
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Convenient synthesis of energetic polynitro materials including (NO2)3CCH2CH2NH3-salts via Michael addition of trinitromethane†‡
Quirin J.
Axthammer
,
Thomas M.
Klapötke
*,
Burkhard
Krumm
,
Regina
Scharf
and
Cornelia C.
Unger
Department of Chemistry, Ludwig-Maximilian University of Munich, Butenandtstr. 5-13 (D), 81377 Munich, Germany. E-mail: tmk@cup.uni-muenchen.de; Fax: +49-89-2180-77492
First published on 4th November 2016
Abstract
The nucleophilic Michael addition of nitroform with acrylamide creates a variety of energetic products. Several interesting compounds with a trinitromethyl group were synthesized, among them salts containing the trinitropropylammonium cation, [(NO2)3CCH2CH2NH3]X. Owing to their positive oxygen balance, the suitability of these compounds as potential high-energy dense oxidizers (HEDOs) in energetic formulations was investigated and discussed. Furthermore, numerous important and reactive compounds for the continuing synthesis of molecules with a high oxygen balance are presented. All compounds were fully characterized, including multinuclear NMR spectroscopy, vibrational analysis (IR, Raman), elemental analysis, as well as single crystal X-ray diffraction. Thermal stabilities were studied using differential scanning calorimetry and sensitivity data against friction, impact and electrostatic discharge were collected. The energies of formation were calculated using Gaussian 09 and energetic properties, such as the specific impulse and detonation velocity, were predicted with the EXPLO5 (V6.02) computer code.
Introduction
The trinitromethane (nitroform) unit is an important building block in the chemistry of high-energy materials, especially in the field of high-energy dense oxidizers (HEDOs).1 This trinitromethane unit can easily be introduced by a nucleophilic addition on electron deficient α,β-unsaturated starting materials. The so-called Michael addition is one of the most important carbon–carbon bond forming reactions in synthetic organic chemistry. Michael donors are substrates with acidic protons which therefore are capable of forming carbanions. This includes anions from nitroform, fluorodinitromethane, primary nitroalkanes, and secondary nitroalkanes.2 The electron deficient alkene in this nucleophilic addition is called the Michael acceptor and includes a wide range of α,β-unsaturated ketones, aldehydes, carboxylic acids, esters, amides and cyanides.3 One such example is reported in the nucleophilic addition of some polynitroalkanes to acrolein oxime.4In this contribution nitroform and the readily available acrylamide are used to build several new oxygen-rich molecules as well as energetic salts containing the (NO2)3CCH2CH2NH3-cation.
Results and discussion
Synthesis
Earlier investigations showed, that with tetranitromethane and various acrylamides, mostly mixtures of 3-nitroisoxazoles and Michael addition products were formed.5 However, the reaction of acrylamide with nitroform resulted exclusively in the formation of the Michael addition product 4,4,4-trinitrobutanamide (1). A similar synthesis of 1 has been reported earlier.6 However, in the herein presented advanced synthesis 1 was obtained without the use of further chemicals, as mentioned in literature procedures from readily available chemicals (Scheme 1).6a,7 A further advantage is the faster conversion without heating, as well as increased yields from 63% to 97%. Due to the almost quantitative conversion of acrylamide, pure 1 without further purification was obtained.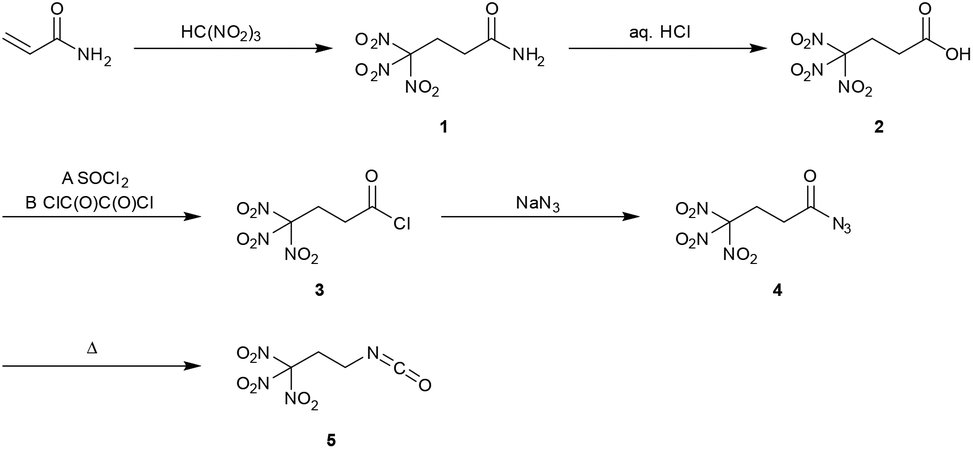 | ||
| Scheme 1 Synthesis of 1,1,1-trinitropropan-3-isocyanate (5) starting from acrylamide and trinitromethane. | ||
The acid 4,4,4-trinitrobutanoic acid (2) was prepared by hydrolysis of the amide 1 in aqueous concentrated hydrochloric acid. The crude material was recrystallized from chloroform to obtain a pure product in 80% yield. Due to their straightforward synthesis with high yields compounds 1 and 2 are excellent starting materials for various compounds containing the trinitromethyl moiety.8 The acid 2 was converted to the corresponding carbonyl chloride by refluxing in excess thionyl chloride (Method A). The reaction time should be longer than 20 hours to ensure complete conversion to the acid chloride and to prevent the formation of the acid anhydride.9 4,4,4-Trinitrobutanoyl chloride (3) was isolated in 88% yield. A more convenient synthesis for the carbonyl chloride 3 is the conversion of acid 2 with a stoichiometric amount of oxalyl chloride and DMF as catalyst (Method B). Compound 3 was obtained in 96% yield while the reaction time was reduced to 4 h. Reaction of compound 3 with sodium azide at ambient temperature yielded the carbonyl azide 4,4,4-trinitrobutanoyl azide (4). To obtain the azide 4 as pure colourless solid, the reaction temperature has to be kept below 30 °C during the whole synthesis and work-up procedure. Due to its high sensitivity, extreme care is required when working with this compound. Heating the azide 4 in an organic inert solvent causes the conversion to 1,1,1-trinitropropan-3-isocyanate (5) via a Curtius rearrangement. A much safer way for the synthesis of 5 is the subsequent in situ conversion of 4 to the isocyanate 5 without isolation of the very sensitive azide 4. The isocyanate 5 is a useful precursor for the synthesis of several energetic carbamates, ureas, amines and salts.8–10 The chloride and nitrate salts 6a and 6b of the 3,3,3-trinitropropyl-1-ammonium cation were obtained by controlled hydrolysis of 5 in diluted mineral acid (Scheme 2).11 The perchlorate, dinitramide and 5,5′-azobistetrazolate salts 6c–e were synthesized by metathesis of the chloride salt 6a with the corresponding silver and potassium salts, respectively (Scheme 2). The salt formation of 6 proceeds in high yields around 90%. The nitrate salt 6b, perchlorate salt 6c and dinitramide salt 6d are air and moisture stable and exhibit high positive oxygen balances ΩCO of +15.6% (6b), +21.7% (6c), and +20.7% (6d).
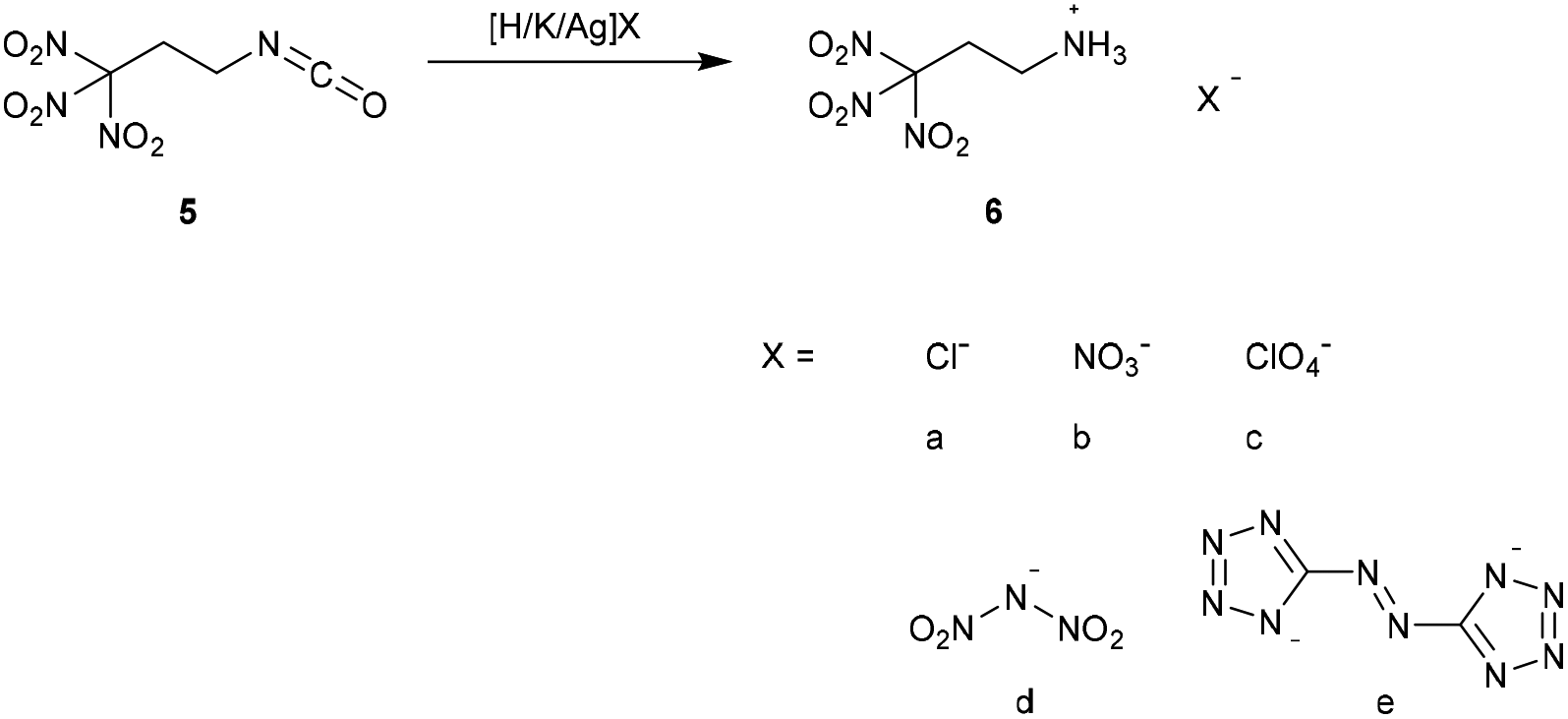 | ||
| Scheme 2 Synthesis of 3,3,3-trinitropropyl-1-ammonium (6) salts. | ||
An interesting combination of Michael addition with Mannich condensation is the one-pot reaction of acrylamide (1 eq.), nitroform (2 eq.) and formaldehylde (1 eq.) to give 4,4,4-trinitro-N-(2,2,2-trinitroethyl)butanamide (7) (Scheme 3).12
 | ||
| Scheme 3 Synthesis of 4,4,4-trinitro-N-(2,2,2-trinitroethyl)butanamide (7). | ||
An oxygen rich molecule was also prepared by the esterification of the amide 1 with the alcohol 2,2,2-trinitroethanol. The reaction was performed in oleum (30% SO3) as strong dehydrating agent.13 After recrystallization from water/methanol the ester 2,2,2-trinitroethyl-4,4,4-trinitrobutanoate (8) was obtained as pure colourless solid (Scheme 4).
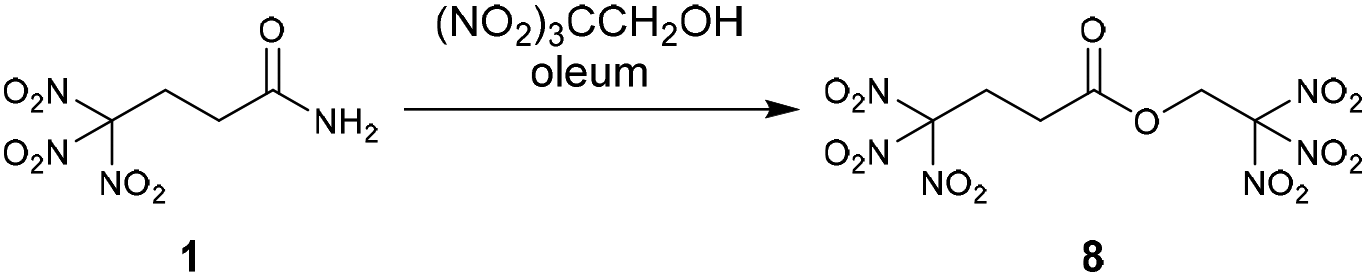 | ||
| Scheme 4 Esterification of the amide 1 to form 2,2,2-trinitroethyl-4,4,4-trinitrobutanoate (8). | ||
Multinuclear NMR spectroscopy
All compounds were thoroughly characterized by 1H, 13C and 14N NMR spectroscopy. In the 1H NMR spectra the two neighboring CH2 groups are within the range of 3.90 to 2.52 ppm. The methylene unit next to the trinitromethyl moiety is mostly shifted to higher field compared to the CH2 groups next to a nitrogen or oxygen atom. The vicinal coupling constants of the hydrogen atoms in the ethylene group are not equivalent due to the rotation around the C–C bond, which causes an AA′XX′ spin system.14 The resonances of the CH2 moiety of the trinitroethyl group is observed at lower field (4.96 ppm (7) and 5.20 ppm (8)) compared to the trinitropropyl group.In the 13C NMR spectra the carbon resonances of the two CH2 groups of the trinitropropyl part are very variable and are found in the range of 40.5 to 27.6 ppm. The carbon resonances of the trinitromethyl moieties are observed as broadened signals. Those of the trinitropropyl unit are located at around 128 ppm whereas the resonances of the trinitroethyl unit of compounds 7 and 8 are slightly upfield shifted to approximately 126 ppm.
In the 14N NMR spectra the resonances for the nitro groups of the trinitromethyl moieties are all quite sharp and found in the range of −13 to −31 ppm. For the ammonium moieties of the salts 6a–e resonances are observed around −355 ppm.
Vibrational spectroscopy
All compounds were also characterized by IR and Raman spectroscopy. The most characteristic frequencies in the compounds are the carbonyl and nitro groups. The characteristic ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching vibration is located in a large range from 1785 to 1676 cm−1. Noticeable is the shift of the carbonyl stretching vibrations to higher wave numbers in molecules which are connected to electron-withdrawing moieties. The maximum is the acid chloride 3 where the ν(CO) is located at 1785 cm−1, while for the two amides 1 and 7 signals at 1695 and 1676 cm−1 are observed. For the trinitromethyl units both the antisymmetric νas(NO2) in the range of 1604–1582 cm−1 and the symmetric stretching vibrations νs(NO2) at 1303–1288 cm−1 are observed. The antisymmetric stretching vibration of the azide moiety of compound 4 is found as the characteristic strong signal at 2148 cm−1.
O) stretching vibration is located in a large range from 1785 to 1676 cm−1. Noticeable is the shift of the carbonyl stretching vibrations to higher wave numbers in molecules which are connected to electron-withdrawing moieties. The maximum is the acid chloride 3 where the ν(CO) is located at 1785 cm−1, while for the two amides 1 and 7 signals at 1695 and 1676 cm−1 are observed. For the trinitromethyl units both the antisymmetric νas(NO2) in the range of 1604–1582 cm−1 and the symmetric stretching vibrations νs(NO2) at 1303–1288 cm−1 are observed. The antisymmetric stretching vibration of the azide moiety of compound 4 is found as the characteristic strong signal at 2148 cm−1.
Single crystal structure analysis
Single crystals suitable for X-ray diffraction measurements were obtained by crystallization at ambient temperature from water (1, 2, 6a, 6b, and 6d), from neat material (4) or from chloroform (8). A full list of the crystallographic refinement parameters and structure data can be found in the ESI.‡The amide 1 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one molecule as asymmetric unit. The density is 1.835 g cm−3 and the molecular structure is shown in Fig. 1. The geometry of the structure has some very typical features of trinitromethyl compounds.1b,c,8 The C–N bond lengths in the trinitromethyl moiety are in the range of 1.54 Å, which is significantly longer than a regular C–N bond (1.47 Å) and results from steric repulsion of the proportionally large nitro groups.1c As expected, the amide unit is nearly planar and shows a shortened C–N bond.
with one molecule as asymmetric unit. The density is 1.835 g cm−3 and the molecular structure is shown in Fig. 1. The geometry of the structure has some very typical features of trinitromethyl compounds.1b,c,8 The C–N bond lengths in the trinitromethyl moiety are in the range of 1.54 Å, which is significantly longer than a regular C–N bond (1.47 Å) and results from steric repulsion of the proportionally large nitro groups.1c As expected, the amide unit is nearly planar and shows a shortened C–N bond.
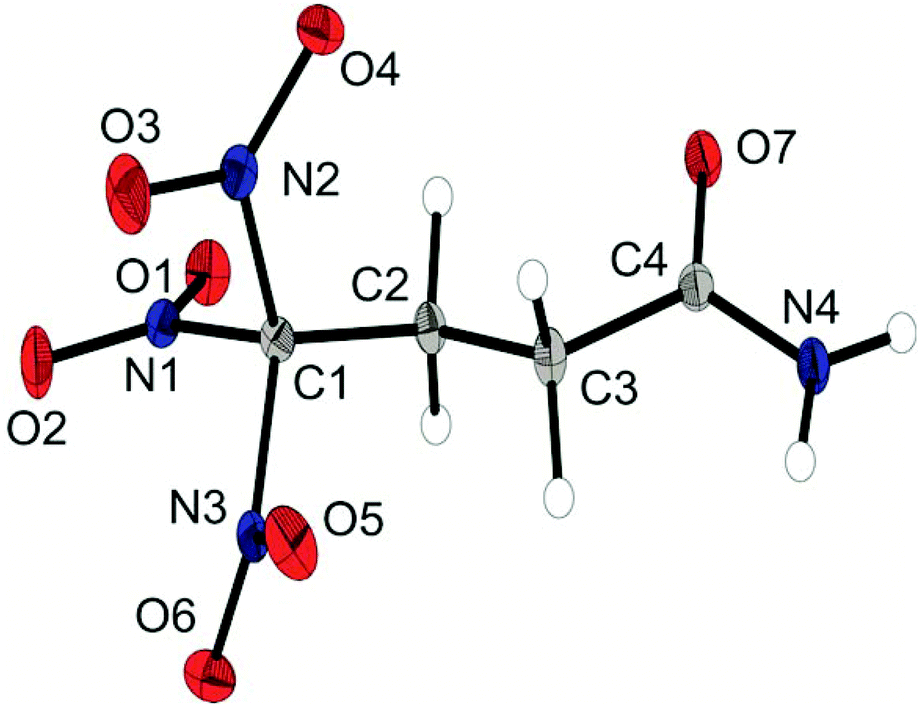 | ||
| Fig. 1 X-ray molecular structure of 4,4,4-trinitrobutanamide (1). Selected atom distances (Å) and angles (°): C1–C2 1.502(2), C1–N1 1.540(2), C1–N2 1.538(2), C1–N3 1.521(1), C2–C3 1.524(2), C3–C4 1.522(2), C4–N4 1.332(2), C4–O7 1.237(1), N1–O1 1.211(1), N4–H5 0.89(2), N4–H6 −0.87(1), C2–C1–N2 114.15(9), C2–C1–N1 112.09(9), C2–C1–N3 110.49(9), H6–N4–C4–C3 −178(1), H5–N4–C4–O7 −177(1), N4–C4–C3–C2 −157.3(1), C3–C2–C1–N1 −175.87(9). | ||
The acid 2 crystallizes in the monoclinic space group P21/n and is shown in Fig. 2. The quite low density of 1.720 g cm−3 can be explained by the strong hydrogen bonds which are formed between two carboxyl moieties with a donor acceptor distance of 2.632 Å (O8–H8⋯O7) and a donor acceptor angle of 176.5° (O8–H8⋯O7).15 In this structure another characteristic structure feature, the propeller-like arrangement of the trinitromethyl group can be observed. The three nitro groups are organized around the carbon in a propeller-like geometry to optimize the non-bonded N⋯O intramolecular attractions (N2⋯O2, O5⋯N1, N3⋯O4). This results in an intramolecular interaction between the partial positive charged nitrogen and the negative charged oxygen in the nitro group. These N⋯O attractions are found with distances in the range of 2.55 Å, which are much shorter than the sum of the van der Waals radii of nitrogen and oxygen (3.07 Å).1c,16
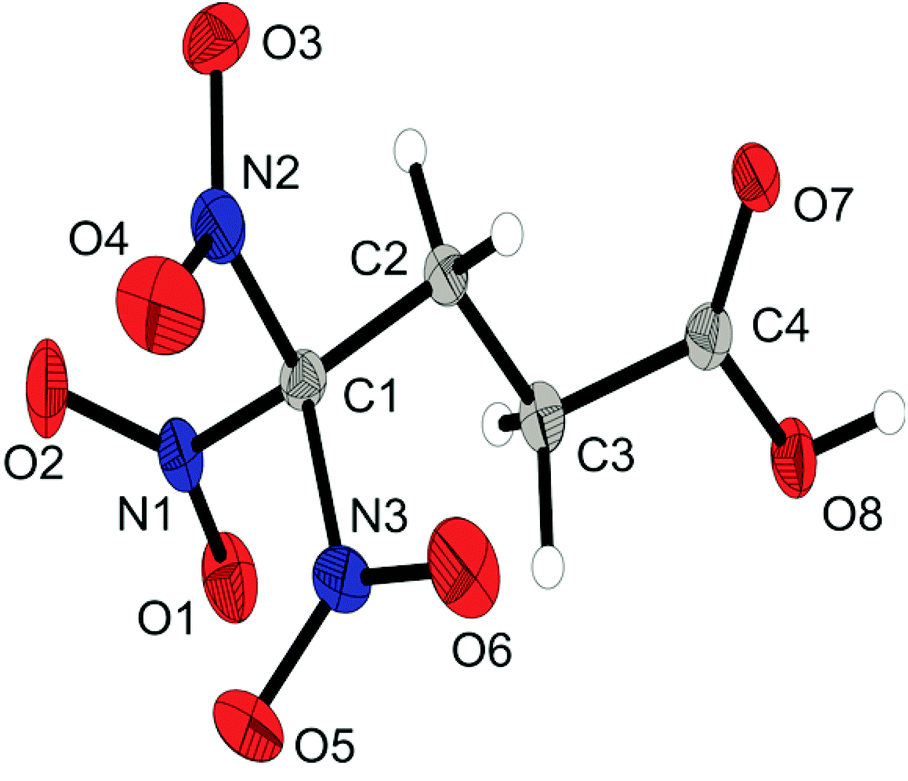 | ||
| Fig. 2 X-ray molecular structure of 4,4,4-trinitrobutanoic acid (2). Selected atom distances (Å) and angles (°): C1–N1 1.523(2), C2–C3 1.528(2), C3–C4 1.509(2), C3–H3 0.99(2), C4–O7 1.218(2), C4–O8 1.311(2), N1–O1 1.216(1), O8–H5 0.86(2), C2–C1–N1 115.2(1), H5–O8–C4–C3 −175(1), O8–C4–C3–C2 179.0(1), C4–C3–C2–C1 −158.4(1), C3–C2–C1–N2 178.3(1), N2–O2 2.557(2), O5–N1 2.571(1), N3–O4 2.550(2). | ||
The carbonyl azide 4 crystallizes in the triclinic space group P with one molecule as an asymmetric unit and shows the propeller-like geometry of the trinitromethyl group. The molecular structure is shown in Fig. 3. The azide, the carbonyl and the carbon backbone inclusively, shows a nearly planar arrangement which is shown by the torsion angle of 1.2(2)° (N5–N4–C4–O7). Typical for carbonyl azides is the slight bending of the azide moiety with an angle of 174.2°. The N4–N5 and N5–N6 bond lengths (1.273(3) and 1.121(3) Å, respectively) are comparable with those in other carbonyl azides.17
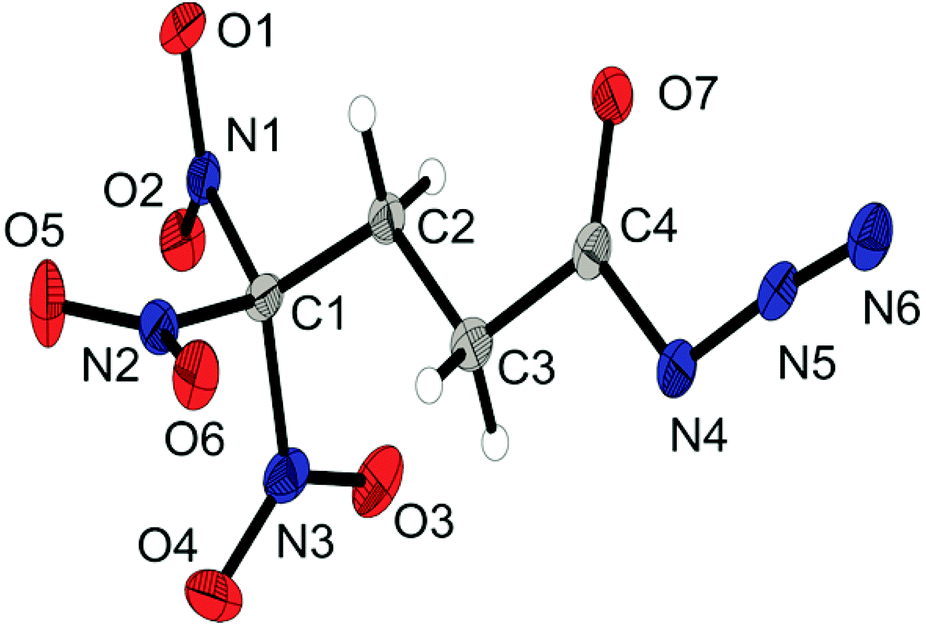 | ||
| Fig. 3 X-ray molecular structure of 4,4,4-trinitrobutanoyl azide (4). Selected atom distances (Å) and angles (°): C1–C2 1.512(2), C1–N1 1.532(2), C2–C3 1.528(3), C3–C4 1.503(2), C4–N4 1.409(2), C4–O7 1.205(2), N4–N5 1.273(2), N5–N6 1.112(2), C2–C1–N3 114.4(1), C4–N4–N5 111.5(1), N4–N5–N6 174.2(2), N6–N5–N4–C4 −176(1), N5–N4–C4–O7 1.2(2), N4–C4–C3–C2 −175.0(1), C4–C3–C2–C1 178.0(1), O2–N2 2.573(2), N1–O5 2.577(2), O4–N3 2.541(1). | ||
The chloride salt 6a crystallizes as a monohydrate in the triclinic space group P and a density of 1.733 g cm−3. The asymmetric unit is shown in Fig. 4. The conformation of the C1, C2, C3 and N4 atoms is almost perfectly staggered. The extended structure involves secondary interactions in terms of classical intermolecular N–H⋯O hydrogen bonds and unusual so-called non-classical hydrogen bonds of the type C–H⋯O; the majority of those can be classified as quite strong.15
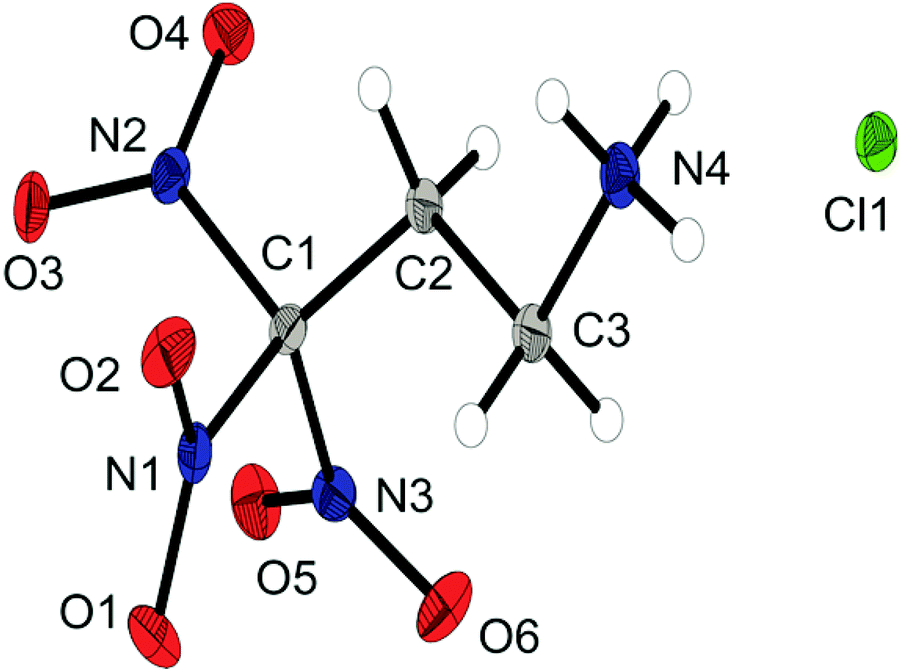 | ||
| Fig. 4 X-ray molecular structure of 3,3,3-trinitropropyl-1-ammonium chloride (6a). Selected atom distances (Å) and angles (°): C1–C2 1.507(2), C1–N1 1.522(2), C2–C3 1.533(2), C3–N4 1.491(2), N1–O1 1.217(1), N4–H6 0.89(2), N4–H7 0.88(2), N4–H8 0.88(2), C2–C1–N3 114.5(1), C3–N4–H7 111(1), C3–N4–H8 107(1), C3–N4–H6 109(1), H7–N4–C3–C2 −178(1), N4–C3–C2–C1 −160.1(1), O5–N2 2.582(2), O1–N3 2.555(2), N1–O3 2.545(2). | ||
The nitrate salt 6b crystallizes in the orthorhombic space group P212121 with a density of 1.804 g cm−3. The asymmetric unit consists of one anion and cation and is illustrated in Fig. 5. The protonated form of the 3,3,3-trinitropropyl-1-amine shows the same structure characteristics as the hydrochloric salt 6a.
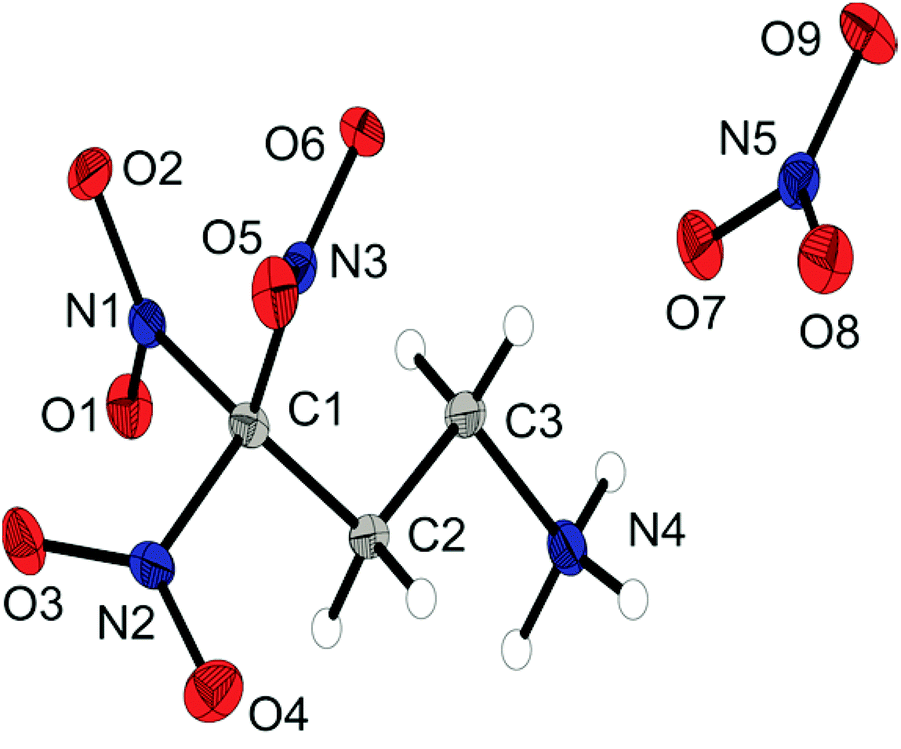 | ||
| Fig. 5 X-ray molecular structure of 3,3,3-trinitropropyl-1-ammonium nitrate (6b). Selected atom distances (Å) and angles (°): C1–C2 1.512(2), C1–N1 1.529(2), C2–C3 1.526(2), C3–N4 1.492(2), N1–O1 1.223(2), N5–O7 1.269(2), N5–O8 1.233(2), N5–O9 1.266(2), N4–C3–C2–C1 −173.7(1), C3–C2–C1–N2 175.8(1), H6–N4–C3–C2 170(1), O8–N5–O7–O9 179.7(3), O5–N2 2.581(2), O2–N3 2.587(2), N1–O3 2.530(2). | ||
The molecular structure of the dinitramide salt 6d is shown in Fig. 6. Compound 6d crystallizes in the monoclinic space group P with two anions and two cations as asymmetric unit and a density of 1.872 g cm−3. The 3,3,3-trinitropropyl-1-ammonium cation shows similar structural features as the ionic structures discussed before. The nitro groups of the dinitramide moiety are slightly twisted out of plane with torsion angles about 20°. The N–N bond lengths with an average distance of 1.37 Å are also slightly shorter than common N–N single bonds.
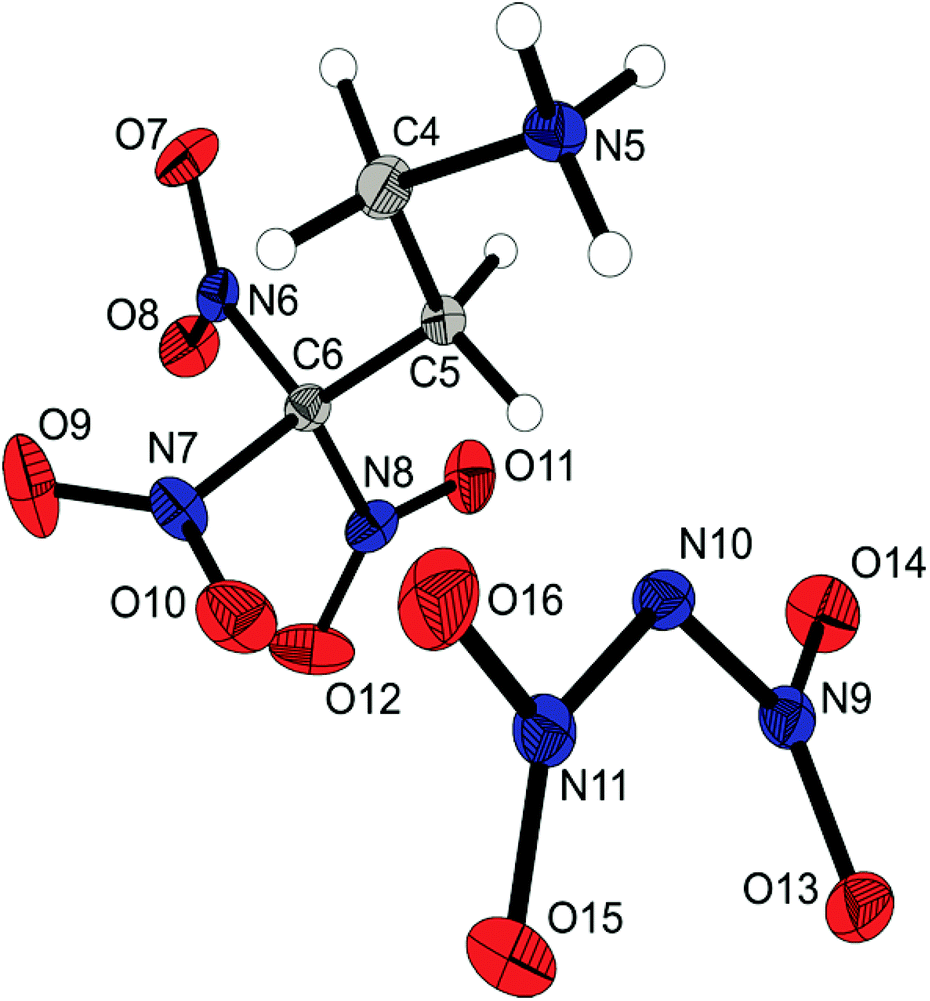 | ||
| Fig. 6 X-ray molecular structure of 3,3,3-trinitropropyl-1-ammonium dinitramide (6d). Selected atom distances (Å) and angles (°): O7–N6 1.216(2), O8–N6 1.209(2), N5–C4 1.484(2), N6–C6 1.529(3), N7–C6 1.523(2), N8–C6 1.528(2), C4–C5 1.530(2), C5–C6 1.505(2), O13–N9 1.220(2), O14–N9 1.239(2), O15–N11 1.233(2), O16–N11 1.243(2), N9–N10 1.380(2), N10–N11 1.357(2), O13–N9–N10 124.2(1), O14–N9–N10 111.7(1), N9–N10–N11 115.4(1), O15–N11–N10 124.7(1), O16–N11–N10 112.8(1), O13–N9–N10–N11 −20.8(2). | ||
The ester 8 crystallizes in the monoclinic space group P21/n with four formula units per unit cell. The asymmetric unit consists of one molecule and is displayed in Fig. 7. The average of the N–O and C–NO2 bond lengths of the trinitromethyl units are all in the same range of 1.21 Å in N–O and 1.52 Å in C–NO2 whereas no distinction between the ethyl and propyl moiety is visible. Also both trinitromethyl groups show independently the propeller-like orientation of the nitro groups. Also the carbon–carbon bonds are virtually identical within a range of 1.50 to 1.52 Å. Although no classical hydrogen bonds are found in the crystal structure a high density of 1.869 g cm−3 was observed. However, non-classical hydrogen bonds of the type C–H⋯O are detected, whereas the majority is classified as quite strong.15
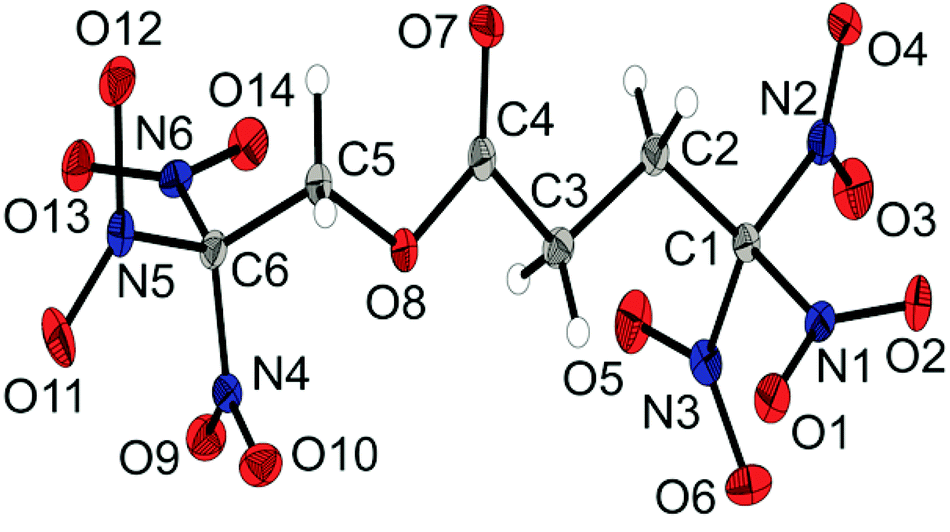 | ||
| Fig. 7 X-ray molecular structure of 2,2,2-trinitroethyl-4,4,4-trinitrobutanoate (8). Selected atom distances (Å) and angles (°): C1–C2 1.515(2), C1–N1 1.528(2), C2–C3 1.514(2), C3–C4 1.497(2), C4–O7 1.200(2), C4–O8 1.363(2), C5–C6 1.520(2), C5–O8 1.424(2), C6–N4 1.525(2), N1–O1 1.222(1), N4–O9 1.213(2), N2–C1–C2–C3 161.5(1), C1–C2–C3–C4 169.4(1), C3–C4–O8–C5 −175.6(1), C4–O8–C5–C6 131.7(1), O8–C5–C6–N5 160.2(1), N3–O3 2.558(2), O6–N1 2.567(2), N2–O2 2.534(1), N4–O11 2.608(2), N5–O13 2.583(2), O9–N6 2.557(1). | ||
Thermal stabilities and energetic properties
Compounds 1, 2, 6a–e, 7, and 8 are stable when exposed to air and moisture. The azide 4 has to be handled very carefully, owning to its high sensitivity towards heat. All manipulations of the isocyanate 5 must be carried out with strict exclusion of moisture. Furthermore it should be stored frozen and is not longtime stable, due to rapid polymerization. The thermal stabilities of all compounds were investigated by performing various DSC measurements with a heating rate of 5 °C min−1. The temperatures at which melting and decomposition occurred, are shown in Table 1 together with other physical properties. A remarkably high decomposition point of 178 °C was observed for compound 6a, likely owing to its stability to form strong hydrogen bonds through the salt structure. Moreover, compounds 7 and 8 (both 155 °C) showed satisfying decomposition points for potential applications as high-energy dense oxidizers based on CHNO compounds. The sensitivities of compounds 2–8 towards impact, friction, and electrostatic discharge were experimentally determined according to the NATO Standardization Agreements;18 the results are displayed in Table 1. All compounds, with exception of the azide 4, the dinitramide salt 6d, and the 5,5′-azobistetrazolate salt 6e showed moderate impact and friction sensitivities.19 For the amide 1 as well as the nitrate salt 6b impact sensitivities of 6 J are found, which are in the range of the well-known explosive Hexogen (RDX).| 1 | 2 | 4 | 6a | 6b | 6c | 6d | 6e | 7 | 8 | AP | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Densities at RT measured by gas pycnometer. b Onset melting point Tm from DSC measurement carried out at a heating rate of 5 °C min−1. c Onset decomposition point Tdec from DSC measurement carried out at a heating rate of 5 °C min−1. d Impact sensitivity. e Friction sensitivity. f Sensitivity toward electrostatic discharge. g Nitrogen content. h Oxygen content. i Sum of nitrogen and oxygen content. j Oxygen balance assuming the formation of CO at the combustion. k Oxygen balance assuming the formation of CO2 at the combustion. l Enthalpy of formation calculated by the CBS-4 M method using Gaussian 09. m Energy of formation calculated by the CBS-4 M method using Gaussian 09. | |||||||||||
| Formula | C4H6N4O7 | C4H5N3O8 | C4H4N6O7 | C3H7N4O6Cl | C3H7N5O9 | C3H7N4O10Cl | C3H7N7O10 | C8H14N18O12 | C6H7N7O13 | C6H6N6O14 | NH4ClO4 |
| MW/g mol−1 | 222.11 | 223.10 | 248.11 | 230.56 | 257.12 | 294.56 | 301.13 | 554.31 | 385.16 | 386.14 | 117.49 |
| Densitya/g cm−3 | 1.78 | 1.67 | 1.71 | 1.76 | 1.77 | 1.97 | 1.84 | 1.67 | 1.84 | 1.83 | 1.95 |
T
m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b/°C b/°C |
93 | 55 | 22 | 161 | 135 | — | — | — | 150 | 92 | — |
|
T
decc/°C |
120 | 176 | 85 | 178 | 138 | 164 | 112 | 120 | 155 | 155 | 240 |
| ISd/J | 6 | 40 | 2 | 20 | 6 | 2.5 | 2 | 2 | 10 | 30 | 15 |
| FSe/N | 360 | 324 | 144 | >360 | 120 | 16 | 30 | 54 | 240 | 240 | >360 |
| ESDf/J | 0.50 | 0.30 | 0.10 | 0.40 | 0.20 | 0.08 | 0.40 | 0.60 | 0.20 | 0.10 | >1.50 |
| Ng/% | 25.2 | 18.8 | 33.9 | 24.3 | 27.2 | 19.0 | 32.6 | 54.6 | 25.5 | 21.8 | 11.9 |
| Oh/% | 50.4 | 57.4 | 45.1 | 41.6 | 56.0 | 54.3 | 53.1 | 26.7 | 54.0 | 58.0 | 54.5 |
| N + Oi/% | 75.6 | 76.2 | 79.0 | 65.9 | 83.2 | 73.3 | 85.7 | 81.3 | 79.5 | 79.8 | 66.4 |
|
Ω
COj/% |
0.0 | +10.1 | +6.5 | 0.0 | +15.6 | +21.7 | +18.6 | −11.1 | +14.5 | +20.7 | +34.6 |
|
Ω
CO2k/% |
−28.1 | −17.9 | −19.4 | −2.4 | −3.1 | +5.4 | +2.7 | −33.4 | −10.4 | −4.1 | +34.6 |
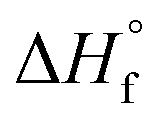 /kJ mol−1
/kJ mol−1 |
−326 | −506 | 54 | −96 | −169 | −119 | 32 | 972 | −330 | −466 | −296 |
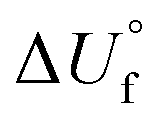 /kJ kg−1
/kJ kg−1 |
−1374 | −2178 | 301 | −318 | −554 | −312 | 205 | 1851 | −770 | −1124 | −2433 |
Predictions of the detonation and combustion parameters by using the EXPLO5 V6.0220 code have been performed based on the heats of formations which were obtained from ab initio calculations. The energetic parameters were calculated with the room temperature densities, which were measured experimentally by gas pycnometer. The resulting heats of detonation Qv, detonation temperatures Tex, detonation pressures p, and detonation velocities Vdet for compounds 1, 2, 4, and 6–8 are shown in Table 2. The dinitramide salt 6d has the highest detonation parameters with a detonation velocity Vdet of 9282 m s−1 and a detonation pressure of 372 kbar and exceeds the high energetic military explosive RDX (8838 m s−1) by far.21
| 1 | 2 | 4 | 6a | 6b | 6c | 6d | 6e | 7 | 8 | AP | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Heat of detonation. b Detonation temperature. c Volume of gaseous products. d Detonation pressure. e Detonation velocity calculated by using the EXPLO5 (version 6.02) program package.20a f Specific impulse of the neat compound using the EXPLO5 (version 6.02) program package20a (70.0 bar chamber pressure, isobaric combustion conditions (1 bar), equilibrium expansion).20a g Specific impulse for compositions with 85% oxidizer/compound and 15% aluminum (70.0 bar chamber pressure, isobaric combustion conditions (1 bar), equilibrium expansion). h Specific impulse for compositions with 71% oxidizer/compound, 15% aluminum, and 14% binder (6% polybutadiene acrylic acid, 6% polybutadiene acrylonitrile and 2% bisphenol A ether) (70.0 bar chamber pressure, isobaric combustion conditions (1 bar), equilibrium expansion). | |||||||||||
| Formula | C4H6N4O7 | C4H5N3O8 | C4H4N6O7 | C3H7N4O6Cl | C3H7N5O9 | C3H7N4O10Cl | C3H7N7O10 | C8H14N18O12 | C6H7N7O13 | C6H6N6O14 | NH4ClO4 |
|
Q
va/kJ kg−1 |
−4956 | −4786 | −5607 | −5281 | −6697 | −6250 | −6671 | −6212 | −5820 | −6121 | −1422 |
|
T
exb/K |
3383 | 3505 | 4071 | 3793 | 4319 | 4309 | 4382 | 4141 | 4009 | 4277 | 1735 |
|
V
0c/L kg−1 |
733 | 731 | 759 | 744 | 821 | 787 | 828 | 814 | 718 | 719 | 885 |
|
P
CJd/kbar |
292 | 246 | 291 | 282 | 335 | 379 | 372 | 299 | 335 | 324 | 158 |
|
V
dete/m s−1 |
8187 | 7624 | 8259 | 8019 | 8913 | 9096 | 9282 | 8541 | 8628 | 8616 | 6368 |
|
I
spf/s |
238 | 241 | 261 | 255 | 274 | 265 | 274 | 271 | 262 | 258 | 157 |
| I sp/s (15% Al)g | 261 | 256 | 270 | 269 | 278 | 270 | 278 | 276 | 267 | 263 | 235 |
| I sp/s (15% Al, 14% binder)h | 244 | 241 | 247 | 247 | 270 | 272 | 275 | 254 | 253 | 255 | 261 |
The specific impulses Isp of compounds 1, 2, 4, and 6–8 were calculated for the neat compounds, for compositions with different amounts of aluminum as fuel, and additional with binder and are also listed in Table 2 (further values are summarized in the ESI‡). These impulses were compared with the calculated impulses of ammonium perchlorate (AP) in an analogous composition. The chosen mixture with AP as an oxidizer provided a specific impulse of 261 s. All compounds show good properties, especially when calculated without binder. The value for the specific impulse of the 5,5′-azobistetrazolate salt 6e exceeds all others; for the neat compound it is calculated to 271 s, with an admixture of 10% aluminum as fuel 282 s could be achieved (see ESI‡). For the nitrate and dinitramide salts 6b and 6d remarkable high specific impulses of 278 s were reached in compositions containing 85% oxidizer and 15% fuel. In composites containing oxidizer, fuel and binder the specific impulses decrease slightly. The best specific impulse is obtained for the dinitramide salt 6d with a calculated value of 275 s in a composite propellant consisting of 15% aluminum and 14% binder. However, also the specific impulses of the nitrate and perchlorate salts 6b and 6c with values of 270 (6b) and 272 s (6c) exceed the specific impulse of the standard optimized mixture of AP (261 s).
Conclusions
Based on the Michael addition of nitroform with acrylamide several energetic polynitro compounds with a positive oxygen balance were synthesized. Although several synthesis steps are needed for most compounds presented herein, only common commercially available chemicals are used and syntheses proceed in high yields. All compounds were comprehensively characterized. Several salts containing the 3,3,3-trinitropropylammonium cation were investigated in terms of their energetic properties. Excellent detonation parameters were found for the 3,3,3-trinitropropylammonium dinitramide 6d with a detonation velocity of 9282 m s−1 and a detonation pressure of 372 kbar. These values are significantly higher than those of TNT, RDX, and PETN.21 With respect to an application as high-energy dense oxidizer in composite solid rocket propellants, the best value was obtained for the corresponding 5,5′-bisazotetrazolate salt 6e; in a mixture comprised of 90% oxidizer and 10% fuel a calculated specific impulse of 282 s was reached. In composites consisting of oxidizer, fuel and binder best values were obtained for the nitrate salt 6b (270 s), the perchlorate salt 6c (272 s) and the dinitramide salt 6d (275 s). All of these exceed the specific impulse of AP in a similar composition (261 s). However, the perchlorate salt 6c, the dinitramide salt 6d, and the 5,5′-bisazotetrazolate salt 6e show low thermal stabilities and/or high sensitivities to external stimuli, and therefore likely will be less considered for practical use.Experimental section
General information
Chemicals were used as supplied (Sigma-Aldrich, Fluka, Acros Organics). Raman spectra were recorded in a glass tube with a Bruker MultiRAM FT-Raman spectrometer with Nd:YAG laser excitation up to 1000 mW (at 1064 nm) in the range between 400 and 4000 cm−1. Infrared spectra were measured with a Perkin-Elmer Spectrum BX-FTIR spectrometer equipped with a Smiths DuraSamplIR II ATR device. All spectra were recorded at ambient (20 °C) temperature. NMR spectra were recorded with JEOL Eclipse 400 instrument and Bruker AV400 and chemical shifts were determined with respect to external standards Me4Si (1H, 399.8 MHz; 13C, 100.5 MHz), MeNO2 (14N, 28.9 MHz; 15N 40.6 MHz), and 1.0 M aqueous NaCl (35Cl, 39.2 MHz). Mass spectrometric data were obtained with a JEOL MStation JMS 700 spectrometer (DCI+, DEI+). Analysis of C/H/N were performed with an Elemental Vario EL Analyzer. Melting and decomposition points were measured with a Perkin-Elmer Pyris6 DSC and an OZM Research DTA 552-Ex with a heating rate of 5 °C min−1 in a temperature range of 15 to 400 °C and checked by a Büchi Melting Point B-540 apparatus (not corrected).X-ray crystallography
The low-temperature single-crystal X-ray diffraction of compounds 1, 2, 4, 6a, 6b, 6d, and 8 were performed on an Oxford XCalibur3 diffractometer equipped with a Spellman generator (voltage 50 kV, current 40 mA) and a Kappa CCD detector operating with MoKα radiation (λ = 0.7107 Å). Data collection was performed using the CRYSALIS CCD software.22 The data reduction was carried out using the CRYSALIS RED software.23 The solution of the structure was performed by direct methods (SIR97)24 and refined by full-matrix least-squares on F2 (SHELXL)25 implemented in the WINGX software package26 and finally checked with the PLATON software.27 All non-hydrogen atoms were refined anisotropically. The hydrogen atom positions were located in a difference Fourier map. ORTEP plots are shown with thermal ellipsoids at the 50% probability level. Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited at the Cambridge Crystallographic Data Centre 1506284–1506290 (1, 2, 4, 6a, 6b, 6d, and 8). Additional crystallographic data and structure refinement parameters are listed in the ESI.‡Computational details
All ab initio calculations were carried out using the program package Gaussian 09 (Rev. A.03)28 and visualized by GaussView 5.08.29 The initial geometries of the structures were taken from the corresponding experimentally determined crystal structures. Structure optimizations and frequency analyses were performed with Becke's B3 three parameter hybrid functional using the LYP correlation functional (B3LYP). For C, H, N and O a correlation consistent polarized double-ξ basis set was used (cc-pVDZ). The structures were optimized with symmetry constraints and the energy is corrected with the zero point vibrational energy.30 The enthalpies (H) and free energies (G) were calculated using the complete basis set (CBS) method in order to obtain accurate values. The CBS models use the known asymptotic convergence of pair natural orbital expressions to extrapolate from calculations using a finite basis set to the estimated complete basis set limit. CBS-4 starts with a HF/3-21G(d) geometry optimization, which is the initial guess for the following SCF calculation as a base energy and a final MP2/6-31+G calculation with a CBS extrapolation to correct the energy in second order. The used CBS-4 M method additionally implements a MP4(SDQ)/6-31+(d,p) calculation to approximate higher order contributions and also includes some additional empirical corrections.31 The enthalpies of the gas-phase species were estimated according to the atomization energy method.32 The liquid (solid) state energies of formation
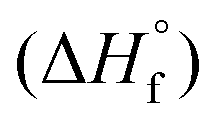 were estimated by subtracting the gas-phase enthalpies with the corresponding enthalpy of vaporization (sublimation) obtained by Trouton's rule.33 All calculations affecting the detonation parameters were carried out using the program package EXPLO5 V6.02 (EOS BKWG-S).20 The detonation parameters were calculated at the Chapman–Jouguet (CJ) point with the aid of the steady-state detonation model using a modified Becker–Kistiakowski–Wilson equation of state for modeling the system. The CJ point is found from the Hugoniot curve of the system by its first derivative. The specific impulses Isp were also calculated with the program package EXPLO5 V6.02 program, assuming an isobaric combustion of a composition of an oxidizer, aluminum as fuel, 6% polybutadiene acrylic acid, 6% polybutadiene acrylonitrile as binder and 2% bisphenol A as epoxy curing agent.20a A chamber pressure of 70.0 bar, an initial temperature of 3300 K and an ambient pressure of 1.0 bar with equilibrium expansion conditions were estimated for the calculations.
were estimated by subtracting the gas-phase enthalpies with the corresponding enthalpy of vaporization (sublimation) obtained by Trouton's rule.33 All calculations affecting the detonation parameters were carried out using the program package EXPLO5 V6.02 (EOS BKWG-S).20 The detonation parameters were calculated at the Chapman–Jouguet (CJ) point with the aid of the steady-state detonation model using a modified Becker–Kistiakowski–Wilson equation of state for modeling the system. The CJ point is found from the Hugoniot curve of the system by its first derivative. The specific impulses Isp were also calculated with the program package EXPLO5 V6.02 program, assuming an isobaric combustion of a composition of an oxidizer, aluminum as fuel, 6% polybutadiene acrylic acid, 6% polybutadiene acrylonitrile as binder and 2% bisphenol A as epoxy curing agent.20a A chamber pressure of 70.0 bar, an initial temperature of 3300 K and an ambient pressure of 1.0 bar with equilibrium expansion conditions were estimated for the calculations.
Synthesis
CAUTION! All prepared compounds are energetic materials with sensitivity towards heat, impact, and friction. No hazards occurred during the preparation and manipulation. However, additional proper protective precautions (face shield, leather coat, earthened equipment and shoes, Kevlar® gloves, and ear plugs) should be used when undertaking work with these compounds.4,4,4-Trinitrobutanamide (1)
An aqueous solution of nitroform (30%, 22.6 g, 45 mmol) was cooled in an ice-bath and acrylamide (3.2 g, 45 mmol) was added. The mixture was stirred 10 minutes at this temperature and 5 h at ambient temperature. The formed precipitate was filtered off and washed several times with cold ethanol and diethyl ether. After drying on air pure 4,4,4-trinitrobutanamide (1) was obtained as colourless solid in 97% yield. DSC (5 °C min−1): 93 °C (mp.), 120 °C (dec.). IR (ATR): ν = 3475 (m), 3368 (w), 3310 (w), 3192 (w), 3010 (w), 2948 (w), 2360 (w), 2340 (w), 1695 (m), 1595 (s), 1567 (vs), 1418 (m), 1364 (w), 1344 (m), 1311 (m), 1299 (m), 1288 (s), 1217 (w), 1155 (w), 1116 (w), 878 (w), 856 (w), 816 (m), 798 (s), 776 (w), 747 (w), 637 (w) cm−1. Raman (500 mW): ν = 3009 (41), 2969 (24), 2938 (78), 1679 (17), 1615 (29), 1599 (43), 1575 (11), 1433 (16), 1417 (43), 1367 (43), 1348 (26), 1307 (31), 1125 (24), 1066 (13), 1055 (14), 967 (13), 904 (12), 881 (43), 858 (60), 811 (20), 546 (18), 441 (71), 390 (100), 364 (73), 312 (69), 274 (17), 208 (66) cm−1. 1H NMR ([D6]DMSO) δ = 7.48 (s, 1H, NH2), 7.09 (s, 1H, NH2), 3.59 (m, 2H, CH2C(NO2)3), 2.52 (m, 2H, OCCH2) ppm. 13C NMR ([D6]DMSO) δ = 170.7 (CO), 131.7 (C(NO2)3), 29.1 (CH2), 29.0 (CH2) ppm. 14N NMR ([D6]DMSO) δ = −28 (C(NO2)3) ppm. MS (DEI+) m/z: 223.2 [(M + H)+]. Elemental analysis C4H6N4O7 (222.11): calc. C 21.63, H 2.72, N 25.22%; found C 21.65, H 2.65, N 25.05%. IS: 6 J (grain size 250–500 μm). FS: 360 N (grain size 250–500 μm). ESD: >0.5 J (grain size 250–500 μm).4,4,4-Trinitrobutanoic acid (2)
4,4,4-Trinitrobutanamide (1) (2.0 g, 9.0 mmol) was added to concentrated hydrochloric acid (37%, 8 mL) and refluxed for 4 hours. The oily layer which formed solidified after standing overnight at 4 °C. The solid was filtered off and recrystallized from chloroform. After drying in the desiccator 4,4,4-trinitrobutanoic acid (2) was obtain as pure colourless product in 70% yield. DSC (5 °C min−1): 55 °C (mp.), 167 °C (dec.). IR (ATR): ν = 3006 (w), 2958 (w), 2880 (w), 2730 (w), 2651 (w), 2527 (w), 1709 (s), 1587 (vs), 1440 (m), 1425 (m), 1312 (s), 1297 (s), 1237 (s), 1153 (m), 1070 (m), 928 (m), 906 (m), 816 (s), 798 (vs), 665 (m) cm−1. Raman (500 mW): ν = 3006 (8), 2987 (11), 2956 (89), 1652 (10), 1605 (33), 1454 (9), 1418 (46), 1380 (25), 1359 (22), 1312 (31), 1226 (8), 1154 (13), 1071 (21), 982 (21), 908 (42), 857 (101), 802 (8), 655 (11), 628 (9), 546 (7), 484 (11), 412 (56), 402 (58), 377 (91), 313 (35), 275 (9) cm−1. 1H NMR ([D6]acetone) δ = 3.71 (m, 2H, CH2C(NO2)3), 2.89 (m, 2H, OCCH2) ppm. 13C NMR ([D6]acetone) δ = 170.4 (CO), 126.3 (C(NO2)3), 29.2 (CH2), 27.6 (CH2) ppm. 14N NMR ([D6]acetone) δ = −29 (C(NO2)3) ppm. MS (DCI+) m/z: 224.1 [(M + H)+]. Elemental analysis C4H5N3O8 (223.10): calc. C 21.53, H 2.26, N 18.83%; found C 21.39, H 2.24, N 18.70%. IS: 40 J (grain size 250–500 μm). Friction tester: 324 N (grain size 250–500 μm). ESD: >0.5 J (grain size 250–500 μm).4,4,4-Trinitrobutanoyl chloride (3)
Method AA mixture of 4,4,4-trinitrobutanoic acid (2) (6.7 g, 30.0 mmol) and thionyl chloride (16.7 mL, 200 mol) was stirred at room temperature for one hour. After this the reaction mixture was refluxed for 24 hours under exclusion of moisture. The excess of thionyl chloride was removed and the remaining oil was distilled (bp. 65 °C, 0.7 mbar) yielding 4,4,4-trinitrobutanoyl chloride as colourless pure product (88%).
Method B
Oxalyl chloride (326 mg, 2.6 mmol) and a catalytical amount of DMF were added to a suspension of 4,4,4-trinitrobutanoic acid (2) (500 mg, 2.2 mmol) in chloroform (10 mL). The reaction mixture was stirred under exclusion of moisture at ambient temperature for 40 min and was refluxed for 3 h. The solvent was removed under reduced pressure yielding 4,4,4-trinitrobutanoyl chloride (3) in 96% yield as pure colourless oil. IR (ATR): ν = 2997 (w), 2957 (w), 2892 (w), 1785 (s), 1585 (vs), 1425 (m), 1411 (w), 1356 (w), 1294 (s), 1216 (w), 1153 (w), 1062 (w), 996 (m), 943 (s), 857 (m), 799 (s), 780 (s), 693 (m) cm−1. Raman (400 mW): ν = 2950 (51), 1792 (16), 1608 (25), 1414 (24), 1381 (22), 1358 (35), 1304 (30), 1224 (11), 1155 (15), 1065 (26), 998 (13), 948 (15), 905 (17), 858 (102), 784 (21), 694 (19), 635 (17), 532 (20), 456 (58), 396 (44), 374 (65), 275 (50), 233 (31) cm−1. 1H NMR (CDCl3) δ = 3.38 (m, 4H, OCCH2, CH2C(NO2)3) ppm. 13C NMR (CDCl3) δ = 171.1 (CO), 127.9 (C(NO2)3), 40.5 (OCCH2), 29.4 (CH2(NO2)3) ppm. 14N NMR (CDCl3) δ = −31 (C(NO2)3) ppm. MS (DEI+) m/z: 206.1 [(M − Cl)+]. Elemental analysis C4H4N3O7Cl (241.54): calc. C 19.89, H 1.67, N 17.40%; found C 19.75, H 1.68, N, 17.80%.
4,4,4-Trinitrobutanoyl azide (4)
To a solution of sodium azide (0.31 g, 4.8 mmol) in water (2 mL) a solution of 4,4,4-trinitrobutanoyl chloride (3) (0.59 g, 2.4 mmol) in acetone (1 mL) was added slowly at 4 °C. After the addition the solution was stirred at 0 °C for two hours. The reaction mixture was extracted with chloroform (3 × 20 mL). The combined organic phases were washed with ice-water (20 mL), an ice-cold sodium bisulfate solution (5%, 20 mL), ice-water (2 × 20 mL) and brine (20 mL). The extracts were dried over magnesium sulfate and the organic solvent was removed at temperatures below 20 °C. The remaining oil solidified in the refrigerator over-night and 4,4,4-trinitrobutanoyl azide (4) was obtained as pure colourless solid in 66% yield. DSC (5 °C min−1): 22 °C (mp.), 85 °C (dec.). IR (ATR): ν = 3000 (w), 2956 (w), 2893 (w), 2148 (s), 1711 (s), 1585 (vs), 1427 (m), 1359 (m), 1296 (s), 1153 (vs), 1098 (s), 1047 (s), 967 (w), 908 (w), 855 (s), 800 (vs), 704 (m) cm−1. Raman (500 mW): ν = 2947 (71), 2156 (26), 2147 (26), 1716 (22), 1608 (26), 1419 (26), 1360 (36), 1305 (30), 1151 (9), 1101 (16), 1050 (14), 968 (12), 910 (31), 857 (101), 788 (10), 669 (26), 543 (9), 502 (29), 373 (69), 279 (39), 262 (36) cm−1. 1H NMR (CDCl3) δ = 3.38 (m, 2H, CH2C(NO2)3), 2.78 (m, 2H, OCCH2) ppm. 13C NMR (CDCl3) δ = 176.2 (CO), 128.6 (C(NO2)3), 30.6 (CH2), 29.3 (CH2) ppm. 14N NMR (CDCl3) δ = −30 (C(NO2)3), −136 (Nβ), −147 (Nγ) ppm. Elemental analysis C4H4N6O7 (248.11): calc. C 19.36, H 1.62, N 33.87%; found C 19.89, H 1.65, N 33.54%. IS: 2 J (grain size 250–500 μm). FS: 144 N (grain size 250–500 μm). ESD 0.3 J (grain size 250–500 μm).1,1,1-Trinitropropan-3-isocyanate (5)
To a solution of sodium azide (0.31 g, 4.8 mmol) in water (2 mL) a solution of 4,4,4-trinitrobutanoyl chloride (3) (0.59 g, 2.4 mmol) in acetone (1 mL) was added slowly at 4 °C. After the addition the solution was stirred at 0 °C for two hours. The reaction mixture was extracted with chloroform (3 × 20 mL). The combined organic phases were washed with ice-water (20 mL), an ice-cold sodium bisulfate solution (5%, 20 mL), ice-water (2 × 20 mL) and brine (20 mL). The extracts were dried over magnesium sulfate. The solution was slowly heated up to 55 °C and kept at this temperature until no more nitrogen evolved (2 h). The organic solvent was removed to obtain 1,1,1-trinitropropan-3-isocyanate (5) as colourless liquid in 68% yield. Raman (500 mW): ν = 2953 (70), 2156 (13), 2147 (13), 1718 (11), 1610 (24), 1451 (18), 1421 (22), 1363 (37), 1304 (30), 1100 (10), 1051 (13), 914 (20), 887 (12), 856 (100), 811 (10), 535 (10), 502 (16), 460 (10), 375 (68), 305 (17), 279 (22), 254 (20) cm−1. 1H NMR (CDCl3) δ = 3.90 (m, 2H, CH2), 3.32 (m, 2H, CH2) ppm. 13C NMR (CDCl3) δ = 127.4 (C(NO2)3), 123.6 (NCO), 37.4 (CH2), 35.0 (CH2) ppm. 14N NMR (CDCl3) δ = −31 (C(NO2)3), −360 (NCO) ppm. Elemental analysis C4H4N4O7 (220.10): calc. C 21.83, H 1.83, N 25.46%; found C 21.31, H 1.80, N 26.07%.3,3,3-Trinitropropyl-1-ammonium chloride (6a)
1,1,1-Trinitropropan-3-isocyanate (5) (1.10 g, 5.0 mmol) was refluxed in hydrochloric acid (6 M, 10 mL) for five hours. The solution was concentrated to dryness and the colourless solid was washed with 1,2-dichloroethane. 3,3,3-Trinitropropyl-1-ammonium chloride (6a) was yielded as colourless solid in 90% yield. DSC (5 °C min−1): 161 °C (mp.), 178 °C (dec.). IR (ATR): ν = 2974 (m), 2884 (m), 2660 (w), 2497 (w), 2305 (w), 1989 (w), 1588 (vs), 1501 (m), 1483 (m), 1458 (m), 1417 (w), 1365 (w), 1292 (s), 1160 (m), 1062 (w), 1032 (w), 995 (w), 931 (w), 911 (w), 855 (w), 846 (w), 836 (w), 796 (s), 768 (w), 734 (w) cm−1. RAMAN (300 mW): ν = 3068 (11), 3022 (18), 2998 (40), 2976 (58), 2938 (68), 2913 (25), 2904 (24), 2878 (18), 2859 (44), 2804 (12), 2083 (7), 1610 (37), 1580 (8), 1548 (17), 1491 (10), 1478 (9), 1460 (17), 1423 (21), 1396 (7), 1367 (39), 1300 (33), 1168 (19), 1119 (6), 1065 (8), 1031 (9), 999 (15), 970 (9), 931 (8), 901 (16), 858 (100), 802 (8), 654 (6), 633 (7), 569 (9), 516 (6), 457 (9), 402 (50), 374 (56), 341 (29), 302 (7) cm−1. 1H NMR ([D6]DMSO) δ = 8.63 (br, 3H, NH3), 3.82 (m, 2H, CH2), 3.19 (m, 2H, CH2) ppm. 13C NMR ([D6]DMSO) δ = 128.8 (C(NO2)3), 33.4 (CH2), 30.3 (CH2) ppm. 14N NMR ([D6]DMSO) δ = –31 (NO2), −356 (NH3) ppm. Elemental analysis C3H9N4O7 (230.56): calc. C 15.63, H 3.06, N 24.30%; found C 16.09, H 3.06, N 24.30%. IS: 20 J (grain size 100–250 μm). FS: 360 N (grain size 100–250 μm). ESD >0.5 J (grain size 100–250 μm).3,3,3-Trinitropropyl-1-ammonium nitrate (6b)
1,1,1-Trinitropropan-3-isocyanate (5) (1.10 g, 5.0 mmol) was refluxed in nitric acid (6 M, 10 mL) for five hours. The solution was concentrated to dryness to give a yellow powder. Recrystallization from ethyl acetate yielded 3,3,3-trinitropropyl-1-ammonium nitrate (6b) as colourless solid in 89% yield. DSC (5 °C min−1): 135 °C (mp.), 138 °C (dec.). IR (ATR): ν = 3120 (m), 3070 (m), 3032 (m), 2977 (m), 2889 (m), 2840 (m), 2763 (w), 2716 (w), 2666 (w), 2588 (w), 2503 (w), 1604 (s), 1506 (w), 1479 (w), 1460 (m), 1425 (w), 1303 (m), 1040 (w), 996 (w), 972 (w), 934 (w), 875 (w), 850 (w), 806 (m), 799 (w), 766 (w), 735 (w), 680 (w) cm−1. RAMAN (1000 mW): ν = 3037 (12), 2984 (47), 3948 (64), 2913 (12), 2859 (52), 2836 (5), 2817 (6), 2083 (11), 2028 (5), 1609 (25), 1465 (13), 1424 (22), 1373 (38), 1305 (27), 1186 (10), 1155 (9), 1035 (99), 1010 (10), 935 (7), 908 (15), 859 (102), 800 (7), 727 (9), 711 (6), 661 (5), 630 (8), 563 (9), 538 (7), 419 (41), 404 (44), 378 (50), 340 (24), 306 (10) cm−1. 1H NMR ([D6]DMSO) δ = 8.10 (br, 3H, NH3), 3.71 (m, 2H, CH2), 3.24 (m, 2H, CH2) ppm. 13C NMR ([D6]DMSO) δ = 128.8 (C(NO2)3), 33.5 (CH2), 30.6 (CH2) ppm. 14N NMR ([D6]DMSO) δ = −4 (NO3−), −30 (NO2), −359 (NH3) ppm. Elemental analysis C3H7N5O9 (257.12): calc. C 14.01, H 2.74, N 27.24%; found C 13.89, H 2.76, N 27.01%. IS: 6 J (grain size 250–500 μm). FS: 120 N (grain size 250–500 μm). ESD 0.3 J (grain size 250–500 μm).3,3,3-Trinitropropyl-1-ammonium perchlorate (6c)
To a solution of 3,3,3-trinitropropyl-1-ammonium chloride (6a) (196 mg, 0.9 mmol) in water (10 mL) was added at 0 °C under exclusion of light a solution of silver perchlorate monohydrate (190 mg, 0.9 mmol) in water (10 mL). The reaction mixture was stirred 1.5 h at 0 °C. The precipitated silver chloride was filtered off, washed with cold water and the filtrate was evaporated to dryness. 3,3,3-Trinitropropyl-1-ammonium perchlorate (6c) was obtained as colourless solid in 90% yield. DSC (5 °C min−1): 164 °C (dec.). IR (ATR): ν = 3259 (w), 3227 (w), 3168 (w), 2992 (w), 2888 (w), 2361 (w), 2333 (w), 1596 (s), 1513 (w), 1501 (w), 1478 (m), 1430 (w), 1368 (w), 1293 (m), 1152 (m), 1067 (vs), 980 (m), 941 (w), 893 (w), 855 (w), 798 (s), 749 (w), 667 (w) cm−1. RAMAN (1000 mW): ν = 3258 (7), 3241 (6), 3214 (6), 3189 (5), 3143 (5), 3133 (6), 3119 (5), 3067 (4), 3028 (9), 2994 (17), 2960 (29), 2821 (6), 1863 (5), 1601 (39), 1467 (10), 1431 (18), 1370 (25), 1356 (15), 1343 (9), 1302 (21), 1162 (16), 1115 (8), 1078 (12), 1019 (17), 982 (14), 942 (101), 858 (88), 803 (10), 664 (5), 634 (5), 628 (25), 563 (13), 467 (25), 453 (22), 417 (38), 405 (38), 382 (49), 341 (32), 300 (8) cm−1. 1H NMR ([D6]DMSO) δ = 8.03 (br, 3H, NH3), 3.70 (m, 2H, CH2), 3.23 (m, 2H, CH2) ppm. 13C NMR ([D6]DMSO) δ = 128.8 (C(NO2)3), 33.5 (CH2), 30.4 (CH2) ppm. 14N NMR ([D6]DMSO) δ = –31 (NO2), −356 (NH3) ppm. 35Cl NMR ([D4]methanol) = −1011 (ClO4−) ppm. Elemental analysis C3H7N4O6Cl (294.56): calc. C 12.23, H 2.40, N 19.02%; found C 12.25, H 2.57, N 18.44%. IS: 2.5 J (grain size <100 μm). FS: 16 N (grain size <100 μm). ESD 0.08 J (grain size <100 μm).3,3,3-Trinitropropyl-1-ammonium dinitramide (6d)
To a solution of 3,3,3-trinitropropyl-1-ammonium chloride (6a) (350 mg, 1.5 mmol) in water (10 mL) was added at 0 °C under exclusion of light a solution of silver dinitramide (320 mg, 1.5 mmol) in water (10 mL). The reaction mixture was stirred 1.5 h at 0 °C. The precipitated silver chloride was filtered off, washed with cold water and the filtrate was evaporated to dryness. 3,3,3-Trinitropropyl-1-ammonium dinitramide (6d) was obtained as colourless solid in 96% yield. DSC (5 °C min−1): 112 °C (dec.). IR (ATR): ν = 3285 (w), 3047 (w), 2981 (m), 2947 (m), 2662 (w), 1989 (w), 1588 (vs), 1520 (m), 1502 (m), 1478 (w), 1448 (m), 1412 (w), 1365 (w), 1295 (m), 1231 (w), 1183 (s), 1160 (s), 1026 (s), 986 (w), 947 (w), 898 (w), 855 (w), 827 (w), 798 (s), 761 (m), 755 (m), 743 (w), 734 (w), 722 (w) cm−1. RAMAN (1000 mW): ν = 3236 (5), 3228 (4), 3208 (4), 3046 (9), 2984 (24), 2948 (30), 2859 (20), 2818 (4), 1609 (19), 1522 (6), 1484 (7), 1469 (9), 1440 (12), 1419 (10), 1368 (40), 1334 (100), 1313 (30), 1300 (21), 1183 (13), 1162 (11), 1143 (22), 1053 (16), 1027 (21), 984 (17), 950 (10), 919 (6), 899 (10), 858 (88), 826 (75), 805 (6), 759 (14), 748 (10), 647 (6), 559 (11), 488 (23), 403 (39), 377 (52), 341 (45), 298 (16), 209 (7) cm−1. 1H NMR ([D4]methanol) δ = 8.31 (br, 3H, NH3), 3.72 (m, 2H, CH2), 3.43 (m, 2H, CH2) ppm. 13C NMR ([D4]methanol) δ = 129.6 (C(NO2)3), 35.4 (CH2), 32.3 (CH2) ppm. 15N NMR ([D4]methanol) δ = −12.6 (N(NO2)), −30.2 (NO2), −59.4 (br, N(NO2)), −352.0 (NH3) ppm. Elemental analysis C3H7N7O10 (301.13): calc. C 11.97, H 2.34, N 32.56%; found C 12.00, H 2.41, N 31.27%. IS: 2 J (grain size <100 μm). FS: 30 N (grain size <100 μm). ESD 0.45 J (grain size <100 μm).Bis(3,3,3-trinitropropyl-1-ammonium) 5,5′-azobistetrazolate (6e)
A solution of 3,3,3-trinitropropylammonium chloride (233 mg, 1.0 mmol) in water (10 mL) was added to a solution of potassium 5,5′-azobistetrazolate (123 mg, 0.5 mmol) in water (2 mL) at 0 °C. Immediately a yellow precipitate was formed. The reaction mixture was stirred 1 h at 0 °C. The precipitate was filtered off, washed with water and dried to yield 49% of bis(3,3,3-trinitropropyl-1-ammonium) 5,5′-azobistetrazolate (6e) as yellow solid. DSC (5 °C min−1): 120 °C (dec.). IR (ATR): ν = 2934 (w), 2757 (w), 2675 (w), 2623 (w), 2512 (w), 2084 (w), 1632 (w), 1603 (s), 1590 (s), 1517 (w), 1458 (w), 1424 (w), 1414 (w), 1394 (w), 1369 (w), 1312 (w), 1297 (w), 1185 (w), 1174 (m), 1148 (w), 1080 (w), 1051 (w), 1042 (w), 1010 (w), 903 (w), 856 (w), 806 (s), 796 (s), 774 (w), 756 (w), 738 (m), 667 (s) cm−1. RAMAN (500 mW): ν = 2935 (2), 1484 (48), 1423 (4), 1390 (100), 1195 (2), 1086 (10), 1058 (40), 927 (8), 857 (3), 341 (2) cm−1. 1H NMR ([D4]methanol) δ = 3.76 (m, 2H, CH2), 3.48 (m, 2H, CH2) ppm. 13C NMR ([D4]methanol) δ = 171.9 (CN4), 128.3 (C(NO2)3), 34.0 (CH2), 31.1 (CH2) ppm. 14N NMR ([D4]methanol) δ = −19 (CN4)), −30 (NO2), −352 (NH3) ppm. Elemental analysis C8H14N18O12 (554.31): calc. C 17.33, H 2.55, N 45.48%; found C 17.48, H 2.49, N 45.28%. IS: 2 J (grain size <100 μm). FS: 54 N (grain size <100 μm). ESD 0.6 J (grain size <100 μm).4,4,4-Trinitro-N-(2,2,2-trinitroethyl)butanamide (7)
To a saturated solution of barium hydroxide in water (10 mL) was added acrylamide (1.30 g, 18.2 mmol) and aqueous formaldehyde (37%, 1.50 g, 18.2 mmol) and stirred for 20 minutes. The solution was treated with solid carbon dioxide (5 g) and the precipitated barium carbonate was filtered off. To the filtrate was added aqueous nitroform solution (30%, 18.3 g, 36.4 mmol), stirred for 20 minutes and refluxed for further 30 minutes. The reaction mixture was cooled in an ice-water bath and the formed precipitate was filtered off. The yellow powder was recrystallized two times from a mixture of methanol/water, to yield 3.93 g (56%) of colourless pure product. DSC (5 °C min−1): 150 °C (mp.), 155 °C (dec.). IR (ATR): ν = 3304 (w), 3071 (w), 3011 (w), 2960 (w), 2892 (w), 1676 (m), 1589 (vs), 1543 (s), 1418 (w), 1363 (w), 1299 (s), 1236 (w), 1217 (w), 1155 (w), 1115 (w), 1092 (w), 1050 (w), 935 (w), 854 (m), 803 (s). Raman (400 mW): ν = 3006 (19), 2957 (39), 1677 (18), 1605 (29), 1421 (24), 1365 (28), 1337 (20), 1305 (36), 1117 (14), 1058 (13), 936 (15), 913 (13), 857 (102), 545 (18), 412 (46), 394 (47), 377 (68), 278 (29) cm−1. 1H NMR (CD3CN) δ = 7.21 (s, 1H, NH), 4.96 (d, 2H, 3J = 6.8 Hz, CH2NH), 3.47 (m, 2H, CH2C(NO2)3), 2.70 (m, 2H, OCCH2) ppm. 13C NMR (CD3CN) δ = 169.9 (CO), 131.4 (C(NO2)3), 127.5 (NHCH2C(NO2)3), 42.2 (NHCH2), 28.8 (CH2), 28.6 (CH2) ppm. 14N NMR (CD3CN) δ = −29 (C(NO2)3), −32 (NHCH2C(NO2)3) ppm. MS (DCI+) m/z: 386.2 [(M + H)+]. Elemental analysis C6H7N7O13 (385.16): calc. C 18.71, H 1.83, N 25.46%; found C 18.83, H 1.81, N 25.49%. IS: 10 J (grain size <100 μm). FS: 240 N (grain size <100 μm). ESD 0.2 J (grain size 100 μm).2,2,2-Trinitroethyl 4,4,4-trinitrobutanoate (8)
To a mixture of fuming sulfuric acid (30% SO3, 4 mL) and concentrated sulfuric acid (8 mL) was added 4,4,4-trinitrobutanoic acid (2) (1.7 g, 7.8 mmol) in small portions with cooling to 4 °C and stirred until complete solution. 2,2,2-Trinitroethanol (1.53 g, 7.8 mmol) dissolved in water (0.5 mL) was added very carefully to the reaction mixture at 4 °C and stirred for further 12 hours at room temperature. The reaction was quenched with ice-water (5 mL) and the colourless precipitate filtered off. The product was washed three times with water (20 mL) and dried to obtain 1.20 g (40%) of pure product. DSC (5 °C min−1): 92 °C (mp.), 155 °C (dec.). IR (ATR): ν = 3007 (w), 2964 (w), 2895 (w), 1761 (s), 1582 (vs), 1441 (w), 1430 (m), 1419 (w), 1400 (w), 1379 (w), 1363 (w), 1299 (s), 1222 (w), 1169 (s), 1100 (w), 1086 (m), 1037 (w), 1015 (w), 913 (w), 873 (w), 855 (m), 799 (vs), 780 (m), 759 (w), 744 (w), 730 (w), 689 (w), 655 (w) cm−1. Raman (500 mW): ν = 3009 (12), 2987 (21), 2953 (49), 1762 (18), 1609 (36), 1442 (10), 1419 (27), 1401 (129), 1364 (38), 1302 (36), 1263 (10), 1154 (9), 1086 (18), 1038 (10), 1015 (13), 972 (8), 915 (21), 873 (13), 857 (105), 798 (11), 781 (9), 744 (6), 647 (11), 539 (17), 487 (11), 404 (70), 373 (95), 341 (13), 326 (12), 269 (28), 232 (21) cm−1. 1H NMR (CDCl3) δ = 5.44 (s, 2H, OCH2), 3.43 (m, 2H, CH2C(NO2)3), 2.90 (m, 2H, OCCH2) ppm. 13C NMR (CDCl3) δ = 167.4 (CO), 128.2 (C(NO2)3), 122.5 (OCH2C(NO2)3), 61.3 (OCCH2), 29.3 (CH2), 28.0 (CH2) ppm. 14N NMR (CDCl3) δ = −31 (C(NO2)3), −35 (C(NO2)3) ppm. MS (DCI+) m/z: 387.1 [(M + H)+]. Elemental analysis C6H6N6O14 (386.14): calc. C 18.66, H 1.57, N 21.76%; found C 18.92, H 1.59, N 21.46%. IS: 30 J (grain size <100 μm). FS: 240 N (grain size <100 μm). ESD 0.1 J (grain size 100 μm).Acknowledgements
Financial support of this work by the Ludwig-Maximilian University of Munich (LMU), the Office of Naval Research (ONR) under grant no. ONR.N00014-16-1-2062, and the Bundeswehr – Wehrtechnische Dienststelle für Waffen und Munition (WTD 91) under grant no. E/E91S/FC015/CF049 is gratefully acknowledged. The authors acknowledge collaborations with Dr Mila Krupka (OZM Research, Czech Republic) in the development of new testing and evaluation methods for energetic materials and with Dr Muhamed Suceska (Brodarski Institute, Croatia) in the development of new computational codes to predict the detonation and propulsion parameters of novel explosives. We are indebted to and thank Drs Betsy M. Rice, Jesse Sabatini and Brad Forch (ARL, Aberdeen, Proving Ground, MD) for many inspired discussions.References
- (a) T. M. Klapötke, Chemistry of High-Energy Materials, de Gruyter, Berlin, 2nd edn, 2012 Search PubMed; (b) Q. J. Axthammer, B. Krumm and T. M. Klapötke, J. Org. Chem., 2015, 80, 6329–6335 CrossRef CAS PubMed; (c) Q. J. Axthammer, T. M. Klapötke, B. Krumm, R. Moll and S. F. Rest, Z. Anorg. Allg. Chem., 2014, 640, 76–83 CrossRef CAS.
- (a) M. Göbel, T. M. Klapötke and P. Mayer, Z. Anorg. Allg. Chem., 2006, 632, 1043–1050 CrossRef; (b) M. Göbel and T. M. Klapötke, Adv. Funct. Mater., 2009, 19, 347–365 CrossRef; (c) M. Göbel and T. M. Klapötke, Z. Anorg. Allg. Chem., 2007, 633, 1006–1017 CrossRef.
- J. P. Agrawal, High Energy Materials Propellants, Explosives and Pyrotechnics, Wiley-VCH, Weinheim, Germany, 1st edn, 2010 Search PubMed.
- A. D. Nikolaeva, I. L. Tsentsiper and V. S. Perekhod'ko, Izv. Vyssh. Uchebn. Zaved., Khim. Khim. Tekhnol., 1980, 23, 152–156 CAS.
- (a) Y. A. Volkova, E. B. Averina, Y. K. Grishin, P. Bruheim, T. S. Kuznetsova and N. S. Zefirov, J. Org. Chem., 2010, 75, 3047–3052 CrossRef CAS PubMed; (b) Y. A. Volkova, O. A. Ivanova, E. M. Budynina, E. B. Averina, T. S. Kuznetsova and N. S. Zefirov, Russ. Chem. Bull., 2008, 57, 2034–2035 CrossRef CAS.
- (a) M. J. Kamlet, J. C. Dacons and J. C. Hoffsommer, J. Org. Chem., 1961, 26, 4881–4886 CrossRef CAS; (b) I. J. Schaffner, US3051743, 1962 Search PubMed.
- D. J. Glover, J. C. Dacons, D. V. Sickman, M. E. Hill and M. J. Kamlet, US3125606, 1964 Search PubMed.
- Q. J. Axthammer, B. Krumm, T. M. Klapötke and R. Scharf, Chem. – Eur. J., 2015, 21, 16229–16239 CrossRef CAS PubMed.
- M. H. Gold, M. B. Frankel, G. B. Linden and K. Klager, J. Org. Chem., 1962, 27, 334–336 CrossRef CAS.
- M. B. Frankel, Tetrahedron, 1963, 19, 213–217 CrossRef CAS.
- M. B. Frankel, US2978509, 1961 Search PubMed.
- (a) H. Feuer and U. E. Lynch-Hart, J. Org. Chem., 1961, 26, 391–394 CrossRef CAS; (b) H. Feuer and U. E. Lynch-Hart, J. Org. Chem., 1961, 26, 587–589 CrossRef CAS; (c) I. J. Schaffner, US3038009, 1962 Search PubMed.
- (a) R. H. Saunders, US2996537, 1961 Search PubMed; (b) M. E. Hill, US3230247, 1966 Search PubMed.
- S. Bienz, L. Bigler, T. Fox, M. Hesse, H. Meier and B. Zeeh, Spektroskopische Methoden in der Organischen Chemie, Thieme, Stuttgart, 8th edn, 2014 Search PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- T. M. Klapötke, B. Krumm, R. Moll, S. F. Rest, Y. V. Vishnevskiy, C. Reuter, H.-G. Stammler and N. W. Mitzel, Chem. – Eur. J., 2014, 20, 12962–12973 CrossRef PubMed.
- (a) T. M. Klapötke, B. Krumm and R. Scharf, Eur. J. Inorg. Chem., 2016, 3086–3093 CrossRef; (b) A. Wolter-Steingrube, B. E. C. Bugenhagen, C. Herrmann and J. Heck, Eur. J. Inorg. Chem., 2014, 4115–4122 CrossRef CAS; (c) A. Baumann, A. Erbacher, C. Evangelisti, T. M. Klapötke, B. Krumm, S. F. Rest, M. Reynders and V. Sproll, Chem. – Eur. J., 2013, 19, 15627–15638 CrossRef CAS PubMed; (d) G. C. Hsu, L. M. Singer, D. B. Cordes and M. Findlater, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, 1298 Search PubMed; (e) J. Lapic, A. Pezerovic, M. Cetina, S. Djakovic and V. Rapic, J. Mol. Struct., 2011, 990, 209–216 CrossRef CAS; (f) G. Laus, V. Kahlenberg, K. Wurst, S. Nerdinger and H. Schottenberger, Z. Naturforsch., 2011, 66B, 479–486 CrossRef; (g) D. Siebler, C. Forster, T. Gasi and K. Heinze, Organometallics, 2011, 30, 313–327 CrossRef CAS; (h) L. Parkanyi and G. Besenyei, J. Mol. Struct., 2004, 691, 97–106 CrossRef CAS; (i) F. M. Menger, J. Bian and V. A. Azov, Angew. Chem., Int. Ed., 2002, 41, 2581–2584 CrossRef CAS; (j) Y. Jiao, E. Valente, S. T. Garner, X. Wang and H. Yu, Tetrahedron Lett., 2002, 43, 5879–5881 CrossRef CAS.
- (a) NATO, Standardization Agreement 4487 (STANAG 4487), Explosives, Friction Sensitivity Tests, 2002; (b) NATO, Standardization Agreement 4489 (STANAG 4489), Explosives, Impact Sensitivity Tests, 1999.
- Test methods according to the UN Manual of Test and Criteria, Recommendations on the Transport of Dangerous Goods, United Nations Publication, New York, Geneva, 4th revised edn, 2003, Impact: insensitive > 40 J, less sensitive ≥ 35 J, sensitive ≥ 4 J, very sensitive ≤ 3 J; friction: insensitive > 360 N, less sensitive = 360 N, sensitive < 360 N > 80 N, very sensitive ≤ 80 N, extremely sensitive ≤ 10 N Search PubMed.
- (a) M. Sućeska, EXPLO5 V.6.02, Zagreb (Croatia), 2013 Search PubMed; (b) M. Sućeska, Propellants, Explos., Pyrotech., 1991, 16, 197–202 CrossRef.
- R. Meyer, J. Köhler and A. Homburg, Explosives, Wiley-VCH, Weinheim, Germany, 7th edn, 2015 Search PubMed.
- CrysAlis CCD, Version 1.171.35. (release 16-05-2011 CrysAlis 171.Net), Oxford Diffraction Ltd, Abingdon, Oxford (U. K.), 2011 Search PubMed.
- CrysAlis RED, Version 1.171.35.11 (release 16-05-2011 CrysAlis 171.NET), Oxford Diffraction Ltd, Abingdon, Oxford (U. K.), 2011 Search PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- (a) G. M. Sheldrick, SHELX-97, Programs for Crystal Structure Determination, 1997 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- A. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. B. G. Scalmani, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT (US), Rev. A.02 edn, 2009 Search PubMed.
- R. D. Dennington, T. A. Keith and J. M. Millam, GaussView, Semichem, Inc., Wallingford CT, Ver. 5.08 edn, 2009 Search PubMed.
- J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 2000, 112, 6532–6542 CrossRef CAS.
- J. W. Ochterski, G. A. Petersson and J. A. Montgomery, J. Chem. Phys., 1996, 104, 2598–2619 CrossRef CAS.
- E. F. C. Byrd and B. M. Rice, J. Phys. Chem., 2005, 110, 1005–1013 CrossRef PubMed.
- (a) F. Trouton, Philos. Mag., 1884, 18, 54–57 CrossRef; (b) M. S. Westwell, M. S. Searle, D. J. Wales and D. H. Williams, J. Am. Chem. Soc., 1995, 117, 5013–5015 CrossRef CAS.
Footnotes |
| † Parts of this study have been presented at the 17th Seminar on New Trends in Research of Energetic Materials, Pardubice, Czech Republic, April 9–11, 2014. |
| ‡ Electronic supplementary information (ESI) available: Crystallographic data, refinement parameters and additional figures. CCDC 1506284–1506290. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt03779h |
| This journal is © The Royal Society of Chemistry 2016 |