DOI:
10.1039/C5EE03316K
(Communication)
Energy Environ. Sci., 2016,
9, 123-129
Single layer graphene encapsulating non-precious metals as high-performance electrocatalysts for water oxidation†
Received
30th October 2015
, Accepted 25th November 2015
First published on 25th November 2015
Abstract
The oxygen evolution reaction (OER) is recognized as a key half-reaction in water electrolysis for clean hydrogen energy, which is kinetically not favored and usually requires precious metal catalysts such as IrO2 and RuO2 to reduce the overpotential. The major challenge in using non-precious metals in place of these precious metal catalysts for OER is their low efficiency and poor stability, which urgently demand new concepts and strategies to tackle this issue. Herein, we report a universal strategy to directly synthesize single layer graphene encapsulating uniform earth-abundant 3d transition-metal nanoparticles such as Fe, Co, Ni and their alloys in a confined channel of mesoporous silica. The single atomic thickness of the graphene shell immensely promotes the electron transfer from the encapsulated metals to the graphene surface, which efficiently optimizes the electronic structure of the graphene surface and thereby triggers the OER activity of the inert graphene surface. We investigated a series of non-precious 3d metals encapsulated within single layer graphene, and found that the encapsulated FeNi alloy showed the best OER activity in alkaline solutions with only 280 mV overpotential at 10 mA cm−2, and also possessed a high durability after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. Both the activity and durability of the non-precious catalyst even exceed that of the commercial IrO2 catalyst, showing great potential to replace precious metal catalysts in the OER.
000 cycles. Both the activity and durability of the non-precious catalyst even exceed that of the commercial IrO2 catalyst, showing great potential to replace precious metal catalysts in the OER.
Broader context
Oxygen evolution reaction (OER), as a key half reaction in water splitting, is kinetically sluggish and largely hinders the overall efficiency of water splitting. Current commercial OER catalysts still rely on the use of ruthenium (Ru) and iridium (Ir) oxides, but they are not suitable for large-scale applications because of their scarcity and high cost. Recently, carbon-based electrodes used as OER catalysts attract great attention due to their low cost and high conductivity, but the activity and durability are still not satisfactory. Herein, we report a universal strategy to directly synthesize single layer graphene encapsulating uniform earth-abundant 3d transition metal nanoparticles, such as Fe, Co, Ni and their alloys. The encapsulated metal nanoparticles immensely optimize the electronic structure of the single atomic thickness of the graphene surface and enhance the OER activity on the graphene surface. More importantly, the OER activity can be adjusted to the optimal value by changing the metal type convinced by the experimental data and theoretical calculations. This work further develops the concept of “Chainmail for Catalyst” by enhancing the electron penetration via reducing the thickness of two dimensional atomic crystals and optimizing the encapsulated metal type.
|
The increasing demand for clean energy and diminishing fossil energy resources are stimulating intense research interest in the development of earth-abundant energy alternatives and designing efficient energy-storage devices.1–5 Among many innovative approaches, the production of hydrogen through electricity-driven water splitting has attracted great attention in recent years. The oxygen evolution reaction (OER), as a key half-reaction involved in water splitting, is kinetically not favored and requires a catalyst to expedite the reaction.6–10 Precious metal catalysts such as IrO2 and RuO2 are the state-of-the-art OER electrocatalysts,11–13 while their applications are hindered due to their high cost and scarcity. Consequently, tremendous research efforts have been devoted to developing non-precious metal catalysts. In particular, earth-abundant 3d transition metals (TMs) like Fe, Co and Ni have been considered promising catalytic materials for the replacement of precious catalysts due to their high theoretical activity and low cost.14–19 However, bare 3d TM catalysts are not stable enough when they are subjected to long-time operation or accelerated degradation measurements under strong alkaline electrolytes and high overpotential.
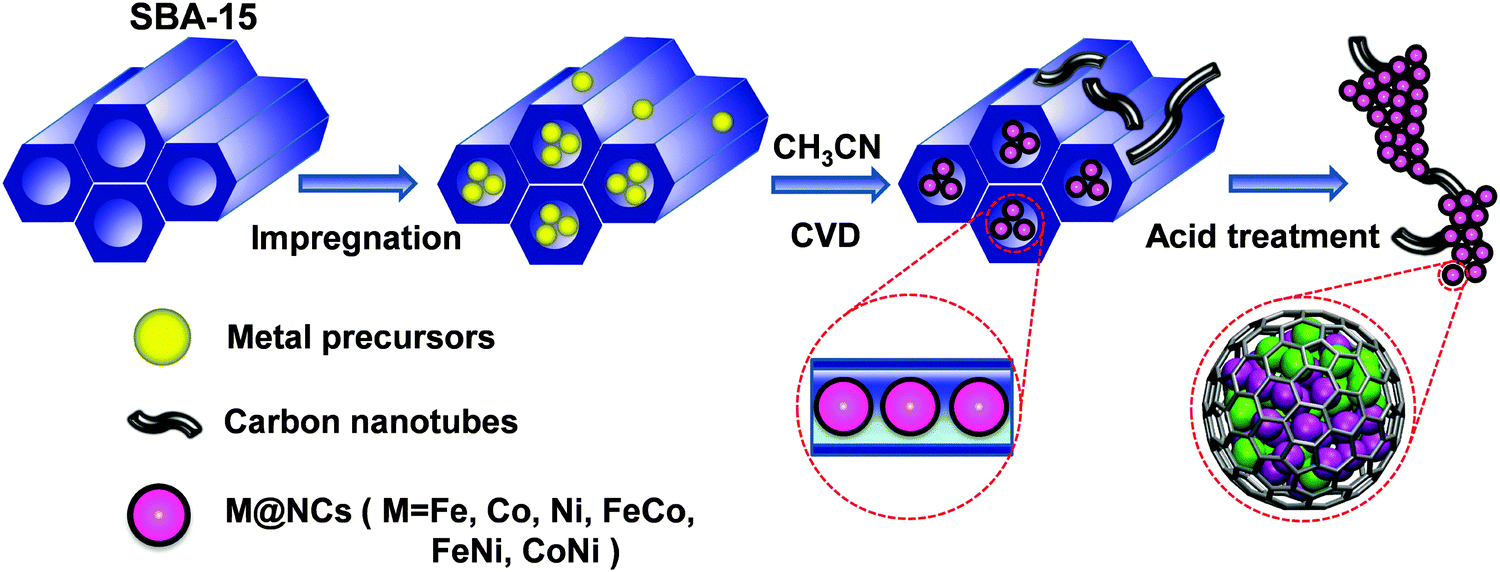
Recently, we proposed a strategy to construct a series of 3d TMs encapsulated into carbon nanotubes or graphitic carbon shells to prevent the corrosion of 3d TMs from a harsh environment and simultaneously promote the catalytic reaction on a carbon surface due to the electron transfer from encapsulated 3d TMs.20–24 This strategy was proved to be efficient by different research groups in a series of reactions, such as the oxygen reduction reaction (ORR)20,24–26 in fuel cells, the hydrogen evolution reaction (HER)21,22,27,28 in water electrolysis and the triiodide reduction reaction in dye-sensitized solar cells (DSSCs)29 as well as catalytic oxidation and reduction reactions in heterogeneous catalysis.30,31 Previous studies indicated that a too thick carbon shell would obstruct the electron transfer from the metals to the carbon and subsequently decrease the catalytic activity on the carbon surface.21,24 Moreover, the electron transfer significantly relies on the work function of different metals and their interaction strength with graphene.20,32–36 Therefore, it is imperative to develop a universal method to reduce the layer number of graphitic carbon and meanwhile achieve the encapsulation of all kinds of metals. Herein, we introduced a facile and universal method for the preparation of single layer graphene encapsulating uniform 3d TMs, including Fe, Co, Ni and their alloy nanoparticles (NPs) via a chemical vapor deposition (CVD) process in the channels of ordered mesoporous silica (SBA-15). As shown in Fig. 1, firstly, metal-containing precursors were filled into the channels of SBA-15 (with a pore size distribution of 7–9 nm) by an impregnation method in methanol solution, and then the samples were heated to 700 °C under H2, followed by CVD of the carbon source (acetonitrile) on the metal NPs formed at high temperature. Due to the confinement of the SBA-15 channels, the metal precursors can firstly form uniform particles under high temperature, and single layer graphene will be formed between the metal nanoparticle surface and the pore walls of SBA-15. Hence, single layer graphene will grow on the surface of the metal NPs in the channels of SBA-15, and a small quantity of multi-walled carbon nanotubes (CNTs) will grow on the residual metal NPs outside SBA-15. The final single layer graphene encapsulating metal NPs on the interlaced CNTs can be obtained after etching SBA-15 and the bare metals using HF solution. Through the method, a series of single layer graphene encapsulating 3d TMs (M@NCs) can be easily prepared, denoted as Fe@NC, Co@NC, Ni@NC, FeCo@NC, FeNi@NC and CoNi@NC, respectively, according to the metal types (see Experimental section in the ESI† for more details).
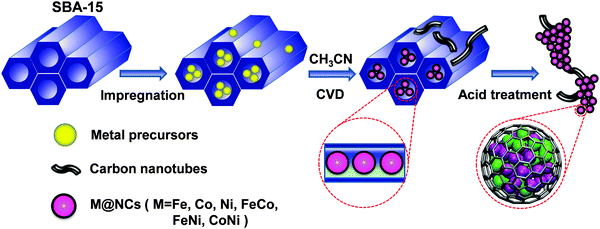 |
| | Fig. 1 Schematic illustration of the synthesis process of M@NCs from metal-containing precursors and SBA-15. | |
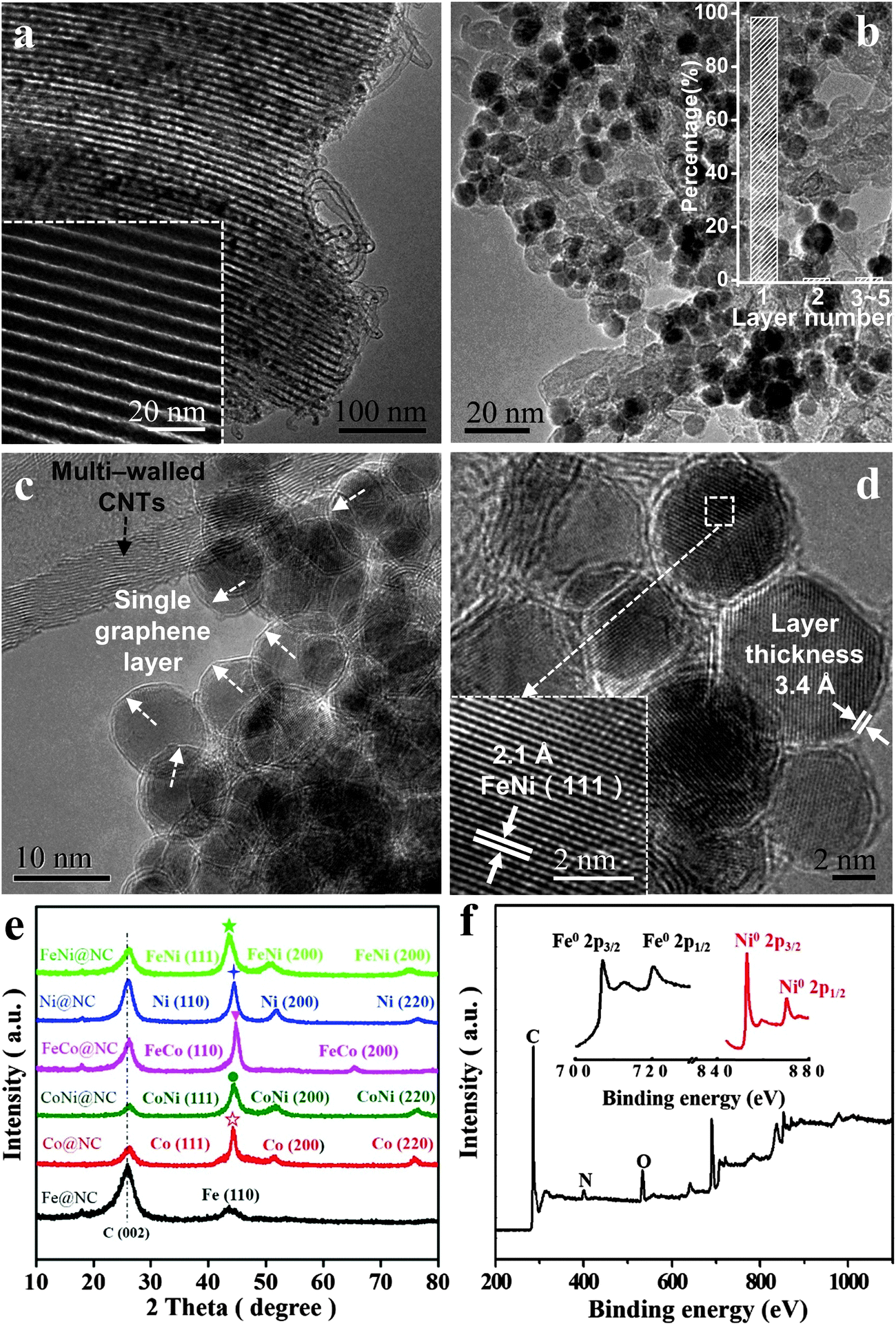
High-resolution transmission electron microscopy (HRTEM), X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) were employed to characterize the structural and electronic properties of the obtained M@NCs. Considering the FeNi@NC sample as an example, the HRTEM image of the non acid treatment sample (FeNi@NC/SBA-15) clearly shows numerous small NPs inside the ordered mesoporous channel of SBA-15 (Fig. 2a) after the CVD process, which should correspond to graphene encapsulating FeNi NPs. After the removal of SBA-15, one can see uniform metal NPs with an average diameter of 6–10 nm distributed on the interlaced CNTs (Fig. 2b and c; Fig. S1 and S2a, ESI†). Further HRTEM analysis (Fig. 2c, d and Fig. S2b, ESI†) indicates that the metal NPs were completely encapsulated by the graphene layer with a layer thickness of around 3.4 Å, which corresponds to single layer graphene. According to the statistical analysis of more than 300 HRTEM images (Fig. 2b), almost 98.5% of the graphene layer on FeNi NPs is a single layer. In addition, these NPs exhibit a d spacing of 2.1 Å, in good agreement with the (111) plane of the FeNi alloy. The crystal structure of FeNi alloy NPs was further confirmed by XRD (Fig. 2e), which shows characteristic peaks at 51.8° and 76.4°, corresponding to the (200) and (220) planes of the FeNi alloy, respectively. Note that the 26.5° peak in the XRD patterns should originate from C (002) of multi-walled CNTs. The chemical states of these NPs by XPS (Fig. 2f) also suggest that both Fe and Ni in FeNi@NC maintain their metallic state, which was consistent with the XRD analysis and HRTEM results. The metallic characteristics of these NPs further prove that FeNi NPs are completely encapsulated within the single layer graphene shell, preventing the oxidation of the FeNi alloy when exposed to air. Likewise, the encapsulation of other metallic Fe, Co, Ni, FeCo and CoNi alloys within the single graphene layer was also confirmed by similar characterization in the ESI† (Fig. S3–S12). These results indicate that a series of uniform 3d NPs and their corresponding alloys have been successfully encapsulated in a single layer graphene shell by using a facile method.
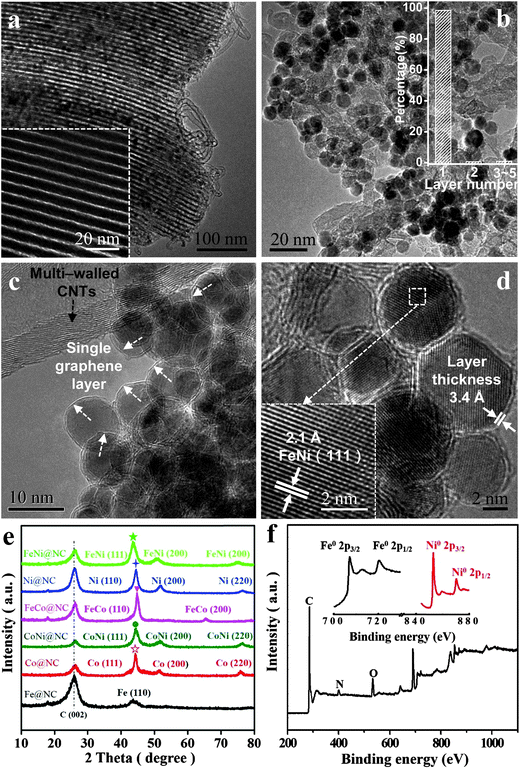 |
| | Fig. 2 Morphology and structural characterizations of M@NCs. (a) TEM images of FeNi@NC/SBA-15, showing the metal NPs inside the channels of SBA-15 and a small quantity of CNTs outside SBA-15. The inset is an image of pure SBA-15. (b–d) HRTEM images of FeNi@NC. The inset in (b) is a statistical analysis of the layer number of the graphene shell on metal nanoparticles. The white arrows in the inset of (c) show the single layer graphene, and the black arrow in the inset of (c) shows the multi-walled carbon nanotubes. The inset in (d) shows the (111) crystal plane of the FeNi alloy and the graphene layer with a layer thickness of 3.4 Å. (e) XRD patterns of different M@NCs. (f) Fe 2p and Ni 2p XPS spectra of FeNi@NC sample. | |
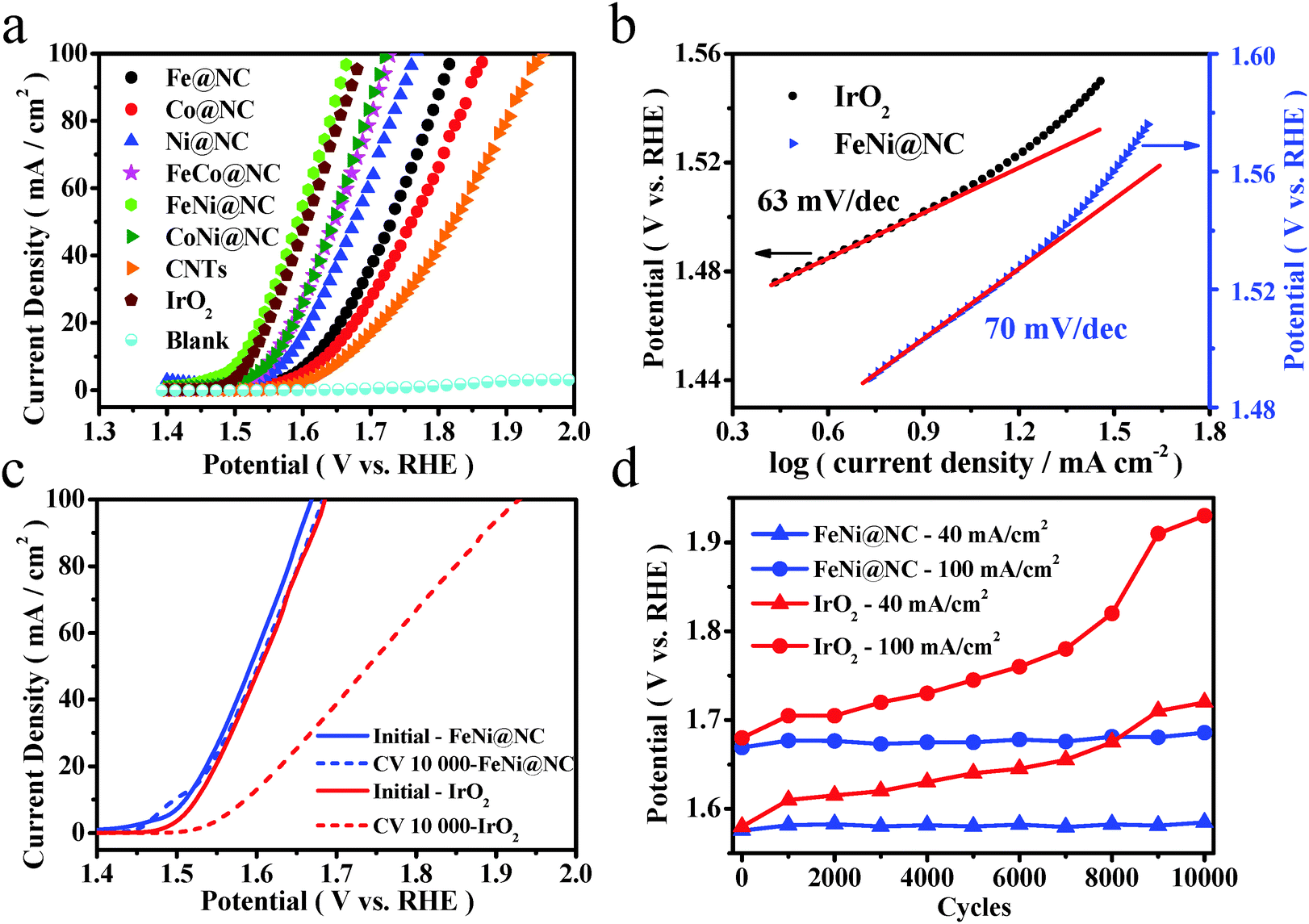
A typical three-electrode electrochemical cell was adopted to evaluate the OER performance of these M@NC catalysts in 1 M NaOH solution. As shown in Fig. 3a, linear sweep voltammograms (LSVs) were carried out to evaluate the OER performance of these M@NC catalysts. As references, we also carried out measurements of pure CNTs, few-layer graphene nanosheets and IrO2. The polarization curves showed that all these M@NC catalysts had much higher OER activity than pristine CNTs and graphene nanosheets (Fig. S15a, ESI†). The trend in the overpotential at the same current density of 10 mA cm−2 is found to be FeNi@NC < CoNi@NC < FeCo@NC < Ni@NC < Fe@NC < Co@NC (Fig. S15c, ESI†), indicating that FeNi@NC possessed the best OER activity among these M@NC catalysts. By analyzing the BET surface area of the M@NC catalysts (Table S1, ESI†), we find that there is not a direct relationship between the activity and the surface area. Besides, the N content in these samples is similar according to the element analysis (Tables S3 and S4, ESI†). Hence, the difference in the activity of these samples should mainly originate from the different metal types. More interestingly, the onset potential of the FeNi@NC sample is only 1.44 V vs. RHE, and the potentials at 10 mA cm−2 and 20 mA cm−2 were 1.51 V vs. RHE and 1.54 V vs. RHE, respectively (Fig. 3a), which is even better than that of commercial IrO2. It is also one of the most active catalysts for OER in the recent reports (Table S9, ESI†). To gain additional insights into the OER process on FeNi@NC, Tafel plots of the potential vs. log (current density) were recorded (Fig. 3b) with their linear portions at low potential fitted to the Tafel equation. The Tafel slope of the FeNi@NC sample was 70 mV dec−1, which is comparable to the value of 63 mV dec−1 of IrO2, showing that the FeNi@NC sample possessed a similar ability to drive the OER process compared to IrO2 at low overpotential. Durability is another critical parameter to evaluate the performance of non-precious-metal electrocatalysts in OER. In this study, an accelerated degradation test was employed to evaluate the durability. The polarization curve for OER was recorded after every 1000 cyclic voltammetric (CV) sweeps between −0.46 and 0.54 V (vs. Hg/HgO) at 100 mV s−1. One can see that the polarization curve of FeNi@NC after 10000 cycles retained an almost similar performance to the initial test (Fig. 3c). A slight peak emerging at around 1.5 V vs. RHE may be ascribed to the oxidation of NiII to NiIII, which is probably caused by a small quantity of exposed FeNi nanoparticles, because some of the graphene layers may be destroyed during the long-term accelerated degradation test in a strong alkaline electrolyte. In contrast, the activity of IrO2 decayed quickly with the same accelerated degradation test. Moreover, from the potential values recorded at different current densities of 40 mA cm−2 and 100 mA cm−2 before and after the test, FeNi@NC exhibited high durability with only a slight increase in the potential (Fig. 3d), obviously superior to the durability of commercial IrO2. Note that both the structure of the single layer graphene and the encapsulating FeNi nanoparticles were still well maintained after the OER durability measurements (5000 cyclic voltammetric sweeps), as confirmed by Raman (Fig. S17, ESI†) and HRTEM (Fig. S18, ESI†) analysis. The high activity and durability of FeNi@NC demonstrate its great potential as a non-precious metal catalyst for OER.
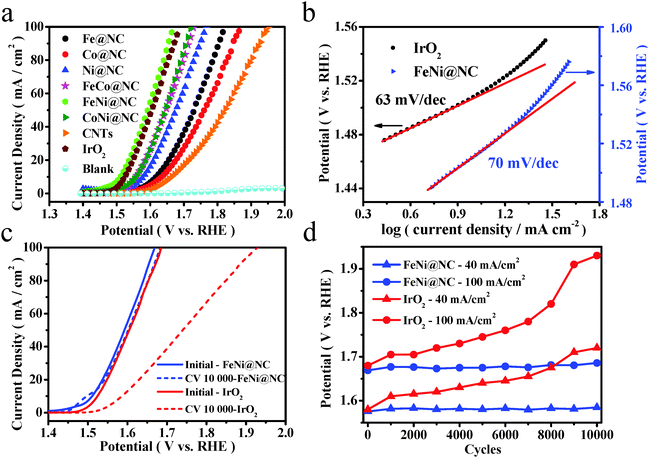 |
| | Fig. 3 Electrocatalytic OER performance test of M@NCs in O2-saturated 1 M NaOH solution at 25 °C. (a) OER polarization curves for M@NCs in comparison with CNTs and IrO2 with the same mass loading. (b) Tafel plots for FeNi@NC and IrO2. (c) Durability test of FeNi@NC in an alkaline electrolyte in contrast to IrO2. (d) The potential changes in current densities at 40 mA cm−2 and 100 mA cm−2 for FeNi@NC and IrO2 during the durability test. | |
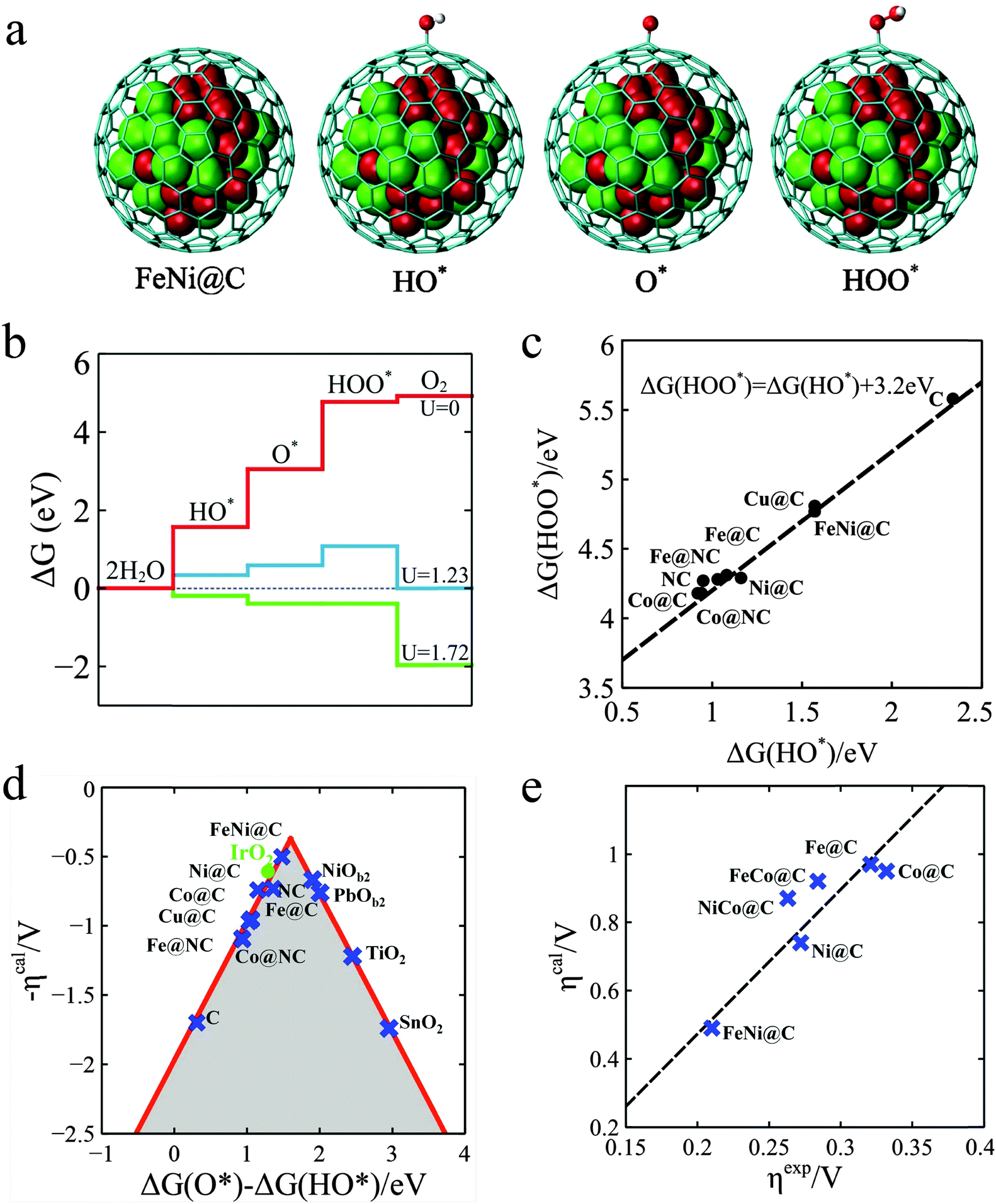
In order to gain further insights into the nature of the M@NCs towards the OER, density functional theory (DFT) calculations were employed. A graphitic carbon cage with 240 atoms encapsulating 55 metal atoms was used as the model of M@C. As shown in Fig. 4a, it is generally agreed that the OER involves the following steps,37 where the symbol “*” represents the active site.
| | | HO* + OH− → H2O + O* + e− | (2) |
| | | HOO* + OH− → O2* + H2O + e− | (4) |
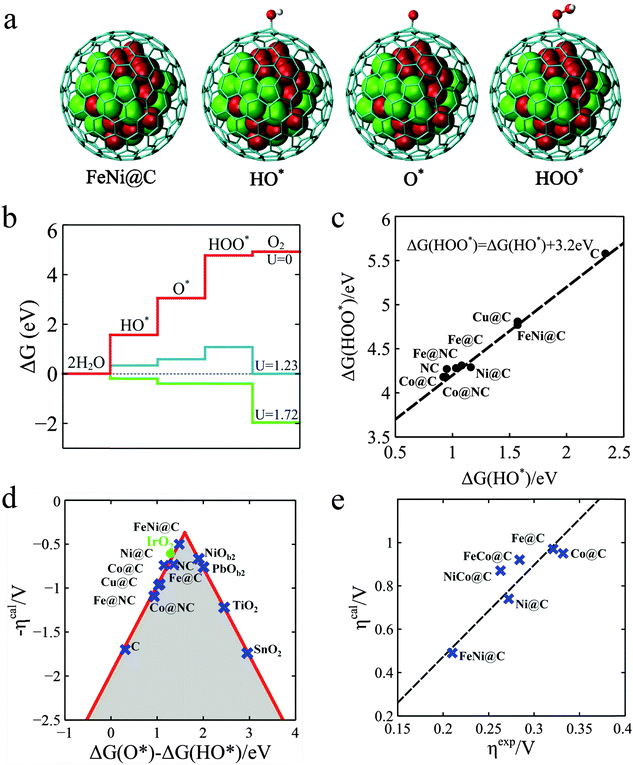 |
| | Fig. 4 Theoretical interpretation of OER over M@Cs. (a) The OER steps on the M@C models, where a graphitic carbon cage encapsulates 55 metal atoms. The grey cage, red and green balls represent C, Fe and Ni atoms, respectively. (b) Free energy profiles for the OER over FeNi@C at zero potential (U = 0), equilibrium potential for oxygen evolution (U = 1.23 V), and minimum potential (U = 1.72 V) where all steps run downhill. (c) Linear relation between the free energy of HO* (ΔG(HO*)) and HOO* species (ΔG(HOO*)) on different catalysts. (d) The calculated negative overpotential (ηcal) against the universal descriptor ΔG(O*) − ΔG(HO*) on different catalysts. The data of oxides were cited from the literature by Man et al.37 (e) Calculated overpotential (ηcal) vs. the experimental overpotential (ηexp) over M@C catalysts. All experimental overpotential (ηexp) values were recorded at 2 mA cm−2 (Table S7, ESI†). | |
Fig. 4b gives a plot of the free energies of various intermediates for the OER catalyzed by FeNi@C at different constant potentials. It is shown that all reaction steps are endothermic at zero potential and the first three reaction steps are also endothermic at an equilibrium potential of 1.23 V, and until the potential increased to 1.72 V, the free energies for all steps run downhill. This means that about 0.49 V overpotential compared with the equilibrium potential is needed for the OER process on FeNi@C. The analysis indicates that the calculated overpotential is determined by the free energies of the reaction intermediates HO*, O* and HOO*. Building a relationship between the free energies of intermediates and the overpotentials of various catalysts is vital to the rational design of catalysts.38,39 Interestingly, as shown in Fig. 4c, there is a good scaling relationship between the free energies of HO* and HOO* with a constant slope and an intercept of 3.2 eV. The slope is equal to one, which implies that HO* and HOO* normally prefer the same bond type to the graphene surface, which is coincident with a wide range of oxides including rutile, perovskite, spinel, rocksalt and bixbyite and is considered a universal scaling relation.37 According to the scaling relation, the overpotential can only be determined by the free energy of O* relative to HO*, which is denoted as ΔG(O*) − ΔG(HO*). With this established activity descriptor, the calculated overpotential exhibits a volcano shape, as shown in Fig. 4d. The ΔG(O*) − ΔG(HO*) of FeNi@C is 1.48 eV, which is closest to the volcano peak among all the catalysts studied. Furthermore, the Fig. 4e plot is a comparison of calculated overpotential with experimental results, which agree quite well in trend, indicating that our theoretical model is able to predict the efficiency of M@C in the OER. The Bader charge analysis38 in Table S5 (ESI†) indicates that the electronic properties of the graphene shell can be significantly altered by electrons transferring from metal NPs, which further tune the binding energies of reaction intermediates on the graphene surface. As a result, the OER descriptor ΔG(O*) − ΔG(HO*) can be adjusted to the optimal value by changing the type of metal, such as Fe, Co, Ni and their alloys as shown in our experiments. Furthermore, introducing nitrogen into the graphene layer can also tune the adsorption of reaction intermediates and the corresponding reaction activity, as Noto et al. reported.40–44 N-doping and enclosed metal clusters will synergistically enhance the adsorption of the intermediates as shown in Table S6 (ESI†). The increase of the OER activity for the M@C samples in experiment (Fig. S15, ESI†) should be a compromise of the contribution of N-doped carbon and N-doped graphene encapsulating metal NPs, which are further discussed in the ESI (Fig. S15 and Table S6, ESI†) for brevity.
In summary, we developed a facile and universal strategy to synthesize single layer graphene encapsulating 3d TMs, such as Fe, Co, Ni and their alloys through a CVD process in the channels of SBA-15. Electrochemical measurements indicated that different encapsulated metals have a different effect on the OER activity, and the optimized FeNi@NC catalyst showed the best activity and a high durability, both of which are superior to those exhibited by commercial IrO2. DFT calculations indicated that the high OER performance originates from the single layer graphene immensely promoting the electron transfer from the encapsulated metals to the graphene surface, which optimizes the electronic structure of the graphene surface and thereby triggers the OER activity on the inert graphene surface. These findings provide an efficient way towards the rational design of high-performance and low-cost OER electrocatalysts, as well as other energy-related catalysts.
Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21321002, 21573220 and 21303191) and the strategic Priority Research Program of the Chinese Academy of Sciences (No. XDA09030100). We thank Dr. Jianping Xiao for fruitful discussions.
References
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- A. Grimaud, K. J. May, C. E. Carlton, Y. L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 2439 Search PubMed.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS PubMed.
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, E28–62 CrossRef CAS PubMed.
- S. Mao, Z. Wen, T. Huang, Y. Hou and J. Chen, Energy Environ. Sci., 2014, 7, 609–616 CAS.
- S. Chen, J. J. Duan, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2013, 52, 13567–13570 CrossRef CAS PubMed.
- X. Long, J. K. Li, S. Xiao, K. Y. Yan, Z. L. Wang, H. N. Chen and S. H. Yang, Angew. Chem., Int. Ed., 2014, 53, 7584–7588 CrossRef CAS PubMed.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- J. Wang, H. X. Zhong, Y. L. Qin and X. B. Zhang, Angew. Chem., Int. Ed., 2013, 52, 5248–5253 CrossRef CAS PubMed.
- S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498–502 CrossRef CAS PubMed.
- X. Lu and C. Zhao, Nat. Commun., 2015, 6, 6616 CrossRef CAS PubMed.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CAS.
- W. Zhou, X.-J. Wu, X. Cao, X. Huang, C. Tan, J. Tian, H. Liu, J. Wang and H. Zhang, Energy Environ. Sci., 2013, 6, 2921 CAS.
- N. D. M. W. Chadwick Ellis, S. Bernhard and T. J. Collins, J. Am. Chem. Soc., 2010, 132, 10990–10991 CrossRef PubMed.
- Q. S. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS PubMed.
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- S. Jin, J. M. Kevin, A. G. Hubert, B. G. John and S. H. Yang, Science, 2011, 334, 1383–1385 CrossRef PubMed.
- Z. Lu, H. Wang, D. Kong, K. Yan, P. C. Hsu, G. Zheng, H. Yao, Z. Liang, X. Sun and Y. Cui, Nat. Commun., 2014, 5, 4345 CAS.
- T. Maiyalagan, K. A. Jarvis, S. Therese, P. J. Ferreira and A. Manthiram, Nat. Commun., 2014, 5, 3949 CAS.
- D. H. Deng, L. Yu, X. Q. Chen, G. X. Wang, L. Jin, X. L. Pan, J. Deng, G. Q. Sun and X. H. Bao, Angew. Chem., Int. Ed., 2013, 52, 371–375 CrossRef CAS PubMed.
- J. Deng, P. J. Ren, D. H. Deng, L. Yu, F. Yang and X. H. Bao, Energy Environ. Sci., 2014, 7, 1919–1923 CAS.
- J. Deng, P. J. Ren, D. H. Deng and X. H. Bao, Angew. Chem., Int. Ed., 2015, 54, 2100–2104 CrossRef CAS PubMed.
- X. Q. Chen, J. P. Xiao, J. Wang, D. H. Deng, Y. F. Hu, J. G. Zhou, L. Yu, T. Heine, X. L. Pan and X. H. Bao, Chem. Sci., 2015, 6, 3262–3267 RSC.
- J. Deng, L. Yu, D. H. Deng, X. Chen, F. Yang and X. Bao, J. Mater. Chem. A, 2013, 1, 14868–14873 CAS.
- Y. Hu, J. O. Jensen, W. Zhang, L. N. Cleemann, W. Xing, N. J. Bjerrum and Q. Li, Angew. Chem., Int. Ed., 2014, 53, 3675–3679 CrossRef CAS PubMed.
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 1922–1926 CrossRef PubMed.
- X. X. Zou, X. X. Huang, A. Goswami, R. Silva, B. R. Sathe, E. Mikmekova and T. Asefa, Angew. Chem., Int. Ed., 2014, 53, 4372–4376 CrossRef CAS PubMed.
- M. Tavakkoli, T. Kallio, O. Reynaud, A. G. Nasibulin, C. Johans, J. Sainio, H. Jiang, E. I. Kauppinen and K. Laasonen, Angew. Chem., Int. Ed., 2015, 54, 4535–4538 CrossRef CAS PubMed.
- X. J. Zheng, J. Deng, N. Wang, D. H. Deng, W. H. Zhang, X. H. Bao and C. Li, Angew. Chem., Int. Ed., 2014, 53, 7143–7147 CrossRef.
- T. Fu, M. Wang, W. M. Cai, Y. M. Cui, F. Gao, L. M. Peng, W. Chen and W. P. Ding, ACS Catal., 2014, 4, 2536–2543 CrossRef CAS.
- C. Wang, P. Zhai, Z. C. Zhang, Y. Zhou, J. Ju, Z. J. Shi, D. Ma, R. P. S. Han and F. Q. Huang, Part. Part. Syst. Charact., 2015, 32, 29–34 CrossRef CAS.
- B. Wang, M. Caffio, C. Bromley, H. Früchtl and R. Schaub, ACS Nano, 2010, 4, 5773–5782 CrossRef CAS PubMed.
- F. Guinea, Phys. Rev. B, 2007, 75, 235433 CrossRef.
- P. A. Khomyakov, G. Giovannetti, P. C. Rusu, G. Brocks, J. van den Brink and P. J. Kelly, Phys. Rev. B, 2009, 79, 195425 CrossRef.
- G. Giovannetti, P. A. Khomyakov, G. Brocks, V. M. Karpan, J. van den Brink and P. J. Kelly, Phys. Rev. Lett., 2008, 101, 026803 CrossRef CAS PubMed.
- T. Olsen, J. Yan, J. J. Mortensen and K. S. Thygesen, Phys. Rev. Lett., 2011, 107, 156401 CrossRef PubMed.
- I. C. Man, H. Y. Su, C. V. Federico, H. A. Hansen, J. I. Martínez, N. G. Inoglu, K. John, T. F. Jaramillo, J. K. Nørskov and R. Jan, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- F. W. B. Richard, T. C. Marshall, J. R. Cheeseman and C. Cheng, J. Am. Chem. Soc., 1987, 109, 7968–7979 CrossRef.
- C. V. Federico, L. David, T. M. K. Marc and S. Philippe, Nat. Chem., 2015, 7, 403–410 CrossRef PubMed.
- E. Negro, S. Polizzi, K. Vezzu, L. Toniolo, G. Cavinato and V. Di Noto, Int. J. Hydrogen Energy, 2014, 39, 2828–2841 CrossRef CAS.
- V. Di Noto, E. Negro, S. Polizzi, F. Agresti and G. A. Giffin, ChemSusChem, 2012, 5, 2451–2459 CrossRef CAS PubMed.
- E. Negro, K. Vezzu, F. Bertasi, P. Schiavuta, L. Toniolo, S. Polizzi and V. Di Noto, ChemElectroChem, 2014, 1, 1359–1369 CrossRef CAS.
- V. Di Noto, E. Negro, S. Polizzi, K. Vezzu, L. Toniolo and G. Cavinato, Int. J. Hydrogen Energy, 2014, 39, 2812–2827 CrossRef CAS.
- V. Di Noto, E. Negro, K. Vezzu, F. Bertasi and G. Nawn, Electrochem. Soc. Interface, 2015, 24, 63–68 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ee03316k |
| ‡ These authors have contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.