
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterization of metal–rich phosphonium polyelectrolytes and their use as precursors to nanomaterials†
Amir
Rabiee Kenaree
and
Joe B.
Gilroy
*
Department of Chemistry and the Centre for Advanced Materials and Biomaterials Research (CAMBR), The University of Western Ontario, 1151 Richmond St. N., London, Ontario, Canada N6A 5B7. E-mail: joe.gilroy@uwo.ca; Tel: +1-519-661-2111 ext. 81561
First published on 31st October 2016
Abstract
Upon efficient quaternization and salt metathesis of stable triethyl ferrocene/ruthenocene phosphines, styrene-based phosphonium triflate monomers with four different stoichiometric ratios of Fe/Ru were synthesized. Free-radical polymerization of the monomers afforded four polyelectrolytes (Mn: 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 650–69100 g mol−1, Đ: 3.16–4.10) that retained many of the spectroscopic and electrochemical properties of the ferrocene/ruthenocene units. TGA studies demonstrated the thermal stability (onset of decomposition: ∼310 °C) and high char yields (33–54% at 1000 °C) of the polyelectrolytes. Pyrolysis in N2/H2 (95/5) (film thickness of ∼6 μm, 1000 °C, 3 h) yielded crystalline, mixed-phase nanomaterials containing iron, ruthenium, and phosphorus with compositions influenced by the structure of the parent polyelectrolytes.
650–69100 g mol−1, Đ: 3.16–4.10) that retained many of the spectroscopic and electrochemical properties of the ferrocene/ruthenocene units. TGA studies demonstrated the thermal stability (onset of decomposition: ∼310 °C) and high char yields (33–54% at 1000 °C) of the polyelectrolytes. Pyrolysis in N2/H2 (95/5) (film thickness of ∼6 μm, 1000 °C, 3 h) yielded crystalline, mixed-phase nanomaterials containing iron, ruthenium, and phosphorus with compositions influenced by the structure of the parent polyelectrolytes.
Introduction
Metallopolymers, which differ from coordination polymers that have dynamic structures, are an intriguing class of materials that benefit from the processability of macromolecules and functional properties of transition metals.1 To date, many metallopolymers have been successfully synthesized and utilized as redox-active, catalytic, emissive, biomedical, and magnetic materials.2 However, examples of heterobimetallic polymers, which can take advantage of the properties of more than one type of metal, are far less common.There are several existing strategies for the incorporation of more than one type of transition metal into polymer structures. For example, post-polymerization functionalization of metallopolymers can be employed for the addition of transition metals to the repeating unit of the polymer backbone.3 However, it can be a challenge to completely functionalize all of the repeating units in the polymer backbone. Manners and co-workers have used their well-established ring-opening polymerization methodology for the synthesis of polyferrocenylsilanes (PFSs) to prepare monometallic acetylide-substituted PFSs, which were further reacted with Co2(CO)8, [MoCp(CO)2]2, and [NiCp(CO)2]2 to produce heterobimetallic polymers 1.3a–c They also showed that reactive ion etching (RIE),3a electron-beam lithography,3b and pyrolysis3c can be used to convert the heterobimetallic polymers produced to the corresponding bimetallic alloy nanoparticles (NPs).
Copolymerization of more than one type of metal-containing monomer is another strategy that can yield heterobimetallic polymers, where the metal ratio can be adjusted by controlling the ratio of the repeating units.4 Following this strategy and starting with two methacrylate-based ferrocene- and cobaltocenium-containing monomers, the Tang group recently performed a reversible addition-fragmentation chain-transfer (RAFT) polymerization and successfully synthesized heterobimetallic diblock copolymers containing cobaltocenium and ferrocene units 2. By pyrolysis of the heterobimetallic copolymer under N2/H2, magnetic nanomaterials comprised of FexCoyP (where x + y = 2) were realized.4e
Starting with a monomer that has more than one metal within its structure is a strategy that affords heterobimetallic polymers with transition metal content equal to that of the monomer.5 For instance, Wong and co-workers synthesized an iron- and platinum-containing heterobimetallic polymer and used nanoimprint lithography to generate nanopatterns of the resulting polymer 3. RIE converted the polymer to nanopatterned magnetic Fe/Pt NPs.5c The Manners group has also reported the synthesis of a palladium-based [1]ferrocenophane 4 that was thermolized at 190 °C under vacuum to directly yield Fe/Pd alloy NPs, presumably via a heterobimetallic polymer.5e
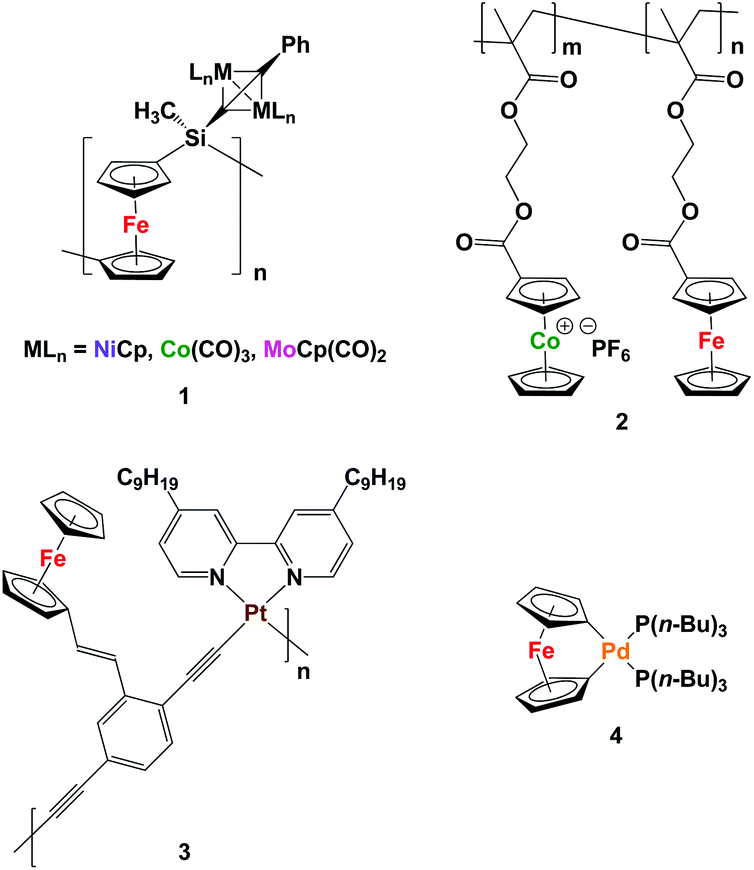
Herein, we describe our efforts to address this deficiency. Specifically, we have prepared polyelectrolytes based on phosphonium scaffolds containing ethylferrocene and ethylruthenocene units (Fe/Ru: 3/0, 2/1, 1/2, 0/3) and explored their preceramic properties by conducting pyrolysis experiments under a reducing atmosphere.
Experimental section
General considerations
Reactions and manipulations were carried out under a nitrogen atmosphere using standard glove box or Schlenk techniques unless otherwise stated. Solvents were obtained from Caledon Laboratories and Fischer Scientific, dried using an Innovative Technologies Inc. solvent purification system, collected under vacuum, and stored under a nitrogen atmosphere over 4 Å molecular sieves. Reagents were purchased from Sigma-Aldrich or Alfa Aesar and used as received, aside from 4-vinylbenzyl chloride which was purified according to a literature procedure and stored under N2 at −35 °C.12 Tertiary phosphines 5a–d were synthesized according to reported protocols.131H, 13C{1H}, 19F and 31P NMR spectra were recorded on a 600 MHz (1H: 599.5 MHz, 13C: 150.8 MHz, 19F: 563.9 MHz and 31P: 242.6 MHz) Varian INOVA instrument. 1H NMR spectra were referenced to residual (CD3)(CD2H)SO (2.50 ppm) and 13C{1H} NMR spectra were referenced to DMSO-d6 (39.5 ppm). 31P NMR spectra were referenced to PPh3 as an internal standard (−6.0 ppm relative to H3PO4). Mass spectrometry data were recorded in positive-ion mode using a Micromass/Waters Q-TOF Ultima LC-MS/MS system. UV-vis absorption spectra were recorded using a Cary 300 Scan instrument. Infrared spectra were recorded using a PerkinElmer Spectrum Two FT-IR spectrometer as thin films on KBr plates. Elemental analyses (C and H) were carried out by Laboratoire d'Analyse Élémentaire de l'Université de Montréal, Montréal, QC, Canada.Cyclic voltammetry
Cyclic voltammograms were collected using a Bioanalytical Systems Inc. (BASi) Epsilon potentiostat and analyzed using BASi Epsilon software. Typical electrochemical cells consisted of a three-electrode setup including a glassy carbon working electrode, platinum wire counter electrode, and silver wire pseudo-reference electrode. 1 mM degassed solutions of monomers 7a–d, combined with supporting electrolyte (0.1 M [n-Bu4N][OTf]), in a CH2Cl2/CH3CN (2/1) solvent mixture were prepared and run at a scan rate of 250 mV s−1 under a blanket of argon. To study the electrochemical behavior of the polymers, different solvents such as THF, DMF, CH3CN, and CH2Cl2 containing 0.1 M [n-Bu4N][OTf] were used to make 0.2 mM solutions of the analytes. In each case, severe plating of the oxidized forms of 8a–d was observed on the glassy carbon working electrode. Therefore, a 2/1 solvent mixture of CH2Cl2 and CH3CN was used as it was the least problematic combination. Degassed solutions of polyelectrolytes 8a–d in a CH2Cl2/CH3CN (2/1) solvent mixture were prepared by stirring the mixture overnight at 40 °C. After addition of the supporting electrolyte, the mixtures were sonicated for 20 s, filtered (Nylon membrane, 0.22 μm) and using these solutions electrochemical studies were performed at a scan rate of 250 mV s−1 under a blanket of argon. Cyclic voltammograms were referenced relative to a decamethylferrocene internal standard (1 mM, −520 mV relative to ferrocene/ferrocenium under identical conditions) and corrected for internal cell resistance using the BASi Epsilon software.X-ray diffraction studies
Single crystals of monomer 7a suitable for X-ray diffraction studies were grown by slow evaporation of a THF solution. The sample was mounted on a MiTeGen polyimide micromount with a small amount of Paratone N oil. X-ray diffraction measurements were made on a Bruker Kappa Axis Apex2 diffractometer at a temperature of 110 K. Initial indexing indicated that the sample crystal was non-merohedrally twinned. The twin law was determined to be:which represents a 179.8° rotation about [100]. The twin fraction was included in the refinement as an adjustable parameter (vide infra). The unit cell dimensions were determined from a symmetry constrained fit of 5386 reflections with 6.58° < 2θ < 47.88°. The data collection strategy was a number of ω and φ scans which collected data up to 53.538° (2θ). The frame integration was performed using SAINT.14 The resulting raw data was scaled and absorption corrected using a multi-scan averaging of symmetry equivalent data using TWINABS.15| 0.99635 | 0.00646 | 0.00848 |
| 0.00835 | −1.00086 | 0.00433 |
| 0.85236 | −0.00198 | −0.99547 |
The structure was solved by using a dual space methodology using the SHELXT program.16 All non-hydrogen atoms were obtained from the initial solution. The hydrogen atoms were introduced at idealized positions and were allowed to ride on the parent atom. The twin fraction refined to a value of 0.465(1). The structural model was fit to the data using full matrix least-squares based on F2. The calculated structure factors included corrections for anomalous dispersion from the usual tabulation. The structure was refined using the SHELXL-2014 program from the SHELX suite of crystallographic software.17 Graphic plots were produced using Mercury software (version 3.3). For additional collection and refinement details, see CCDC 1476067, Table 1 and Fig. 1.
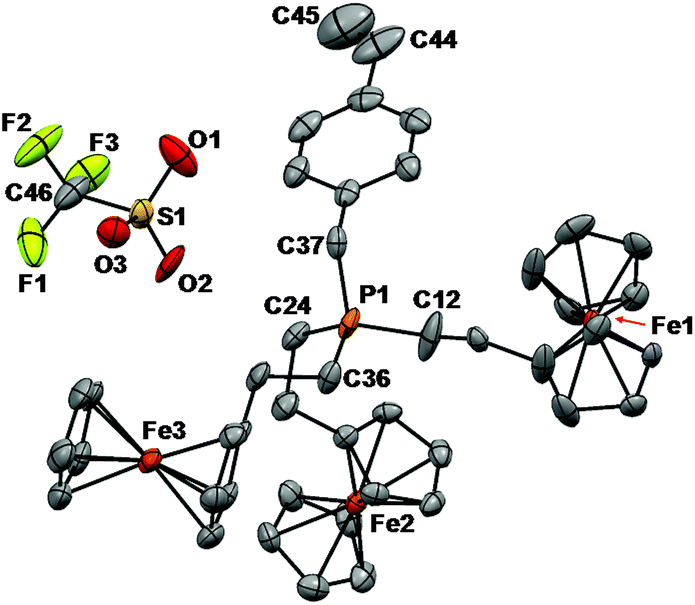 | ||
| Fig. 1 Solid-state structure of monomer 7a. Anisotropic displacement ellipsoids are shown at 50% probability and hydrogen atoms have been omitted for clarity. Selected bond lengths (Å): P1–C12 1.782(6), P1–C24 1.803(5), P1–C36 1.798(5), P1–C37 1.791(6), C44–C45 1.162(9). Selected bond angles (degrees): C12–P1–C24 111.7(3), C12–P1–C36 108.5(3), C12–P1–C37 110.9(3), C24–P1–C36 110.0(3), C24–P1–C37 106.2(2), and C36–P1–C37 109.6(3). | ||
| 7a | |
|---|---|
| R 1 = ∑(|Fo| − |Fc|)/∑Fo, wR2 = [∑(w(Fo2 − Fc2)2)/∑(wFo4)]1/2; GOF = [∑(w(Fo2 − Fc2)2)/(No. of reflns. − No. of params.)]1/2. | |
| Chemical formula | C46H48F3Fe3O3PS |
| FW (g mol−1) | 936.42 |
| Temp (K) | 110 |
| Crystal syst. | Triclinic |
| Crystal habit | Yellow plate |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| λ (Å) | 0.71073 |
| a (Å) | 12.378(6) |
| b (Å) | 12.619(5) |
| c (Å) | 14.252(7) |
| α (°) | 79.298(7) |
| β (°) | 68.524(10) |
| γ (°) | 89.953(7) |
| V (Å3) | 2030.1(17) |
| Z | 2 |
| ρ (g cm−3) | 1.532 |
| μ (cm−1) | 1.204 |
| R 1 [I > 2σ(I)] | 0.0534 |
| wR2 [I > 2σ(I)] | 0.1278 |
| R 1 (all data) | 0.0917 |
| wR2 (all data) | 0.1463 |
| GOF | 1.042 |
Powder X-ray diffraction (PXRD) data for nanomaterial films deposited on silicon wafers were acquired using an Inel CPS powder diffractometer with an Inel XRG 3000 generator and Inel CPS 120 detector using a CuKα radiation source. For diffractograms, see Fig. 5 and S36−S39.†
Gel permeation chromatography (GPC)
GPC experiments were performed by PolyAnalytik Inc. Canada (London, Ontario). Molecular weights and dispersities (Đ = Mw/Mn) were obtained using a Viscotek TDA302/GPCmax gel permeation chromatograph equipped with automatic sampler, isocratic pump, injector, in-line degasser, column and detector oven (60 °C), refractive index detector, and Viscotek Inert Series Columns: 1× Mixed Bed Low Molecular Weight (I-MBLMW, exclusion limit of 20 kDa PS) and 1× Mixed Bed High Molecular Weight (I-MBHMW, exclusion limit of 10000 g mol−1 PS). The eluent employed was DMF (60 °C) containing 0.02 M [n-Bu4N][OTf] at a flow rate of 0.7 mL min−1. Samples were dissolved in the eluent (5 mg mL−1), heated for 1.5 h, and filtered (Nylon membrane, 0.22 μm) before analysis. Conventional calibration of the refractive index detector was performed using a series of monodisperse polystyrene standards (PolyAnalytik). All data were processed using Viscotek's OmniSEC v4.6.2 software.
Thermal analysis
Thermal degradation studies were performed using a TA Instruments Q50 TGA instrument under an atmosphere of N2. Samples were placed in a platinum pan and heated at a rate of 10 °C min−1 from 20 °C to 1000 °C under a flow of N2 (60 mL min−1). Glass transition temperatures were determined under an atmosphere of N2 using differential scanning calorimetry (DSC) on a TA Instruments DSC Q20. The polymer samples were placed in an aluminum Tzero pan and heated from room temperature to 300 °C at 10 °C min−1 under a flow of N2 (50 mL min−1) and cooled down to 0 °C at 5 °C min−1, before they underwent two more heat/cool cycles. The glass transitions were determined from the second heat/cool cycle.Pyrolysis studies and scanning electron microscopy
Films of polyelectrolytes 8a–d were prepared by drop-casting 250 μL of a 20 mg mL−1 chlorobenzene solution of each polyelectrolyte onto a silicon wafer (A = 2.5 cm2). The samples were dried in air, transferred into a vacuum oven, and allowed to dry at 50 °C for 16 h before they were heated at a rate of 10 °C min−1 to a temperature of 1000 °C under a gentle flow (ca. 60 mL min−1) of a N2/H2 (95/5) gas mixture in a Lindberg Blue M tube furnace. The temperature was maintained at 1000 °C for an additional 3 h before the furnace was cooled to room temperature at a rate of 10 °C min−1. Polymer film thickness and the surface morphologies of thermally deposited nanomaterials on silicon wafers were assessed directly using scanning electron microscopy (SEM) at 1 keV beam energy using LEO/Zeisss 1530 and LEO/Zeisss 1540XB instruments. Energy dispersive X-ray spectroscopy (EDX) experiments were performed at 10 keV beam energy on the 1540XB with the equipped Oxford X-sight X-ray detector and INCA analysis software.Representative procedure for the preparation of 6a–d
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.86 (d, 3JHH,trans = 18 Hz, 1H, ArCHCH2), 5.26 (d, 3JHH,cis = 11 Hz, 1H, ArCHCH2), 4.17 (pseudo-t, 3JHH = 2 Hz, 6H, β-C5H4R), 4.12 (s, 15H, C5H5), 4.10 (pseudo-t, 3JHH = 2 Hz, 6H, α-C5H4R), 4.06 (d, 2JHP = 15 Hz, 2H, PCH2Ar), 2.59–2.43 (m, 12H, C5H4CH2CH2 and C5H4CH2CH2, overlaps with residual CD3CD2HSO signal). 13C{1H} NMR (DMSO-d6): δ 137.0 (d, JCP = 3 Hz), 135.9 (d, JCP = 2 Hz), 130.4 (d, JCP = 6 Hz), 128.8 (d, JCP = 8 Hz), 127.0 (d, JCP = 3 Hz), 115.1 (s), 86.8 (d, JCP = 17 Hz), 68.5 (s), 67.7 (s), 67.4 (s), 25.4 (d, JCP = 43 Hz), 20.8 (d, JCP = 2 Hz), 19.3 (d, JCP = 44 Hz). 31P{1H} NMR (DMSO-d6): δ 31.1 (s). FT-IR: 3092 (w), 3009 (w), 2922 (m), 2455 (w), 1629 (w), 1511 (w), 1410 (w), 1219 (w), 1105 (m), 1000 (m), 922 (w), 820 (m), 753 (s), 660 (w) cm−1. UV-vis (THF): λmax 285 nm (ε = 2620 M−1 cm−1), 295 nm (ε = 1380 M−1 cm−1), 325 nm (ε = 290 M−1 cm−1), 436 nm (ε = 300 M−1 cm−1). Mass Spec. (ESI, +ve mode): exact mass calculated for [C45H4856Fe3P]+: 787.1542; exact mass found: 787.1563; difference: +2.7 ppm.
CH2), 5.90 (d, 3JHH,trans = 18 Hz, 1H, ArCHCH2), 5.30 (d, 3JHH,cis = 11 Hz, 1H, ArCHCH2), 4.65 [s, 2H, β-C5H4R(Ru)], 4.57 [s, 5H, C5H5(Ru)], 4.51 [s, 2H, α-C5H5R(Ru)], 4.21 [s, 4H, β-C5H4R(Fe)], 4.16 [s, 10H, C5H5(Fe)], 4.13 [s, 4H, α-C5H5R(Fe)], 4.10 (d, 2JHP = 16 Hz, 2H, PCH2Ar), 2.65–2.30 (m, 12H, C5H4CH2CH2 and C5H4CH2CH2, overlaps with residual CD3CD2HSO signal). 13C{1H} NMR (DMSO-d6): δ 137.0 (d, JCP = 3 Hz), 135.9 (s), 130.4 (d, JCP = 5 Hz), 128.8 (d, JCP = 9 Hz), 126.9 (d, JCP = 3 Hz), 115.1 (s), 91.2 (d, JCP = 18 Hz), 86.8 (d, JCP = 17 Hz), 70.6 (s), 70.4 (s), 69.6 (s), 68.4 (s), 67.7 (s), 67.3 (s), 25.3 (d, JCP = 43 Hz), 20.8 (s), 20.2 (s), 20.0 (d, JCP = 45 Hz), 19.3 (d, JCP = 45 Hz). 31P{1H} NMR (DMSO-d6): δ 30.9 (s). FT-IR: 3091 (w), 3008 (w), 2922 (m), 2455 (w), 1629 (w), 1511 (w), 1410 (w), 1221 (w), 1104 (m), 999 (m), 918 (w), 811 (m), 753 (s), 661 (w) cm−1. UV-vis (THF): λmax 287 nm (ε = 2170 M−1 cm−1), 295 nm (ε = 1290 M−1 cm−1), 320 nm (ε = 420 M−1 cm−1), 431 nm (ε = 220 M−1 cm−1). Mass Spec. (ESI, +ve mode): exact mass calculated for [C45H4856Fe2P102Ru]+: 833.1236; exact mass found: 833.1259; difference: +2.8 ppm.
CH2), 5.92 (d, 3JHH,trans = 18 Hz, 1H, ArCHCH2), 5.31 (d, 3JHH,cis = 11 Hz, 1H, ArCHCH2), 4.65 [s, 4H, β-C5H4R(Ru)], 4.56 [s, 10H, C5H5(Ru)], 4.52 [s, 4H, α-C5H5R(Ru)], 4.21 [pseudo-t, 3JHH = 2 Hz, 2H, β-C5H4R(Fe)], 4.16 [s, 5H, C5H5(Fe)], 4.14 [pseudo-t, 3JHH = 2 Hz, 2H, α-C5H5R(Fe)], 4.06 (d, 2JHP = 16 Hz, 2H, PCH2Ar), 2.60–2.30 (m, 12H, C5H4CH2CH2 and C5H4CH2CH2, overlaps with residual CD3CD2HSO signal). 13C{1H} NMR (DMSO-d6): δ 137.0 (d, JCP = 5 Hz), 135.9 (s), 130.3 (d, JCP = 5 Hz), 128.7 (d, JCP = 9 Hz), 126.9 (d, JCP = 3 Hz), 115.1 (s), 91.1 (d, JCP = 17 Hz), 86.7 (d, JCP = 17 Hz), 70.6 (s), 70.4 (s), 69.7 (s), 68.4 (s), 67.7 (s), 67.4 (s), 25.3 (d, JCP = 43 Hz), 20.8 (s), 20.3 (d, JCP = 3 Hz), 20.0 (d, JCP = 44 Hz), 19.3 (d, JCP = 45 Hz). 31P{1H} NMR (DMSO-d6): δ 30.8 (s). FT-IR: 3095 (w), 3010 (w), 2923 (m), 2455 (w), 1630 (w), 1511 (w), 1409 (w), 1219 (w), 1101 (m), 998 (m), 918 (w), 810 (m), 753 (s), 666 (w) cm−1. UV-vis (THF): λmax 287 nm (ε = 2520 M−1 cm−1), 295 nm (ε = 1600 M−1 cm−1), 320 nm (ε = 680 M−1 cm−1), 434 nm (ε = 150 M−1 cm−1). Mass Spec. (ESI, +ve mode): exact mass calculated for [C45H4856FeP96Ru99Ru]+: 870.0978; exact mass found: 870.1002; difference: +2.8 ppm.
CH2), 5.90 (d, 3JHH,trans = 18 Hz, 1H, ArCHCH2), 5.31 (d, 3JHH,cis = 11 Hz, 1H, ArCHCH2), 4.64 (pseudo-t, 3JHH = 2 Hz, 6H, β-C5H4R), 4.55 (s, 15H, C5H5), 4.51 (pseudo-t, 3JHH = 2 Hz, 6H, α-C5H4R), 3.98 (d, 2JHP = 15 Hz, 2H, PCH2Ar), 2.27–2.47 (m, 12H, C5H4CH2CH2 and C5H4CH2CH2). 13C{1H} NMR (DMSO-d6): δ 137.1 (d, JCP = 3 Hz), 135.9 (s), 130.3 (d, JCP = 5 Hz), 128.6 (d, JCP = 9 Hz), 127.0 (s), 115.3 (s), 91.1 (d, JCP = 18 Hz), 70.6 (s), 70.5 (s), 69.8 (s), 25.4 (d, JCP = 44 Hz), 20.4 (s), 20.0 (d, JCP = 45 Hz). 31P{1H} NMR (DMSO-d6): δ 30.8 (s). FT-IR: 3093 (w), 3010 (w), 2919 (m), 2460 (w), 1670 (w), 1511 (w), 1409 (m), 1218 (w), 1100 (m), 997 (m), 917 (w), 809 (s), 752 (s), 660 (w) cm−1. UV-vis (THF): λmax 287 nm (ε = 2060 M−1 cm−1), 297 nm (ε = 1560 M−1 cm−1), 315 nm (ε = 890 M−1 cm−1). Mass Spec. (ESI, +ve mode): exact mass calculated for [C45H4896Ru99Ru100RuP]+: 914.0671; exact mass found: 914.0678; difference: +0.8 ppm.
CH2), 5.86 (d, 3JHH,trans = 18 Hz, 1H, ArCHCH2), 5.26 (d, 3JHH,cis = 11 Hz, 1H, ArCHCH2), 4.17 (pseudo-t, 3JHH = 2 Hz, 6H, β-C5H4R), 4.12 (s, 15H, C5H5), 4.10 (pseudo-t, 3JHH = 2 Hz, 6H, α-C5H4R), 4.06 (d, 2JHP = 15 Hz, 2H, PCH2Ar), 2.59–2.43 (m, 12H, C5H4CH2CH2 and C5H4CH2CH2, overlaps with residual CD3CD2HSO signal). 13C{1H} NMR (DMSO-d6): δ 137.0 (d, JCP = 3 Hz), 135.9 (d, JCP = 2 Hz), 130.4 (d, JCP = 6 Hz), 128.8 (d, JCP = 8 Hz), 127.0 (d, JCP = 3 Hz), 115.1 (s), 86.8 (d, JCP = 17 Hz), 68.5 (s), 67.7 (s), 67.4 (s), 25.4 (d, JCP = 43 Hz), 20.8 (d, JCP = 2 Hz), 19.3 (d, JCP = 44 Hz). 31P{1H} NMR (DMSO-d6): δ 31.1 (s). FT-IR: 3092 (w), 3009 (w), 2922 (m), 2455 (w), 1629 (w), 1511 (w), 1410 (w), 1219 (w), 1105 (m), 1000 (m), 922 (w), 820 (m), 753 (s), 660 (w) cm−1. UV-vis (THF): λmax 285 nm (ε = 2620 M−1 cm−1), 295 nm (ε = 1380 M−1 cm−1), 325 nm (ε = 290 M−1 cm−1), 436 nm (ε = 300 M−1 cm−1). Mass Spec. (ESI, +ve mode): exact mass calculated for [C45H4856Fe3P]+: 787.1542; exact mass found: 787.1563; difference: +2.7 ppm.
CH2), 5.90 (d, 3JHH,trans = 18 Hz, 1H, ArCHCH2), 5.30 (d, 3JHH,cis = 11 Hz, 1H, ArCHCH2), 4.65 [s, 2H, β-C5H4R(Ru)], 4.57 [s, 5H, C5H5(Ru)], 4.51 [s, 2H, α-C5H5R(Ru)], 4.21 [s, 4H, β-C5H4R(Fe)], 4.16 [s, 10H, C5H5(Fe)], 4.13 [s, 4H, α-C5H5R(Fe)], 4.10 (d, 2JHP = 16 Hz, 2H, PCH2Ar), 2.65–2.30 (m, 12H, C5H4CH2CH2 and C5H4CH2CH2, overlaps with residual CD3CD2HSO signal). 13C{1H} NMR (DMSO-d6): δ 137.0 (d, JCP = 3 Hz), 135.9 (s), 130.4 (d, JCP = 5 Hz), 128.8 (d, JCP = 9 Hz), 126.9 (d, JCP = 3 Hz), 115.1 (s), 91.2 (d, JCP = 18 Hz), 86.8 (d, JCP = 17 Hz), 70.6 (s), 70.4 (s), 69.6 (s), 68.4 (s), 67.7 (s), 67.3 (s), 25.3 (d, JCP = 43 Hz), 20.8 (s), 20.2 (s), 20.0 (d, JCP = 45 Hz), 19.3 (d, JCP = 45 Hz). 31P{1H} NMR (DMSO-d6): δ 30.9 (s). FT-IR: 3091 (w), 3008 (w), 2922 (m), 2455 (w), 1629 (w), 1511 (w), 1410 (w), 1221 (w), 1104 (m), 999 (m), 918 (w), 811 (m), 753 (s), 661 (w) cm−1. UV-vis (THF): λmax 287 nm (ε = 2170 M−1 cm−1), 295 nm (ε = 1290 M−1 cm−1), 320 nm (ε = 420 M−1 cm−1), 431 nm (ε = 220 M−1 cm−1). Mass Spec. (ESI, +ve mode): exact mass calculated for [C45H4856Fe2P102Ru]+: 833.1236; exact mass found: 833.1259; difference: +2.8 ppm.
CH2), 5.92 (d, 3JHH,trans = 18 Hz, 1H, ArCHCH2), 5.31 (d, 3JHH,cis = 11 Hz, 1H, ArCHCH2), 4.65 [s, 4H, β-C5H4R(Ru)], 4.56 [s, 10H, C5H5(Ru)], 4.52 [s, 4H, α-C5H5R(Ru)], 4.21 [pseudo-t, 3JHH = 2 Hz, 2H, β-C5H4R(Fe)], 4.16 [s, 5H, C5H5(Fe)], 4.14 [pseudo-t, 3JHH = 2 Hz, 2H, α-C5H5R(Fe)], 4.06 (d, 2JHP = 16 Hz, 2H, PCH2Ar), 2.60–2.30 (m, 12H, C5H4CH2CH2 and C5H4CH2CH2, overlaps with residual CD3CD2HSO signal). 13C{1H} NMR (DMSO-d6): δ 137.0 (d, JCP = 5 Hz), 135.9 (s), 130.3 (d, JCP = 5 Hz), 128.7 (d, JCP = 9 Hz), 126.9 (d, JCP = 3 Hz), 115.1 (s), 91.1 (d, JCP = 17 Hz), 86.7 (d, JCP = 17 Hz), 70.6 (s), 70.4 (s), 69.7 (s), 68.4 (s), 67.7 (s), 67.4 (s), 25.3 (d, JCP = 43 Hz), 20.8 (s), 20.3 (d, JCP = 3 Hz), 20.0 (d, JCP = 44 Hz), 19.3 (d, JCP = 45 Hz). 31P{1H} NMR (DMSO-d6): δ 30.8 (s). FT-IR: 3095 (w), 3010 (w), 2923 (m), 2455 (w), 1630 (w), 1511 (w), 1409 (w), 1219 (w), 1101 (m), 998 (m), 918 (w), 810 (m), 753 (s), 666 (w) cm−1. UV-vis (THF): λmax 287 nm (ε = 2520 M−1 cm−1), 295 nm (ε = 1600 M−1 cm−1), 320 nm (ε = 680 M−1 cm−1), 434 nm (ε = 150 M−1 cm−1). Mass Spec. (ESI, +ve mode): exact mass calculated for [C45H4856FeP96Ru99Ru]+: 870.0978; exact mass found: 870.1002; difference: +2.8 ppm.
CH2), 5.90 (d, 3JHH,trans = 18 Hz, 1H, ArCHCH2), 5.31 (d, 3JHH,cis = 11 Hz, 1H, ArCHCH2), 4.64 (pseudo-t, 3JHH = 2 Hz, 6H, β-C5H4R), 4.55 (s, 15H, C5H5), 4.51 (pseudo-t, 3JHH = 2 Hz, 6H, α-C5H4R), 3.98 (d, 2JHP = 15 Hz, 2H, PCH2Ar), 2.27–2.47 (m, 12H, C5H4CH2CH2 and C5H4CH2CH2). 13C{1H} NMR (DMSO-d6): δ 137.1 (d, JCP = 3 Hz), 135.9 (s), 130.3 (d, JCP = 5 Hz), 128.6 (d, JCP = 9 Hz), 127.0 (s), 115.3 (s), 91.1 (d, JCP = 18 Hz), 70.6 (s), 70.5 (s), 69.8 (s), 25.4 (d, JCP = 44 Hz), 20.4 (s), 20.0 (d, JCP = 45 Hz). 31P{1H} NMR (DMSO-d6): δ 30.8 (s). FT-IR: 3093 (w), 3010 (w), 2919 (m), 2460 (w), 1670 (w), 1511 (w), 1409 (m), 1218 (w), 1100 (m), 997 (m), 917 (w), 809 (s), 752 (s), 660 (w) cm−1. UV-vis (THF): λmax 287 nm (ε = 2060 M−1 cm−1), 297 nm (ε = 1560 M−1 cm−1), 315 nm (ε = 890 M−1 cm−1). Mass Spec. (ESI, +ve mode): exact mass calculated for [C45H4896Ru99Ru100RuP]+: 914.0671; exact mass found: 914.0678; difference: +0.8 ppm.
Representative procedure for the preparation of phosphonium salts 7a–d
CH2), 5.89 (d, 3JHH,trans = 18 Hz, 1H, ArCHCH2), 5.29 (d, 3JHH,cis = 11 Hz, 1H, ArCHCH2), 4.19 (s, 6H, β-C5H4R), 4.14 (s, 15H, C5H5), 4.13 (s, 6H, α-C5H4R), 3.96 (d, 2JHP = 15 Hz, 2H, PCH2Ar), 2.60–2.42 (m, 12H, C5H4CH2CH2 and C5H4CH2CH2, overlaps with residual CD3CD2HSO signal). 13C{1H} NMR (DMSO-d6): δ 137.1 (d, JCP = 5 Hz), 135.9 (s), 130.4 (d, JCP = 3 Hz), 128.6 (d, JCP = 9 Hz), 127.0 (s), 120.7 (q, JCF = 322 Hz), 115.2 (s), 86.8 (d, JCP = 18 Hz), 68.4 (s), 67.7 (s), 67.4 (s), 25.4 (d, JCP = 44 Hz), 20.8 (s), 19.2 (d, JCP = 45 Hz). 19F NMR (DMSO-d6): δ −77.6 (s). 31P{1H} NMR (DMSO-d6): δ 31.2 (s). FT-IR: 3095 (w), 3010 (w), 2913 (w), 1512 (w), 1410 (w), 1262 (s), 1157 (m), 1124 (w), 1105 (w), 1030 (s), 1000 (w), 922 (w), 821 (m), 754 (m), 637 (s) cm−1. UV-vis (THF): λmax 285 nm (ε = 2470 M−1 cm−1), 295 nm (ε = 1290 M−1 cm−1), 325 nm (ε = 260 M−1 cm−1), 436 nm (ε = 320 M−1 cm−1). Mass Spec. (ESI, +ve mode): exact mass calculated for [C45H4856Fe3P]+: 787.1542; exact mass found: 787.1564; difference: +2.7 ppm. Anal. Calcd (%) for C46H48F3Fe3O3PS: C, 59.00; H, 5.17. Found: C, 58.94; H, 5.37.
CH2), 5.91(d, 3JHH,trans = 18 Hz, 1H, ArCHCH2), 5.31 (d, 3JHH,cis = 11 Hz, 1H, ArCHCH2), 4.64 [s, 2H, β-C5H4R(Ru)], 4.56 [s, 5H, C5H5(Ru)], 4.52 [s, 2H, α-C5H5R(Ru)], 4.20 [s, 4H, β-C5H4R(Fe)], 4.15 [s, 10H, C5H5(Fe)], 4.14 [s, 4H, α-C5H5R(Fe)], 3.96 (d, 2JHP = 15 Hz, 2H, PCH2Ar), 2.57–2.32 (m, 12H, C5H4CH2CH2 and C5H4CH2CH2, overlaps with residual CD3CD2HSO signal). 13C{1H} NMR (DMSO-d6): δ 137.2 (d, JCP = 3 Hz), 135.9 (s), 130.4 (d, JCP = 6 Hz), 128.6 (d, JCP = 9 Hz), 127.1 (d, JCP = 2 Hz), 120.7 (q, JCF = 322 Hz), 115.3 (d, JCP = 164 Hz), 91.2 (d, JCP = 18 Hz), 86.8 (d, JCP = 17 Hz), 70.6 (s), 70.5 (s), 69.8 (s), 68.5 (s), 67.8 (s), 67.5 (s), 25.4 (d, JCP = 43 Hz), 20.9 (s), 20.3 (s), 20.1 (d, JCP = 45 Hz), 19.3 (d, JCP = 45 Hz). 19F NMR (DMSO-d6): δ −77.8 (s). 31P{1H} NMR (DMSO-d6): δ 31.0 (s). FT-IR: 3089 (w), 3011 (w), 2912 (w), 1630 (w), 1512 (w), 1410 (w), 1262 (s), 1224 (m), 1158 (m), 1104 (w), 1030 (s), 999 (w), 919 (w), 813 (m), 755 (s), 637 (s) cm−1. UV-vis (THF): λmax 284 nm (ε = 2860 M−1 cm−1), 295 nm (ε = 1390 M−1 cm−1), 320 nm (ε = 440 M−1 cm−1), 431 nm (ε = 240 M−1 cm−1). Mass Spec. (ESI, +ve mode): exact mass calculated for [C45H4856Fe2P96Ru]+: 827.1268; exact mass found: 827.1274; difference: +0.7 ppm. Anal. Calcd (%) for C46H48F3Fe2O3PRuS: C, 56.28; H, 4.93. Found: C, 56.22; H, 5.11.
CH2), 5.92 (d, 3JHH,trans = 18 Hz, 1H, ArCHCH2), 5.32 (d, 3JHH,cis = 11 Hz, 1H, ArCHCH2), 4.65 [s, 4H, β-C5H4R(Ru)], 4.56 [s, 10H, C5H5(Ru)], 4.52 [s, 4H, α-C5H5R(Ru)], 4.21 [s, 2H, β-C5H4R(Fe)], 4.16 [s, 5H, C5H5(Fe)], 4.15 [s, 2H, α-C5H5R(Fe)], 3.96 (d, 2JHP = 15 Hz, 2H, PCH2Ar), 2.57–2.21 (m, 12H, C5H4CH2CH2 and C5H4CH2CH2, overlaps with residual CD3CD2HSO signal). 13C{1H} NMR (DMSO-d6): δ 137.1 (d, JCP = 3 Hz), 135.9 (s), 130.3 (d, JCP = 5 Hz), 128.5 (d, JCP = 8 Hz), 127.0 (d, JCP = 2 Hz), 115.2 (s), 91.0 (d, JCP = 18 Hz), 86.7 (d, JCP = 17 Hz), 70.6 (s), 70.4 (s), 69.7 (s), 68.4 (s), 67.6 (s), 67.4 (s), 25.3 (d, JCP = 44 Hz), 20.8 (s), 20.2 (s), 19.9 (d, JCP = 45 Hz), 19.2 (d, JCP = 45 Hz). 19F NMR (DMSO-d6): δ −77.7 (s). 31P{1H} NMR (DMSO-d6): δ 30.9 (s). FT-IR: 3093 (w), 3013 (w), 2911 (w), 1630 (w), 1512 (w), 1410 (w), 1262 (s), 1224 (s), 1159 (s), 1101 (m), 1030 (s), 998 (w), 918 (w), 811 (m), 756 (s), 637 (s) cm−1. UV-vis (THF): λmax 283 nm (ε = 3260 M−1 cm−1), 295 nm (ε = 1770 M−1 cm−1), 320 nm (ε = 730 M−1 cm−1), 434 nm (ε = 140 M−1 cm−1). Mass Spec. (ESI, +ve mode): exact mass calculated for [C45H4856FeP96Ru99Ru]+: 870.0978; exact mass found: 870.1000; difference: +2.5 ppm. Anal. Calcd (%) for C46H48F3FeO3PRu2S: C, 53.80; H, 4.71. Found: C, 53.94; H, 4.77.
CH2), 5.92 (d, 3JHH,trans = 18 Hz, 1H, ArCHCH2), 5.32 (d, 3JHH,cis = 11 Hz, 1H, ArCHCH2), 4.64 (pseudo-t, 3JHH = 2 Hz, 6H, β-C5H4R), 4.56 (s, 15H, C5H5), 4.52 (pseudo-t, 3JHH = 2 Hz, 6H, α-C5H4R), 3.94 (d, 2JHP = 15 Hz, 2H, PCH2Ar), 2.49–2.25 (m, 12H, C5H4CH2CH2 and C5H4CH2CH2). 13C{1H} NMR (DMSO-d6): δ 137.1 (s), 135.8 (s), 130.3 (s), 128.5 (d, JCP = 8 Hz), 127.0 (s), 120.6 (q, JCF = 321 Hz), 115.2 (s), 91.0 (d, JCP = 17 Hz), 70.6 (s), 70.4 (s), 69.7 (s), 25.3 (d, JCP = 44 Hz), 20.3 (s), 20.0 (d, JCP = 45 Hz). 19F NMR (DMSO-d6): δ −77.7 (s). 31P{1H} NMR (DMSO-d6): δ 30.8 (s). FT-IR: 3094 (w), 3013 (w), 2911 (w), 1630 (w), 1512 (w), 1409 (w), 1261 (s), 1159 (m), 1101 (w), 1030 (s), 997 (w), 917 (w), 810 (m), 755 (s), 637 (s) cm−1. UV-vis (THF): λmax 286 nm (ε = 1970 M−1 cm−1), 296 nm (ε = 1510 M−1 cm−1), 315 nm (ε = 910 M−1 cm−1). Mass Spec. (ESI, +ve mode): exact mass calculated for [C45H4896Ru99Ru100RuP]+: 914.0671; exact mass found: 914.0664; difference: −0.8 ppm. Anal. Calcd (%) for C46H48F3O3PRu3S: C, 51.53; H, 4.51. Found: C, 51.84; H, 4.62.
Representative procedure for the preparation of polyelectrolytes 8a–d
900, Mw = 148000 g mol−1, Đ = 3.16.
100, Mw = 184900 g mol−1, Đ = 4.10.
100, Mw = 278100 g mol−1, Đ = 4.02.
650, Mw = 143450 g mol−1, Đ = 3.71.
Results
Synthesis and characterization
Reaction of phosphines 5a–d13 with a slight excess of 4-vinylbenzyl chloride and heating at 75 °C afforded phosphonium chloride monomers 6a–d in quantitative yields (Scheme 1). Due to the poor solubility of phosphines 5a–d in DMF, they were initially dissolved in a minimum amount of THF and later combined with DMF, an effective solvent for the quaternization reaction. Butylated hydroxytoluene (BHT) was added to prevent the undesired polymerization of the styrene groups during the quaternization reaction. To improve the solubility of the phosphonium salts in organic solvents and to prevent metallocene degradation,2k the chloride counter anions were exchanged with triflate anions to quantitatively afford monomers 7a–d. The structure and purity of the monomers were confirmed using multinuclear NMR spectroscopy, IR and UV-vis absorption spectroscopy, mass spectrometry, elemental analysis, and X-ray crystallography (Fig. 1, S1−S16† and Table 2).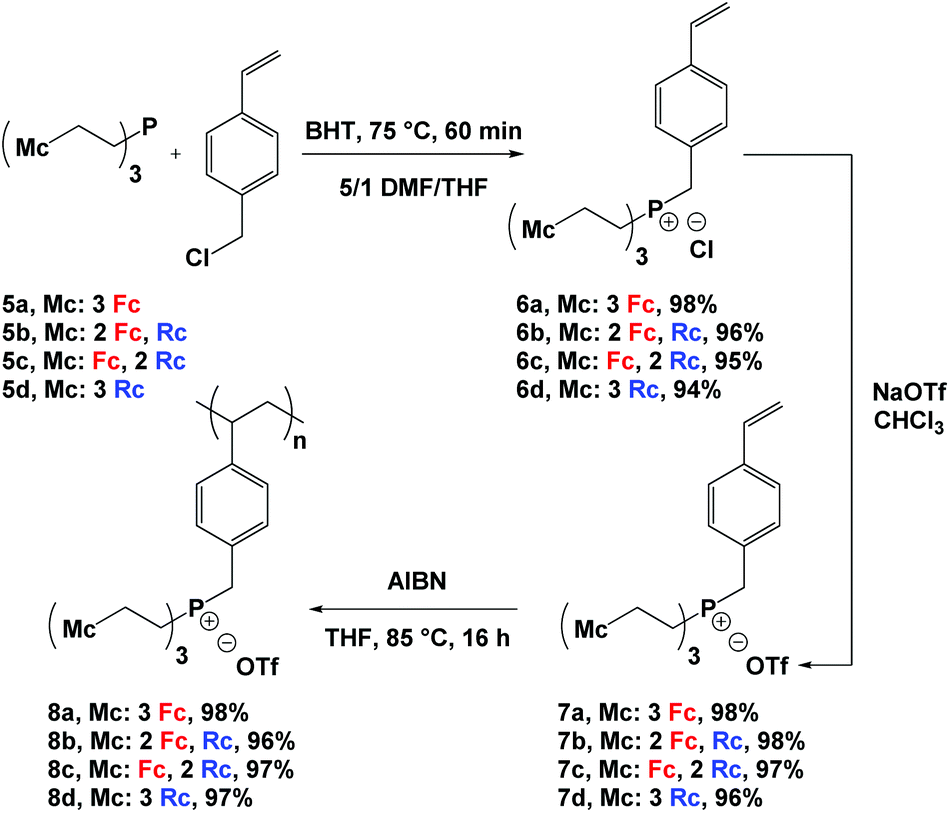 | ||
| Scheme 1 Synthetic pathway for the preparation of polyelectrolytes 8a–d. | ||
| Compound | 31P NMR shifta (δ) | ε, 320 nmb (M−1 cm−1) | ε, 434 nmb (M cm−1) | E pa,Rc , (mV) | E 1/2,Fcc (mV) | M n (g mol−1) | DPne | Đ |
|---|---|---|---|---|---|---|---|---|
| a Recorded in DMSO-d6 and referenced relative to triphenylphosphine internal standard. b Recorded in THF. c Recorded at a scan rate of 250 mV s−1 in degassed CH2Cl2/CH3CN (2/1) solutions of the analyte containing 0.1 M [n-Bu4N][OTf] as supporting electrolyte and referenced relative to the ferrocene/ferrocenium redox couple. d Irreversible process; anodic peak potential reported. e Determined by conventional calibration GPC vs. polystyrene standards in DMF (60 °C) containing 0.02 M [n-Bu4N][OTf]. | ||||||||
| 7a | 31.2 | 260 | 320 | — | 10 | — | — | — |
| 7b | 31.0 | 440 | 240 | 465 | 15 | — | — | — |
| 7c | 30.9 | 730 | 140 | 510 | 20 | — | — | — |
| 7d | 30.8 | 910 | — | 555 | — | — | — | — |
| 8a | 31.1 | 260 | 330 | — | 20 | 46900 |
50 | 3.16 |
| 8b | 30.9 | 440 | 230 | — | 20 | 45100 |
46 | 4.10 |
| 8c | 30.8 | 620 | 150 | 410 | 35 | 69100 |
67 | 4.02 |
| 8d | 30.8 | 870 | — | 450 | — | 38650 |
36 | 3.71 |
Phosphonium triflate monomers 7a–d gave rise to a singlet in their 19F NMR spectra at δ ∼ −77 and a singlet in their 31P{1H} NMR spectra at δ ∼31.0. Single crystals of monomer 7a suitable for X-ray crystallography were grown by slow evaporation of the solvent from a THF solution (Fig. 1 and Table 1). In the solid-state structure, each tetrahedral phosphonium cation was in close proximity to a triflate anion (shortest contact: P1–O2 3.958 Å). The C–P bond lengths ranged from 1.782(6) to 1.803(5) Å, and are shorter than those of the parent phosphine 5a [1.843(1) to 1.855(1) Å].2k The average C–P–C angle was 109.5(3)° and the C44–C45 bond length for the vinyl group was 1.162(9) Å.
Using azobisisobutyronitrile (AIBN) as an initiator, the phosphonium triflate monomers 7a–d were polymerized in THF before they were precipitated into diethyl ether to yield the corresponding polyelectrolytes 8a–d (Scheme 1). At room temperature, these polyelectrolytes gave rise to very broad 1H, 31P{1H}, and 19F NMR spectra. However, upon heating to 125 °C the spectra sharpened (Fig. S17−S20†). Disappearance of the vinyl proton resonances and the observation of broad peaks associated with the unsaturated polyelectrolyte backbone (δ 0.75–2.15) in the 1H NMR spectra of the polyelectrolytes confirmed successful polymerization. The presence of the aromatic (δ 5.75–7.50), metallocene (δ 4.00–5.00), methylene bridge (δ 3.40–4.20), and ethylene bridge (δ 2.10–3.25) proton resonances in the 1H NMR spectra; a peak for each polyelectrolyte (δ ∼ 31.0) in the 31P{1H} NMR spectra; and a peak for each polyelectrolyte (δ ∼ −77) in the 19F NMR spectra further supported the proposed structures of the polyelectrolytes.
Gel permeation chromatography (GPC) experiments were carried out to evaluate the molecular weight distributions for polyelectrolytes 8a–d. To overcome common issues regarding strong interactions between polyelectrolytes and GPC columns, a 60 °C DMF solution containing 0.02 M [n-Bu4N][OTf] was used as an eluent in tandem with Teflon-treated size-exclusion columns. The results confirmed the macromolecular nature of the polyelectrolytes 8a–d (Mn: 38650–69100 gmol−1, Đ: 3.16–4.10, Tables 2 and S1, and Fig. S21†).
UV-vis absorption spectroscopy and cyclic voltammetry
Similar to phosphines 5a–d,13 the ferrocene units in polyelectrolytes 8a–d gave rise to two absorption maxima at ca. 434 nm and 320 nm while the ruthenocene units exhibited a single absorption maximum at ca. 320 nm. The relative intensities of each peak varied with the number of ferrocene/ruthenocene units present (Table 2 and Fig. S22−S24†).The electrochemical properties of the phosphonium triflate monomers 7a–d and the corresponding polyelectrolytes 8a–d were examined by cyclic voltammetry (CV) in a CH2Cl2/CH3CN (2/1) solvent mixture containing 0.1 M [n-Bu4N][OTf] as supporting electrolyte. Consistent with the electrochemical properties of the parent phosphines,13 the ferrocene units of the monomers and polyelectrolytes were oxidized reversibly, while the ruthenocene moieties gave rise to an irreversible oxidation wave. The observed irreversible behavior was consistent with the ability of ruthenocenium cations to rapidly engage in electrochemically-induced reactions.5d,18 Due to the presence of the cationic phosphonium centers in the monomers, the recorded E1/2,Fc and Epa,Rc values were slightly more positive than those of the parent phosphines (Table 2, Fig. S25 and S26†).13
Thermal analysis and pyrolysis studies
Differential scanning calorimetry (DSC) studies of polyelectrolytes 8a–d revealed glass transition temperatures (Tg) between 165 and 177 °C (Fig. S27† and Table 3). TGA studies demonstrated that polyelectrolytes 8a–d have high thermal stability, with the onset of decomposition observed at ca. 310 °C and char yields ranging from 33 to 54% (Fig. 2 and Table 3).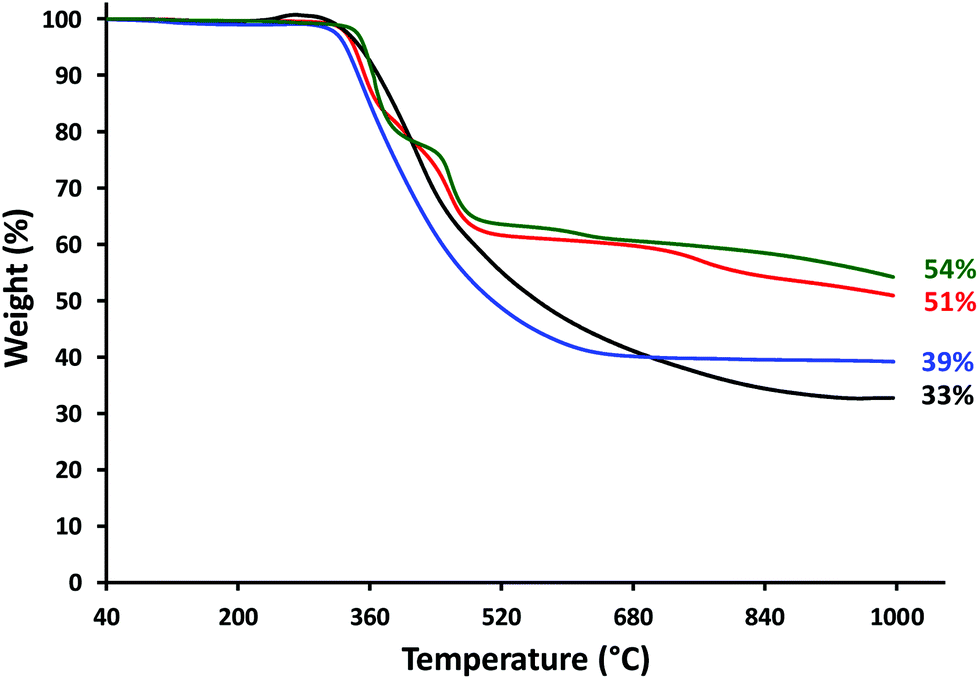 | ||
| Fig. 2 TGA data obtained for polyelectrolytes: 8a (3 × Fc, black), 8b (2 × Fc, 1 × Rc; red), 8c (1 × Fc, 2 × Rc; blue), and 8d (3 × Rc, green). | ||
| Polymer |
T
gb (°C) |
Onset of decompositionc (°C) | Char yieldb (%) | Regiond | Atomic ratiod,e | |||
|---|---|---|---|---|---|---|---|---|
| P | Fe | Ru | Fe + Ru | |||||
| a Carried out at 1000 °C under N2/H2 (95/5) gas mixture as the carrier gas at an approximate flow rate of ca. 60 mL min−1. b Determined using DSC. c Determined using TGA. d Atomic ratios determined using EDX spectroscopy for dense regions of relatively large particles (‘bulk’) and less dense regions of relatively small particles (‘particles’). See ESI for additional details. e Phosphorus stoichiometry fixed at 1. | ||||||||
| 8a | 177 | 310 | 33 | Bulk | 1 | 1.9 ± 0.3 | — | 1.9 ± 0.3 |
| Particles | 1 | 2.1 ± 0.5 | — | 2.1 ± 0.5 | ||||
| 8b | 165 | 311 | 51 | Bulk | 1 | 1.7 ± 0.7 | 1.1 ± 0.4 | 2.8 ± 0.8 |
| Particles | 1 | 1.5 ± 0.3 | 0.9 ± 0.1 | 2.4 ± 0.3 | ||||
| 8c | 173 | 312 | 39 | Bulk | 1 | 0.5 ± 0.1 | 1.6 ± 0.1 | 2.1 ± 0.1 |
| Particles | 1 | 0.6 ± 0.3 | 1.8 ± 0.5 | 2.4 ± 0.6 | ||||
| 8d | 171 | 330 | 54 | Bulk | 1 | — | 2.1 ± 0.0 | 2.1 ± 0.0 |
| Particles | 1 | — | 1.5 ± 0.1 | 1.5 ± 0.1 | ||||
Based on the high char yields observed and growing interest in the catalytic and electrochemical properties of nano-structured metal phosphides,19 we decided to explore the preceramic behavior of polyelectrolytes 8a–d. Polyelectrolyte films with approximate thickness of 6 μm (Fig. S28†) were prepared by drop casting and pyrolyzed at 1000 °C under a flow of N2/H2 (95/5) for 3 h. Each pyrolysis experiment was repeated in triplicate, and representative SEM images and relevant data are shown in Fig. 3 and Table 3. It is worth noting that we were unable to employ transmission electron microscopy (TEM) for our studies as our thorough attempts to dislodge the nanomaterials produced by pyrolysis from silicon substrates using physical scraping (razor blade), ultrasonication, and solvent rinsing were unsuccessful. However, our SEM analyses provided significant insight into the structures of the nanomaterials produced. In each case, pyrolysis of polyelectrolyte films resulted in the formation of large particles and/or continuous materials surrounded by numerous smaller particles. In the case of the nanomaterials produced from 8b and 8c, the multi-faceted appearance of the imaged materials hinted to the fact that they may be crystalline (vide infra).
 | ||
| Fig. 3 SEM images of the nanomaterials prepared via the pyrolysis of films of polyelectrolytes (a) 8a, (b) 8b, (c) 8c, and (d) 8d. Scale bars = 1 μm. | ||
Elemental maps (EDX spectroscopy) revealed that the nanomaterials produced were comprised of C, O, P, Fe, and/or Ru and that the inorganic components were distributed throughout the nanomaterials produced (Fig. 4, S29−S31†). The presence of carbon was attributed to incomplete volatilization of the polystyrene backbone and oxidation during brief (and unavoidable) exposure of the samples to air prior to SEM analysis accounts for the presence of oxygen. Unfortunately, the elemental maps obtained provide little quantitative information about the composition of the nanomaterials produced. With this in mind, at least five different EDX data sets (elemental analyses) were collected for regions (ca. 1 μm2) of the nanomaterial surfaces densely populated with relatively large particles and/or continuous material (‘bulk’) and with relatively small particles (‘particles’) for each of the samples (Fig. S32−S35† and Table 3). For each sample, the smaller particles produced were clearly embedded within a carbon-rich matrix, and the overall phosphorus/metal content was significantly lower than those observed for areas densely populated with bulk material.
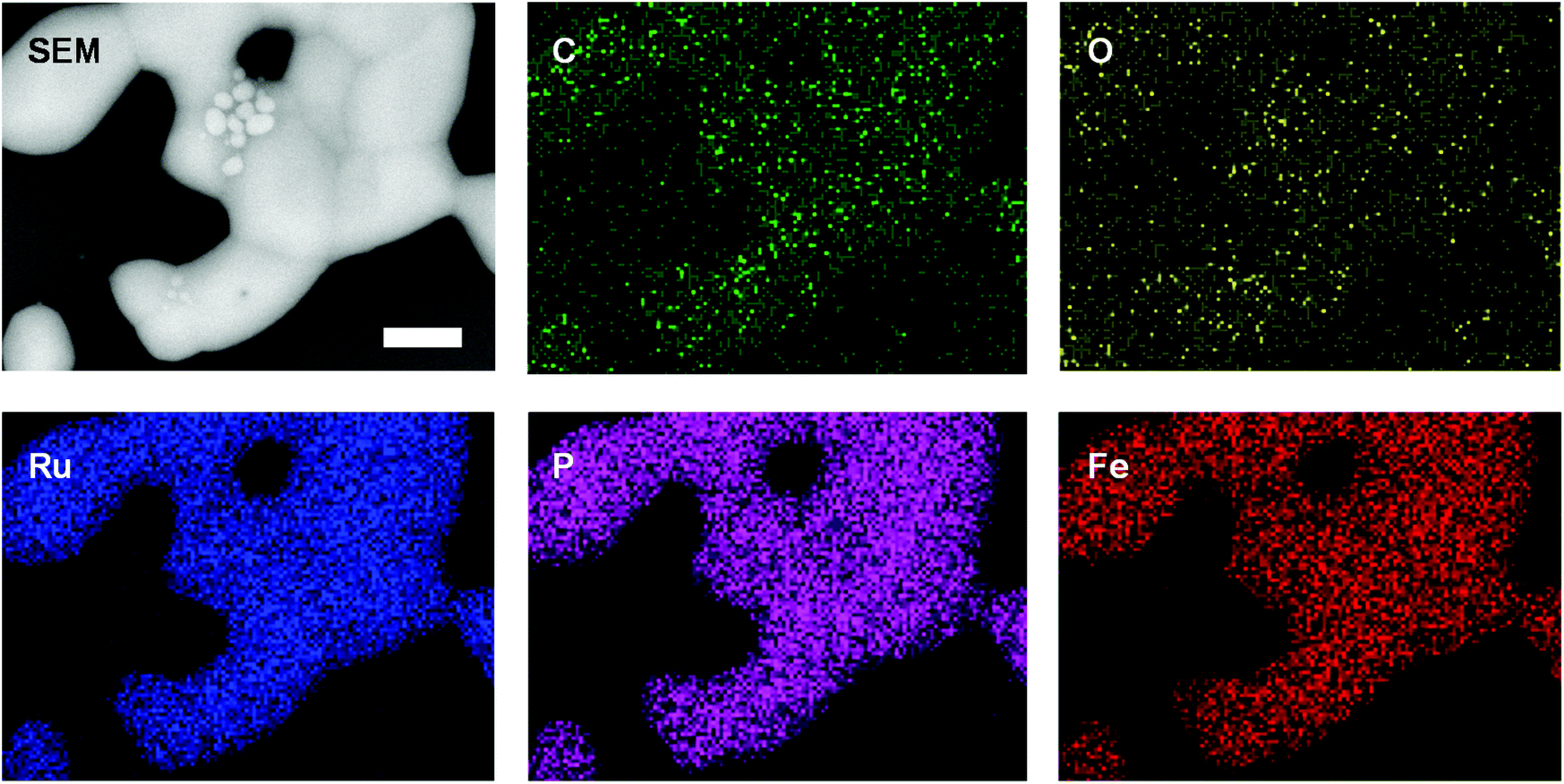 | ||
| Fig. 4 SEM images and elemental maps (O, Ru, Fe, P, C) for the nanomaterials prepared via the pyrolysis of a film of polyelectrolyte 8b. Scale bar = 1 μm. | ||
Powder X-ray diffraction studies
Powder X-ray diffraction studies of the nanomaterials produced by pyrolysis of films of polyelectrolytes 8a–d are shown in Fig. 5. In each case, the patterns produced confirmed the presence of crystalline materials. Qualitative phase identification studies were performed through careful comparison of our diffraction data with those of a PXRD database,20 which included data for known mono- and bimetallic phosphides (i.e., M4P, M3P, M2P, and MP; where M: Fe and/or Ru), metal carbides, pure metals, etc. (Fig. S36−S39†). The PXRD data for the films produced from polyelectrolytes 8a–c were not closely matched with those of any known phases, and were indicative of the presence of multiple crystalline phases, which prevented us from indexing the data. Conversely, the PXRD data collected for the film produced from polyelectrolyte 8d was consistent with that of Ru2P (Fig. 5d and S39†).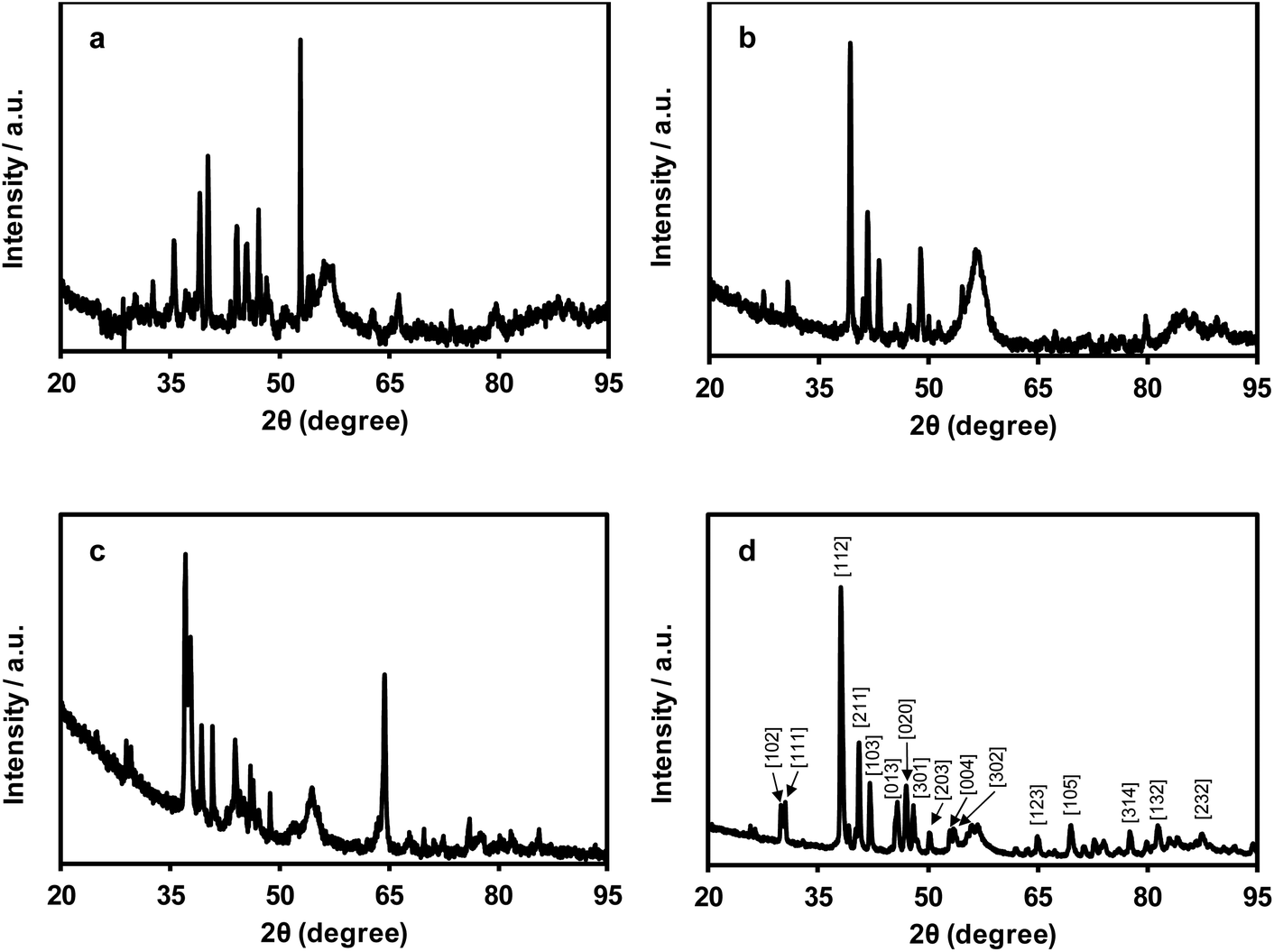 | ||
| Fig. 5 Powder X-ray diffractograms for the nanomaterials prepared via the pyrolysis of films of polyelectrolytes (a) 8a, (b) 8b, (c) 8c, and (d) 8d. Miller indices corresponding to Ru2P are shown in panel (d). | ||
Discussion
Considering the PXRD, EDX spectroscopy, and elemental mapping results obtained for the nanomaterials produced from pyrolyzed films of polyelectrolytes 8a–c, two points become immediately obvious. First, given the large standard deviations associated with the atomic ratios determined from the EDX data collected, it is clear that phosphorus, iron, and/or ruthenium are not distributed uniformly throughout the nanomaterials produced. It is therefore probable that multiple different materials/phases have been produced. For the heterobimetallic materials derived from 8b and 8c, PXRD data confirmed the absence of simple Fe2P/Ru2P/Fe3P/Ru3P phases and supported this hypothesis. Second, within a single standard deviation, the average composition of the small particles and bulk materials analyzed were the same. Conversely, for the nanomaterials derived from 8d, the relatively smaller standard deviations calculated point toward the uniform distribution of phosphorus and ruthenium throughout regions containing bulk material and relatively small particles. Furthermore, this was the only case where the composition of the bulk (P:Ru, 1:2.1 ± 0.0) and small-particle-rich (P:Ru, 1:1.5 ± 0.1) were statistically different. When these results are combined with the PXRD data collected and indexed for this sample, it becomes clear that the bulk phase produced from the pyrolysis of films of polyelectrolyte 8d is comprised of Ru2P.
Conclusion
In conclusion, starting from stable tertiary phosphines containing all possible combinations of ethylferrocene and ethylruthenocene substituents, styrene-based phosphonium triflate monomers with four different stoichiometric ratios of Fe/Ru were synthesized and fully characterized. Via free-radical polymerization of the triflate salts, four polyelectrolytes were prepared and analyzed. Due to the presence of ferrocene/ruthenocene, these materials exhibited redox properties and gave rise to UV-vis absorption maxima consistent with the number of each metallocene present. GPC and DSC results confirmed the macromolecular nature of the polyelectrolytes and TGA studies confirmed their stability up to ∼310 °C. Studies of the nanomaterials that resulted from the pyrolysis of polyelectrolytes 8a–d using SEM and PXRD showed that they can be used as precursors to crystalline nanomaterials. EDX spectroscopy and elemental mapping data indicated that the crystalline nanomaterials contained Fe, Ru, and P distributed throughout, with Fe/Ru/P ratios influenced by the polyelectrolyte structures.Acknowledgements
The authors acknowledge the University of Western Ontario, the Natural Science and Engineering Research Council (NSERC) of Canada (DG, 435675), the Ontario Ministry of Research and Innovation (ERA, ER14-10-147) and the Canada Foundation for Innovation (JELF, 33977) for funding this work. We thank Prof. Paul Ragogna and Mr Tyler Cuthbert for assistance with phosphine experiments, use of equipment, and helpful discussions. We thank Mr Tim Goldhawk and Dr Todd Simpson of the Western Nanofabrication Facility for assistance with SEM and EDX spectroscopy studies, and Dr Paul Boyle, Ms Stephanie Barbon, Dr Anastasia Colomba, and Mr Joe Paquette for assistance with X-ray diffraction experiments and helpful discussions. We also appreciate the time and effort that Mr Eric Landry and Mr Ahmad Abu Romeh (PolyAnalytik Canada) spent collecting the GPC data presented.Notes and references
- Reviews: (a) C.-L. Ho and W.-Y. Wong, Coord. Chem. Rev., 2011, 255, 2469–2502 CrossRef CAS; (b) G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 CrossRef CAS PubMed; (c) D. Astruc, Nat. Chem., 2012, 4, 255–267 CrossRef CAS PubMed; (d) C. Friebe, M. D. Hager, A. Winter and U. S. Schubert, Adv. Mater., 2012, 24, 332–345 CrossRef CAS PubMed; (e) C. G. Hardy, L. Ren, J. Zhang and C. Tang, Isr. J. Chem., 2012, 52, 230–245 CrossRef CAS; (f) H. Bhattacharjee and J. Müller, Coord. Chem. Rev., 2016, 314, 114–133 CrossRef CAS; (g) Y. Yan, J. Zhang, L. Ren and C. Tang, Chem. Soc. Rev., 2016, 45, 5232–5263 RSC.
- For example: (a) T. J. Peckham, A. J. Lough and I. Manners, Organometallics, 1999, 18, 1030–1040 CrossRef CAS; (b) B. F. G. Johnson, K. M. Sanderson, D. S. Shephard, D. Ozkaya, W. Zhou, H. Ahmed, M. D. R. Thomas, L. Gladden and M. Mantle, Chem. Commun., 2000, 1317–1318 RSC; (c) Y. Ma, W.-F. Dong, M. A. Hempenius, H. Mohwald and G. Julius Vancso, Nat. Mater., 2006, 5, 724–729 CrossRef CAS PubMed; (d) X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644–647 CrossRef CAS PubMed; (e) C. Ornelas, J. Ruiz, C. Belin and D. Astruc, J. Am. Chem. Soc., 2009, 131, 590–601 CrossRef CAS PubMed; (f) H. Zhan, S. Lamare, A. Ng, T. Kenny, H. Guernon, W.-K. Chan, A. B. Djurišić, P. D. Harvey and W.-Y. Wong, Macromolecules, 2011, 44, 5155–5167 CrossRef CAS; (g) Z. M. Al-Badri, R. R. Maddikeri, Y. Zha, H. D. Thaker, P. Dobriyal, R. Shunmugam, T. P. Russell and G. N. Tew, Nat. Commun., 2011, 2, 482 CrossRef PubMed; (h) L. Ren, J. Zhang, C. G. Hardy, S. Ma and C. Tang, Macromol. Rapid Commun., 2012, 33, 510–516 CrossRef CAS PubMed; (i) R. H. Staff, M. Gallei, M. Mazurowski, M. Rehahn, R. Berger, K. Landfester and D. Crespy, ACS Nano, 2012, 6, 9042–9049 CrossRef CAS PubMed; (j) X. Wang, K. Cao, Y. Liu, B. Tsang and S. Liew, J. Am. Chem. Soc., 2013, 135, 3399–3402 CrossRef CAS PubMed; (k) A. Rabiee Kenaree, B. M. Berven, P. J. Ragogna and J. B. Gilroy, Chem. Commun., 2014, 50, 10714–10717 RSC; (l) R. Guterman, A. Rabiee Kenaree, J. B. Gilroy, E. R. Gillies and P. J. Ragogna, Chem. Mater., 2015, 27, 1412–1419 CrossRef CAS; (m) J. Zhang, J. Yan, P. Pageni, Y. Yan, A. Wirth, Y.-P. Chen, Y. Qiao, Q. Wang, A. W. Decho and C. Tang, Sci. Rep., 2015, 5, 11914 CrossRef CAS PubMed; (n) M. Hadadpour, J. Gwyther, I. Manners and P. J. Ragogna, Chem. Mater., 2015, 27, 3430–3440 CrossRef CAS.
- (a) S. B. Clendenning, S. Han, N. Coombs, C. Paquet, M. S. Rayat, D. Grozea, P. M. Brodersen, R. N. S. Sodhi, C. M. Yip, Z. H. Lu and I. Manners, Adv. Mater., 2004, 16, 291–296 CrossRef CAS; (b) W. Y. Chan, S. B. Clendenning, A. Berenbaum, A. J. Lough, S. Aouba, H. E. Ruda and I. Manners, J. Am. Chem. Soc., 2005, 127, 1765–1772 CrossRef CAS PubMed; (c) K. Liu, S. B. Clendenning, L. Friebe, W. Y. Chan, X. Zhu, M. R. Freeman, G. C. Yang, C. M. Yip, D. Grozea, Z.-H. Lu and I. Manners, Chem. Mater., 2006, 18, 2591–2601 CrossRef CAS; (d) M. Zamora, S. Bruña, B. Alonso and I. Cuadrado, Macromolecules, 2011, 44, 7994–8007 CrossRef CAS.
- (a) J. B. Gilroy, S. K. Patra, J. M. Mitchels, M. A. Winnik and I. Manners, Angew. Chem., Int. Ed., 2011, 50, 5851–5855 CrossRef CAS PubMed; (b) Y. Zha, H. D. Thaker, R. R. Maddikeri, S. P. Gido, M. T. Tuominen and G. N. Tew, J. Am. Chem. Soc., 2012, 134, 14534–14541 CrossRef CAS PubMed; (c) J. Zhang, L. Ren, C. G. Hardy and C. Tang, Macromolecules, 2012, 45, 6857–6863 CrossRef CAS; (d) J. Zhang, Y. Yan, M. W. Chance, J. Chen, J. Hayat, S. Ma and C. Tang, Angew. Chem., Int. Ed., 2013, 52, 13387–13391 CrossRef CAS PubMed; (e) J. Zhang, Y. Yan, J. Chen, W. M. Chance, J. Hayat, Z. Gai and C. Tang, Chem. Mater., 2014, 26, 3185–3190 CrossRef CAS; (f) R. Ciganda, H. Gu, P. Castel, P. Zhao, J. Ruiz, R. Hernández and D. Astruc, Macromol. Rapid Commun., 2016, 37, 105–111 CrossRef CAS PubMed.
- (a) K. Liu, C.-L. Ho, S. Aouba, Y.-Q. Zhao, Z.-H. Lu, S. Petrov, N. Coombs, P. Dube, H. E. Ruda, W.-Y. Wong and I. Manners, Angew. Chem., Int. Ed., 2008, 47, 1255–1259 CrossRef CAS PubMed; (b) B. Bagh, J. B. Gilroy, A. Staubitz and J. Müller, J. Am. Chem. Soc., 2010, 132, 1794–1795 CrossRef CAS PubMed; (c) Q. Dong, G. Li, C.-L. Ho, M. Faisal, C.-W. Leung, P. W.-T. Pong, K. Liu, B.-Z. Tang, I. Manners and W.-Y. Wong, Adv. Mater., 2012, 24, 1034–1040 CrossRef CAS PubMed; (d) M. Erhard, K. Lam, M. Haddow, G. R. Whittell, W. E. Geiger and I. Manners, Polym. Chem., 2014, 5, 1264–1274 RSC; (e) A. D. Russell, G. R. Whittell, M. F. Haddow and I. Manners, Organometallics, 2014, 33, 5349–5357 CrossRef CAS; (f) Q. Dong, G. Li, H. Wang, P. Wing-Tat Pong, C.-W. Leung, I. Manners, C.-L. Ho, H. Li and W.-Y. Wong, J. Mater. Chem., 2015, 3, 734–741 CAS; (g) H. Braunschweig, A. Damme, S. Demeshko, K. Dück, T. Kramer, I. Krummenacher, F. Meyer, K. Radacki, S. Stellwag-Konertz and G. R. Whittell, J. Am. Chem. Soc., 2015, 137, 1492–1500 CrossRef CAS PubMed; (h) Z. Meng, K. Sato, T. Sukegawa, K. Oyaizu, C.-L. Ho, J. Xiang, Y.-H. Feng, Y. H. Lo, H. Nishide and W.-Y. Wong, J. Organomet. Chem., 2016, 812, 51–55 CrossRef CAS.
- (a) J. A. Rodriguez and D. W. Goodman, Science, 1992, 257, 897–903 CAS; (b) S. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 287, 1989–1992 CrossRef CAS PubMed; (c) D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed; (d) A.-H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed; (e) K. J. Major, C. De and S. O. Obare, Plasmonics, 2009, 4, 61–78 CrossRef CAS; (f) S. Shylesh, V. Schünemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428–3459 CrossRef CAS PubMed; (g) H. Goesmann and C. Feldmann, Angew. Chem., Int. Ed., 2010, 49, 1362–1395 CrossRef CAS PubMed; (h) U. Banin, Y. Ben-Shahar and K. Vinokurov, Chem. Mater., 2014, 26, 97–110 CrossRef CAS; (i) P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef CAS PubMed.
- (a) B. Li, J. Wang, Y. Yuan, H. Ariga, S. Takakusagi and K. Asakura, ACS Catal., 2011, 1, 1521–1528 CrossRef CAS; (b) V. Kelsen, A. Meffre, P.-F. Fazzini, P. Lecante and B. Chaudret, ChemCatChem, 2014, 6, 1714–1720 CrossRef CAS; (c) M. Kaushik, H. M. Friedman, M. Bateman and A. Moores, RSC Adv., 2015, 5, 53207–53210 RSC; (d) R. Hudson, V. Chazelle, M. Bateman, R. Roy, C.-J. Li and A. Moores, ACS Sustainable Chem. Eng., 2015, 3, 814–820 CrossRef CAS.
- J.-Q. Du, Y. Zhang, T. Tian, S.-C. Yan and H.-T. Wang, Mater. Res. Bull., 2009, 44, 1347–1351 CrossRef CAS.
- (a) G. L. Ott, T. Fleisch and W. N. Delgass, J. Catal., 1979, 60, 394–403 CrossRef CAS; (b) M. C. Bahome, L. L. Jewell, K. Padayachy, D. Hildebrandt, D. Glasser, A. K. Datye and N. J. Coville, Appl. Catal., A, 2007, 328, 243–251 CrossRef CAS.
- (a) J. P. Wilcoxon and B. L. Abrams, Chem. Soc. Rev., 2006, 35, 1162–1194 RSC; (b) N. Semagina and L. Kiwi-Minsker, Catal. Rev.: Sci. Eng., 2009, 51, 147–217 CrossRef CAS.
- M. Zaheer, T. Schmalz, G. Motz and R. Kempe, Chem. Soc. Rev., 2012, 41, 5102–5116 RSC.
- T. Nishikubo, T. Iizawa, K. Kobayashi and M. Okawara, Tetrahedron Lett., 1981, 22, 3873–3874 CrossRef CAS.
- A. Rabiee Kenaree, T. J. Cuthbert, S. M. Barbon, P. D. Boyle, E. R. Gillies, P. J. Ragogna and J. B. Gilroy, Organometallics, 2015, 34, 4272–4280 CrossRef.
- S. v. Bruker-AXS, 2013, Bruker-AXS, Madison, WI 53711, USA Search PubMed.
- T. v. Bruker-AXS, 2012, Bruker-AXS, Madison, WI 53711, USA Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- (a) S. Trupia, A. Nafady and W. E. Geiger, Inorg. Chem., 2003, 42, 5480–5482 CrossRef CAS PubMed; (b) J. C. Swarts, A. Nafady, J. H. Roudebush, S. Trupia and W. E. Geiger, Inorg. Chem., 2009, 48, 2156–2165 CrossRef CAS PubMed.
- (a) H. Teller, O. Krichevski, M. Gur, A. Gedanken and A. Schechter, ACS Catal., 2015, 5, 4260–4267 CrossRef CAS; (b) Y. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC; (c) A. Berenguer, T. M. Sankaranarayanan, G. Gómez, I. Moreno, J. M. Coronado, P. Pizarro and D. P. Serrano, Green Chem., 2016, 18, 1938–1951 RSC; (d) Y. Lu, T. Wang, X. Li, G. Zhang, H. Xue and H. Pang, RSC Adv., 2016, 6, 87188–87212 RSC; (e) J. F. Callejas, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Chem. Mater., 2016, 28, 6017–6044 CrossRef CAS.
- The PXRD patterns were compared using the ICSD database and PDF4+ software.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1476067. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt02541b |
| This journal is © The Royal Society of Chemistry 2016 |