at the onset potential of −0.45 V under photoelectrochemical conditions. At −0.7 V, the yield rate of formate reaches approximately 900 μmol cm−2. Considering the reduction time span further, the yield rate of formate reaches nearly 110 μmol cm−2 h−1, even at a reduction potential of −0.6 V. The yield rate of formate on Ru(bpy)2dppz-Co3O4/CA is higher than most reported values, e.g., nearly 29-, 5.2-, and 2-fold higher than that on Ru(II)–Re(I) multinuclear complexes,22 Ru(II) electro-polymerized p-InP–Zn,24 and Cu2ZnSnS4,35 respectively. The estimated Faradaic efficiency for the yield of formate is approximately 86%, which is comparable to that on semiconductor–molecular photocatalytic systems as well as on other semiconductor-based photocatalytic systems, such as Mg-doped CuFeO23 and Au/ZnO/ZnTe.5 Notably, formate yield rates on the Ru(bpy)2dppz/CA were lower than that on the Co3O4/CA at each investigated potential, the reason is described below.
To confirm the carbon source for the reduction product of formate, isotopic measurements were conducted using 13CO2-saturated NaH13CO3 (0.1 M) electrolyte solution.36,37 Control experiments were also performed in N2-purged Na2SO4 (0.1 M) solutions. These related 1H-NMR spectra are shown as Fig. 2c. The spectrum obtained from the N2-purged Na2SO4 (0.1 M) solutions is featureless (Fig. 2c-I) in the chemical shift range of 9.2 to 7.7, indicating that no formate is produced when no carbonaceous species (e.g., CO2, HCO3−) are present. In Fig. 2c-II, a singlet peak at the chemical shift value of 8.42 is detected, likely resulting from the yield of HCOO− from CO2-saturated NaHCO3. When 13CO2-saturated NaH13CO3 solution is used, a doublet peak is shown at the chemical shift values of 8.64 and 8.25 (Fig. 2c-III). This doublet peak indicates the production of H13COO−.3 In other words, formate is obtained from the 13C species in the electrolyte rather than from the impurities in the electrolyte or on the surface of the photocathode. Photoelectrochemical CO2 reduction thus occurs on the adsorption-enhanced molecular catalyst–semiconductor hybrid interface.
To investigate the energy conversion efficiency, the turnover number (TON) and turnover frequency (TOF) for photoelectrochemical CO2 reduction were estimated at different potentials on this interface. These values are tabulated in Tables S2 and S3 (ESI†). Co3O4/CA has a 3- to 5-fold increment in both TON and TOF compared to Co3O4/FTO, resulting from the effect of improved CO2 adsorption. This result further demonstrates the importance of the primary CO2 fixation process. On the Ru(bpy)2dppz molecular catalyst immobilized interface, the magnitudes of both TON and TOF were increased by approximately 1 order. This trend was also proven by the results from [Zn(II)TRP]4+/[SiW12O40]4−38 and COF-367-Co,21 both of which possess a high surface area, thus resulting in a high TON value. The TON value was increased with the negative shift of the potential. For example, at −0.6 V vs. NHE, the TON value already reached 978.7, with a TOF value of 122. In Table 1, these results are further compared with those shown in the literature. The higher TON value (978.7) in our case compared to those reported (including those of similar semiconductor–molecular complex model systems (e.g., InP/[MCE2-A + MCE4]–TiO2/Pt,39 the NiO–RuRe complex40 and Ru(dcbpy)/N-Ta2O541)) demonstrates the distinguished photoelectrochemical performance of the concurrent Ru(bpy)2dppz-Co3O4/CA system.
| Catalyst | Condition | Product | TON (TOF) | Ref. |
|---|---|---|---|---|
| Ru(bpy)2dppz-Co3O4/CA | −0.6 V vs. NHE, 9 mW cm−2 Xe lamp, 8 h | HCOO− | 978.7 (122) | This work |
| Ru(bpy-H2PO3)2(CO)2Cl2–C3N4 | 400 W Hg lamp (λ > 400 nm), 20 h | HCOOH | 1100 (55) | 42 |
| Ru(bpy)3–(CH2)2–Re(CH3-bpy)(CO)3Cl | Hg lamp (λ > 500 nm), 24 h | HCOOH | 25 (1.04) | 43 |
| Ru(H4P2O6–C2H4-bpy)(CO)2Cl2-mpg C3N4 | 450 W Hg lamp (λ > 400 nm), 20 h | HCOOH | ∼210 (10.5) | 44 |
| InP/[MCE2-A + MCE4] | AM 1.5G, 24 h | HCOO− | >17 (0.7) | 39 |
| Ru(dcbpy)/N-Ta2O5 | Xe lamp (410 < λ < 750 nm), 20 h | HCOO− | 90 (4.5) | 45 |
| NiO–RuRe complex | −1.2 V vs. Ag/Ag+, 300 W Xe lamp, 5 h | CO | 32 (6.4) | 40 |
| Zn–TPP–Re complex | 200 W Hg lamp, λ > 375 nm, 50 h | CO | 12.8 (0.26) | 46 |
| Re(bpy)(CO)3Cl | −1.25 V vs. SHE, 14 h | CO | 300 (22) | 47 |
| [Co(CR)Cl2](ClO4) | LED light strip (λ > 460 nm), 22 h | CO | 268 (12.18) | 48 |
Hydrogen, as the only side product detected, only occupied 0.05% of the total products, which is indicative of the high selectivity of Ru(bpy)2dppz-Co3O4/CA toward CO2 reduction. On Co3O4/CA, the hydrogen production is 3 times (Fig. S12b, ESI†) higher, but the yield rate of formate is approximately 1.5 times lower (Fig. 2b). On Ru(bpy)2dppz/CA (Fig. S12c, ESI†), lower amounts of H2 were detected after applying a potential of −0.9 V. On Co3O4/FTO (Fig. S12d, ESI†), the yield rate of formate is the lowest, although no hydrogen is detected. The selectivity for Ru(bpy)2dppz-Co3O4/CA is higher than those of Ru(II)-electropolymerized p-InP–Zn24 and Cu2ZnSnS4,35 in which non-negligible amounts of H2 and CO were generated. Therefore, the selectivity of the CO2 conversion to formate is extremely high on Ru(bpy)2dppz-Co3O4/CA (99.95%).
The hydrogen evolution potentials were further estimated based on the potential-dependent hydrogen evolution profiles shown in Fig. 2b and Fig. S12a–d (ESI†). The potentials on Ru(bpy)2dppz-Co3O4/CA, Co3O4/CA, Ru(bpy)2dppz/CA and Co3O4/FTO are −0.6 V, −0.7 V and −0.9 V vs. NHE, respectively. However, there are no clear cathodic peaks shown in Fig. 1 for hydrogen evolution because the Ru(II) centre on the molecular catalyst Ru(bpy)2dppz has fully coordinated to the nitrogen atoms from two 2,2′-bipyridine and dppz ligands. Then, both the bipyridine ligand and dppz are not able to dissociate from Ru(II) during the electrochemical processes. In other words, there will be no opportunity to generate any catalytic wave for hydrogen evolution. These results indicate that this unwanted reaction has not been involved in our system during CO2 reduction. Thus, an improved conversion efficiency and high selectivity are expected.
The stability of the proposed photoelectrocatalytic interface was examined by recording the XRD patterns and cyclic voltammograms of Ru(bpy)2dppz-Co3O4/CA before and after a long-term photoelectrochemical CO2 reduction. The XRD patterns of both CA and Co3O4 do not vary after the 8 h photoelectrochemical reduction of CO2 (Fig. S13, ESI†). The peak current density of Ru(bpy)2dppz-Co3O4/CA is slightly enhanced after 100 cycles of cyclic voltammetry (Fig. S14, ESI†). These facts confirm the high stability of our molecular catalyst–semiconductor-assembled photocathode.
The underlying mechanism of such a synergic effect, the high selectivity for formate and its relation to the photoelectrochemical CO2 reduction is discussed. Prior to this discussion, the interfacial energetics between the two components were analysed. The bandgap (Fig. S15, ESI†) and the flat-band potential (Fig. S16, ESI†) of Co3O4/CA were estimated to be 1.87 V and 0.37 V, respectively. Provided that the valence band of Co3O4 is 0.1 V more positive than the flat band potential, the valence band of Co3O4 was estimated to be 0.47 V. So the conduction band of Co3O4/CA was estimated to be −1.40 V vs. NHE (Eg = |ECB − EVB|). In contrast, the energy difference between the HOMO and the LUMO for Ru(bpy)2dppz is approximately 2.29 eV, as calculated using the intersection wavelength at 540.8 nm from its normalized UV-vis and fluorescence spectra in acetonitrile (Fig. S17a, ESI†). According to the Ru(III)/Ru(II) redox potential (Fig. S17b, ESI†), the LUMO energy level of Ru(bpy)2dppz was calculated to be −0.71 V. Ru(bpy)2dppz's LUMO energy level is then 0.69 V more positive than the conduction band of Co3O4/CA. Therefore, the photo-induced electrons on Co3O4 transfer to the LUMO of the molecular catalyst, as confirmed by various reports.24,32,35,40,41 Although the value of the conduction band and the redox potential of the molecular complex vary with the pH value of the electrolyte, the shift of the redox potential of a similar molecular complex [Ru(phen)2(ptpbβ)]2+ by a rate of −62 mV pH−149 is in line with the typical −59 mV pH−1 for semiconductors.
Electrochemical characterization data can also support such an electron transfer process. The onset potential of electrochemical CO2 reduction using Ru(bpy)2dppz was at −0.4 V vs. NHE, which is similar to that for photoelectrochemical CO2 reduction using Ru(bpy)2dppz-Co3O4 (−0.45 V vs. NHE). Moreover, the cathodic current density of electrochemical CO2 reduction on Ru(bpy)2dppz (ca. 0.63 mA cm−2) was far smaller than that on Ru(bpy)2dppz-Co3O4 (ca. 15.0 mA cm−2). From in situ IR spectroelectrochemical spectra (Fig. 3a), the upward IR peaks at 1419 and 1446 cm−1, assigned to the A1 mode of the C–C–H deformation bending vibration on bpy˙− and dppz˙−,50,51 validate such a statement. A relatively strong broad upward peak at 1716 cm−1 displays the same trend. This peak likely arises from the stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O from the as-formed formate.52,53 These facts suggest that the electrons are transferred from the excited Co3O4 to Ru(bpy)2dppz, and then take part in photoelectrochemical CO2 reduction. As a result of such a photo-induced electron transfer to the LUMO of Ru(bpy)2dppz, CO2 conversion/reduction occurs in the adsorptive substrate CA, the photocatalyst Co3O4 and the molecular catalyst Ru(bpy)2dppz-Co3O4/CA, exactly as illustrated in Scheme 1. Such an electron transfer process is the core of such an interface. Note that Ru(bpy)2dppz in its activated form is capable of reducing CO2 into formate with a two-electron process. The potential of its singly reduced form, Ru(bpy)2(dppz˙−), is −0.95 V vs. NHE in MeCN.30 Its doubly reduced state, Ru(bpy)(bpy˙−)(dppz˙−), is possible to be obtained by merging Ru(bpy)2dppz and Co3O4. This is because the electrons at the conduction band of Co3O4 bear only an energy of −1.40 V. In the presence of Co3O4, the formation of a band-alignment interface based on Ru(bpy)2dppz and Co3O4 allows the direct injection of photo-induced electrons to the LUMO of Ru(bpy)2dppz, which greatly suppresses the quenching of the molecular catalyst in the pure water phase.49 Eventually, the photocatalytic activity of Ru(bpy)2dppz is remarkably improved. This is further supported by the fact that the rate yield of formate, along with the TON/TOF values of the Ru(bpy)2dppz-Co3O4/CA after 8 h of reduction was almost 20 times higher than that on the Ru(bpy)2dppz/CA (Fig. 2b). And on the basis of the Marcus–Gerischer model,54,55 the negative bias applied to the Ru(bpy)2dppz-Co3O4 system suppresses the back electrons transferred from Ru(bpy)2dppz to Co3O4. The utilization of the electrons is then greatly improved, and a high heterogeneous electron transfer rate of 2.94 × 10−3 cm s−1 is reached.
O from the as-formed formate.52,53 These facts suggest that the electrons are transferred from the excited Co3O4 to Ru(bpy)2dppz, and then take part in photoelectrochemical CO2 reduction. As a result of such a photo-induced electron transfer to the LUMO of Ru(bpy)2dppz, CO2 conversion/reduction occurs in the adsorptive substrate CA, the photocatalyst Co3O4 and the molecular catalyst Ru(bpy)2dppz-Co3O4/CA, exactly as illustrated in Scheme 1. Such an electron transfer process is the core of such an interface. Note that Ru(bpy)2dppz in its activated form is capable of reducing CO2 into formate with a two-electron process. The potential of its singly reduced form, Ru(bpy)2(dppz˙−), is −0.95 V vs. NHE in MeCN.30 Its doubly reduced state, Ru(bpy)(bpy˙−)(dppz˙−), is possible to be obtained by merging Ru(bpy)2dppz and Co3O4. This is because the electrons at the conduction band of Co3O4 bear only an energy of −1.40 V. In the presence of Co3O4, the formation of a band-alignment interface based on Ru(bpy)2dppz and Co3O4 allows the direct injection of photo-induced electrons to the LUMO of Ru(bpy)2dppz, which greatly suppresses the quenching of the molecular catalyst in the pure water phase.49 Eventually, the photocatalytic activity of Ru(bpy)2dppz is remarkably improved. This is further supported by the fact that the rate yield of formate, along with the TON/TOF values of the Ru(bpy)2dppz-Co3O4/CA after 8 h of reduction was almost 20 times higher than that on the Ru(bpy)2dppz/CA (Fig. 2b). And on the basis of the Marcus–Gerischer model,54,55 the negative bias applied to the Ru(bpy)2dppz-Co3O4 system suppresses the back electrons transferred from Ru(bpy)2dppz to Co3O4. The utilization of the electrons is then greatly improved, and a high heterogeneous electron transfer rate of 2.94 × 10−3 cm s−1 is reached.
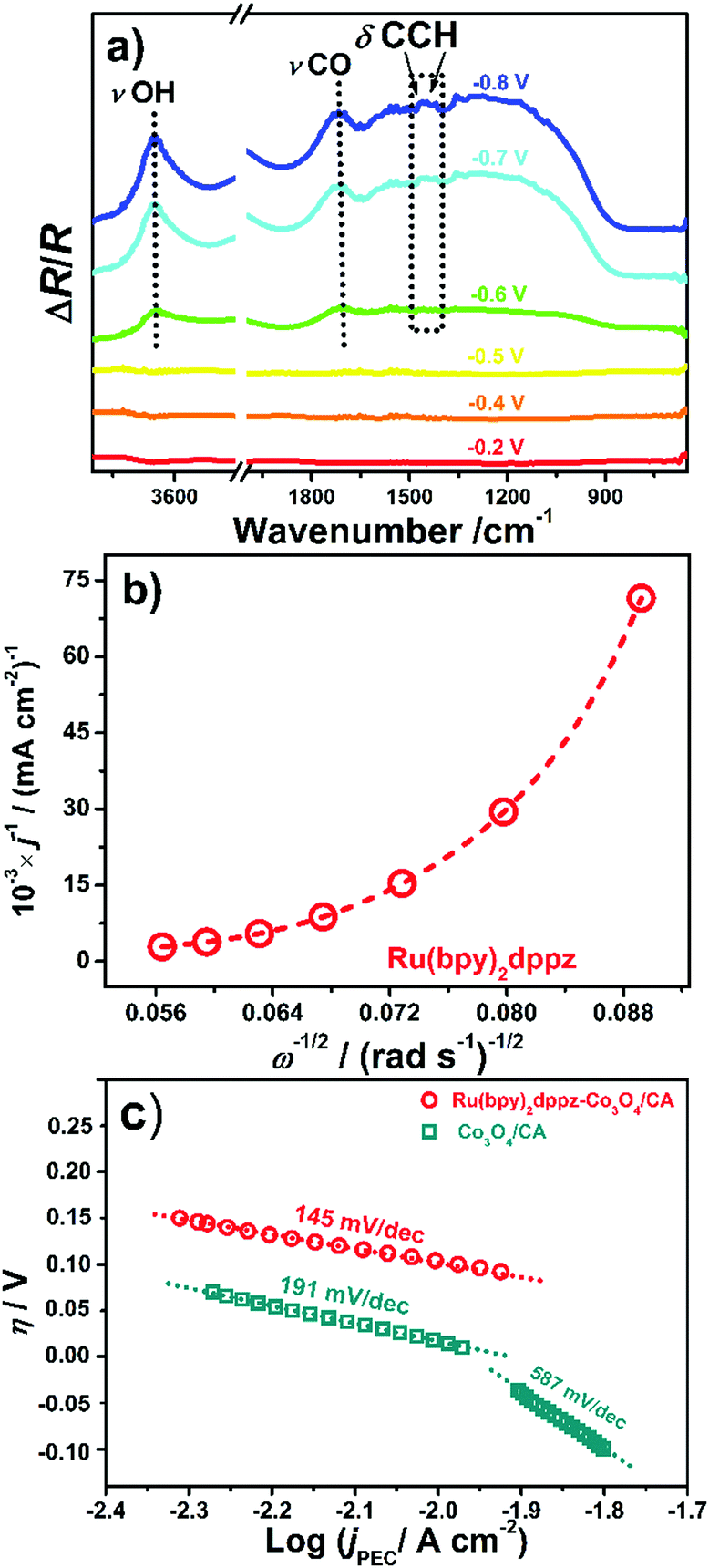 | ||
| Fig. 3 (a) In situ photoelectrochemical IR spectra of Ru(bpy)2dppz within the wavenumber ranges of 4000–3400 cm−1 and 2000–650 cm−1; (b) Koutecky–Levich fitting of the steady-state current on a Ru(bpy)2dppz-coated glassy carbon electrode. (c) Tafel plots of Ru(bpy)2dppz-Co3O4/CA (circles) and Co3O4/CA (squares) at a scan rate of 50 mV s−1. All of these electrochemical measurements were conducted in CO2-saturated 0.1 M NaHCO3. | ||
It also noteworthy that the absence of the stretching vibrations of Ru–H, N–H, and C–H in the IR spectra indicates that the protons are not directly bonded to Ru(bpy)2dppz.56,57 A broad peak at approximately 3691 cm−1 emerges from the featureless IR spectra when the applied potential is higher than −0.6 V vs. NHE (Fig. 3a). This newly emerged upward peak is assigned to the stretching vibration of a non-hydrogen bonded hydroxyl. Water molecules are attracted and nearly dissociate from their bulky form due to the existence of localized electrons on the bpy ligand in Ru(bpy)2dppz, thus causing the appearance of the peak at 3691 cm−1. Hydrodynamic voltammograms using rotating glassy carbon disk electrodes were recorded using the electrode coated with Ru(bpy)2dppz (Fig. S18a, ESI†) and Co3O4 (Fig. S18b, ESI†). Fig. 3b shows the related Koutecky–Levich plot for the electrode coated with Ru(bpy)2dppz, indicating its non-linear features. This non-linearity is quite helpful for CO2 conversion in that it indicates that CO2 and the surrounding water fix into the outer sphere of Ru(bpy)2dppz, apart from its diffusion and surface reaction with Ru(bpy)2dppz under hydrodynamic conditions.58 Taking a Langmuir–Hinshelwood bimolecular reaction mechanism as a model,59 these results clearly confirm the enzyme-mimicking role of the molecular catalyst, Ru(bpy)2dppz, for CO2 conversion. In contrast, a linear reaction is observed on the electrode coated with Co3O4 (Fig. S18c, ESI†), indicating different electrokinetics between Co3O4 and Ru(bpy)2dppz.
The rate-determining step of the CO2 conversion was estimated through Tafel analysis (Fig. 3c). For Ru(bpy)2dppz-Co3O4/CA, a Tafel slope of 145 mV dec−1 is determined. Due to the porous feature of CA, it is slightly deviated from its classical value of 118 mV dec−1.60,61 Therefore the rate-determining step of photoelectrochemical CO2 conversion on Ru(bpy)2dppz-Co3O4/CA is the single electron transfer step rather than the proton transfer step. This result is in agreement with our previous study regarding the photoelectrochemical CO2 conversion on hierarchical Co3O4.4 This finding also supports the notion that the protonation of the reaction intermediate occurs rapidly in CO2-saturated solution. Namely, faster electrode kinetics for CO2 conversion were obtained with the help of Ru(bpy)2dppz. In contrast, a Tafel slope of 191 mV dec−1 is observed for Co3O4/CA, which originates from the porous structure of CA and is indicative of its slow photoelectrochemical kinetics in the absence of Ru(bpy)2dppz. In addition, the activation energy of the CO2 reduction in our system, defined as the energy required to activate CO2 into its radicals,62–64 was calculated to be 1.047 kJ mol−1 (see the ESI† for details). This value is presented for the first time in this study and therefore further density functional-based theoretical calculations are required to confirm this value.
Based on the above analysis, the activation of Ru(bpy)2dppz is accomplished only by the photoelectrochemical process from the irradiated Co3O4/CA. Ru(bpy)2dppz is actually “catalysed” by Co3O4/CA rather than merely an electron conductor. Therefore, the reduction performance is markedly enhanced with the assistance of the photoelectrocatalyst of Co3O4. Therefore, as schematically shown in Scheme 1, the possible pathways for CO2 conversion on Ru(bpy)2dppz-Co3O4/CA involve the transfer and mediation of electrons, protons, and the activated form of Ru(bpy)2dppz.
Conclusions
In summary, an adsorption-enhanced Co3O4/Ru(bpy)2dppz hybrid photoelectrocatalytic interface has been established. The CO2 adsorption is remarkably increased through the utilization of a high-surface-area and micropore-dominated carbon aerogel substrate. The epitaxial growth of high-index-facet Co3O4 along with the intrinsic highly conductive network of the carbon aerogel enables an elevated electron transfer rate with the assistance of a molecular catalyst of Ru(bpy)2dppz. Eventually, such an interface bears a unique proton-coupled electron-transfer reactivity and rapid electron kinetics toward CO2 reduction. Through the simultaneous activation of the bpy and dppz ligands of the Ru(II) molecular catalyst, the photoelectrochemical conversion of CO2 to formate has been realized on such a photocathode with an onset potential as low as −0.45 V vs. NHE. At a potential of −0.6 V vs. NHE, the yield rate of formate reaches approximately 110 μmol cm−2 h−1, with a selectivity of 99.95% and a Faradaic efficiency of 86%. The mechanism of such a rapid photoelectrochemical electron transfer process is explained as the synergistic effect of the photoelectrochemical activation of bpy and dppz inside Ru(bpy)2dppz as well as the remarkably promoted CO2 adsorption on Co3O4/CA. Although future work on the effect of the morphology of Co3O4/CA (e.g., the size, facets, and shape of Co3O4 on CA) and the loading amount of Co3O4/CA (e.g., distribution and density) as well as the light density on the efficiency of the CO2 conversion must be conducted, such an adsorption-enhanced molecular catalytic photoelectrocatalytic interface has potential for application in the production of useful chemicals from CO2 carbon stock in the future.Acknowledgements
The authors acknowledge Dr Sanjun Zhang (State Key Laboratory of Precision Spectroscopy, East China Normal University) for his assistance with spectral data analysis and discussion on the electron transfer mechanism. G. Z. acknowledges the financial support from the National Natural Science Foundation of China (NSFC) under projects No. 21537003 and 21477085. N. Y. acknowledges the financial support from the German Research Foundation (DFG) under project YA344/1-1.Notes and references
- N. J. Brown, C. A. Newell, S. Stanley, J. E. Chen, A. J. Perrin, K. Kajala and J. M. Hibberd, Science, 2011, 331, 1436 CrossRef CAS PubMed.
- Q. Shen, Z. Chen, X. Huang, M. Liu and G. Zhao, Environ. Sci. Technol., 2015, 49, 5828 CrossRef CAS PubMed.
- J. Gu, A. Wuttig, J. W. Krizan, Y. A. Hu, Z. M. Detweiler, R. J. Cava and A. B. Bocarsly, J. Phys. Chem. C, 2013, 117, 12415 CAS.
- X. F. Huang, T. C. Cao, M. C. Liu and G. H. Zhao, J. Phys. Chem. C, 2013, 117, 26432 CAS.
- J.-W. Jang, S. Cho, G. Magesh, Y. J. Jang, J. Y. Kim, W. Y. Kim, J. K. Seo, S. Kim, K.-H. Lee and J. S. Lee, Angew. Chem., Int. Ed., 2014, 53, 5852 CrossRef CAS PubMed.
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888 CrossRef CAS PubMed.
- L. I. Bendavid and E. A. Carter, J. Phys. Chem. C, 2013, 117, 26048 CAS.
- Y. K. Chen, N. S. Lewis and C. X. Xiang, Energy Environ. Sci., 2015, 8, 3663 CAS.
- N. Kornienko, Y. B. Zhao, C. S. Kley, C. H. Zhu, D. H. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. D. Yang, J. Am. Chem. Soc., 2015, 137, 14129 CrossRef CAS PubMed.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364 CrossRef CAS PubMed.
- H.-Q. Xu, J. H. Hu, D. K. Wang, Z. H. Li, Q. Zhang, Y. Luo, S.-H. Yu and H. L. Jiang, J. Am. Chem. Soc., 2015, 137, 13440 CrossRef CAS PubMed.
- P. Li, H. Hu, J. Xu, H. Jing, H. Peng, J. Lu, C. Wu and S. Ai, Appl. Catal., B, 2014, 147, 912 CrossRef CAS.
- P. Li, X. Sui, J. Xu, H. Jing, C. Wu, H. Peng, J. Lu and H. Yin, Chem. Eng. J., 2014, 247, 25 CrossRef CAS.
- E. E. Barton, D. M. Rampulla and A. Bocarsly, J. Am. Chem. Soc., 2008, 130, 6342 CrossRef CAS PubMed.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983 CrossRef CAS PubMed.
- J. Agarwal, E. Fujita, H. F. Schaefer and J. T. Muckerman, J. Am. Chem. Soc., 2012, 134, 5180 CrossRef CAS PubMed.
- Z. Chen, C. Chen, D. R. Weinberg, P. Kang, J. J. Concepcion, D. P. Harrison, M. S. Brookhart and T. J. Meyer, Chem. Commun., 2011, 47, 12607 RSC.
- B. Kumar, J. M. Smieja and C. P. Kubiak, J. Phys. Chem. C, 2010, 114, 14220 CAS.
- Y. Matsubara, S. E. Hightower, J. Z. Chen, D. C. Grills, D. E. Polyansky, J. T. Muckerman, K. Tanaka and E. Fujita, Chem. Commun., 2014, 50, 728 RSC.
- Y. Hayashi, S. Kita, B. S. Brunschwig and E. Fujita, J. Am. Chem. Soc., 2003, 125, 11976 CrossRef CAS PubMed.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208 CrossRef CAS PubMed.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673 CrossRef CAS PubMed.
- K. Sekizawa, K. Maeda, K. Domen, K. Koike and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 4596 CrossRef CAS PubMed.
- T. Arai, S. Sato, K. Uemura, T. Morikawa, T. Kajino and T. Motohiro, Chem. Commun., 2010, 46, 6944 RSC.
- J. D. Hong, W. Zhang, J. Ren and R. Xu, Anal. Methods, 2013, 5, 1086 RSC.
- T. Yamamoto, D. A. Tryk, K. Hashimoto, A. Fujishima and M. Okawa, J. Electrochem. Soc., 2000, 147, 3393 CrossRef CAS.
- S. Perez-Rodriguez, N. Rillo, M. J. Lazaro and E. Pastor, Appl. Catal., B, 2015, 163, 83 CrossRef CAS.
- M. F. Wu, Y. N. Jin, G. H. Zhao, M. F. Li and D. M. Li, Environ. Sci. Technol., 2010, 44, 1780 CrossRef CAS PubMed.
- Y. N. Zhang, Y. F. Jin, X. F. Huang, H. J. Shi, G. H. Zhao and H. Y. Zhao, Electrochim. Acta, 2014, 130, 194 CrossRef CAS.
- J. Fees, W. Kaim, M. Moscherosch, W. Matheis, J. Klima, M. Kerjclk and S. Zalls, Inorg. Chem., 1993, 31, 166 CrossRef.
- J. L. Qiao, Y. Y. Liu, F. Hong and J. J. Zhang, Chem. Soc. Rev., 2014, 43, 631 RSC.
- M. Schreier, P. Gao, M. T. Mayer, J. S. Luo, T. Moehl, M. K. Nazeeruddin, S. D. Tilley and M. Gratzel, Energy Environ. Sci., 2015, 8, 855 CAS.
- J. G. Velasco, Electroanalysis, 1997, 9, 880 CrossRef.
- H. Zhong, K. Fuji, Y. Nakano and F. M. Jin, J. Phys. Chem. C, 2015, 119, 55 CAS.
- T. Arai, S. Tajima, S. Sato, K. Uemura, T. Morikawa and T. Kajino, Chem. Commun., 2011, 47, 12664 RSC.
- X. Q. An, K. F. Li and J. W. Tang, ChemSusChem, 2014, 7, 1086 CrossRef CAS PubMed.
- L. H. Zhang, D. Zhu, G. M. Nathanson and R. J. Hamers, Angew. Chem., Int. Ed., 2014, 53, 9746 CrossRef CAS PubMed.
- M. Garcia, M. J. Aguirre, G. Canzi, C. P. Kubiak, M. Ohlbaum and M. Issacs, Electrochim. Acta, 2014, 115, 146 CrossRef CAS.
- S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240 CrossRef CAS PubMed.
- G. Sahara, R. Abe, M. Higashi, T. Morikawa, K. Maeda, Y. Ueda and O. Ishitani, Chem. Commun., 2015, 51, 10722 RSC.
- S. Sato, T. Morikawa, S. Saeki, T. Kajino and T. Motohiro, Angew. Chem., Int. Ed., 2010, 49, 5101 CrossRef CAS PubMed.
- R. Kuriki, K. Sekizawa, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2015, 54, 2406 CrossRef CAS PubMed.
- A. Nakada, K. Koike, T. Nakashima, T. Morimoto and O. Ishitani, Inorg. Chem., 2015, 54, 1800 CrossRef CAS PubMed.
- K. Maeda, K. Sekizawa and O. Ishitani, Chem. Commun., 2013, 49, 10127 RSC.
- S. Sato, T. Morikawa, S. Saeki, T. Kajino and T. Motohiro, Angew. Chem., Int. Ed., 2010, 49, 5101 CrossRef CAS PubMed.
- C. Matlachowski, B. Braun, S. Tschierlei and M. Schwalbe, Inorg. Chem., 2015, 54, 10351 CrossRef CAS PubMed.
- J. Hawecker, J. M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1984, 328 RSC.
- L. J. Chen, Z. G. Guo, X. G. Wei, C. Gallenkamp, J. Bonin, E. A. Mallart, K. C. Lau, T. C. Lau and M. Robert, J. Am. Chem. Soc., 2015, 137, 10918 CrossRef CAS PubMed.
- D. J. Boston, Y. M. Franco Pachon, R. O. Lezna, N. R. de Tacconi and F. M. MacDonnell, Inorg. Chem., 2014, 53, 6544 CrossRef CAS PubMed.
- P. K. Mallick, G. D. Danzer, D. P. Strommen and J. R. Kincaid, J. Phys. Chem., 1988, 92, 5628 CrossRef CAS.
- K. M. Omberg, J. R. Schoonover, J. A. Treadway, R. M. Leasure, R. B. Dyer and T. J. Meyer, J. Am. Chem. Soc., 1997, 119, 7013 CrossRef CAS.
- K. Jiang, K. Xu, S. Zou and W.-B. Cai, J. Am. Chem. Soc., 2014, 136, 4861 CrossRef CAS PubMed.
- J. Y. Wang, H. X. Zhang, K. Jiang and W. B. Cai, J. Am. Chem. Soc., 2011, 133, 14876 CrossRef CAS PubMed.
- B. H. Farnum, Z. A. Morseth, M. K. Brennaman, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 15869 CrossRef CAS PubMed.
- N. S. Lewis, J. Phys. Chem. B, 1998, 102, 4843 CrossRef CAS.
- Q. Wang, X. Wang and L. Andrews, J. Phys. Chem. A, 2011, 115, 12194 CrossRef CAS PubMed.
- K. Toyohara, K. Tsuge and K. Tanaka, Organometallics, 1995, 14, 5099 CrossRef CAS.
- R. Vargas, C. Borras, J. Mostany and B. R. Scharifker, Electrochim. Acta, 2012, 80, 326 CrossRef CAS.
- Y. Kiya, O. Hatozaki, N. Oyama and H. D. Abruna, J. Phys. Chem. C, 2007, 111, 13129 CAS.
- Y. H. Chen and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 1986 CrossRef CAS PubMed.
- J. N. Soderberg, A. C. Co, A. H. C. Sirk and V. I. Birss, J. Phys. Chem. B, 2006, 110, 10401 CrossRef CAS PubMed.
- S. Antonello, M. Musumeci, D. D. M. Wayner and F. Maran, J. Am. Chem. Soc., 1997, 119, 9541 CrossRef CAS.
- S. Antonello and F. Maran, J. Am. Chem. Soc., 1999, 121, 9668 CrossRef CAS.
- C. Ji, M. Ahmida, M. Chahma and A. Houmam, J. Am. Chem. Soc., 2006, 128, 15423 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental methods, SEM and TEM characterization, chronocoulometry, EQCM results, BET and BJH results, XRD patterns, CVs, UV-DRS, Tauc plots, Mott–Schottky plot, and RDE results. See DOI: 10.1039/c6ee00968a |
| This journal is © The Royal Society of Chemistry 2016 |