
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Different manifestations of enhanced π-acceptor ligation at every redox level of [Os(9-OP)L2]n, n = 2+, +, 0, − (9-OP− = 9-oxidophenalenone and L = bpy or pap)†
Arijit Singha
Hazari
a,
Alexa
Paretzki
b,
Jan
Fiedler
c,
Stanislav
Zalis
c,
Wolfgang
Kaim
*b and
Goutam Kumar
Lahiri
*a
aDepartment of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai 400076, India. E-mail: lahiri@chem.iitb.ac.in
bInstitut für Anorganische Chemie, Universität Stuttgart, Pfaffenwaldring 55, D-70550 Stuttgart, Germany. E-mail: kaim@iac.uni-stuttgart.de
cJ. Heyrovský Institute of Physical Chemistry, v.v.i., Academy of Sciences of the Czech Republic, Dolejskova 3, CZ-18223 Prague, Czech Republic
First published on 17th October 2016
Abstract
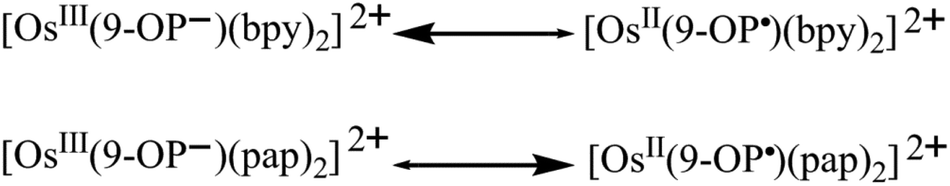
The title complexes were isolated as structurally characterised compounds [OsII(9-OP)L2]ClO4, L = 2,2′-bipyridine (bpy) or 2-phenylazopyridine (pap), and were compared with ruthenium analogues. A reversible one-electron oxidation and up to three reduction processes were observed by voltammetry (CV, DPV) and spectroelectrochemistry (UV-vis-NIR, partially EPR). Supporting calculations (DFT, TD-DFT) were used to assess the oxidation state combinations of the different redox active ligands and of the metal, revealing the effects of Os versus Ru exchange and of bpy versus pap acceptor ligation. Several unexpected consequences of these variations were observed for members of the new osmium-containing redox series. Remarkably, the EPR results exhibit a clear dichotomy between the complex ion [OsIII(9-OP−)(bpy)2]2+ and the radical species [OsII(9-OP˙)(pap)2]2+, which has not been similarly observed for the analogous [RuIII(9-OP−)L2]2+ systems. This difference, unprecedented for 5dn systems, is attributed to the superior stabilisation of the OsII state by the strongly π-accepting pap ligands. The reduced forms [OsII(9-OP−)(pap˙−)(pap)] and [OsII(9-OP−)(pap˙−)2]− exhibit strong inter-ligand interactions, leading to spin isomers and electron hopping.
Introduction
9-Oxidophenalenone (9-OP−, A), a special β-diketonate chelate ligand, has been studied with respect to its formation of a dianionic radical (B) via reduction. Complexes of a 9-oxidophenalenone anion or a dianion have been described with positively charged main group ions such Be2+, B3+, Al3+, Si4+ and Ge4+.1 Our earlier research on ruthenium complexes [Ru(9-OP)n (L)3−n]m has provided evidence that both the transition metal and (9-OP)x can exhibit redox reactivity,2 the latter with the formation of a dianionic (B) or also of a neutral radical (C). The (9-OP)x system may thus be included in the group of redox-active and potentially “non-innocent” ligands,3 involving a rather uncommon situation4 with six-membered chelate rings. Recently, Mandal et al. have reported iron(III) complexes [Fe(9-OP)3]x and their functioning as electroactive materials for H2O2 fuel cell application.5To complement the series with group 8 metals, we are now describing two osmium compounds [1]ClO4 and [2]ClO4 (Scheme 2) in contrast to the ruthenium analogues [Ru(9-OP)(bpy)2]ClO4 ([3]ClO4)2a and [Ru(9-OP)(pap)2]ClO4 ([4]ClO4).2b While the corresponding osmium and ruthenium compounds are frequently similar, there are distinct differences in certain complexes such as mixed-valent materials.6,7 In comparison to ruthenium, osmium has a preference for higher oxidation states (lower redox potentials) and larger spin–orbit coupling parameters, manifest through the absorption spectral and magnetic effects (EPR g factors, spin–spin coupling).6,7
It is shown in this contribution that the contrast between complexes 1n with the moderate π-acceptor bpy and system 2n with the much stronger π-accepting2a,b pap can result in frequently variable oxidation state situations involving (9-OP)x and the metal in their respective accessible charge states.
Results and discussion
Synthesis, general characterisation and crystal structures
The mononuclear complexes [OsII(bpy)2(9-OP)]ClO4 ([1]ClO4) and [OsII(pap)2(9-OP)]ClO4 ([2]ClO4) (bpy = 2,2′-bipyridine, pap = 2-phenylazopyridine, 9-OP− = 9-oxidophenalenone) have been obtained via the reactions of H(9-OP) (9-hydroxyphenalenone) with the respective metal precursors cis-[Os(bpy)2Cl2] and ctc-[Os(pap)2Cl2] (ctc = cis–trans–cis configurations of chlorides, pyridine and azo nitrogen atoms, respectively) in the presence of Et3N in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 EtOH–H2O solvent mixture under a dinitrogen atmosphere, followed by precipitation using a saturated aqueous NaClO4 solution. The complexes were purified by column chromatography using a neutral alumina column (Experimental) and characterized by their microanalytical data, electrical conductivity, mass spectrometry (Fig. S1†), IR and 1H NMR spectroscopy (Fig. S2,† and the Experimental section).
:1 EtOH–H2O solvent mixture under a dinitrogen atmosphere, followed by precipitation using a saturated aqueous NaClO4 solution. The complexes were purified by column chromatography using a neutral alumina column (Experimental) and characterized by their microanalytical data, electrical conductivity, mass spectrometry (Fig. S1†), IR and 1H NMR spectroscopy (Fig. S2,† and the Experimental section).
The identities of the complexes [1]ClO4 and [2]ClO4 have been authenticated by their single crystal X-ray structures (Fig. 1, 2 and Tables 1, 2). In contrast to [1]ClO4·C6H6, the pap containing complex [2]ClO4 (in ctc configuration) is not isostructural with the ruthenium analogue which crystallises as dichloromethane solvated [4]ClO4·CH2Cl2.2a,b The oxygen donors of 9-OP− form a six-membered chelate with the {OsII(bpy)2} or {OsII(pap)2} fragments in [1]ClO4 or [2]ClO4, respectively. The appreciably shorter average trans (172.59°) and cis-angles (9-OP: 88.83°, bpy, 78.96°, pap: 77.3°) around the Os centre in the complexes illustrate a distorted octahedral situation. The shorter OsII–N(pap) bond distance (average: 1.997 Å) compared to the OsII–N(bpy) distance (average: 2.028 Å)8 reveals the stronger π-accepting feature of pap. Furthermore, the average OsII–N(azo) distance is 0.06 Å shorter than the average OsII–N(pyridine) distance of pap due to OsII → azo(pap) back-bonding as has also been reflected in the lengthening of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond from 1.25 Å in free pap9 to about 1.32 Å in [2]ClO4. A remarkable amount of osmium(II)-to-pap π-back donation is evident from the rather short (<2.00 Å) metal-(azo)nitrogen distances and from the lengthened N–N bonds in the coordinated pap ligands in 2+ (Table 2). Using a distance/oxidation state correlation for azo ligands10a one can imply a certain degree of contributions from pap radical anion ligands antiferromagnetically coupled with osmium(III). The σ and π electron donation from the (9-OP)− ligand serves to enhance the electron density at the metal. The bond lengths within the (9-OP)− ligand are in agreement with the standard description (including short C2C3 and C9C10 bonds, Scheme 1). Notably, the DFT calculations (Table 2) do not fully reproduce the experimental structural effects of the strong Os/pap π back donation (underestimation of N–N bond lengthening) which probably leads to the later discussed discrepancy of the spin distribution (Scheme 3).
N bond from 1.25 Å in free pap9 to about 1.32 Å in [2]ClO4. A remarkable amount of osmium(II)-to-pap π-back donation is evident from the rather short (<2.00 Å) metal-(azo)nitrogen distances and from the lengthened N–N bonds in the coordinated pap ligands in 2+ (Table 2). Using a distance/oxidation state correlation for azo ligands10a one can imply a certain degree of contributions from pap radical anion ligands antiferromagnetically coupled with osmium(III). The σ and π electron donation from the (9-OP)− ligand serves to enhance the electron density at the metal. The bond lengths within the (9-OP)− ligand are in agreement with the standard description (including short C2C3 and C9C10 bonds, Scheme 1). Notably, the DFT calculations (Table 2) do not fully reproduce the experimental structural effects of the strong Os/pap π back donation (underestimation of N–N bond lengthening) which probably leads to the later discussed discrepancy of the spin distribution (Scheme 3).
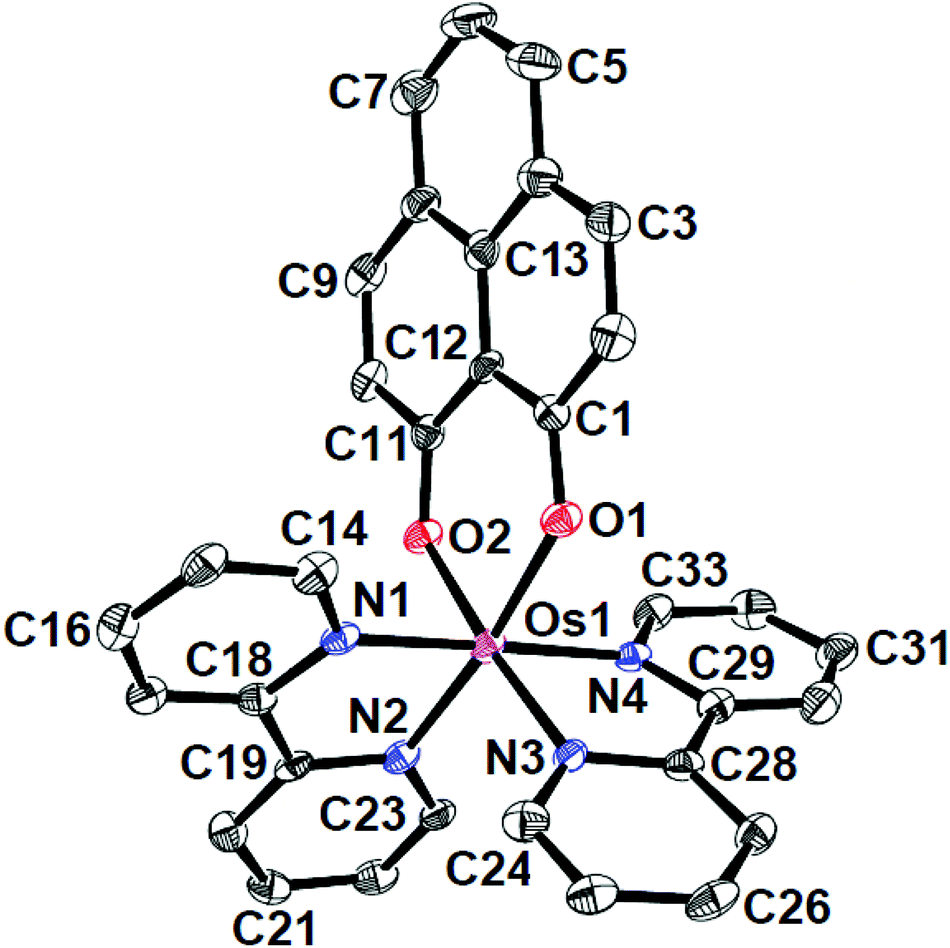 | ||
| Fig. 1 ORTEP diagram of the cationic part of [1]ClO4·C6H6. Ellipsoids are drawn at the 50% probability level. Hydrogen atoms and solvent molecules are omitted for clarity. | ||
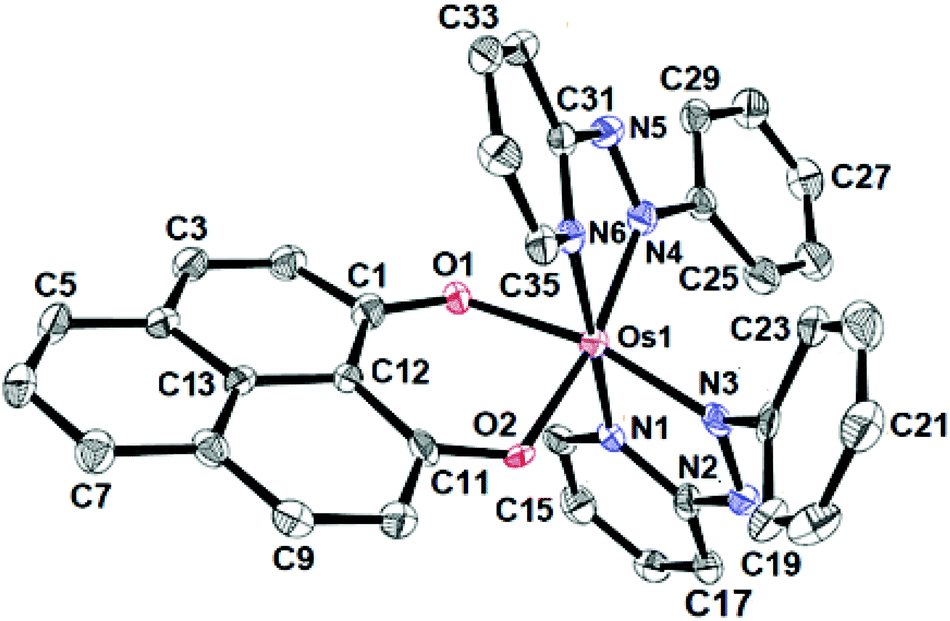 | ||
| Fig. 2 ORTEP diagram of the cationic part of [2]ClO4. Ellipsoids are drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. | ||
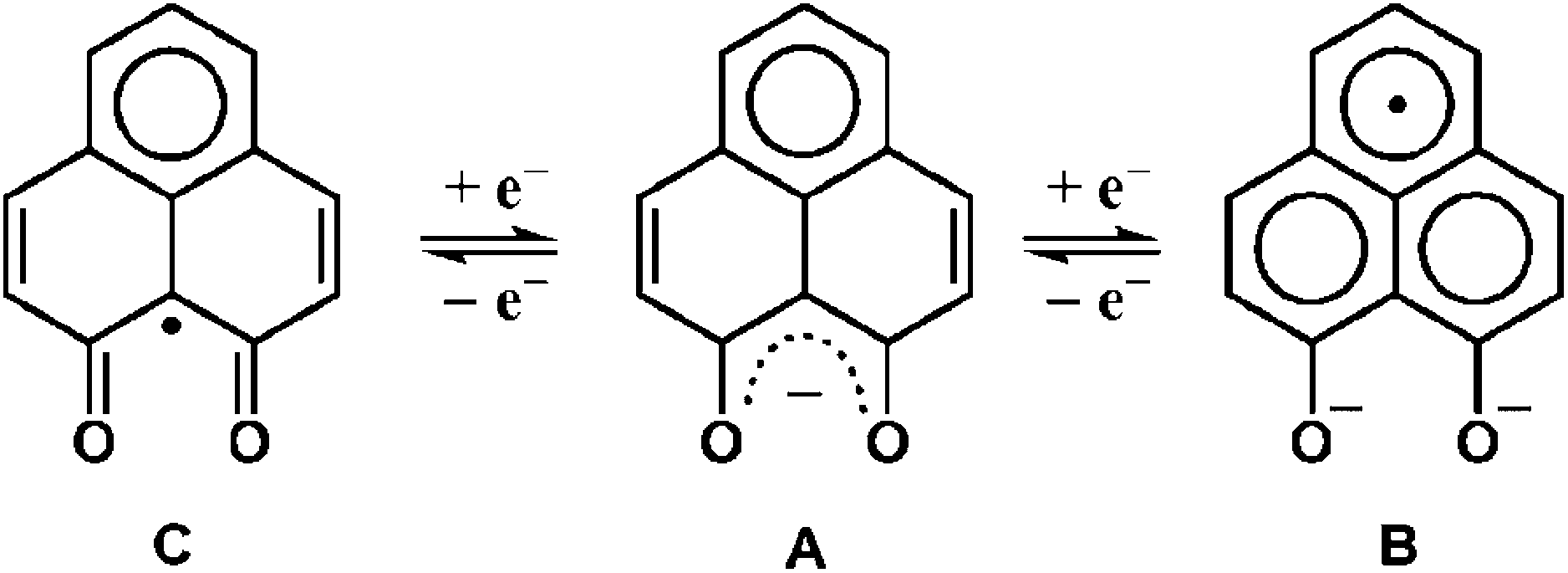 | ||
| Scheme 1 Two-step redox series of (9-OP)x. | ||
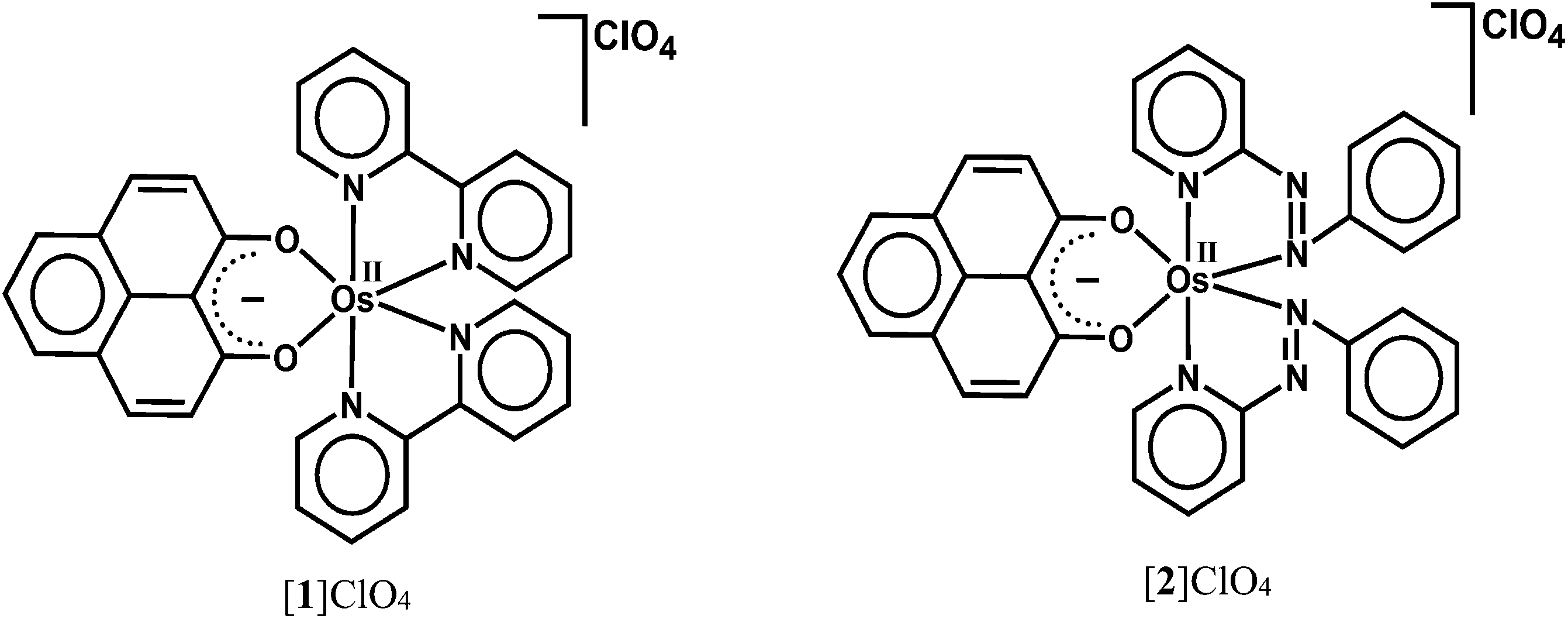 | ||
| Scheme 2 Representation of complexes. | ||
 | ||
| Scheme 3 Variable electronic structural situations in 12+ and 22+. | ||
| Complex | [1]ClO4·C6H6 | [2]ClO4 |
|---|---|---|
| Empirical formula | C39H29ClN4O6Os | C35H25ClN6O6Os |
| Formula weight | 875.35 | 851.26 |
| Radiation | MoKα | MoKα |
| Crystal system | Triclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
| a/Å | 11.694(3) | 10.552(2) |
| b/Å | 12.469(4) | 19.785(3) |
| c/Å | 13.505(4) | 15.902(2) |
| α (°) | 110.800(3) | 90.00 |
| β (°) | 97.5790(10) | 109.0190(10) |
| γ (°) | 112.346(3) | 90.00 |
| V/Å3 | 1619.5(8) | 3138.6(9) |
| Z | 2 | 4 |
| μ/mm−1 | 4.072 | 4.207 |
| T/K | 100(2) | 100(2) |
| ρ calcd/g cm−3 | 1.795 | 1.801 |
| F(000) | 804 | 1672 |
| θ range (°) | 3.04 to 25.00 | 3.37 to 25.00 |
| Data/restraints/parameters | 5606/0/460 | 5441/0/442 |
| R 1, wR2 [I > 2σ(I)] | 0.0290, 0.0651 | 0.0448,0.1035 |
| R 1, wR2 (all data) | 0.0317, 0.0668 | 0.0528, 0.1115 |
| GOF on F2 | 1.047 | 0.991 |
| Largest difference in peak and hole (e Å−3) | 0.90/−0.68 | 2.68/−2.20 |
| Bond length (Å) | [1]ClO4·C6H6 | [2]ClO4 | ||
|---|---|---|---|---|
| X-ray | DFT | X-ray | DFT | |
| Os1–N1 | 2.037(3) | 2.044 | 2.022(5) | 2.031 |
| Os1–N2 | 2.014(4) | 2.017 | — | — |
| Os1–N3 | 2.012(3) | 2.017 | 1.956(6) | 1.981 |
| Os1–N4 | 2.048(3) | 2.044 | 1.974(5) | 1.976 |
| Os1–N6 | — | — | 2.035(5) | 2.046 |
| Os1–O1 | 2.037(3) | 2.037 | 2.030(4) | 2.029 |
| Os1–O2 | 2.042(3) | 2.037 | 2.026(4) | 2.032 |
| C1–O1 | 1.297(5) | 1.284 | 1.305(7) | 1.286 |
| C11–O2 | 1.292(5) | 1.284 | 1.308(7) | 1.288 |
| N2–N3 | — | — | 1.335(7) | 1.294 |
| N4–N5 | — | — | 1.312(6) | 1.288 |
The Os–N(bpy/pap) distance trans to the O donor of 9-OP− is shorter than that of trans to N(bpy/pap) in [1]ClO4/[2]ClO4 due to the predominating σ-donating effect of the O donors. The average OsII–O(9-OP) distances of 2.040 Å and 2.032 Å in [1]ClO4 and [2]ClO4 match fairly well with those of the reported analogous diketonate complexes of osmium(II).10b The average C–O bond distance in coordinated 9-OP of 1.29 Å in the complexes refers to its delocalized β-diketonate form.11
The experimental bond parameters of [1]ClO4 and [2]ClO4 (Table 2) are well reproduced by the DFT calculations (Table 2). These calculations also describe the bond length variations such as the shortening of Os–N bonds when going from [1]ClO4 to [2]ClO4. The bond length variations in the course of the redox processes are presented in Tables S1–S4, Fig. S3.†
Electrochemistry
The compounds [1]ClO4 and [2]ClO4 exhibit multiple oxidation (O1/O2) and reduction (R1–R3) processes within a potential window of ±2.0 V versus SCE in CH3CN (Fig. 3, Table 3). The second oxidation wave (O2) is found to be irreversible in each case as is the third reduction of 1+. An appreciable anodic shift of the redox potentials takes place on moving from the bpy containing [1]ClO4 to [2]ClO4 with the more π-accepting pap ligands. The effect of stronger π-donating OsII in [1]ClO4/[2]ClO4 compared to RuII containing [3]ClO4/[4]ClO4 is similarly reflected by the change of the redox potentials (Table 3). The potentials for the first reversible oxidation decrease from the systems with π-accepting pap to the more electron rich bpy complexes, especially for the 1+/12+ transition with the π electron richer osmium. Conversely, the first reduction is the easiest for the pap complexes, especially for the 2+/2 transition, suggesting reduction of the acceptor ligands as will be shown later by EPR. The following reduction waves are more split in the case of 2n, reflecting the stronger ligand–ligand interaction. The potential separations between successive redox processes of [1]ClO4 and [2]ClO4 are quantified by the comproportionation constants (RTlnKc = nF(ΔE)).
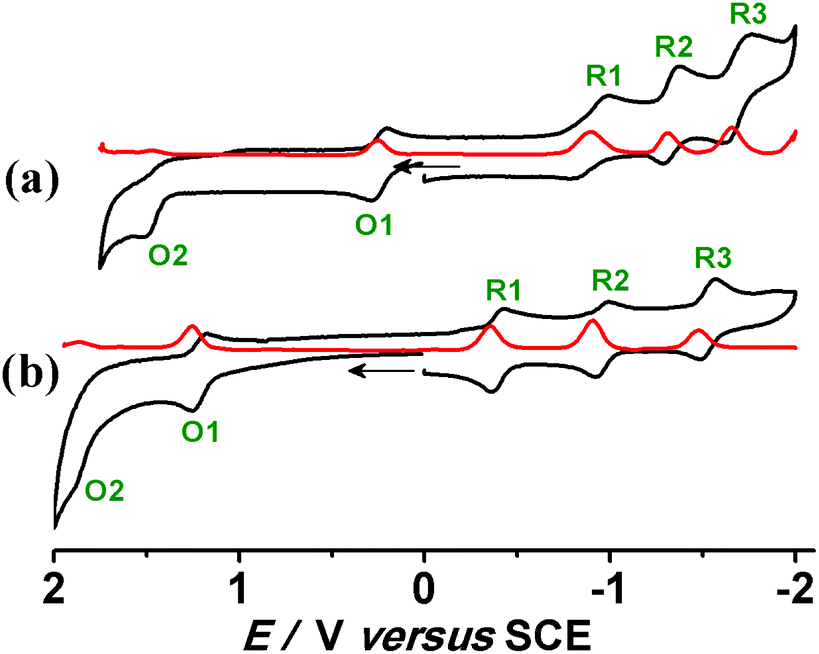 | ||
| Fig. 3 Cyclic (black) and differential pulse (red) voltammograms in CH3CN/0.1 M Et4NClO4 for (a) [1]ClO4 and (b) [2]ClO4 at 298 K. | ||
| Complex |
|
K
cc |
Ref. | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| O2d | O1 | R1 | R2 | R3 |
K
c1e |
K
c2f |
K
c3g |
K
c4h |
||
|
a From cyclic voltammetry in CH3CN/0.1 M Et4NClO4 at 100 mV s−1.
b Potential in V versus SCE; peak potential differences ΔEp/mV (in parentheses).
c Comproportionation constant from RTlnKc = nF(ΔE).
d Irreversible.
e
K
c1 between O2 and O1.
f
K
c2 between O1 and R1.
g
K
c3 between R1 and R2.
h
K
c4 between R2 and R3.
|
||||||||||
| [1]ClO4 | 1.49 | 0.24 (80) | −0.89 (150) | −1.32 (80) | −1.68 (110) | 1.4 × 1021 | 1.4 × 1019 | 1.9 × 107 | 1.2 × 106 | This work |
| [2]ClO4 | 1.89 | 1.21 (60) | −0.39 (60) | −0.95 (60) | −1.52 (70) | 3.3 × 1011 | 1.3 × 1027 | 3.1 × 109 | 4.5 × 109 | This work |
| [3]ClO4 | 1.78 | 0.50 (70) | −1.48 (70) | −1.74 (80) | — | 4.9 × 1021 | 3.6 × 1033 | 2.5 × 104 | — | 2a |
| [4]ClO4 | — | 1.22 (70) | −0.50 (60) | −1.00 (70) | — | — | 1.4 × 1029 | 2.9 × 104 | — | 2b |
The electronic structures of the redox states of 1n or 2n (3+, 2+, 1+, 0, 1−, 2−) have been ascertained by MO calculations (Tables S5–S17, Fig. S3†), EPR (Fig. 5, Tables 5 and S4†), Mulliken spin density distributions at the paramagnetic intermediate states (Table 4, Fig. 4), spectroelectrochemistry (Fig. 6) and TD-DFT calculations (Table 6).
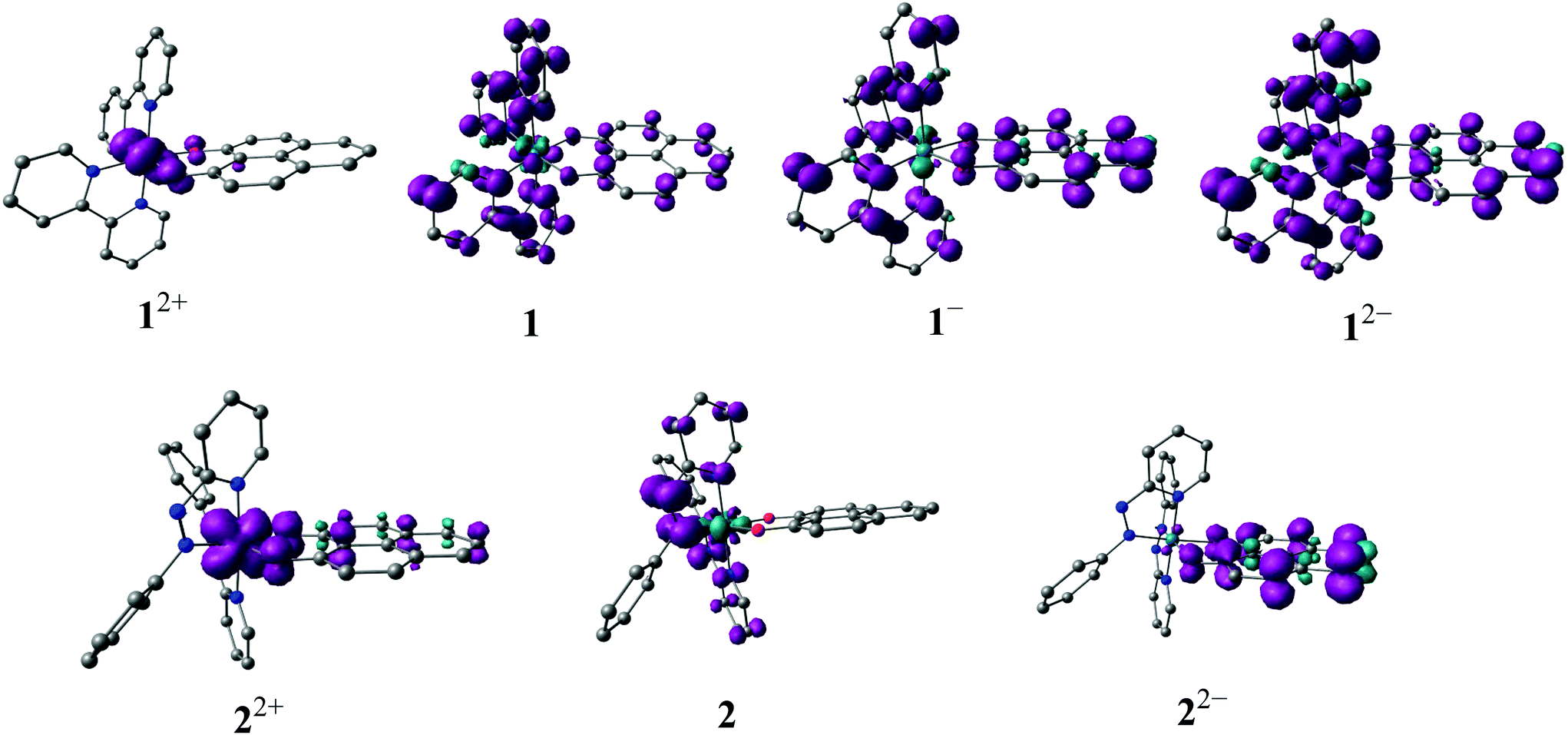 | ||
| Fig. 4 DFT calculated Mulliken spin density plots for 1n and 2n. | ||
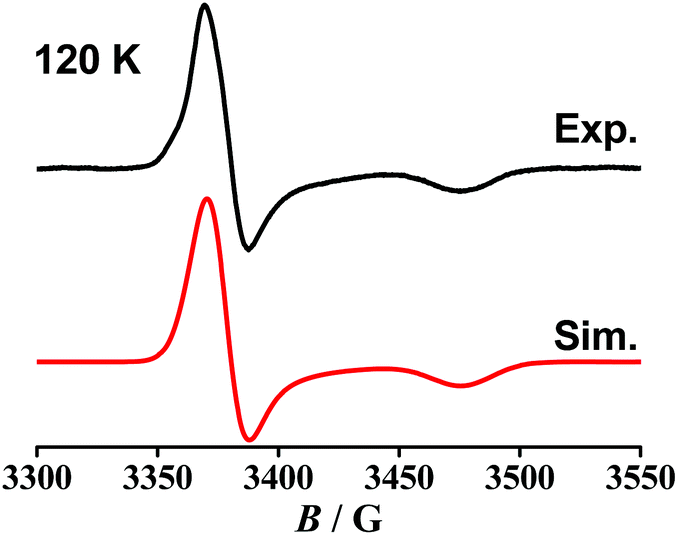 | ||
| Fig. 5 EPR spectrum of 22+ after in situ oxidation of [2]ClO4 in CH2Cl2/0.1 M Bu4NPF6. | ||
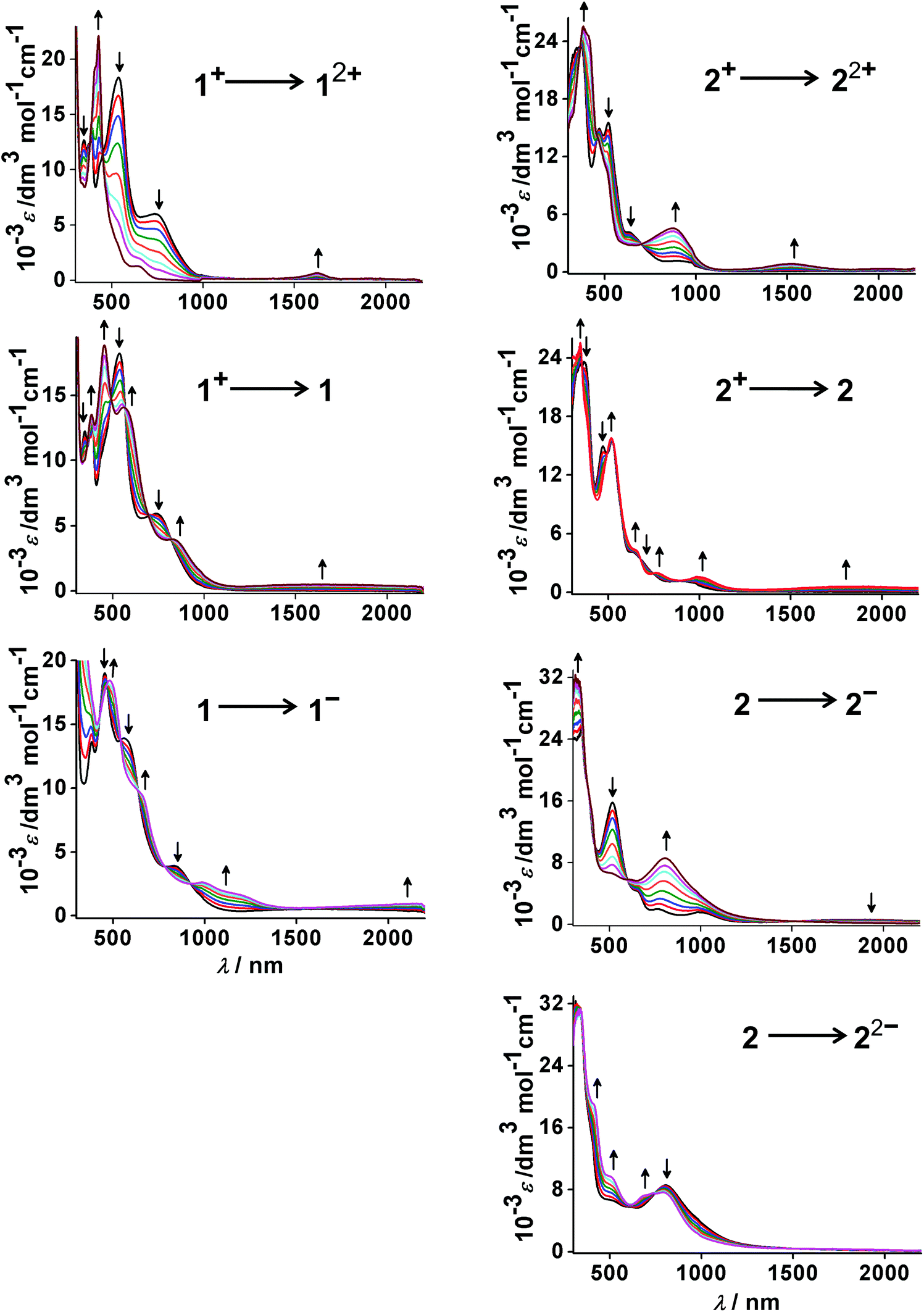 | ||
| Fig. 6 UV-vis-NIR spectroelectrochemistry of 1n and 2n (n = 2+, 1+, 0, 1−, 2−) in CH3CN/0.1 M Bu4NPF6. | ||
| Complex | Os | bpy | pap | 9-OP |
|---|---|---|---|---|
| 1 2+ (S = 1/2) | 0.790 | 0.023 | — | 0.187 |
| 1 (S = 1/2) | 0.000 | 0.844 | — | 0.188 |
| 1 − (S = 1) | −0.062 | 1.346 | — | 0.764 |
| 1 2− (S = 3/2) | 0.372 | 1.674 | — | 1.028 |
| 2 2+ (S = 1/2) | 0.655 | — | 0.072 | 0.262 |
| 2 (S = 1/2) | −0.108 | — | 1.113 | −0.003 |
| 2 2− (S = 1/2) | 0.029 | — | −0.008 | 1.032 |
| Complex | g iso (298 K) | g 1 (120 K) | g 2 | g 3 | <g>c | Δgd |
|---|---|---|---|---|---|---|
| a From in situ electrolysis in CH2Cl2/0.1 M Bu4NPF6. b α iso(189Os) = 8.0 G, calculated αiso(189Os) = 7.7 G. c <g> = {(1/3)(g12 + g22 + g32)}1/2. d Δg = g1 − g3. | ||||||
| 1 (Exp.) | 2.006b | 2.022 | 2.005 | 1.986 | 2.004 | 0.036 |
| 1 (Calc.) | 1.991 | 2.008 | 2.003 | 1.964 | 1.991 | 0.044 |
| 2 | n.o. | 1.98 (4 K) | 1.94 | 1.89 | 1.937 | 0.09 |
| 2 2+ | 1.986 | 2.007 | 2.004 | 1.946 | 1.985 | 0.061 |
|
λ
max
,
(λTDDFTb) |
(ε/dm3 mol−1 cm1)a,c (f)d | Transitions | Character |
|---|---|---|---|
| a Experimental absorption maxima (λmax > 300 nm) from OTTLE spectroelectrochemistry in CH3CN/0.1 M Bu4NPF6. b In nm. c Molar extinction coefficients in dm3 mol−1 cm1. d Calculated oscillator strengths. | |||
| 1 2+ (S = 1/2) | |||
| 1622 (2100) | 650 (0.002) | HOMO−1(β) → LUMO(β)(90) | 9-OP(π) → Os(dπ) |
| 635 (652) | 1290 (0.026) | HOMO−2(β) → LUMO(β)(73) | Os(dπ) → Os(dπ) |
| 426 (434) | 22130 (0.066) |
HOMO(α) → LUMO+3(α)(78) | 9-OP(π) → 9-OP(π*) |
| 410 (408) | 820 (0.047) | HOMO−1(α) → LUMO+1(α)(86) | 9-OP(π) → bpy(π*) |
| 288 (287) | 28670 (0.017) |
HOMO−1(α) → LUMO+6(α)(51) | 9-OP(π) → bpy(π*) |
| 242 (256) | 29590 (0.031) |
HOMO−3(α) → LUMO+3(α)(86) | 9-OP(π)/bpy(π) → bpy(π*) |
| 1 + (S = 0) | |||
| 739 (625) | 5920 (0.012) | HOMO → LUMO(52) | Os(dπ) → bpy(π*) |
| 537 (557) | 18230 (0.023) |
HOMO−1 → LUMO+1(54) | Os(dπ) → bpy(π*) |
| 391 (361) | 12720 (0.051) |
HOMO−3 → LUMO(67) | 9-OP(π) → bpy(π*) |
| 347 (342) | 12240 (0.095) |
HOMO−3 → LUMO+2(64) | 9-OP(π) → 9-OP(π*) |
| 294 (296) | 41790 (0.024) |
HOMO−5 → LUMO(66) | 9-OP(π) → bpy(π*) |
| 241 (239) | 29210 (0.061) |
HOMO−5 → LUMO+3(60) | 9-OP(π) → bpy(π*) |
| 1 (S = 1/2) | |||
| 1650 (1057) | 480 (0.0013) | HOMO(α) → LUMO+2(α)(98) | 9-OP(π) → 9-OP(π*) |
| 825 (869) | 3950 (0.004) | HOMO−1(α) → LUMO+1(α)(51) | Os(dπ) → 9-OP(π*) |
| 557 (552) | 14070 (0.048) |
HOMO−2(β) → LUMO+1(β)(59) | Os(dπ) → bpy(π*) |
| 454 (444) | 18840 (0.144) |
HOMO−2(β) → LUMO+2(β)(64) | Os(dπ) → bpy(π*) |
| 384 (380) | 13520 (0.019) |
HOMO−3(β) → LUMO+1(β)(63) | 9-OP(π) → bpy(π*) |
| 296 (295) | 33940 (0.004) |
HOMO−5(β) → LUMO+1(β)(64) | bpy(π) → bpy(π*) |
| 243 (255) | 29700 (0.014) |
HOMO−5(β) → LUMO+4(β)(64) | 9-OP(π) → bpy(π*) |
| 1 − (S = 1) | |||
| 2200 (2519) | 950 (0.037) | HOMO−1(α) → LUMO(α)(71) | 9-OP(π)/bpy(π) → bpy(π*) |
| 1150 (992) | 590 (0.001) | HOMO−1(α) → LUMO+1(α)(74) | 9-OP(π)/bpy(π) → bpy(π*) |
| 986 (934) | 2630 (0.014) | HOMO−1(α) → LUMO(α)(85) | Os(dπ) → bpy(π*)/9-OP(π*) |
| 635 (650) | 8330 (0.004) | HOMO−1(β) → LUMO+1(β)(55) | Os(dπ) → bpy(π*) |
| 480 (470) | 18440 (0.002) |
HOMO−2(α) → LUMO+1(α)(71) | Os(dπ) → bpy(π*) |
| 306 (303) | 26950 (0.040) |
HOMO−5(β) → LUMO(β)(60) | bpy(π) → 9-OP(π*) |
| 2 2+ (S = 1/2) | |||
| 1515 (1961) | 830 (0.004) | HOMO−1(β) → LUMO(β)(83) | 9-OP(π) → Os(dπ) |
| 872 (839) | 4540 (0.035) | HOMO−3(β) → LUMO(β)(50) | pap(π)/Os(dπ) → Os(dπ)/9-OP(π*) |
| 512 (526) | 10670 (0.002) |
HOMO−9(β) → LUMO(β)(76) | pap(π)/9-OP(π) → Os(dπ)/9-OP(π*) |
| 383 (383) | 25580 (0.045) |
HOMO−2(β) → LUMO+2(β)(57) | pap(π) → 9-OP(π*) |
| 2 + (S = 0) | |||
| 896 (699) | 1130 (0.006) | HOMO−1 → LUMO(65) | Os(dπ)/9-OP(π) → pap(π*) |
| 635 (637) | 4120 (0.008) | HOMO → LUMO+1(59) | 9-OP(π)/Os(dπ) → pap(π*) |
| 520 (590) | 15500 (0.011) |
HOMO−1 → LUMO+1(62) | Os(dπ)/9-OP(π) → pap(π*)/9-OP(π*) |
| 469 (471) | 14950 (0.012) |
HOMO → LUMO+2(69) | 9-OP(π)/Os(dπ) → 9-OP(π*) |
| 367 (354) | 23620 (0.039) |
HOMO−3 → LUMO+2(65) | pap(π)/9-OP(π) → 9-OP(π*) |
| 343 (322) | 23240 (0.074) |
HOMO−4 → LUMO+2(69) | pap(π) → 9-OP(π*) |
| 2 (S = 1/2) | |||
| 1880 (1650) | 580 (0.019) | HOMO(α) → LUMO+1(α)(95) | pap(π) → pap(π*)/Os(dπ) |
| 996 (1078) | 1560 (0.001) | HOMO(α) → LUMO(α)(95) | pap(π) → P(π*) |
| 762 (722) | 1990 (0.014) | HOMO(β) → LUMO(β)(77) | Os(dπ)/pap(π) → 9-OP(π*) |
| 642 (603) | 4040 (0.001) | HOMO−2(α) → LUMO(α)(58) | Os(dπ)/9-OP(π) → 9-OP(π*) |
| 517 (513) | 15780 (0.047) |
HOMO−1(β) → LUMO+1(β)(79) | Os(dπ)/pap(π) → pap(π*) |
| 346 (348) | 25460 (0.014) |
HOMO−4(β) → LUMO+1(β)(86) | 9-OP(π)/pap(π) → pap(π*) |
| 308 (302) | 23990 (0.018) |
HOMO−7(β) → LUMO+2(β)(81) | pap(π) → pap(π*) |
| 2 − (S = 0) | |||
| 804 (637) | 8570 (0.032) | HOMO−2 → LUMO(66) | Os(dπ)/pap(π) → 9-OP(π*) |
| 320 (331) | 31850 (0.044) |
HOMO−5 → LUMO+1(51) | pap(π) → pap(π*) |
| 2 2− (S = 1/2) | |||
| 790 (850) | 7650 (0.008) | HOMO(β) → LUMO+1(β)(98) | pap(π)/Os(dπ) → 9-OP(π*) |
| 738 (706) | 7530 (0.005) | HOMO−3(α) → LUMO(α)(55) | Os(dπ)/pap(π) → pap(π*) |
| 690 (632) | 7250 (0.020) | HOMO−4(α) → LUMO(α)(65) | Os(dπ)/pap(π) → pap(π*) |
| 519 (538) | 9520 (0.027) | HOMO−2(β) → LUMO+1(β)(71) | Os(dπ)/pap(π) → 9-OP(π*) |
| 415 (415) | 18970 (0.007) |
HOMO(α) → LUMO+7(α)(64) | 9-OP(π) → 9-OP(π*) |
| 335 (361) | 31090 (0.038) |
HOMO−5(β) → LUMO+1(β) (61) | 9-OP(π) → 9-OP(π*) |
EPR spectroscopy and spin density calculations
The odd-electron forms 1/2 and 12+/22+ of the redox series [Os(9-OP)L2]n have been studied by in situ EPR spectroscopy. Electrolytic reduction of 1+ at room temperature in CH2Cl2/0.1 M Bu4NPF6 produces an unresolved signal of 1 at giso = 2.006 (Fig. S4†), a typical value for complexes between the bpy˙− anion radical ligand12 and diamagnetic metal complex fragment.13,14 An oxidation state description [OsII(9-OP−)(bpy˙−)(bpy)] is thus formulated for 1. At 120 K in frozen solution, a slight g factor anisotropy can be detected (Table 5, Fig. S4†) which is in agreement with a π type radical ion species.12 A metal isotope coupling from 189Os (16.1% nat. abundance, I = 3/2)15 is detected at Aiso = 8 G (Fig. S4†) which confirms8d the calculated minor contribution from the metal to the spin distribution (Table 4, Fig. 4).Oxidation to 12+ did not produce an EPR signal at ambient or low temperature, suggesting predominant spin location at the metal with its rather high spin–orbit coupling constant of about 3000 cm−1.15 Osmium(III) species are thus frequently EPR silent due to widely spread g components and rapid relaxation.7 DFT spin density calculations confirm a bpy-based unpaired electron in 1 and an osmium(III) species 12+ (Table 4, Fig. 4).
Reduction of [OsII(9-OP−)(pap)2]+ to 2 in the EPR cavity did not result in a detectable EPR signal at 298 or 120 K, although a pap-based unpaired electron is expected, as predicted also by DFT spin density calculations (Table 4, Fig. 4). As in previous studies16 we assume facile electron hopping between equivalent (degenerate) sites for spin accommodation in [OsII(9-OP−)(pap˙−)(pap)] which can result in severe EPR line broadening.17 Apparently, the barrier for such intramolecular electron transfer [OsII(9-OP−)(L˙−)(L°)] ⇌ [OsII(9-OP−)(L°)(L˙−)] is very different in 1 and 2. However, the expected pap based free radical EPR of 2 has been detected at 4 K which in effect corroborates the aforesaid rationale for its absence at higher temperature. Unexpectedly, the one-electron oxidation of 2+ in CH2Cl2/0.1 M Bu4NPF6 gave an EPR signal at room temperature (giso = 1.986, Fig. S4†) which displayed a nearly axial g anisotropy in the frozen state at 125 K (Fig. 5, Table 5).
The signal detection even at 298 K, the isotropic g and the anisotropy Δg = g1 − g3 = 0.061 suggests6,18 a predominantly ligand-based spin with minor metal contribution. The oxidizable ligand would be 9-OP− which has been proven to exhibit a two-way non-innocent behaviour (9-OP˙2−/9-OP−/9-OP˙);2a,b the resulting species must then be formulated as [OsII(9-OP˙)(pap)2]2+. This experimental result is in full agreement with the notion19 that strongly π-accepting pap stabilises the lower valent state (here OsII) which in turn leaves only the ligand remaining for oxidation (Scheme 3). Similar strategies have been employed for ruthenium complexes of triazenides or amidines which are oxidised to triazenyl or amidinyl complexes when the metal is made electron-poor through the coordination of π acceptors.4c,20
The bpy containing system 1n is obviously more susceptible to metal-based oxidation in 12+ due to the less pronounced π-acceptor strength of the 2,2′-bipyridine co-ligands (Scheme 3).
The experimental results illustrated in Scheme 3 could not be satisfactorily reproduced by DFT calculations. For instance, a metal centred spin was calculated for both 12+ and 22+. However, the g parameters and Os hyperfine coupling constant for neutral 1 were reasonably reproduced by DFT. Attempts to obtain the optimised structure corresponding to the state with a spin localised at the ligand in 22+ were unsuccessful. This discrepancy may be caused by the existence of two closely lying minima with a low energy barrier and by the failure of one-determinant DFT to fully reproduce the pronounced structural effects of strong osmium-to-pap π back donation in order to reach the appropriate minimum.
The ambivalent situation as illustrated in Scheme 3, i.e. the generation of an intramolecular oxidation state shift through the variation of the ancillary ligands can be similarly observed e.g. for copper/quinone21 and iron/quinone compounds22 (Scheme 4) where such redox isomer alternatives can have biochemical implications.23
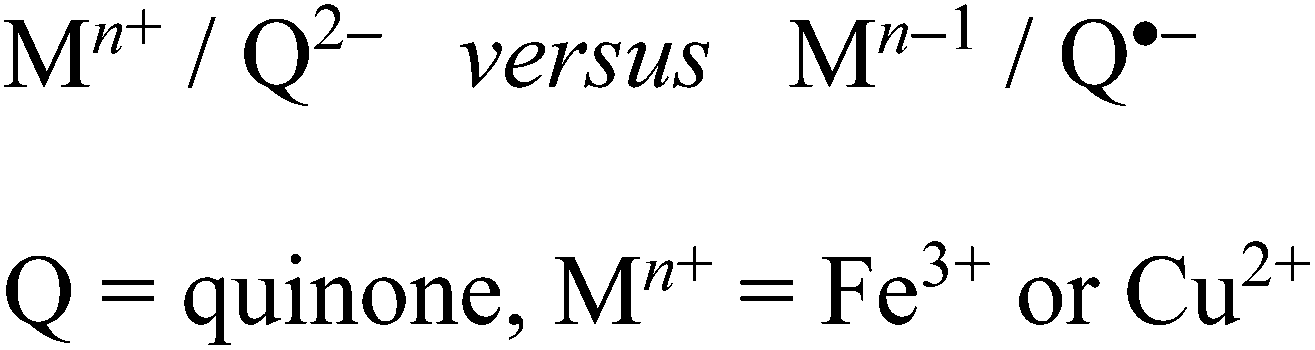 | ||
| Scheme 4 Metal/quinone oxidation state ambivalence. | ||
For a heavy 5dn transition metal from the platinum metal group, this kind of redox isomerism is reported here for the first time.
Other oxidation states accessible by spectroelectrochemistry were EPR silent. The precursor complexes 1+ and 2+ are S = 0 species as is the spin ground state of 2− (Scheme 5) while 1− is a triplet species (Scheme 5). For 22− the DFT calculations suggest an S = 1/2 spin ground state with two antiferromagnetically coupled pap˙− ligands whereas 12− was calculated to have an S = 3/2 ground state; however, this latter form could not be generated reversibly. The DFT calculated structures and MO compositions (Tables S5–S17†) confirm the following assignments.
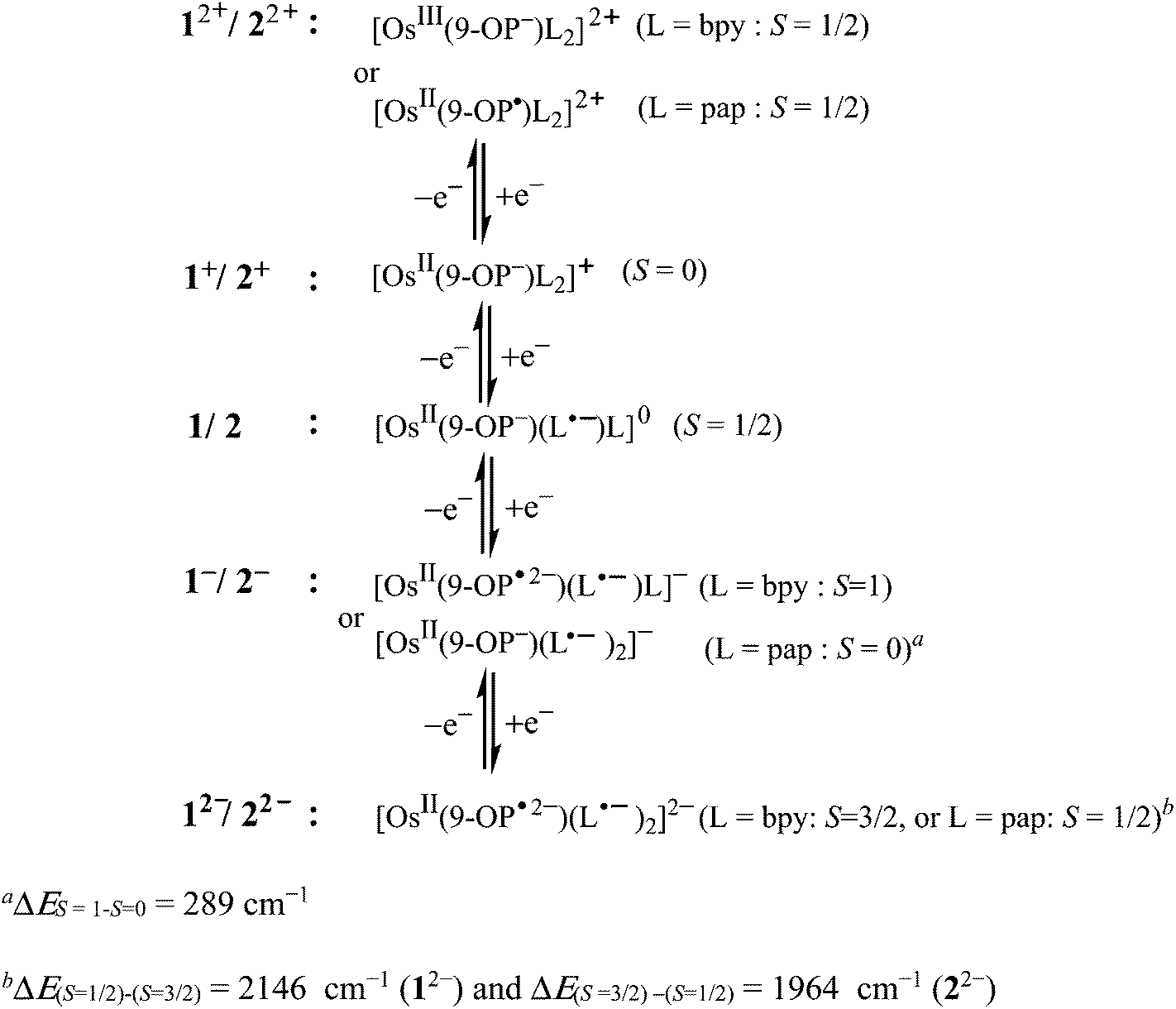 | ||
| Scheme 5 Assignments of most appropriate oxidation states within the redox series 1n and 2n. | ||
UV-vis-NIR spectroelectrochemistry and TD-DFT calculations
According to UV-vis-NIR spectroelectrochemistry, using an optically transparent thin-layer electrolysis (OTTLE) cell,24 reversible electron transfer processes could be monitored for transitions 12+–1− and 22+–22−. This approach allowed us to study the EPR active and EPR silent species as well as diamagnetic states. The spectra are shown in Fig. 6 while the absorption data are summarised in Table 6 together with the results from time-dependent DFT (TD-DFT) calculations.The structurally characterised precursor cations [OsII(9-OP−)L2]+ exhibit the expected metal-to-ligand charge transfer (MLCT) transitions in the visible, with the π* orbitals of the acceptors bpy or pap as the target. Compared with the ruthenium analogues 3+ and 4+ (ref. 2a,b) the osmium compounds exhibit decreased MLCT transition energy, in agreement with the more destabilised metal d orbitals of the heavier homologue. Ligand-to-ligand charge transfer (LLCT) and intra-ligand (IL) transitions occur at higher energies than the MLCT absorptions (Table 6).
The ligand (L)-based reduction of the compounds results in shifted MLCT bands and in typically25 weak “intra-radical” bands in the near infrared regions.26 While the spectral response is similar for 1 and 2, the two-electron reduced forms 1− and 2− differ considerably, both spectroscopically and according to the TD-DFT calculation results. Whereas spin–spin coupling between the two pap˙− ligands leads to a diamagnetic species 2− (S = 0) with only one major band in the visible region (metal-to-(9-OP) ligand charge transfer), the corresponding system 1− with two bpy˙− ligands is computed with an S = 1 spin ground state and various LLCT and MLCT transitions in the near infrared and visible regions (Fig. 6, Table 6). The 2− form, reversibly accessible only for the pap-containing series, exhibits several absorptions in the visible, mostly of MLCT character.
The oxidised forms 12+ and 22+ were identified as rather different (Scheme 5) through EPR spectroscopy, although the DFT calculations did not well reproduce the experimentally observed (9-OP) ligand-centred spin distribution. Therefore, the TD-DFT results must also be used with caution although the absorption spectral data appear generally well reproduced (Table 6). However, a look at the spectra (Fig. 6) illustrates a rather different appearance of 12+ and 22+ in the visible region, which suggests qualitatively different electronic structures as deduced already from the EPR studies (Scheme 5).
Conclusion
The present study is complementary to previous reports2,5 on iron and ruthenium complexes with a non-innocent 9-oxidophenalenone ligand. The use of the heavier group 8 metal osmium produces a remarkable array of differences for all four members of the redox series [Os(9-OP) L2]n, n = 2+, +, 0, −, when the co-ligands L are changed from bpy to the more π-accepting pap.The combination between the third-row transition metal osmium and two different kinds of non-innocently behaving ligands in complexes [Os(9-OP)L2]n (L = bpy or pap) produced a redox series with remarkably diverse oxidation state arrangements.
(i) Even the structurally characterised precursor cations [OsII(9-OP−)L2]+ exhibit a notable difference between the more conventional 1+ and the pap-containing ion 2+ with significant structural effects of very strong d(Os) → π*(pap) back donation.
(ii) Starting from there, the spectroelectrochemical studies and partially successful calculations revealed the expected reduction of one of the acceptor ligands L. However, the absence of an EPR signal for [OsII(9-OP−)(pap˙−)(pap)]0 in contrast to [OsII(9-OP−)(bpy˙−)(bpy)]0 (and [RuII(9-OP−)L2]0)2a,b points to different dynamics (barrier) for intramolecular electron exchange (“spin hopping”) between L˙− and L.
(iii) Furthermore surprising is the dichotomous behaviour on oxidation of the precursors to [Os(9-OP)L2]2+: whereas the EPR silence of the bpy containing a complex ion suggests a metal-oxidised form [OsIII(9-OP−)(bpy)2]2+, similar to both ruthenium species [RuIII(9-OP−)L2]2+ (L = bpy2a and pap2b), the stabilisation of the divalent metal state through strong π*(pap) to d(Os) back donation leads to an EPR-spectroscopically evidenced oxidation of the β-diketonate ligand 9-oxidophenalenone to the corresponding neutral radical ligand (Schemes 3 and 5).
This unexpected ambivalence, the first recognized here for a 5dn transition metal situation, represents another case of “hidden” noninnocence4b and may be related to similar observations in the biochemically relevant metal/quinone series (Fe, Cu; Scheme 4). Computational approaches to such ambivalent arrangements are difficult27 as confirmed here, and more sophisticated methods will have to be applied.
(iv) Although not accessible by EPR but supported by spectroelectrochemistry there is a further dichotomy suggested by DFT calculations for the two-electron reduction products [OsII(9-OP−)(L˙−)2]−, with antiferromagnetically coupled pap˙− ligands (S = 0) in contrast to the case with L˙− = bpy (S = 1).
Summarising, in the series of Ru/Os and bpy/pap combinations 1n–4n the osmium(II)-pap π backbonding is clearly distinguished by its strength, causing structural effects, dichotomous metal–ligand spin shifts, and strong inter-ligand interactions resulting in spin isomerism and variable electron hopping.
Experimental section
Materials
The starting metal precursors, cis-[Os(bpy)2Cl2]28 and ctc-[Os(pap)2Cl2]29 and the ligand precursor, 9-hydroxy-1H-phenalen-1-one (H(9-OP)),30 were prepared according to the reported procedures. Other chemicals and solvents were of reagent grade and used as received. For spectroscopic and electrochemical studies, HPLC grade solvents were used.Physical measurements
The electrical conductivity of solutions was checked by using an autoranging conductivity meter (Toshcon Industries, India). 1H NMR spectra were recorded on a Bruker Avance III 500 MHz spectrometer. The EPR measurements were made in a two electrode capillary tube31 with an X-band (9.5 GHz) Bruker system ESP300 spectrometer; for better formation of glassy frozen solutions the measurements were performed in CH2Cl2/0.1 M Bu4NPF6. Cyclic voltammetric and differential pulse voltammetric measurements of the complexes were done using a PAR model 273A electrochemistry system under a dinitrogen atmosphere. A glassy carbon-working electrode, a platinum wire auxiliary electrode, and a saturated calomel reference electrode (SCE) were used in a standard three-electrode configuration with tetraethylammonium perchlorate (TEAP) as the supporting electrolyte and a scan rate of 100 mV s−1. UV-vis-NIR spectroelectrochemical experiments were performed in CH3CN/0.1 M Bu4NPF6 at 298 K using an optically transparent thin-layer electrode cell (OTTLE) mounted in the sample compartment of a J&M TIDAS spectrophotometer.24 All spectroelectrochemical experiments were performed under a dinitrogen atmosphere. Elemental analyses were performed on a Perkin Elmer 240C elemental analyser. IR spectra of the complexes as KBr pellets were recorded on a Nicolet spectrophotometer. Electrospray mass spectra (ESI-MS) were recorded on a Bruker Maxis Impact instrument (282001.00081). Program easyspin-4.5.5 with Matlab R2014b was used for EPR simulation.32
Preparation of complexes
:1 ethanol:water and refluxed under a dinitrogen atmosphere for 72 h. The initial light brown colour of the solution gradually changed to dark violet. The reaction mixture was evaporated to dryness, the crude product was redissolved in a minimum volume of CH3CN, and 10 cm3 saturated NaClO4 solution was added. The resulting violet precipitate was filtered off and washed twice with ice-cold distilled water and dried under vacuum. The crude product was purified by using a neutral alumina column. The pure violet complex [1]ClO4 was eluted by a 5:1 solvent mixture of dichloromethane–acetonitrile. Evaporation of the solvent under reduced pressure yielded pure [1]ClO4.
[1]ClO4: yield: 75 mg, 61%. Anal. calcd for C33H23ClN4O6Os: C, 49.72; H, 2.91; N, 7.03; found: C, 50.01; H, 3.02; N, 6.98. ΛM (Ω−1 cm2 M−1) in acetonitrile at 298 K: 95. ESI-MS(+) in CH3CN, m/z calcd for {1+}: 697.79; found: 697.13. 1H-NMR (400 MHz, CDCl3): δ(ppm, J(Hz)): 8.46 (d, 2H, 8.04), 8.34 (d, 1H, 8.0), 8.22 (d, 2H, 7.7), 8.16 (d, 2H, 7.5), 8.06 (d, 2H, 9.3), 7.65 (t, 2H, 7.7), 7.36–7.45 (m, 6H), 7.17 (d, 2H, 7.4), 6.93 (t, 2H, 6.7), 6.88 (d, 2H, 8.2). ν(ClO4−, cm−1): 1090, 622.
:1 ethanol:water was refluxed under a dinitrogen atmosphere for 72 h. The initial violet colour of the solution gradually turned to dark red. The reaction mixture was evaporated to dryness. It was redissolved in a minimum volume of CH3CN and a saturated solution of NaClO4 (10 cm3) was added. The resulting red precipitate was filtered off, washed twice with ice-cold distilled water and dried under vacuum. It was purified further by using a neutral alumina column. The pure red complex [2]ClO4 was eluted by a 8:1 dichloromethane–acetonitrile solvent mixture. Evaporation of the solvent under reduced pressure yielded pure [2]ClO4.
[2]ClO4: yield: 80 mg, 67%. Anal. calcd for C35H25ClN6O6Os: C, 49.38; H 2.96; N 9.87; found: C, 49.51; H, 3.05; N, 9.89. ΛM (Ω−1 cm2 M−1) in acetonitrile at 298 K: 106. ESI-MS(+) in CH3CN, m/z calcd for {2+}: 751.84; found: 751.17. 1H-NMR (400 MHz, CDCl3): δ(ppm, J(Hz)):8.69 (d, 2H, 8.2), 8.05 (m, 4H), 7.89 (t, 2H, 7.7), 7.64–7.68 (m, 3H), 7.34 (t, 2H, 7.5), 7.15–7.28 (m, 8H), 6.71 (d, 4H, 8.1). ν(ClO4−, cm−1): 1093, 619.
(Caution! Perchlorate salts are explosive and should be handled with care).
Crystal structure determination
Single crystals of [1]ClO4 and [2]ClO4 were grown by slow evaporation of 1:1 CH2Cl2–benzene and 1:2 CH3CN–benzene solutions, respectively. X-ray crystal data were collected on a RIGAKU SATURN-724+ CCD single crystal X-ray diffractometer using Mo-Kα radiation. Data collection was evaluated by using the CrystalClear-SM Expert software. The data were collected by the standard ω-scan technique. The structure was solved by direct methods using SHELXS-97 and refined by full matrix least squares with SHELXL-97, refining on F2.33 All non-hydrogen atoms were refined anisotropically. The hydrogen atoms were placed in geometrically constrained positions and refined with isotropic temperature factors, generally 1.2Ueq of their parent atoms. Hydrogen atoms were included in the refinement process as per the riding model. CCDC no. 1465137 and 1465138 contain the supplementary crystallographic data for [1]ClO4 and [2]ClO4, respectively.
Computational details
Calculations were performed with Gaussian0934 and ADF 2016.0135 program packages. DFT geometry optimization of 1+ and 2+ utilized the Perdew, Burke, Ernzerhof hybrid functional36 (PBE0) together with quasi-relativistic effective core pseudopotentials and a correspondingly optimised set of basis functions for Os37 and the polarized triple ζ basis set 6-311 g(d)38 for the remaining atoms. In order to rationalise the computation time, geometry optimisations within the whole series were carried out by using the density functional theory method at the (R)B3LYP level for 1+, 2+, 2− and the (U)B3LYP level39 for 13+, 12+, 1, 1−, 12− and 23+, 22+, 2, 22− with the Gaussian 09 program package.34 Except osmium all other elements were assigned the 6-31G* basis set. The LANL2DZ basis set with an effective core potential was employed for the osmium atom.37 The vibrational frequency calculations were performed to ensure that the optimised geometries represent the local minima and there are only positive eigenvalues. Vertical electronic excitations based on (R)B3LYP/(U)B3LYP optimized geometries were computed for 1n (n = +2, +1, 0, −1) and 2n (n = +2, +1, 0, −1, −2) using the time-dependent density functional theory (TD-DFT) formalism40 in acetonitrile using the conductor-like polarisable continuum model (CPCM).41A and g tensors were obtained by first-order perturbation theory from a ZORA Hamiltonian42,43 in the presence of a time-independent magnetic field.44 The g tensor was obtained from a spin-non-polarised wave function after incorporating the spin–orbit (SO) coupling using the PBE0 functional. Chemissian 1.745 was used to calculate the fractional contributions of various groups to each molecular orbital. All calculated structures were visualised with ChemCraft.46Acknowledgements
Financial support received from the Department of Science and Technology, University Grant Commission (fellowship to A. H.), New Delhi (India) and the Land Baden-Württemberg (Germany) is gratefully acknowledged.References
- (a) S. K. Mandal, S. Samanta, M. E. Itkis, D. W. Jensen, R. W. Reed, R. T. Oakley, F. S. Tham, B. Donnadieu and R. C. Haddon, J. Am. Chem. Soc., 2006, 128, 1982–1994 CrossRef CAS PubMed; (b) S. K. Pal, F. S. Tham, R. W. Reed, R. T. Oakley and R. C. Haddon, Polyhedron, 2005, 24, 2076–2083 CrossRef CAS; (c) T. K. Sen, A. Mukherjee, A. Modak, P. K. Ghorai, D. Kratzert, M. Granitzka, D. Stalke and S. K. Mandal, Chem. −Eur. J., 2012, 18, 54–58 CrossRef CAS PubMed.
- (a) A. Das, T. Scherer, S. M. Mobin, W. Kaim and G. K. Lahiri, Inorg. Chem., 2012, 51, 4390–4397 CrossRef CAS PubMed; (b) A. Das, T. Scherer, P. Mondal, S. M. Mobin, W. Kaim and G. K. Lahiri, Chem. – Eur. J., 2012, 18, 14434–14443 CrossRef CAS PubMed; (c) H. Agarwala, T. Scherer, S. M. Mobin, W. Kaim and G. K. Lahiri, Dalton Trans., 2014, 43, 3939–3948 RSC; (d) A. Das, T. K. Ghosh, A. D. Chowdhury, S. M. Mobin and G. K. Lahiri, Polyhedron, 2013, 52, 1130–1137 CrossRef CAS.
- W. Kaim, Inorg. Chem., 2011, 50, 9752–9765 CrossRef CAS PubMed.
- (a) A. Mandal, A. Grupp, B. Schwederski, W. Kaim and G. K. Lahiri, Inorg. Chem., 2015, 54, 7936–7944 CrossRef CAS PubMed; (b) M. M. Khusniyarov, E. Bill, T. Weyhermüller, E. Bothe and K. Wieghardt, Angew. Chem., Int. Ed., 2011, 50, 1652–1655 ( Angew. Chem. , 2011 , 123 , 1690–1693 ) CrossRef CAS PubMed; (c) F. Ehret, M. Bubrin, S. Záliš and W. Kaim, Z. Anorg. Allg. Chem., 2014, 640, 2781–2787 CrossRef CAS.
- A. Pariyar, G. Vijaykumar, M. Bhunia, S. K. Dey, S. K. Singh, S. Kurungot and S. K. Mandal, J. Am. Chem. Soc., 2015, 137, 5955–5960 CrossRef CAS PubMed.
- W. Kaim and G. K. Lahiri, Angew. Chem., Int. Ed., 2007, 46, 1778–1796 ( Angew. Chem. , 2007 , 119 , 1808–1828 ) CrossRef CAS PubMed.
- W. Kaim and B. Sarkar, Coord. Chem. Rev., 2013, 257, 1650–1659 CrossRef CAS.
- (a) P. Ghosh, R. Ray, A. Das and G. K. Lahiri, Inorg. Chem., 2014, 53, 10695–10707 CrossRef CAS PubMed; (b) A. Das, S. M. Mobin and G. K. Lahiri, Dalton Trans., 2015, 44, 13204–13219 RSC; (c) A. Das, H. Agarwala, T. Kundu, P. Ghosh, S. Mondal, S. M. Mobin and G. K. Lahiri, Dalton Trans., 2014, 43, 13932–13947 RSC; (d) S. Ye, B. Sarkar, C. Duboc, J. Fiedler and W. Kaim, Inorg. Chem., 2005, 44, 2843–2847 CrossRef CAS PubMed.
- P. Majumdar, S.-M. Peng and S. Goswami, J. Chem. Soc., Dalton Trans., 1998, 1569–1574 RSC.
- (a) A. Das, T. Scherer, S. M. Mobin, W. Kaim and G. K. Lahiri, Chem. – Eur. J., 2012, 18, 11007–11018 CrossRef CAS PubMed; (b) K. J. H. Young, O. A. Mironov, R. J. Nielsen, M.-J. Cheng, T. Stewart, W. A. Goddard and R. A. Periana, Organometallics, 2011, 30, 5088–5094 CrossRef CAS.
- (a) A. Mandal, A. Grupp, B. Schwederski, W. Kaim and G. K. Lahiri, Inorg. Chem., 2015, 54, 10049–10057 CrossRef CAS PubMed; (b) A. Das, P. Ghosh, S. Plebst, B. Schwederski, S. M. Mobin, W. Kaim and G. K. Lahiri, Inorg. Chem., 2015, 54, 3376–3386 CrossRef CAS PubMed.
- W. Kaim, Coord. Chem. Rev., 1987, 76, 187–235 CrossRef CAS.
- W. Kaim, Chem. Ber., 1981, 114, 3789–3800 CrossRef CAS.
- C. Wolff, A. Gottschlich, J. England, K. Wieghardt, W. Saak, D. Haase and R. Beckhaus, Inorg. Chem., 2015, 54, 4811–4820 CrossRef CAS PubMed.
- J. A. Weil, J. R. Bolton and J. E. Wertz, Electron Paramagnetic Resonance, Wiley, New York, 1994, p. 532–536 Search PubMed.
- M. Heilmann, F. Baumann, W. Kaim and J. Fiedler, J. Chem. Soc., Faraday Trans., 1996, 92, 4227–4231 RSC.
- D. E. Morris, K. W. Hanck and M. K. DeArmond, J. Am. Chem. Soc., 1983, 105, 3032–3038 CrossRef CAS.
- S. Patra, B. Sarkar, S. M. Mobin, W. Kaim and G. K. Lahiri, Inorg. Chem., 2003, 42, 6469–6473 CrossRef CAS PubMed.
- D. Das, B. Sarkar, T. K. Mondal, S. M. Mobin, J. Fiedler, W. Kaim and G. K. Lahiri, Inorg. Chem., 2011, 50, 7090–7098 CrossRef CAS PubMed.
- (a) F. Ehret, M. Bubrin, S. Záliš and W. Kaim, Angew. Chem., Int. Ed., 2013, 52, 4673–4675 ( Angew. Chem. , 2013 , 125 , 4771–4773 ) CrossRef CAS PubMed; (b) F. Ehret, M. Bubrin, S. Záliš and W. Kaim, Chem. – Eur. J., 2015, 21, 12275–12278 CrossRef CAS PubMed.
- J. Rall and W. Kaim, J. Chem. Soc., Faraday Trans., 1994, 90, 2905–2908 RSC.
- N. Shaikh, S. Goswami, A. Panja, X.-Y. Wang, S. Gao, R. J. Butcher and P. Banerjee, Inorg. Chem., 2004, 43, 5908–5918 CrossRef CAS PubMed.
- W. Kaim, B. Schwederski and A. Klein, Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life, John Wiley & Son, Chichester, 2nd edn, 2013 Search PubMed.
- M. Krejcik, M. Danek and F. Hartl, J. Electroanal. Chem., 1991, 317, 179–187 CrossRef CAS.
- (a) P. S. Braterman, J.-I. Song, F. M. Wimmer, S. Wimmer, W. Kaim, A. Klein and R. D. Peacock, Inorg. Chem., 1992, 31, 5084–5088 CrossRef CAS; (b) W. Kaim, R. Reinhardt and M. Sieger, Inorg. Chem., 1994, 33, 4453–4459 CrossRef CAS.
- W. Kaim, Coord. Chem. Rev., 2011, 255, 2503–2513 CrossRef CAS.
- C. Remenyi and M. Kaupp, J. Am. Chem. Soc., 2005, 127, 11399–11413 CrossRef CAS PubMed.
- P. A. Lay, M. Sargeson and H. Taube, Inorg. Synth., 1986, 24, 291–299 CrossRef CAS.
- B. K. Ghosh, S. Goswami and A. Chakravorty, Inorg. Chem., 1983, 22, 3358–3360 CrossRef CAS.
- R. C. Haddon, R. Rayford and A. M. Hirani, J. Org. Chem., 1981, 46, 4587–4588 CrossRef CAS.
- W. Kaim, S. Ernst and V. Kasack, J. Am. Chem. Soc., 1990, 112, 173–178 CrossRef CAS.
- S. Stoll and A. Schweiger, EasySpin, a comprehensive software package for spectral simulation and analysis in EPR, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr, 2008, 64, 112–122 CrossRef CAS PubMed; (b) Program for Crystal Structure Solution and Refinement, University of Goettingen, Goettingen, Germany, 1997 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision A.02), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- ADF2016.01, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com Search PubMed.
- (a) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed; (b) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- (a) D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS; (b) P. Fuentealba, H. Preuss, H. Stoll and L. von Szentpaly, Chem. Phys. Lett., 1989, 89, 418–422 CrossRef; (c) J. M. L. Martin and A. Sundermann, J. Chem. Phys., 2001, 114, 3408 CrossRef CAS.
- K. Raghavachari, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- (a) R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS; (b) R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8225 CrossRef CAS; (c) M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4450 CrossRef CAS.
- (a) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS; (b) M. Cossi and V. Barone, J. Chem. Phys., 2001, 115, 4708–4718 CrossRef CAS; (c) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- E. V. Lenthe, A. E. Ehlers and E. J. Baerends, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef.
- E. V. Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef.
- (a) E. V. Lenthe, A. van der Avoird and P. E. S. Wormer, J. Chem. Phys., 1997, 107, 2488 CrossRef; (b) E. V. Lenthe, A. van der Avoird and P. E. S. Wormer, J. Chem. Phys., 1998, 108, 4783 CrossRef.
- S. Leonid, Chemissian 1.7, 2005–2010. Available at http://www.chemissian.com Search PubMed.
- D. A. Zhurko and G. A. Zhurko, ChemCraft 1.5, Plimus, San Diego, CA. Available at http://www.chemcraftprog.com Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic file for [1]ClO4 and [2]ClO4 in CIF format, mass spectrometry (Fig. S1), NMR spectra (Fig. S2), DFT optimised structures (Fig. S3), bond parameters (Tables S1–S4), DFT data (Tables S5–S17). CCDC 1465137 and 1465138. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt03764j |
| This journal is © The Royal Society of Chemistry 2016 |