DOI:
10.1039/C6DT03880H
(Paper)
Dalton Trans., 2016,
45, 19361-19367
Ruthenium water oxidation catalysts containing the non-planar tetradentate ligand, biisoquinoline dicarboxylic acid (biqaH2)†
Received
7th October 2016
, Accepted 16th November 2016
First published on 17th November 2016
Abstract
Two ruthenium complexes containing the tetradentate ligand [1,1′-biisoquinoline]-3,3′-dicarboxylic acid, and 4-picoline or 6-bromoisoquinoline as axial ligands have been prepared. The complexes have been fully characterised and initial studies on their potential to function as molecular water oxidation catalysts have been performed. Both complexes catalyse the oxidation of water in acidic media with CeIV as a stoichiometric chemical oxidant, although turnover numbers and turnover frequencies are modest when compared with the closely related Ru-bda and Ru-pda analogues. Barriers for the water nucleophilic attack and intermolecular coupling pathways were obtained from density functional theory calculations and the crucial influence of the ligand framework in determining the most favourable reaction pathway was elucidated from a combined analysis of the theoretical and experimental results.
Introduction
As society strives to wean itself off its reliance on carbon-based fuels, alternative new technologies need to be discovered and developed to facilitate this revolution.1–5 Researchers in the field of Artificial Photosynthesis aim to achieve this by using sunlight to drive the production of so called solar fuels (e.g. H2, MeOH) from the oxidation of water and the reduction of Earth-abundant substrates (e.g. H+, CO2).6–8 Of the key steps involved in this process (light harvesting, charge separation, substrate reduction and water oxidation),9 water oxidation is considered to be a major bottle neck and new catalysts capable of facilitating this reaction are therefore constantly in high demand.10–13 The group of Licheng Sun has been particularly active in this area and has published a number of molecular water oxidation catalysts (WOCs), including the Ru-bda catalysts (bdaH2 = bipyridine dicarboxylic acid),14–19 some of the fastest and most stable molecular WOCs reported to date. Much work was carried out on varying the axial ligands of these catalysts, which resulted in catalysts with increased turnover numbers (TONs) and turnover frequencies (TOFs).20–23 However, less work has been done with respect to studying equatorial ligand derivatives.24–28 In the particular example where the bdaH2 ligand was exchanged for the pdaH2 ligand (pdaH2 = phenanthroline dicarboxylic acid), a profound effect on the water oxidation mechanism was observed, resulting in a switch from an intermolecular coupling mechanism (I2M) to water nucleophilic attack (WNA). The reasoning behind this switch was proposed to be caused by changes in the reorganisation energies throughout the catalytic cycle as a result of the increased rigidity of the ligand. Intrigued by these results, we decided to study the complementary effect of enforced non-planarity as opposed to enforced planarity and rigidity in the equatorial ligand plane.
Herein, we report a new variation of the Ru-bda-type WOCs, based on the non-planar ligand [1,1′-biisoquinoline]-3,3′-dicarboxylic acid (biqaH2). The synthesis of the new equatorial ligand and two of its ruthenium complexes along with their characterisation and evaluation as WOCs is presented forthwith.
Results and discussion
Synthesis
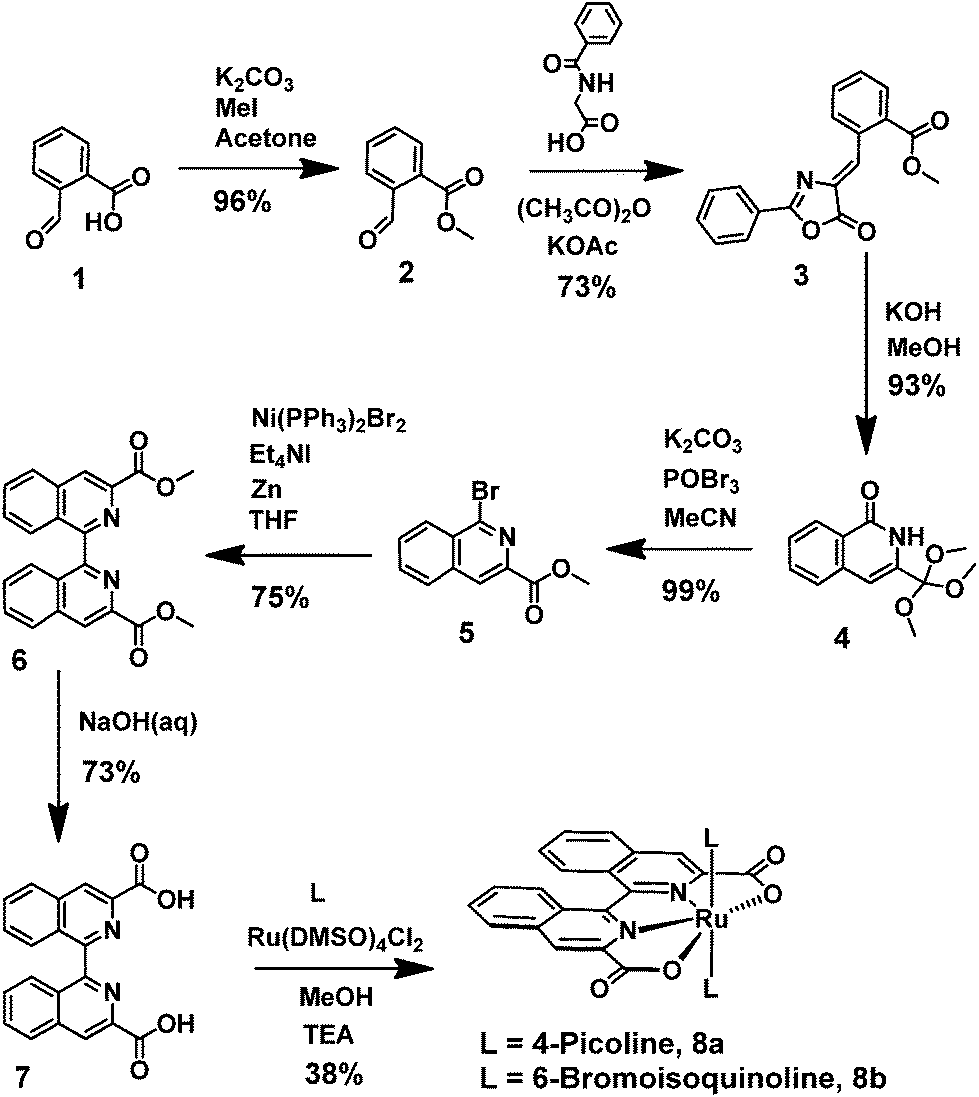
The biqaH2 ligand 7 was prepared in good overall yield over six steps starting from cheap and readily available precursors (Scheme 1). The first four steps followed previously reported procedures,29–31 whilst the conditions for the final two steps were adapted from reported procedures of analogous compounds.32,33 An initial five step synthesis of the biqaH2 ligand 7, starting from 3-methylisoquinoline and forming the diacid via the chromic acid oxidation of 3,3′-dimethyl-1,1′-biisoquinoline, was originally planned,34 however, this route proved to be unsuccessful due to the inability to selectively oxidise the methyl groups without affecting the isoquinoline moiety. The six step procedure without the need for the harsh oxidation step was consequently adopted (Scheme 1). Coordination of the BiqaH2 ligand to ruthenium and substitution of the axial ligands with either 4-picoline (pic) or 6-bromoisoquinoline (Br-isoq) was accomplished in a single step to afford complexes 8a and 8b respectively. A mixture of compounds was formed under these conditions but the desired complexes were always the major product and were isolable via simple precipitation or column chromatography. Attempts to coordinate the ruthenium in a 2-step sequence and first isolate the Ru(biqa)(DMSO)2 intermediate were unsuccessful and tended to yield mixtures of compounds, none of which were the desired intermediate compound as analysed by HPLC, MS and 1H NMR. The isolated complexes 8a and 8b were fully characterised by standard analytical techniques (HPLC, 1D/2D NMR, MS, UV-vis, EA and voltammetry).
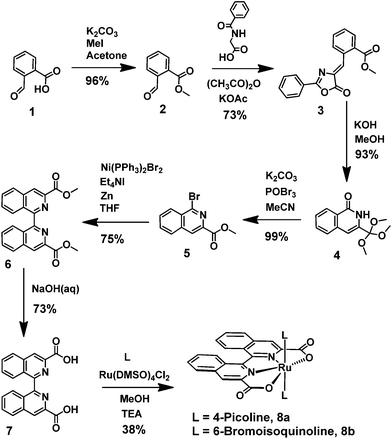 |
| | Scheme 1 Synthesis of the biqaH2 ligand 7 and subsequent ruthenium complexes 8a and 8b. | |
X-ray structure
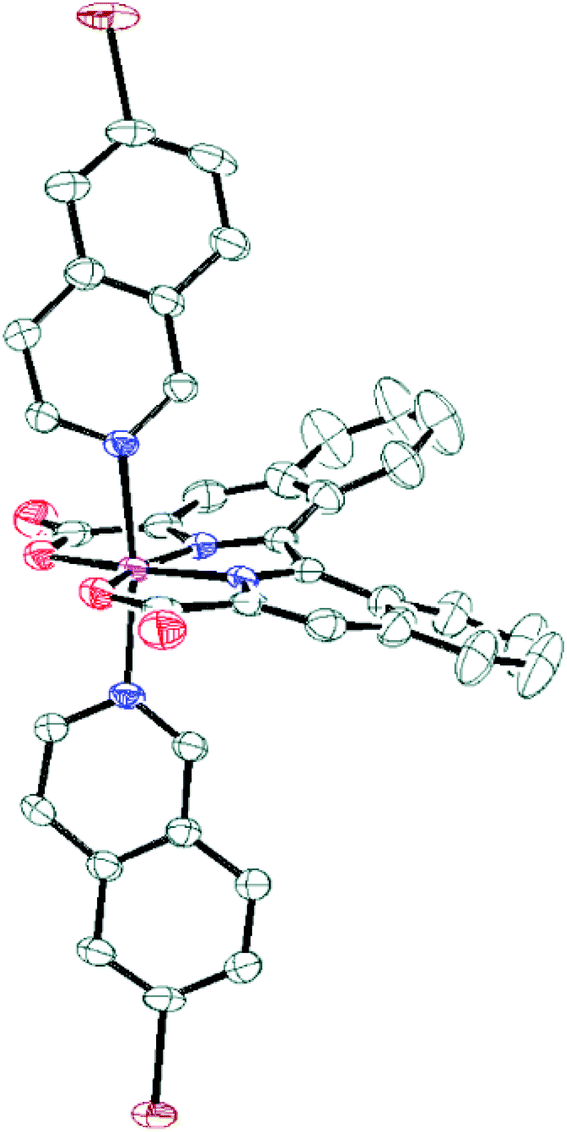
Additionally for complex 8b, single crystals suitable for X-ray diffraction were grown and the absolute structural configuration was confirmed (see Fig. 1 and ESI†). Interestingly, the coordination environment of the Ru centre in complex 8b is very similar to analogous Ru-bda-type structures previously reported,15,16,35 despite the obvious distortion of the biisoquinoline moiety. The strain imposed by the inherent non-planarity of the biisoquinoline, combined with an almost planar coordination of the donor atoms to ruthenium, is therefore manifested in a bending of the isoquinoline carboxylates rather than a twisted coordination geometry. This is thought to occur because the octahedral geometry is already distorted due to the widening of the O–Ru–O angle from the ideal 90° to ca. 120°, similar to Ru-bda structures, and therefore does not permit any further distortion of the primary coordination sphere that may have been expected as a result of the non-planarity of the biqa ligand backbone.
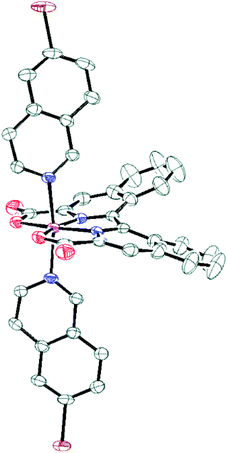 |
| | Fig. 1 ORTEP plot at 50% probability for compound 8b. Colour code: C, grey; Br, brown; N, blue; O, red; Ru, magenta. Hydrogens have been omitted for clarity. | |
Electrochemistry
Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) in both organic and aqueous solutions were used to probe the electrochemical properties of compounds 8a and 8b. Not surprisingly, the electrochemical behaviour of the Ru-biqa complexes was found to be very similar to the analogous Ru-bda complexes. The CVs and DPVs in DCM (0.1 M TBA(PF6)) + 20% trifluoroethanol (TFE) indicated a single reversible redox process for the RuIII–II couple at 0.87 V and 0.92 V (vs. NHE)36 for 8a and 8b respectively (Table 1, Fig. 2 and ESI†). In aqueous solvent (0.1 M HOTf(aq) + TFE) three redox couples were observed due to the coordination of water permitting access to higher oxidation states through proton coupled electron transfer (PCET)37,38 and a large current increase above ca. 1.3 V was attributed to catalytic water oxidation. E1/2 values for RuIII–II, RuIV–III, and RuV–IV are given in Table 1 and their shifts as a function of changes in pH are presented in the corresponding Pourbaix diagrams in the ESI (Fig. S7 and S8†). Previous work by Sun et al. showed that small quantities of TFE had little or no effect on the electrochemistry of Ru-bda complexes in aqueous solutions at pH 1 but at higher concentrations the catalytic currents were suppressed.16 TFE coordination to ruthenium has also been observed for the Ru(Mebimpy)(bpy)(OH2)2+ WOC previously reported by Meyer et al.39 In our electrochemical studies we also found that high TFE quantities could significantly influence the electrochemical behaviour of the Ru-biqa complexes. Due to the poor solubility of 8b in 0.1 M aqueous HOTf, 33% TFE was required to obtain a 0.3 mM solution but no effect on the electrochemical response was observed. In the case of 8a only 20% of TFE was required to fully dissolve the complex, again without disturbing the electrochemical response, however, the RuIV-III and RuV–IV waves that were initially observed for catalyst 8a were greatly diminished at TFE concentrations of 33% and above (ESI Fig. S6†). These results indicated an equilibrium between H2O and TFE coordination for the Ru-biqa complexes but as the initial Ru-biqa complexes are coordinatively saturated this would require prior decoordination of one of the biqa ligand donor atoms. A potentially related observation made during the electrochemical experiments was a change in colour of the acidic aqueous solutions of the catalysts from blue to yellow. UV-vis spectroscopy was used to probe this further.
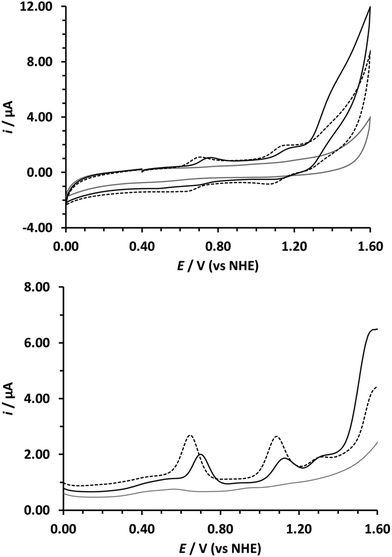 |
| | Fig. 2 Cyclic voltammograms (top) and differential pulse voltammograms (bottom) of 8a (dashed black) and 8b (solid black). [8a] = 0.3 mM in 0.1 M HOTf(aq) with 20% TFE, [8b] = 0.3 mM in 0.1 M HOTf(aq) with 33% TFE. Electrolyte only blank scan (solid grey). CV scan rate = 100 mV s−1. DPV sweep rate = 20 mV s−1. GC disc as working electrode, Pt rod as counter electrode, Ag/AgCl as reference electrode (values converted to NHE using E1/2 Ag/AgCl = 0.2 V vs. NHE). | |
Table 1 Summary of electrochemical data for complexes 8a and 8ba
| Complex |
E
1/2 RuIII–II (ΔEp) |
E
1/2 RuIV–III (ΔEp) |
E
1/2 Ru2IV–V |
|
E
1/2 values in V vs. NHE (ref. 36) and ΔE in mV.
0.5 mM in 0.1 M TBA(PF6) DCM with 20% TFE.
0.3 mM in 0.1 M HOTf(aq) with 20% TFE.
0.3 mM in 0.1 M HOTf(aq) with 33% TFE.
Values taken from DPV.
|
|
8a
|
0.87 (80) |
— |
— |
|
8a
|
0.67 (50) |
1.11 (60) |
1.30e |
|
8b
|
0.92 (80) |
— |
— |
|
8b
|
0.73 (50) |
1.15 (40) |
1.31e |
UV-vis
Changes in the absorbance spectra and a number of other observations were recorded after dissolving catalysts 8a and 8b in TFE–0.1 M HOTf(aq) mixed solvent (see ESI†): the rate of decay at 600 nm was identical for both catalysts, the spectra of the resulting pale yellow solutions contained very similar absorbance features for both compounds, no change in absorbance was observed for either compound when the TFE–0.1 M HOTf(aq) mixed solvent was replaced with a TFE–water mix, and finally, the process could be reversed by the addition of a reducing agent like sodium ascorbate. The conclusions that can be drawn from these results are; (i) the process involves a reversible oxidation event, (ii) the oxidation is pH dependent, and (iii) the axial ligands have little effect on the conversion. The oxidation of the RuII complexes to the RuIII–OH2 species by oxygen is consistent with each of these statements. The final proof was obtained by carrying out redox titrations of the catalyst solutions with the strong oxidant, ceric ammonium nitrate (CAN). The same spectra as those observed after one hour standing in air were obtained after the addition of 1.0 eq. of CAN, thus confirming the 1-electron oxidation of the catalysts as the reason for the observed colour changes.
Chemical water oxidation
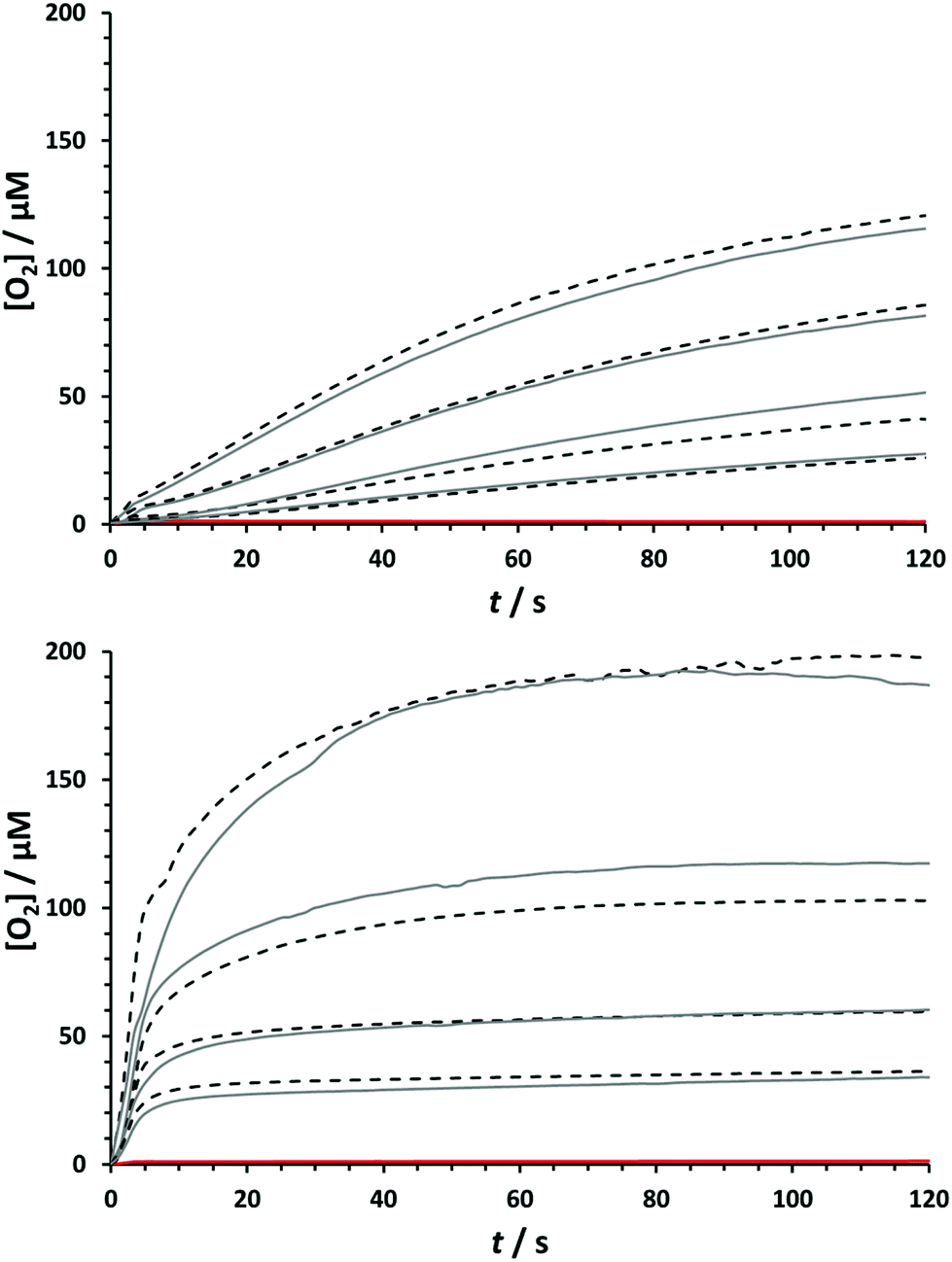
Given the structural similarities between the Ru-biqa WOCs and the Ru-bda WOCs, conditions for assessing the chemical water oxidation were chosen to give the closest possible comparison. Catalysts 8a and 8b were therefore added to a solution of excess CAN oxidant in 0.1 M HOTf(aq) and the oxygen concentration was measured in the aqueous phase using a fluorescence-based oxygen probe. Fig. 3 shows the oxygen evolution profiles at various catalyst concentrations of 8a and 8b. What is evident from these plots is that catalyst 8a and catalyst 8b behave quite differently under the same reaction conditions: catalyst 8a has a fairly smooth oxygen evolution profile that gradually slows down over time due to catalyst deactivation, whereas for catalyst 8b there appears to be a fast initial phase, abruptly followed by a slow second phase. Despite these differences, the TONs of both catalysts were very similar; 87 for 8a and 80 for 8b (see ESI† for all TON and TOF values). The absorbance decay of CeIV, which was proportional to oxygen production, was measured under the same reaction conditions and used to indirectly estimate the reaction rates (see ESI†). Linear fitting of the data from 4–20 s gave almost identical first order rate constants of 2.51 s−1 for 8a and 2.27 s−1 for 8b, corresponding to TOFs40 of 0.63 s−1 and 0.57 s−1 respectively. This data fits well with the previous studies of Sun et al., where they compared the Ru-bda catalyst framework to the more rigid phenanthroline analogue, Ru-pda, and found that the increased rigidity shifted the preference from the I2M mechanism for the Ru-bda's to WNA for the Ru-pda's.24 Considering the reaction is first order in Ru-biqa catalyst and the TOFs are closer to the (pic)2Ru(pda) analogue than the (pic)2Ru(bda) analogue (0.1 s−1 and 3.0 s−1 respectively) measured under the same conditions, a similar shift towards WNA was indicated here.15,24 The rates recorded for catalyst 8b over the initial period (0–4 s) after catalysts injection were visibly faster than the data recorded after 4 s but this range is not as accurate because it includes the errors for manual injection of the catalyst solution and so no reliable correlations to rate order could be extracted from this data. It is possible that the energy barriers for the two possible pathways (I2M vs. WNA) are perhaps so close for the Ru-biqa compounds that both pathways are energetically feasible and that compound 8b begins catalysis via I2M but quickly switches to WNA after only a few cycles. Density functional theory (DFT) calculations were performed on Ru-biqa catalyst models to determine the energy barriers of the two pathways and the results are presented in the following computational section.
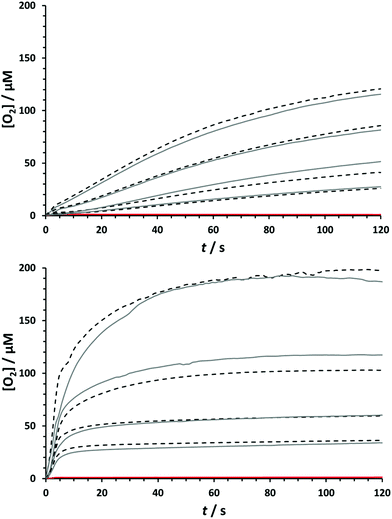 |
| | Fig. 3 Oxygen evolution profiles after addition of 8a (top) and 8b (bottom) to CAN measured with fluorescence probe. [CAN] = 1.5 mM in 0.1 M HOTf(aq) (pH = 1). [8a] = [8b] = 4.0, 2.0, 1.0, 0.5 μM. T = 25 °C. Reaction volume = 4.0 mL. Experiments for each [cat.] were carried out in duplicate (1st experiment, solid grey; 2nd experiment, dashed black). The red line is the blank experiment for the addition of solvent with no catalyst to the CAN solution. | |
Another possible explanation is that the initial complex is converted to another active species during the initial phase of catalysis, thus resulting in the more complex behaviour. HPLC analysis of the catalysis reaction solutions for both compounds 8a and 8b supported this hypothesis to some extent (see ESI†): for catalysis solutions of 8b (16 μM) and CAN (1.5 mM, ca. 94 eq.), both the starting complex and free biqa ligand were identified in the reaction mixture after catalysis. For catalysis solutions of 8b (8 μM) and CAN (1.5 mM, ca. 188 eq.), both the starting complex and free biqa ligand were again present in the reaction mixture but the relative amount of free ligand had increased. No starting complex was observed at the end of catalysis reactions with 8a and CAN, free biqa ligand was the only species identified at both catalyst concentrations. Hence, both catalysts 8a and 8b are not stable under the reaction conditions and the majority of the original catalyst has been converted after only 23–47 cycles. What is not entirely clear, is what the other degradation products are, however, the fact that CeIV consumption does not continue after the original catalysts are gone would indicate that their activity is negligible in comparison to the original molecular species. Work to determine the fate of the axial ligands and the ruthenium and to identify the exact nature of other reaction by-products is still ongoing in our labs.
Computational studies
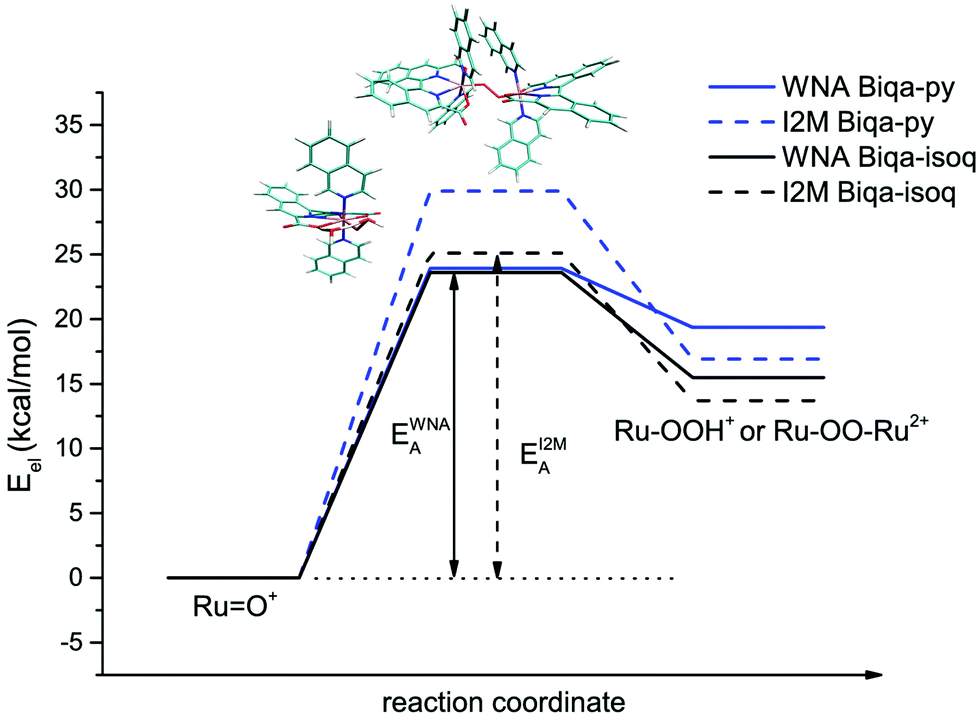
Assuming that the molecular catalysts are responsible for catalysis in the initial phases of the reaction with CAN and that the O–O bond formation is the rate limiting step, as was previously observed for the analogous Ru-bda compounds,20,41 the activation barriers of the two well-known I2M and WNA mechanisms24,42 were compared for model complexes of the new Ru-biqa compounds 8a and 8b in an exhaustive DFT study (see ESI† for more details). For both model complexes, Ru-biqa-py and Ru-biqa-isoq (py = pyridine, isoq = isoquinoline as axial ligands), the barriers for the WNA pathway were found to be lower than those of the I2M pathway (see Table 2 and Fig. 4). The flexibility of the bda ligand allows it to adapt to the various transition state and intermediate structures of the I2M pathway.24 Both the pda and biqa ligands are restrained in their movement and are therefore less adaptable, thus increasing the I2M barriers beyond those of WNA. The axial ligand identity did not appear to play an important role in determining the transition state geometries or the activation energies for the WNA mechanism (see Table 2, Fig. 5 and S33†). Transition state (TS) structures calculated using either one or two explicit water molecules highlighted the importance of an accurate description of the solvation shell. In particular, the hydrogen bonding network which shuttles the proton off the attacking water molecule to the carboxylate of the biqa ligand plays a crucial role. Overall, our findings are in good agreement with previous studies by Tong et al., where the necessity of an additional water molecule was discussed in detail.24
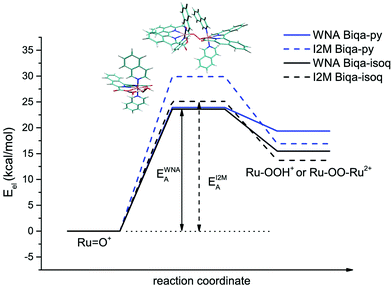 |
| | Fig. 4 Energy diagram comparing the energy barriers of oxygen bond formation for the WNA and I2M mechanisms for Ru-biqa model complexes. | |
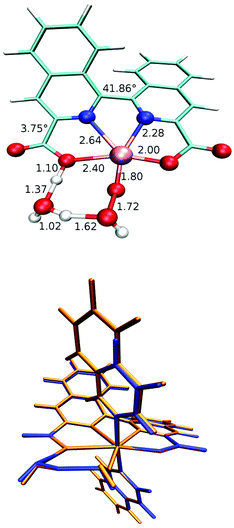 |
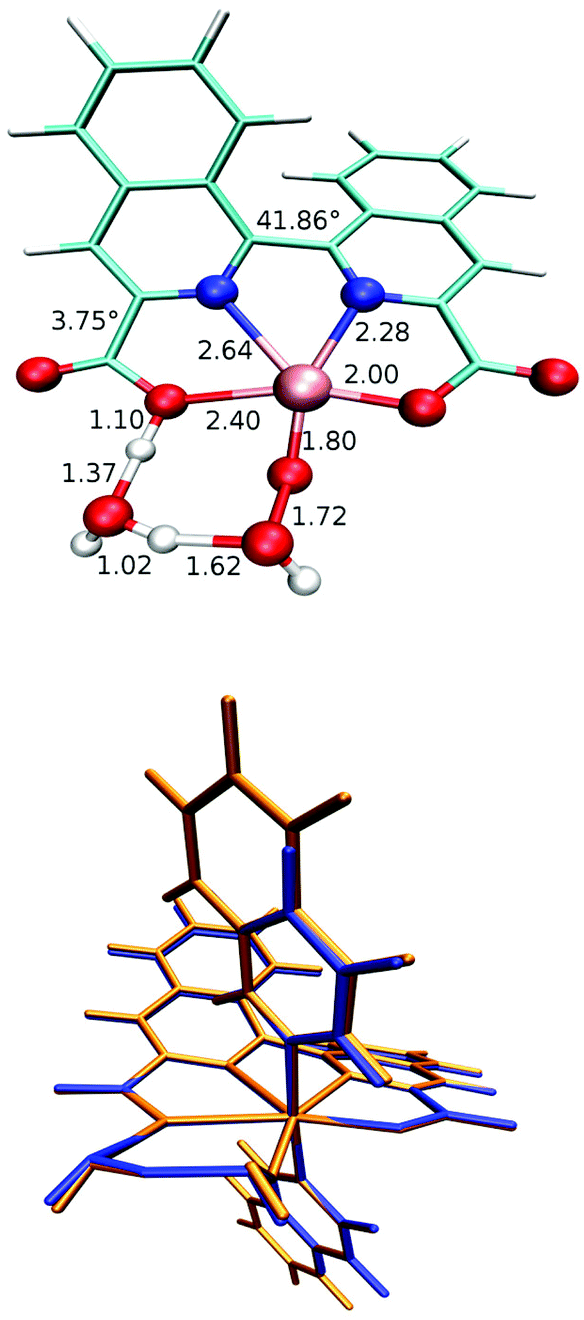
| | Fig. 5 Top: WNA transition state for Ru-biqa-py calculated with two explicit water molecules. Selected bond lengths (in Å) and dihedral angles are also given. Axial ligands are not shown for clarity. Bottom: Overlay of TS structure for Ru-biqa-py (blue) and Ru-biqa-isoq (orange) highlighting the fact that the axial ligand does not influence the WNA-TS structure. | |
Table 2 Activation energies for the I2M and WNA mechanisms
| Ligands |
E
a(l2M-HS) (kcal mol−1) |
E
a(WNA-H2O) (kcal mol−1) |
|
Present work.
Ref. 24.
Ref. 43.
|
| Ru-bda-py |
22.5a (15.0b) |
26.9a (18.0b/32.7c) |
| Ru-biqa-py |
30.7a |
23.9a |
| Ru-biqa-isoq |
25.1a |
23.6a |
For the I2M mechanism, we observed a significant contribution of π–π stacking that leads to a lower barrier for Ru-biqa-isoq compared to Ru-biqa-py. This π-stacking effect is in line with observations made by Sun and co-workers for Ru-bda complexes with pyridine and isoquinoline axial ligands.20 However, this effect would only become relevant in situations where the I2M was favoured over WNA.
The barriers obtained from the DFT calculations help to explain the observations made during the catalysis experiments, which showed regular oxygen evolution curves and first order kinetics for the Ru-biqa-pic complex (8a) but a more complex behaviour for the Ru-biqa-Br-isoq complex (8b): while the activation energy difference between the two mechanisms for Ru-biqa-py is 6.8 kcal mol−1, for Ru-biqa-isoq it is only 1.5 kcal mol−1. This clear preference for the WNA mechanism for Ru-biqa-py agrees well with the experimentally observed first order kinetics (in catalyst) for compound 8a, whereas for the Ru-biqa-isoq compound the energy difference of the two pathways is negligible, which indicates that both pathways are equally energetically feasible. Thus, for compound 8b both mechanisms could potentially operate, resulting in a more complex kinetic behaviour as the mechanism switches or the compound begins to degrade. It is also worth pointing out that other mechanisms for O–O bond formation with Ru-bda complexes have recently been proposed,44 which could offer an alternative theoretical explanation as well and would be worth considering in future studies.
Conclusions
In summary, the novel biqaH2 ligand framework was successfully prepared and complexed with ruthenium to give a new family of molecular water oxidation catalysts, the Ru-biqa's. The ruthenium complexes were shown to be active for chemically induced water oxidation catalysis when added to CAN oxidant at pH 1, however, their activity was greatly inferior to the analogous Ru-bda compounds tested under similar experimental conditions.15,20,22,35 DFT studies revealed that for Ru-biqa complexes, in contrast to Ru-bda analogues, the oxygen–oxygen bond formation is more favourable via the WNA pathway than the I2M pathway. The slow catalytic rates and relatively lower barriers for WNA pathway were also in better agreement with previous studies of ruthenium complexes containing the rigid pda ligand framework, which were proposed to catalyse water oxidation via the WNA pathway.24 It therefore appears that inducing non-planarity in the equatorial coordinating ligand framework has the same negative effect as inducing rigidity; resulting in a switch from the I2M mechanism to the WNA mechanism and consequently slower catalysis. Furthermore, the stability (indicated by TON) of the Ru-biqa complexes was lower than both the Ru-bda's and the Ru-pda's. Replacement of the bda ligand by biqa therefore offers no improvement with regards to catalytic performance, however, these findings help to further consolidate our understanding of the mechanisms surrounding catalytic water oxidation and will be invaluable in the design of future molecular catalysts. For example, future work that may look to investigate substitution effects on the bda ligand should avoid substituents positioned at the 3 and 3′ positions as they may also lead to similarly induced non-planarity and a consequent drop in catalytic performance.
Acknowledgements
We would like to thank Peter Deglmann for fruitful discussions concerning the calculations. We also thank the Swiss National Supercomputing Centre for computational time (project s502) as well as the University of Zurich for a Forschungskredit (FK-15-101) and the University of Zurich and the LightChEC program for funding.
References
- N. S. Lewis, Science, 2016, 351(6271) DOI:10.1126/science.aad1920.
- S. H. Jensen, C. Graves, M. Mogensen, C. Wendel, R. Braun, G. Hughes, Z. Gao and S. A. Barnett, Energy Environ. Sci., 2015, 8, 2471–2479 CAS.
- Q. Yi, W. Li, J. Feng and K. Xie, Chem. Soc. Rev., 2015, 44, 5409–5445 RSC.
- M. Z. Jacobson, M. A. Delucchi, G. Bazouin, Z. A. F. Bauer, C. C. Heavey, E. Fisher, S. B. Morris, D. J. Y. Piekutowski, T. A. Vencilla and T. W. Yeskooa, Energy Environ. Sci., 2015, 8, 2093–2117 CAS.
- N. Armaroli and V. Balzani, Chem. – Eur. J., 2015, 22, 32–57 CrossRef PubMed.
- D. Kim, K. K. Sakimoto, D. Hong and P. Yang, Angew. Chem., Int. Ed., 2015, 54, 3259–3266 CrossRef CAS PubMed.
- J. A. Herron, J. Kim, A. A. Upadhye, G. W. Huber and C. T. Maravelias, Energy Environ. Sci., 2015, 8, 126–157 CAS.
- S. A. Bonke, M. Wiechen, D. R. MacFarlane and L. Spiccia, Energy Environ. Sci., 2015, 8, 2791–2796 CAS.
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
-
L. Francàs, R. Bofill, J. García-Antón, L. Escriche, X. Sala and A. Llobet, Molecular Water Oxidation Catalysis: A Key Topic for New Sustainable Energy Conversion Schemes, John Wiley & Sons Ltd, Chichester, UK, 2014 Search PubMed.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS PubMed.
- R. Cao, W. Lai and P. Du, Energy Environ. Sci., 2012, 5, 8134–8157 CAS.
- H. Inoue, T. Shimada, Y. Kou, Y. Nabetani, D. Masui, S. Takagi and H. Tachibana, ChemSusChem, 2011, 4, 173–179 CAS.
- L. Duan, L. Wang, F. Li, F. Li and L. Sun, Acc. Chem. Res., 2015, 48, 2084–2096 CrossRef CAS PubMed.
- L. Duan, A. Fischer, Y. Xu and L. Sun, J. Am. Chem. Soc., 2009, 131, 10397–10399 CrossRef CAS PubMed.
- L. Duan, L. Wang, A. K. Inge, A. Fischer, X. Zou and L. Sun, Inorg. Chem., 2013, 52, 7844–7852 CrossRef CAS PubMed.
- L. Wang, L. Duan, B. Stewart, M. Pu, J. Liu, T. Privalov and L. Sun, J. Am. Chem. Soc., 2012, 134, 18868–18880 CrossRef CAS PubMed.
- L. Duan, Y. Xu, P. Zhang, M. Wang and Li. Sun, Inorg. Chem., 2010, 49, 209–215 CrossRef CAS PubMed.
- Y. Jiang, F. Li, B. Zhang, X. Li, X. Wang, F. Huang and L. Sun, Angew. Chem., Int. Ed., 2013, 52, 3398–3401 CrossRef CAS PubMed.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- L. Duan, C. M. Araujo, M. S. G. Ahlquist and L. Sun, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15584–15588 CrossRef CAS PubMed.
- L. Wang, L. Duan, Y. Wang, M. S. G. Ahlquist and L. Sun, Chem. Commun., 2014, 50, 12947–12950 RSC.
- C. J. Richmond, R. Matheu, A. Poater, L. Falivene, J. Benet-Buchholz, X. Sala, L. Cavallo and A. Llobet, Chem. – Eur. J., 2014, 20, 17282–17286 CrossRef CAS PubMed.
- L. Tong, L. Duan, Y. Xu, T. Privalov and L. Sun, Angew. Chem., Int. Ed., 2011, 50, 445–449 CrossRef CAS PubMed.
- T.-T. Li, W.-L. Zhao, Y. Chen, F.-M. Li, C.-J. Wang, Y.-H. Tian and W.-F. Fu, Chem. – Eur. J., 2014, 20, 13957–13964 CrossRef CAS PubMed.
- Z. Liu, Y. Gao, Z. Yu, M. Zhang and J. Liu, Chin. J. Cat., 2015, 36, 1742–1749 CrossRef CAS.
- R. Matheu, M. Z. Ertem, J. Benet-Buchholz, E. Coronado, V. S. Batista, X. Sala and A. Llobet, J. Am. Chem. Soc., 2015, 137, 10786–10795 CrossRef CAS PubMed.
- D. W. Shaffer, Y. Xie, D. J. Szalda and J. J. Concepcion, Inorg. Chem., 2016 DOI:10.1021/acs.inorgchem.6b02193 , asap.
-
E. F. Cotter, Patent WO2010/125343A1, 2010 Search PubMed.
- E. T. Stiller, J. Chem. Soc., 1937, 473–476 RSC.
- Y. L. Janin, E. Roulland, A. Beurdeley-Thomas, D. Decaudin, C. Monneret and M.-F. Poupon, J. Chem. Soc., Perkin Trans. 1, 2002, 529–532 RSC.
- M. Hapke, H. Staats, I. Wallmann and A. Lützen, Synthesis, 2007, 2711–2719 CAS.
- M. Slangy and P. Stang, Synthesis, 1996, 1019–1028 CrossRef.
- G. Bozoklu, C. Marchal, C. Gateau, J. Pécaut, D. Imbert and M. Mazzanti, Chem. – Eur. J., 2010, 16, 6159–6163 CrossRef CAS PubMed.
- C. J. Richmond, R. Matheu, A. Poater, L. Falivene, J. Benet-Buchholz, X. Sala, L. Cavallo and A. Llobet, Chem. – Eur. J., 2014, 20, 17282–17286 CrossRef CAS PubMed.
- Values recorded vs. Fc/Fc+ internal standard and converted to vs. NHE by addition of 630 mV.
- M. H. V. Huynh and T. J. Meyer, Chem. Rev., 2007, 107, 5004–5064 CrossRef CAS PubMed.
- C. J. Gagliardi, B. C. Westlake, C. A. Kent, J. J. Paul, J. M. Papanikolas and T. J. Meyer, Coord. Chem. Rev., 2010, 254, 2459–2471 CrossRef CAS.
- Z. Chen, J. J. Concepcion, H. Luo, J. F. Hull, A. Paul and T. J. Meyer, J. Am. Chem. Soc., 2010, 132, 17670–17673 CrossRef CAS PubMed.
- TOF values calculated by dividing the rate of oxygen production (μM s−1) by the ruthenium catalyst concentration (μM).
- R. Staehle, L. Tong, L. Wang, L. Duan, A. Fischer, M. S. G. Ahlquist, L. Sun and S. Rau, Inorg. Chem., 2014, 53, 1307–1319 CrossRef CAS PubMed.
- S. Romain, L. Vigara and A. Llobet, Acc. Chem. Res., 2009, 42, 1944–1953 CrossRef CAS PubMed.
- R. Kang, K. Chen, J. Yao, S. Shaik and H. Chen, Inorg. Chem., 2014, 53, 7130–7136 CrossRef CAS PubMed.
- J. J. Concepcion, D. K. Zhong, D. J. Szalda, J. T. Muckerman and E. Fujita, Chem. Commun., 2015, 51, 4105–4108 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1508531. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt03880h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a