DOI:
10.1039/C6DT03571J
(Paper)
Dalton Trans., 2016,
45, 19012-19023
Systematic molecular engineering of Zn-ketoiminates for application as precursors in atomic layer depositions of zinc oxide†
Received
14th September 2016
, Accepted 24th October 2016
First published on 25th October 2016
Abstract
Molecular engineering of seven closely related zinc ketoiminates, namely, [Zn(dapki)2], [Zn(daeki)2], [Zn(epki)2], [Zn(eeki)2], [Zn(mpki)2], [Zn(meki)2], and [Zn(npki)2], leads to the optimisation of precursor thermal properties in terms of volatilisation rate, onset of volatilisation, reactivity and thermal stability. The influence of functional groups at the imine side chain of the ligands on the precursor properties is studied with regard to their viability as precursors for atomic layer deposition (ALD) of ZnO. The synthesis of [Zn(eeki)2], [Zn(epki)2] and [Zn(dapki)2] and the crystal structures of [Zn(mpki)2], [Zn(eeki)2], [Zn(dapki)2] and [Zn(npki)2] are presented. From the investigation of the physico-chemical characteristics, it was inferred that all compounds are monomeric, volatile and exhibit high thermal stability, all of which make them promising ALD precursors. Compound [Zn(eeki)2] is in terms of thermal properties the most promising Zn-ketoiminate. It is reactive towards water, possesses a melting point of 39 °C, is stable up to 24 days at 220 °C and has an extended volatilisation rate compared to the literature known Zn-ketoiminates. It demonstrated self-saturated, water assisted growth of zinc oxide (ZnO) with growth rates in the order of 1.3 Å per cycle. Moreover, it displayed a broad temperature window from TDep = 175–300 °C and is the first report of a stable high temperature (>200 °C) ALD process for ZnO returning highly promising growth rates.
Introduction
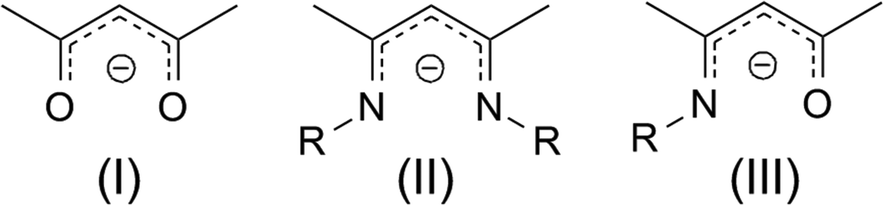
Precursor chemistry plays a crucial role in atomic layer deposition (ALD) which is a preferred method for the fabrication of high quality thin films on complex structures.1 The properties of ALD precursors can in principle be tuned by ligand engineering to suit the demands of the process. Nevertheless, it is not so trivial to achieve this and therefore a thorough understanding of the chemistry is needed. Until now, β-diketonates, amides, alkyls, halides, and cyclopentadienyls have been predominantly used for the ALD of functional metal oxides.2 From the library of standard Zn precursors available, the freedom to tune the precursor properties is not really high. By selecting ketoiminates as a ligand system, the choice to tune the physico-chemical properties is enhanced especially by varying the ligand moieties.3 β-Diketonates(I), β-diketiminates(II) and β-ketoiminates(III) (Fig. 1) are known as chelating ligands for metals throughout the complete periodic table of the elements. As a general observation, metal oxygen bonds are stronger than metal nitrogen bonds and therefore the respective metal complexes tend to be more stable when coordinated to β-diketonates compared to β-diketiminates. A compromise in reactivity and stability between these two ligands is the β-ketoiminate. The bidentate mixed oxygen and nitrogen system can be thought of as a mix of the generally stable metal β-diketonates (e.g. M(acac)2 acac = acetylacetone) and the thermally unstable but highly reactive metal β-diketiminates. The β-ketoiminate complexes should be worthwhile candidates which provide the necessary thermal stability, reactivity and volatility for atomic layer deposition. Metal-ketoiminates are applied as catalysts in lactone polymerization4 and in some cases used as precursors for the CVD of metal oxide thin films5 and nanostructures.6 More recently Cosham et al. have synthesised a series of fluorinated Zn-ketoiminates for atmospheric pressure MOCVD.7 Besides their favourable stability and reactivity, in contrast to β-diketonates and β-diketiminates, β-ketoiminates offer the opportunity to vary the side chain at the R-position easily. β-ketoimines are easily synthesized in high yields, and are therefore available with countless variations. Homoleptic Zn-ketoiminates have been used as precursors by the groups of Devi8 (R = EtOMe, PrOMe) and Matthews9 (R = n-Bu, n-Pr, i-Pr) for the chemical vapour deposition (CVD) of ZnO. Furthermore, Schulz et al.10 claim the suitability of Zn-ketoiminates, where R = EtN(Me)2, for the application as CVD precursors. Surprisingly, the thermal properties of Zn-ketoiminates are significantly different, although the variation between these complexes remains only minor. More importantly, the performance of Zn-ketoiminates in the deposition process changes. Complexes bearing ligands with R = alkyl form carbon rich zinc oxide films9 while those with a methoxy substituted side chain can grow films with minimal carbon incorporation.8 In order to identify the origin of the variable thermal properties of different Zn-ketoiminates, we adopted the principle of molecular engineering and systematically varied the side chain of ketoimine ligands [(RN)-2-penten-2-ol-4-imine] at the R position. Seven different Zn-ketoiminates, namely, bis(N-(3′-dimethylaminopropyl)-2-penten-2-on-4-iminate) zinc(II) [Zn(dapki)2], bis(N-(3′-dimethylaminoethyl)-2-penten-2-on-4-iminate) zinc(II) [Zn(daeki)2],10 bis(N-(3′-ethoxypropyl)-2-penten-2-on-4-iminate) zinc(II) [Zn(epki)2], bis(N-(2′-ethoxyethyl)-2-penten-2-on-4-iminate) zinc(II) [Zn(eeki)2], bis(N-(3′-methoxypropyl)-2-penten-2-on-4-iminate) zinc(II) [Zn(mpki)2],8 bis(N-(2′-methoxyethyl)-2-penten-2-on-4-iminate) zinc(II) [Zn(meki)2],8 and bis(N-(npropyl)-2-penten-2-on-4-iminate) zinc(II) [Zn(npki)2]9 were synthesized. The solid state structures were analysed and correlated with the thermal properties in solution and as solids. After thorough characterization we applied the most promising compound as an ALD precursor for the deposition of ZnO with water as an oxidation source. Zinc oxide ALD precursors are few and far between and those which have been synthesised include Zn acetate,11 Zn metal12 and ZnCl2.12 This scarcity is the result of the ALD of ZnO being dominated by zinc alkyls, namely, dimethyl13 and diethyl zinc14 (DEZ) which not only provide very high growth rates (∼1.8 Å per cycle) but also pose a safety hazard in that they are immensely pyrophoric. The challenge with DEZ is that its thermal window tends to extend from around 110–170 °C or thereabouts15 so it is not reliable to provide the means for a stable high temperature ALD process above 200 °C. Processes can be performed outside of this window but the thickness of the layer is then subjected to temperature fluctuations which can result in condensation or reactivity limited depositions at lower temperatures and decomposition or desorption at higher temperatures. This makes the incorporation of organic molecules in a molecular layer deposition (MLD) process for hybrid systems and metal organic framework (MOF) thin films, which can be grown in the gaseous phase using the aforementioned approach,16 rather difficult. It also makes the implementation of a consistent doping process for ternary systems problematic. Recently there has been an increased tendency in the literature to identify materials of both inorganic and organic nature using zinc oxide as the source of inorganic material.17 For these hybrid systems, compatible ALD windows of the ZnO inorganic and organic systems are essential and until now diethyl zinc has been the precursor of choice in most of these applications.18 The problem with this is that common organic molecules include carboxylic acids, other organic anions such as phosphonates and sulfonates as well as aromatic amines.19 Due to the robust, solid and aromatic nature of these molecules, they tend to possess rather high melting points & boiling points (∼250 °C), limiting their integration in both MLD and a gaseous MOF synthesis process with zinc oxide. Therefore it can be seen that as volatilisation of the organic part increases, diethyl zinc may lead to problems at higher temperatures. The instability of DEZ at high temperatures has been shown in the zincone MLD process carried out by Yoon et al.20 whereby, along with double reactions of ethylene glycol, the diffusion of DEZ is partly responsible for a decrease in the growth rate because at higher temperatures less DEZ may diffuse into the film and more DEZ may desorb at a faster rate. Herein, we report on the assessment of the structural and thermal properties of a range of new and known zinc ketoiminate precursors. We also establish a precursor that is able to grow polycrystalline thin films of ZnO with good growth rates (∼1.3 Å per cycle) at temperatures in the range from 175–300 °C. This precursor will extend the library of known zinc precursors as well as enlarge the cross-over window of ternary and quaternary systems for doping applications, while also increasing the potential variety of organic co-reactant molecules for MLD21 and gaseous MOF synthesis.22 It provides a solution to the known high temperature inconsistency of DEZ by allowing a stable ZnO ALD process to be carried out at high temperatures with promising growth rates.
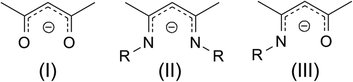 |
| | Fig. 1 Skeleton structures of β-diketonates(I), β-diketiminates(II) and β-ketoiminates(III). | |
Results and discussion
Synthesis
The synthesis of ketoimine ligands was realised by a condensation reaction of acetylacetone, H(acac), with the respective primary amines in diethylether and subsequent distillation. For the reaction of H(acac) with N-alkyl amines the Lewis acidic catalyst ytterbium triflate, [Yb(OTf)3],23 was added to the reaction solution to enhance the yield. All zinc ketoiminate complexes were synthesized by the addition of neutral ketoimines to a solution of diethylzinc in hexane at −20 °C. The proton NMR shifts are very similar for all complexes. The new compounds [Zn(eeki)2], [Zn(epki)2] and [Zn(dapki)2] show characteristic 1H-NMR chemical shifts of the pentane moiety at 4.81 ppm, 2.00 ppm and 1.65 ppm which match the literature reported values for other zinc ketoiminates.8–10 Crystals suitable for single crystal XRD were obtained from concentrated hexane reaction solutions in the cases of [Zn(mpki)2], [Zn(eeki)2] and [Zn(dapki)2] and stored at −30 °C. [Zn(npki)2] precipitates during the reaction as a white powder. Crystals were obtained from THF/hexane mixtures at −30 °C. In contrast to all other reported zinc ketoiminates, [Zn(epki)2] is the first liquid zinc ketoiminate under ambient conditions. The solid state structures of [Zn(mpki)2], [Zn(eeki)2], [Zn(dapki)2] and [Zn(npki)2] with an atomic numbering scheme are depicted in Fig. 2. All compounds possess a tetrahedral coordination sphere with a zinc cation surrounded by two nitrogen and two oxygen atoms from two bidentate, homoleptic ketoimines. In contrast, the solid state structure of zinc ketoiminate [Zn(daeki)2] reported by Schulz et al.10 showed an octahedral coordination sphere where the nitrogens of the EtN(Me)2 side chains complete the octahedral geometry surrounding the zinc. Selected bond lengths, bond angles and torsion angles of the solid state molecular structures are displayed in Table 1. All compounds crystallised were monoclinic and a full characterisation of the solid state structures can be found in the crystallographic data at the end of this paper. All compounds are monomeric and the ketoiminato ligands are orthogonal to each other. The main bonding atoms of the chelate ring ZnONC2–4 are creased slightly and the Zn–O bonds, for each compound, are shorter than the corresponding Zn–N bond distances. All bond lengths, angles and torsions are very similar to those previously reported.8 The bite angles of the chelate ring are all ∼97° while the bond angles of the binding oxygens and nitrogens with the Zn metal centre range from ∼106° to ∼122°, which differs from those expected for an ideal tetrahedron. The torsion angles illustrate that the chelating rings of the ligands are nonplanar. The C2–C3 bond lengths are closer to the ethylene bond length of 1.33 Å while the C3–C4 bond lengths are slightly longer than, but still less than, a regular carbon–carbon single bond of 1.54 Å. In all cases the side chains are located far from the coordination sphere of the zinc centre. The compound [Zn(npki)2] is a special case in that the molecule is chiral and both enantiomers are present making it a kind of racemate. However the group here is not centrosymmetric but still chiral which is known as a kryptoracemate.24
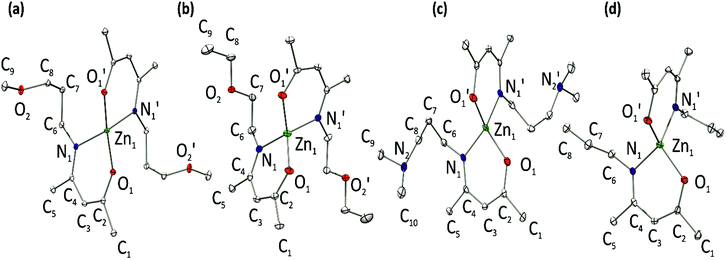 |
| | Fig. 2 Molecular solid-state structures of (a) [Zn(mpki)2], (b) [Zn(eeki)2], (c) [Zn(dapki)2] and (d) [Zn(npki)2]. Only one enantiomer of each racemic mixture is depicted, respectively. Thermal ellipsoids are shown at 50% probability. Hydrogen atoms are omitted for clarity. | |
Table 1 Selected bond lengths, bond angles and torsion angles of the solid state molecular structures. For [Zn(npki)2] only length and angles of one of the two enantiomers is given. The second enantiomer is described in Table S1 ESI
| Bond lengths (Å) |
[Zn(eeki)2] |
[Zn(mpki)2] |
[Zn(dapki)2] |
[Zn(npki)2] |
| Zn1–O1 |
1.9492(11) |
1.9484(9) |
1.9460(10) |
1.951(3) |
| Zn1–N1 |
1.9979(12) |
2.0020(11) |
1.9764(11) |
1.974(3) |
| O1–C2 |
1.2926(19) |
1.2924(15) |
1.2874(17) |
1.294(4) |
| N1–C4 |
1.311(2) |
1.3121(16) |
1.3067(18) |
1.305(4) |
| C2–C3 |
1.380(2) |
1.3826(18) |
1.377(2) |
1.375(5) |
| C3–C4 |
1.428(2) |
1.4323(18) |
1.429(2) |
1.424(4) |
| Bond angles (°) |
| O1–Zn1–N1 |
97.36(5) |
96.49(4) |
97.47(4) |
97.09(10) |
| O1–Zn1–N1′ |
122.35(5) |
120.43(4) |
115.68(4) |
118.84(11) |
| O1–Zn1–O1′ |
106.32(6) |
106.55(6) |
108.17(6) |
109.95(18) |
| N1–Zn1–N1′ |
112.84(7) |
117.61(6) |
122.60(7) |
116.29(17) |
| Torsion angles (°) |
| Zn1–N1–C4–C3 |
−6.0(2) |
−12.54(17) |
0.67(18) |
0.2(4) |
| Zn1–O1–C2–C3 |
1.9(2) |
11.94(18) |
0.6(2) |
−6.9(5) |
Thermal properties and solubility
To be considered as precursors for ALD gas phase depositions, compounds require a certain level of thermal and chemical stability in order to guarantee that surface reactions, as opposed to decomposition, take place at the surface. In order to bring a precursor into the gas phase without decomposition and to ensure a constant flux of the molecule at a specific temperature, the thermal properties of the precursor need to be assessed. Therefore each complex underwent thermogravimetric (TG), isothermal-TG (iso-TG), differential thermal analysis (DTA) and melting point analyses. All compounds are relatively low melting solids with the exception of [Zn(epki)2] which is liquid at room temperature. As can be seen from the TG of the compounds in Fig. 3a, all compounds are volatile and show similar behaviour in that they have a one-step mass loss with a relatively large temperature window for vaporisation (170–270 °C) where the majority of the compound is volatilised. The two exceptions to this are [Zn(npki)2] which has a steeper mass loss at lower temperatures (130–230 °C) and [Zn(epki)2] which has a broader mass loss at higher temperatures (170–305 °C). A closer analysis of the melting points in Table 2 illustrates the effect that the functional group of the imine has on the overall thermal properties. Firstly, by looking at the influence of the chain length with identical functional species, it can be deduced that increasing the alkyl chain length was found to bring about a reduction of the melting point and generally an increase in the residual masses of the compounds. This can be observed for all compounds except the methoxy terminated species, as can be seen from Table 2. Long and low branched alkyl chains tend to become highly mobile upon moving towards the liquid state which makes melting more entropically favoured.25 Looking at the influence of the different functional groups, it can be seen that the ethoxy terminated species have the lowest melting points with 39 °C for [Zn(eeki)2] and [Zn(epki)2] being a liquid at room temperature. These values are much lower than their methoxy counterparts [Zn(meki)2] and [Zn(mpki)2] (69 °C and 73 °C, respectively). The substitution of the ether moiety for a dimethyl amido group also had an influence upon the melting point with [Zn(dapki)2] having a melting point of 62 °C which is slightly lower than the methoxy terminated compound of the same chain length. However this is still much higher compared to the corresponding ethoxy terminated compound [Zn(epki)2]. The highest melting point belongs to [Zn(npki)2] (103 °C), the only ketoiminate in this series without a functional group. To evaluate the suitability of a metal–organic compound as an ALD precursor, additional thermal properties need to be considered such as onset of volatilisation (∼1% weight loss), one-step weight loss and low residual mass. The onsets of volatilisation show the same trend for the ethoxy terminated ketoiminates compared to the methoxy terminated species. With [Zn(eeki)2] and [Zn(epki)2], volatilisation begins around 120 °C while for [Zn(meki)2] and [Zn(mpki)2], it starts much higher at 145 °C. Despite the fact that [Zn(epki)2] is a liquid, the volatilisation behaviour is not significantly influenced compared to the solid [Zn(eeki)2]. Although, the dimethylamido terminated compounds have melting points in a relatively similar range as the methoxy terminated species, their onsets are much lower. Interestingly the volatilisation of [Zn(npki)2] begins at 94.6 °C, the lowest temperature compared to the other complexes. It undergoes a one-step mass loss between 130 °C and 230 °C which is similar to that reported by Holmes et al. (140–240 °C).9 The residual masses of all precursors are ∼2%, much lower than the literature known fluorinated ketoiminates8 and far below the percentage weight value of ZnO and therefore a sign of their volatility. [Zn(eeki)2] and [Zn(epki)2] are the outliers here with higher residual masses than the others of 4.9% and 7.1%, respectively. This could be most likely due to residual carbon contamination associated with the mechanism of decomposition of the ethoxy terminated species. In the case of [Zn(epki)2] the increased value can be justified by the longer chain of the side arm compared to [Zn(eeki)2]. The influence of molecular engineering on the compounds is greatly highlighted by the comparison of [Zn(eeki)2] and [Zn(mpki)2]. These two compounds have the same molecular mass and differ only in the placement of the oxygen within the side chain, however, their thermal properties are drastically different. What can be derived from this is, ethoxy terminated species result in low melting points, reduced volatilisation onsets but with higher residual masses, whereas methoxy terminated side chains give moderate melting points with increased volatilisation onsets but with lower residual masses. To compare, dimethyl amido terminated compounds provide more moderate melting points with reduced volatilisation onsets than the methoxy species and lower residual masses than the ethoxy species. The shortest and only unfunctionalised ketoiminate, namely [Zn(npki)2] results in a high melting point in conjunction with the lowest onset and a normal residual mass for this series of zinc ketoiminates. The sublimation behaviour as well as thermal stability of the precursors were also assessed by iso-TG experiments carried out at 130 °C. As depicted in Fig. 3b, a linear weight loss was observed in each case highlighting that the compounds can sublime at the pre-programmed temperature with constant evaporation rates for an extended duration of time, except for [Zn(daeki)2] and [Zn(npki)2]. A slight curve at the beginning of the measurement is indicative of partial decomposition. High volatilisation rates are also associated with these compounds. The sublimation rates are listed in the analysis data of Table 2.
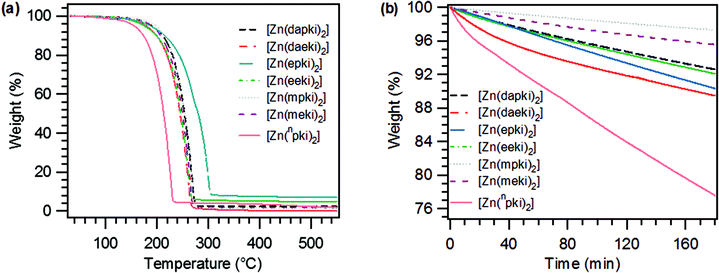 |
| | Fig. 3 (a) TG and (b) isothermal-TG analysis of all zinc ketoiminate compounds synthesised. | |
Table 2 Thermal analyses of all synthesised ketoiminates including melting point, volatilisation onsets, residual masses and evaporation rates at 130 °C
| Precursor |
Side chain |
Melting point (°C) |
Onset of volatilisation (°C) |
Residual mass (%) |
Evaporation rate (130 °C – μg min−1) |
| [Zn(dapki)2] |
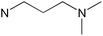
|
62.2 |
121.7 |
2.4 |
5.1 |
| [Zn(daeki)2] |
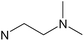
|
69.7 |
135.8 |
0.1 |
10.8 |
| [Zn(epki)2] |

|
<RT |
125.6 |
7.1 |
5.4 |
| [Zn(eeki)2] |
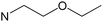
|
39.4 |
120.2 |
4.9 |
5.4 |
| [Zn(mpki)2] |

|
73.2 |
145.3 |
1.5 |
1.8 |
| [Zn(meki)2] |

|
69.1 |
145.7 |
1.8 |
2.8 |
| [Zn(npki)2] |

|
103.4 |
94.6 |
2.1 |
21.3 |
The linear behaviour of the isothermal curves of the other precursors demonstrates that the complexes are capable of vaporising cleanly without the possibility of premature decomposition. The thermal stabilities of all ketoiminate complexes were further investigated by NMR decomposition experiments26 whereby concentrated solutions of the compounds, sealed in heavy walled NMR tubes, were heated in an oven at 220 °C for a long period of time. Intermittently, the compounds were cooled to room temperature and a 1H NMR study was carried out. Through the comparison of the integral areas of the NMR peaks which are normalised to the solvent peak, the amount of undecomposed precursor was assessed. Using linear fits of the obtained curves, the half-life of the compound was calculated. The results for the [Zn(meki)2] compound is taken here as a representative example in Fig. 4. For the entire series of compounds, decompositional studies data are illustrated in Table 3. The data illustrates that all zinc ketoiminates are thermally stable complexes, which is a pre-requisite for good ALD precursors. The example, [Zn(meki)2] shown here has a half-life of ∼11 days at 220 °C and highlights that this particular compound is quite thermally resilient. See Fig. S1–S6 (ESI†) for other spectra. Although, it must be noted that the method is solution based, it gives us a good idea of the general thermostability of the complex, through which we must infer that this provides an insight into how it will behave in the gas phase.
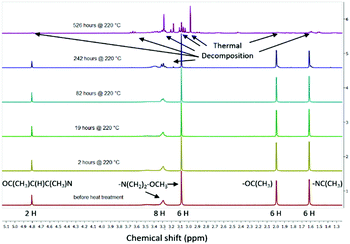 |
| | Fig. 4 NMR decomposition study of [Zn(meki)2] carried out at 220 °C. | |
Table 3 Half-life decompositional values for all synthesized zinc ketoiminate compounds
| Precursor |
Half-life (@220 °C) |
| [Zn(dapki)2] |
5.6 days |
| [Zn(daeki)2] |
2.2 days |
| [Zn(epki)2] |
8.4 days |
| [Zn(eeki)2] |
23.8 days |
| [Zn(mpki)2] |
35 days |
| [Zn(meki)2] |
11 days |
| [Zn(npki)2] |
<12 hours |
ZnO process development
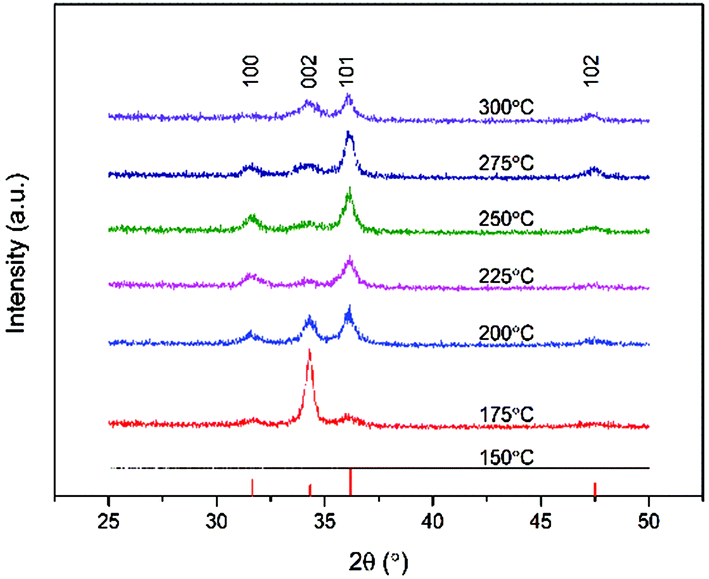
The mix of a low melting point in conjunction with high volatility is a desirable combination for the implementation of a precursor for ALD. Therefore the deposition of ZnO thin films by ALD was studied using [Zn(eeki)2] as the metal and water as the oxygen sources, respectively, over a temperature range of 150–300 °C. This precursor has an intramolecular metal oxygen and an intramolecular metal nitrogen bond within the molecular framework and therefore shows great reactivity towards water under ambient conditions. As seen above, the thermostability of the compound is immensely high. One of the main aims of this work was to develop a water assisted ALD process for zinc oxide at high temperatures and so a series of experiments were designed to assess the characteristic features of ALD such as, the ALD window, self-saturated growth and the linearity. Fig. 5a shows the growth rate per cycle as a function of deposition temperature. An ALD window can be observed from Tdep = 175–300 °C with an average growth rate of ∼1.3 Å per cycle. Growth rates were minimal at low temperatures highlighting that the activation energy of the process was not sufficient to form a film as it was reactivity limited. Depositions were not carried out below 150 °C as a result, while none were carried out over 300 °C due to limitations imposed by the reactor. Depositions in this set of experiments were carried out using [Zn(eeki)2] and H2O pulses of 4 s and 4 s, respectively, with purge lengths of 10 s each between pulses and 250 cycles in total. All films deposited were uniform and faint light brown in appearance. The broad window provides a means of lengthening the window for stable high temperature ALD processes and is the first zinc precursor to do so above 200 °C with reasonable growth rates. This should be of interest for MLD processes as it widens the precursor cross-over window for organic molecules as well as dopants27 for high temperature, ternary and quaternary systems. Fig. 5b shows a plot of the film thickness against the number of cycles. From the graph it is evident that the thickness increases linearly with the increasing number of cycles within the ALD window (225 °C). This was carried out with [Zn(eeki)2] and H2O pulses of 4 s and 4 s, respectively, with 2 intervening 10 s purges. Films were grown at 250, 500 and 1000 cycles. To verify the self- limiting growth of the process, the saturation behaviours of both the [Zn(eeki)2] and H2O precursors were investigated at a deposition temperature of 225 °C. Samples were deposited holding the H2O pulse, purges and number of cycles constant at 4 s, 10 s and 250, respectively, while the precursor pulse was systematically increased from 0.1, 0.25, 0.5, 1, 2 to 4 s. This same pulse sequence was then repeated for water whilst keeping the above process parameters constant. From Fig. 5c, it is clear that for [Zn(eeki)2] pulses <1 s result in lower growth rates due to subsaturative growth while pulses ≥1 s enable saturated self-limiting growth. A similar result was obtained for water saturation but at 2 s as illustrated in Fig. 5d. A full list of the deposition parameters used in this work can be found in Table 4. The crystallinity of the ALD grown ZnO thin films (thickness ∼30 nm) deposited on the Si(100) was analysed using grazing incidence X-ray diffraction (GI-XRD). As expected the deposition temperature plays a major role in the crystallisation of the films. Fig. 6 shows that all films were found to be polycrystalline at temperatures ranging from 175 to 300 °C and were indexed according to the ZnO wurtzite structure (PDF no. 00-036-1451, zincite phase). Intensities of the reflections are low due to the thickness of the films. No reflections were found at 150 °C. For films grown at 175 °C, only a single reflection at 34.4° is observed corresponding to the 002 orientation (c-axis) with minute satellite contributions from the encompassing 100 and 101 reflections at 31.8° and 36.3°, respectively. Upon increasing the temperature further to 200 °C, there is a marked decrease in the 002 peak and a notable increase in the 101 reflection. Both reflections are of similar intensity and there is also a slight contribution from the a-axis 100 reflection. The decrease in the 002 signal continues with a further increase of the temperature to 225 °C and 250 °C where [100] and [101] become the dominant orientations.
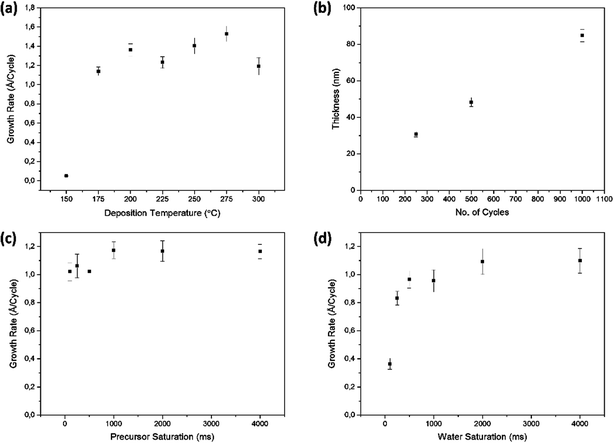 |
| | Fig. 5 (a) Growth rate as a function of deposition temperature. An ALD window is observed between 175 and 300 °C. (b) Film thickness as a function of the number of deposition cycles. Data were collected using 4 s and 4 s pulse lengths for [Zn(eeki)2] and H2O, respectively, at 225 °C. (c) Precursor saturation and (d) water saturation both carried out at 225 °C. | |
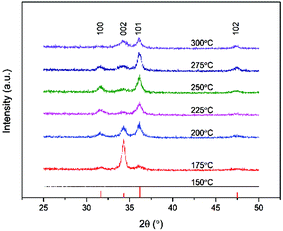 |
| | Fig. 6 GI-XRD patterns of ZnO thin films as-deposited by ALD on Si(100) at various stated deposition temperatures (thicknesses ∼30 nm). Reference pattern PDF no. 00-036-1451, zincite phase. | |
Table 4 Deposition parameters used for the thermal ALD of [Zn(eeki)2]
| Precursor |
[Zn[eeki)2] |
| Deposition temperature (°C) |
150–300 |
| Evaporation temperature (°C) |
100 |
| Reactor pressure (mbar) |
0–3 |
| Mass of precursor (g) |
0.4–1 |
| Precursor pulse (ms) |
100–4000 |
| Water pulse (ms) |
100–4000 |
| Purge times (ms) |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| Number of cycles |
250–1000 |
| Pulse sequence (s) |
4–10–4–10 |
| Wafers |
Si(100), quartz glass |
At higher temperatures again (275 °C) we see a slight decrease of the 100 reflection combined with a minor resurgence of the 002 peak with 101 as the dominant reflection. Finally, at 300 °C the 002 and 101 reflections are of similar intensity once again. There is also a weak contribution from the 102 reflection at 47.6° which increases modestly at higher temperatures. These orientations are in contrast to those made with diethyl zinc which have a strong emphasis on the a-axis between 160 and 200 °C28 and then increasing the temperature above 220 °C switches the predominant orientation to the c-axis29 which is also favoured at low deposition temperatures. The chemical compositions of the films deposited at each temperature were investigated using Rutherford backscattering (RBS) and nuclear reaction analysis (NRA) experiments as illustrated in Fig. 7. For all films measured, the zinc and the oxygen signals are evident apart from that of 150 °C which shows a reduced signal due to low surface coverage. The metal to oxygen ratios in the ZnO films were obtained by fitting the RBS curves. Selected results of RBS/NRA have been shown in Table 5. As is evident, the films are slightly oxygen rich with the ratio moving closer towards 1:1 at higher temperatures. See Table S2 (ESI†) for the full list of data. The films grown at lower temperatures tend to have a higher oxygen content. This was also observed by Guziewicz et al.30 The carbon content in the films is moderately high, especially at lower temperatures, however it is unclear as to whether these contaminations were on the surface or in the films and therefore complimentary XPS measurements were carried out to devise a greater knowledge of the chemical composition at the surface. XPS analyses were carried out on selected ZnO films deposited at 175 °C, 225 °C and 275 °C to further investigate the stoichiometry of the films. Fig. 8 shows the complete survey spectra before and after sputtering of the film deposited at 225 °C. The samples were analysed ‘as-introduced’ and after one minute of sputtering. The composition and purity remain similar for the whole temperature range. The samples ‘as introduced’ contain anywhere between ∼18 and 25 at% of adventitious carbon depending on the temperature. Despite the relatively high carbon content on the surface, it is worth noting that the C 1s photoelectron signals were reduced to below the atomic percentage detection limit after 1 min Ar+ sputtering and all Zn/O ratios were calculated to be ∼1.08. Thus it is concluded that the bulk is practically free of carbon. Overall it can be stated that within the measurement uncertainty of both compositional analysis methods, the films were determined to be stoichiometric for zinc oxide (1:1). These observations, along with the absence of any nitrogen peaks, suggest a negligible incorporation of precursor residuals into the obtained thin films. As a consequence, we conclude that the applied precursor underwent clean reactions to ZnO under the processing conditions. The signal shapes and position of the Zn 2p3/2 and O 1s peaks indicate the presence of different species as displayed in Fig. 8(b) and (c), respectively, and are also in good agreement with the previous reports for ZnO.31 The O 1s signal was composed of three distinct peaks at 530.1 eV, 531.4 eV, and 532.3 eV. The O 1s component at 530.1 eV corresponds to zinc oxide, the peak at 531.4 eV belongs to organic contaminants while the remaining signal at 532.3 eV is attributed to the hydroxyl groups of zinc hydroxide32. The Zn 2p3/2 signal was fitted with two components at 1021.7 eV and 1023.1 eV assigned to ZnO and Zn(OH)2, respectively.33 In order for ZnO to be implemented for potential TCO applications such as, as a transparent electrode or for solar cell devices with aluminium, transmission values should be at least 80% to be feasible.34 With this in mind the optical band gap of the as-deposited ZnO material was determined using UV-Vis spectroscopy. Fig. 9(a) illustrates the transmission plotted against the wavelength measurement while (b) shows the corresponding Tauc plot. As can be seen, the transmission of the sample is high (∼96%) and consequently the absorbance is low throughout the entire visible region. This highlights that the transmission value is eligible to be implemented in TCO applications. Interestingly, transmission values in this study are slightly higher than those reported in the literature (∼90%) which were grown using diethyl zinc and water and are of a similar thickness.35 The band gap derived from the Tauc plot was calculated to be 3.29 eV which was found to be in accordance with the literature values according to Viezbicke et al.36
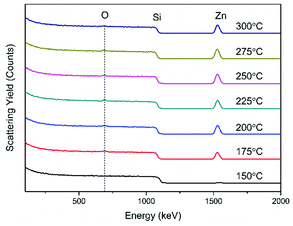 |
| | Fig. 7 RBS data of ZnO thin films as-deposited by ALD on Si(100) at deposition temperatures 150–300 °C. Spectra are vertically shifted for clarity. | |
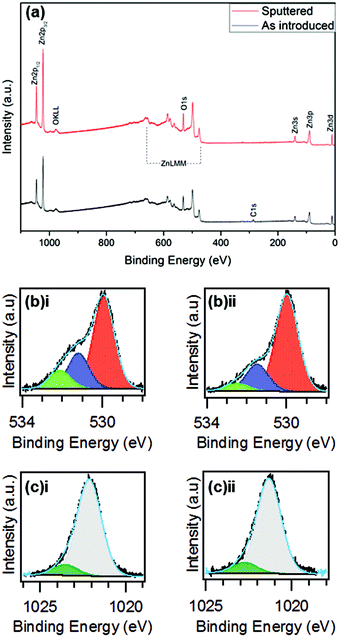 |
| | Fig. 8 (a) XPS survey spectra of a ZnO film deposited on Si(100) at 225 °C both as introduced and after 1 min sputtering. (b) O 1s binding energy peaks (i) as introduced & (ii) after sputtering. (c) Zn 2p3/2 binding energy peaks (i) as introduced & (ii) after sputtering. | |
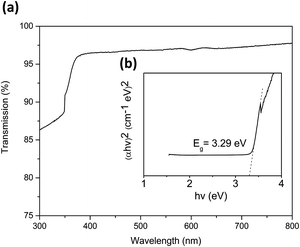 |
| | Fig. 9 (a) Optical transmission and (b) the derived Tauc plot of a ZnO film deposited at 225 °C on quartz glass. The fluctuation seen within the Tauc plot corresponds to the changing of the lamps in the spectrometer at low wavelengths. | |
Table 5 RBS/NRA data for selected ZnO samples deposited at 175 °C, 225 °C and 275 °C. The zinc to oxygen ratio determined by RBS and the amount of carbon impurities derived from NRA are featured below. For a full set of data see Table S2 (ESI)
| Deposition temperature |
175 °C |
225 °C |
275 °C |
| Zn/O ratio |
0.86 |
0.90 |
0.91 |
| C (%) |
19.1 |
5.6 |
9.3 |
Conclusions
A series of zinc ketoiminates were molecularly engineered in order to optimise the physico-chemical properties of the ketoiminate precursor class, in terms of melting point, onset of volatilisation rate, volatilisation rate, reactivity and thermal stability. The influence of the functional groups at the imino side chain and thermostability properties were investigated as each precursor was assessed for their suitability as a precursor for gas phase depositions. In terms of use as an ALD precursor, the ethoxy species provided the best thermal properties with low melting points, reduced onsets and stable fluxes in comparison to the other species. The dimethyl amido compound [Zn(dapki)2] does have a similar onset and flux with respect to the ethoxy species however it has a higher melting point associated with it also. The only unfunctionalised compound [Zn(npki)2] was found to have the lowest onset but the highest melting point and evaporation rate. Due to the low thermal stability and high evaporation rates of this compound and also [Zn(daeki)2], these compounds might be good candidates for CVD. All compounds are easily synthesised with good yields, are inferentially monomeric, exhibit high thermal stability and are sufficiently volatile making each a viable candidate for ALD or CVD. Their varied oxygen and nitrogen coordination displays a decent agreement between the highly reactive and unstable β-diketiminates and the stable β-diketonates and so allows their reaction with water. The most promising candidate was selected for ALD testing, namely [Zn(eeki)2], where it demonstrated self-saturated, water assisted growth of ZnO with growth rates in the range of 1.3 Å per cycle. The low melting solid compound, in contrast to commercially available precursors, is capable of depositing stoichiometric and polycrystalline ZnO, in a stable manner over a very broad range at high temperatures for ALD, from 175–300 °C. The crystallographic orientations of the layers were found to be temperature dependent and go against the literature trend of c-axis dominant ZnO thin films mainly grown using diethyl zinc. UV-Vis measurements revealed that transparency is high in the visible range and exhibits a band gap of 3.29 eV. This precursor, as far as the authors are aware, is the first of its kind to carry out stable high temperature ALD for ZnO above 200 °C with good growth rates. It extends the known library of precursors for ZnO while also contributing to MLD and gaseous MOF synthesis by widening the potential organic co-reactants available for the aforementioned procedures with its enlarged temperature window. Preliminary studies in this area are now being investigated within the group.
Experimental
Precursor synthesis and characterisation
Syntheses of the ketoimines were carried out via a condensation reaction of acetylacetone with the respective primary amine in diethylether. Spectroscopically pure products were obtained via distillation of the crude product in a vacuum. The subsequent formation of zinc complexes was carried out following a modified procedure reported by Bekermann et al.8
Crystals suitable for single crystal X-ray diffraction were selected from the crude product with the aid of an optical microscope. The crystal was coated with perfluoropolyether, picked up with a glass fibre ([Zn(mpki)2], and [Zn(dapki)2]) or a cryo loop ([Zn(eeki)2] and [Zn(npki)2]) and immediately mounted in the nitrogen cold gas stream of a diffractometer. The diffraction data were processed with CrysAlisPro.37 An absorption correction based on multiple-scanned reflections was carried out with ABSPACK in CrysAlisPro. The crystal structures were solved by direct methods using SHELXS-9738 and refined with SHELXLe-2013.39 The X-ray intensity data of [Zn(mpki)2], and [Zn(dapki)2] were collected on an Oxford Diffraction Xcalibur2 diffractometer with a Sapphire2 CCD and Mo Kα radiation. The X-ray intensity data for [Zn(eeki)2] and [Zn(npki)2] were collected on an Agilent Technologies SuperNova diffractometer with an Atlas CCD detector and Cu Kα radiation from a microfocus X-ray source with multilayer X-ray optics. Crystal data and details of structure determination can be found in Table 6. CCDC numbers for the compounds [Zn(mpki)2], [Zn(npki)2], [Zn(eeki)2] and [Zn(dapki)2], are 1502611, 1502612, 1502613 and 1502614 respectively. The NMR spectra of moisture and air sensitive samples were obtained in degassed, deuterated solvents, which were stored over activated molecular sieves. Preparation of air and moisture sensitive samples was carried out under an argon atmosphere in glove boxes (MBraun, Labmaster). 1H- and 13C-NMR spectra were recorded on a Bruker Advance DPX 200 (automation mode) instrument and referenced to internal solvents (residual proton signals) and corrected to the TMS (tetramethylsilane) standard values. Decomposition NMR experiments were recorded manually on a Bruker Advance DPX 250 instrument. Heavy walled NMR tubes (Wilmad-LabGlass, thickness 1.4 mm) were connected to a full length joint by RUB glass blowing facility. The compound was dissolved in deuterated, freshly degassed benzene. The samples were frozen in liquid nitrogen, evacuated and NMR tubes were ablated. 1H-NMR spectra were recorded before heat treatment as reference spectra. For all compounds, one NMR tube was kept at 220 °C, respectively. The receiver gain and the number of scans were kept constant for all experiments. The integrals of the deuterated solvents were taken as a reference for the decomposition of the compound. Elemental analysis (EURO EA, EURO VECTOR) and atomic absorption spectroscopy (M-Series, Thermo Electron) were performed by the lab of microanalytics and thermoanalysis, University Duisburg-Essen. Electron ionization mass spectra (EI-MS) were recorded at 70 eV by using a Varian MAT spectrometer. Simultaneous thermogravimetric and differential thermal analyses (TG/DTA) were carried out by using a Seiko TG/DTA 6200/SII at ambient pressure (sample size ≈ 10 mg), at a heating rate of 5 K min−1 (N2 flow rate = 300 mL min−1). Compounds [Zn(meki)2],8 [Zn(mpki)2],8 [Zn(npki)2],9 and [Zn(daeki)2]10 were all synthesised using a modified procedure reported by Bekermann et al.8
Table 6 Crystallographic data for the compounds [Zn(mpki)2], [Zn(eeki)2], [Zn(dapki)2] and [Zn(npki)2]
| |
[Zn(mpki)2] |
[Zn(eeki)2] |
[Zn(dapki)2] |
[Zn(npki)2] |
| Formula |
C18H32N2O4Zn |
C18H32N2O4Zn |
C20H38N4O2Zn |
C16H28N2O2Zn |
|
M
r (g mol−1) |
405.82 |
405.82 |
431.93 |
345.77 |
| Temperature (K) |
106 |
106 |
113 |
110 |
| Wavelength (Å) |
0.71073 |
1.54178 |
0.71073 |
1.54178 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P2/n |
P2/n |
P2/c |
C2 |
|
a (Å) |
10.1081(2) |
13.2557(4) |
10.6251(2) |
21.5697(5) |
|
b (Å) |
5.45455(12) |
5.89050(10) |
5.40880(10) |
5.67097(8) |
|
c (Å) |
17.4938(4) |
13.9479(6) |
20.3670(3) |
17.9276(4) |
|
α (°) |
90 |
90 |
90 |
90 |
|
β (°) |
95.081814(19) |
106.615(3) |
102.906(2) |
127.823(4) |
|
γ (°) |
90 |
90 |
90 |
90 |
| Volume (Å3) |
960.73(4) |
1043.62(6) |
1140.90(3) |
1732.20(10) |
|
Z
|
2 |
2 |
2 |
4 |
|
D
calc. (g cm−3) |
1.403 |
1.291 |
1.257 |
1.326 |
|
μ (mm−1) |
1.302 |
1.819 |
1.097 |
2.009 |
|
F(000) |
432 |
432 |
464 |
736 |
| Crystal size (mm) |
0.15 × 0.13 × 0.05 |
0.14 × 0.1 × 0.07 |
0.19 × 0.15 × 0.11 |
0.37 × 0.067 × 0.044 |
| 2Θ range (°) |
7.472–57.734 |
8.116–148.122 |
6.288–49.998 |
8.2–152.34 |
| Reflections collected |
15499 |
7792 |
20128 |
31521 |
| Unique reflections |
2338 (Rint = 0.0291) |
2093 (Rint = 0.0298) |
2016 (Rint = 0.0250) |
3572 (Rint = 0.0333) |
| Completeness to Θ max |
99.1 (Θ max = 27.494) |
98.7 (Θ max = 74.061) |
99.7 (Θ max = 24.99) |
99.0 (Θ max = 76.17) |
| Data/restraints/parameters |
2338/0/117 |
2093/0/117 |
2016/0/127 |
3572/1/197 |
| GOF on F2 |
1.107 |
1.066 |
1.144 |
1.047 |
| Final R indices [I > 2σ (I)] |
R
1 = 0.0235 |
R
1 = 0.0301 |
R
1 = 0.0222 |
R
1 = 0.0366 |
|
|
wR2 = 0.0580 |
wR2 = 0.0802 |
wR2 = 0.0557 |
wR2 = 0.0994 |
|
R indices (all data) |
R
1 = 0.0266 |
R
1 = 0.0308 |
R
1 = 0.0234 |
R
1 = 0.0369 |
|
|
wR2 = 0.0592 |
wR2 = 0.0810 |
wR2 = 0.0563 |
wR2 = 0.0997 |
| Largest diff. peak and hole (e Å−3) |
0.45/−0.20 |
0.33/−0.29 |
0.33/−0.13 |
1.23/−0.44 |
Synthesis of N-(3′-dimethylamidopropyl)-2-penten-2-ol-4-imine [H(dapki)].
To a solution of 150 mmol (15.4 ml) acetyl acetone in 40 ml diethylether, 151 mmol (21.05 ml) 3-dimethyamidopropyl amine were added. The mixture was refluxed for one hour and stirred overnight at room temperature. The mixture was dried over magnesium sulphate and filtered before the removal of diethylether via vacuum. The resulting yellow solution was distilled under reduced pressure (Tb = 69–75 °C, p = 4–6 × 10−2 mbar) Yield: 28.3 g (95.7%) of a clear colourless solution. 1H NMR (200 MHz, C6D6) δ 11.49–10.84 (s, 1H), 4.87 (s, 1H), 3.26 (qui., 1H), 3.15 (t, J = 6.1 Hz, 2H), 2.92 (q, J = 6.4 Hz, 2H), 2.00 (s, 3H), 1.45 (m, 6H), 1.02 (s, 3H), 0.99 (s, 3H). 13C NMR (50 MHz, C6D6) δ 194.07, 162.11, 95.24, 56.35, 45.34, 40.52, 28.95, 28.42, 18.36.
Synthesis of N-(2′-ethoxyethyl)-2-penten-2-ol-4-imine [H(eeki)].
To a solution of 150 mmol (15.4 ml) acetyl acetone in 40 ml diethylether, 151 mmol (21.05 ml) 2-ethoxyethyl amine were added. The mixture was refluxed for one hour and stirred overnight at room temperature. The mixture was dried over magnesium sulphate and filtered before the removal of diethylether via vacuum. The resulting yellow solution was distilled under reduced pressure (Tb = 61–65 °C, p = 4–6 × 10−2 mbar). Yield: 28.3 g (95.7%). 1H NMR (200 MHz, C6D6) δ 11.23 (s, 1H), 4.89 (s, 1H), 3.14 (q, J = 7.0 Hz, 2H), 3.01 (t, J = 3.0 Hz, 2H), 2.83 (q, J = 5.5 Hz, 2H), 2.02 (s, 3H), 1.48 (s, 3H), 1.02 (t, J = 7.0 Hz, 3H). 13C NMR (50 MHz, C6D6) δ 194.33, 162.05, 95.60, 69.96, 66.66, 43.00, 28.98, 18.57, 15.27.
Synthesis of N-(3′-ethoxypropyl)-2-penten-2-ol-4-imine [H(epki)].
To a solution of 240 mmol (24.7 ml) acetylacetone in 60 ml diethylether, 238.4 mmol (25 ml) 2-ethoxypropyl amine were added. The mixture was refluxed for one hour and stirred overnight at room temperature. The mixture was dried over magnesium sulphate and filtered before the removal of diethylether via vacuum. The resulting yellow liquid was distilled under reduced pressure (Tb = 70–75 °C, p = 4–6 × 10−2 mbar). Yield: 39.5 g (96.8%) of a clear colourless liquid. 1H NMR (200 MHz, C6D6) δ 11.15 (s, 1H), 4.87 (s, 1H), 3.15 (m, 4H), 2.90 (q, J = 6.4 Hz, 2H), 2.01 (s, 3H), 1.55–1.31 (s + qui, 5H), 1.04 (t, J = 7.0 Hz, 3H). 13C NMR (50 MHz, C6D6) δ 194.11, 162.28, 95.30, 67.14, 66.26, 39.79, 30.75, 28.89, 18.28, 15.36.
Synthesis of bis(N-(3′-dimethylaminopropyl)-2-penten-2-on-4-iminate) zinc(II) [Zn(dapki)2].
After a solution of 29.4 mmol (3 ml) diethylzinc in 100 ml hexane was stirred overnight, 56.2 mmol (10.4 g) N-(3′-dimethylaminopropyl)-2-penten-2-ol-4-imine [H(dapki)] were added via syringe at −20 °C. The solution was allowed to heat to room temperature and stirring was continued overnight. The solution was concentrated and the product was crystallised at −20 °C. Pale yellow crystals were collected by filtration and dried via vacuum. Yield 11.2 g (92.6%). Found: C, 54.9%; H, 9.00%; N, 12.8%; Zn 14.9%. C20H38N4O2Zn requires: C, 55.62%; H, 8.87%; N 12.97%; Zn 15.14%. 1H NMR (200 MHz, C6D6) δ 4.81 (s, 1H), 3.28 (t, J = 6.8 Hz, 2H), 2.20–1.92 (m, 11H), 1.74 (m, 2H), 1.65 (s, 3H). 13C NMR (50 MHz, C6D6) δ 183.15, 172.33, 96.95, 57.34, 49.09, 45.40, 29.37, 27.81, 21.48. EI-MS [m/z]: 430.4 [M+], 247.1 [(M − L)+], 176.1 [C6H9NOZn], 58.0 [NMe3].
Synthesis of bis(N-(2′-ethoxyethyl)-2-penten-2-on-4-iminate) zinc(II) [Zn(eeki)2].
After a solution of 29.4 mmol (3 ml) diethylzinc in 150 ml hexane was stirred overnight, 58.8 mmol (10.1 g) N-(2′-ethoxyethyl)-2-penten-2-ol-4-imine [H(eeki)] were added via a syringe at −20 °C. The solution was allowed to heat to room temperature and stirring was continued overnight. The solution was concentrated and the product was crystallised at −20 °C. Pale yellow needle-like crystals were collected by filtration. The crystals were then washed with cold pentane and dried via vacuum. Yield 11.1 g (93.4%). Found: C, 52.4%; H, 7.95%; N, 6.7%; Zn 14.9%. C18H32N2O4Zn requires: C, 53.27%; H, 7.95%; N 6.9%; Zn 16.11%. 1H NMR (200 MHz, C6D6) δ 4.81 (s, 1H), 3.64–3.21 (m, 6H), 2.00 (s, 3H), 1.65 (s, 3H), 1.06 (t, J = 7.0 Hz, 3H). 13C NMR (50 MHz, C6D6) δ 183.48, 172.96, 97.06, 70.32, 66.57, 50.95, 27.83, 21.90, 15.31. EI-MS [m/z]: 405.1 [M+], 234.0 [(M − L)+], 188.0 [(M − L − OEt)+].
Synthesis of bis(N-(3′-ethoxypropyl)-2-penten-2-on-4-iminate) zinc(II) [Zn(epki)2].
After a solution of 12.74 mmol (1.3 ml) diethylzinc in 100 ml hexane was stirred overnight, 25.6 mmol (4.72 g) N-(3′-ethoxypropyl)-2-penten-2-ol-4-imine [H(epki)] were added via syringe at −20 °C. The solution was allowed to heat to room temperature and afterwards refluxed for 1.5 hours. It was cooled to room temperature and stirring was continued overnight. Hexane was removed and the resulting orange liquid was dried in a vacuum. Yield 5.3 g (96.3%). Found: C, 61.2%; H, 8.87%; N, 5.4%; Zn 15.8%. C20H36N2O4Zn requires: C, 55.36%; H, 8.36%; N 6.46%; Zn 15.07%. 1H NMR (200 MHz, C6D6) δ 4.80 (s, 1H), 3.46–3.08 (m, 6H), 2.00 (s, 3H), 1.95–1.78 (m, 2H), 1.65 (s, 3H), 1.05 (t, J = 7.0 Hz, 3H). 13C NMR (50 MHz, C6D6) δ 183.20, 172.68, 97.01, 68.07, 66.10, 48.15, 31.54, 27.79, 21.45, 15.47. EI-MS [m/z]: 432.2 [M+], 248.1 [(M − L)+], 176.0 [C6H9NOZn].
ALD of ZnO
ALD experiments were carried out in an F-120 SAT ALCVD reactor (ASM Microchemistry Ltd) on Si(100) substrates (Siegert Consulting, 2′′ diameter). Substrates were rinsed with 2-propanol and sonicated for 30 minutes in water. The 2-propanol and water used were of Baker HPLC analysed quality. The substrates were dried in an argon flow; the native oxide was not removed. The precursor boat was filled inside a glove box, but during the transport to the reactor the precursor was in short contact with air for a maximum time of one minute. Water used as a precursor during depositions was again of Baker HPLC analysed quality. A constant nitrogen flow (alphagaz™ 2, 99.9999%, Air Liquide) was used to transport the precursor into the reactor. Film growth occurred at a pressure of 0–3 mbar for the duration of the process. The ALD growth of ZnO was studied at substrate temperatures ranging from 150 to 300 °C using the following standard pulsing sequence (ALD growth cycle): 4 s of zinc precursor pulse, followed by 10 s of nitrogen purge, 4 s of water pulse, and finally 10 s of nitrogen purge.
Thin film characterisation
Thickness measurements were carried out with a Filmetrics F20 system. The crystallinity of the films was investigated by grazing-incidence X-ray diffraction (GI-XRD) analyses using an X′ Pert PRO PANalytical diffractometer (Bragg–Brentano θ − 2θ geometry) with automatical divergence slits, position sensitive detector, continuous mode, room temperature, Cu-Kα radiation, and Ni-filter, in the range of 2θ = 25–50° (step size 0.01°, time per step – 1 s). RBS measurements using 2.0 MeV 4He+ ions were performed using an instrument from the Dynamitron Tandem Laboratory (DTL) in Bochum. A beam intensity of about 20–40 nA incident to the sample at a tilt angle of 7° was used. The backscattered particles were measured at an angle of 160° by using a Si detector with a resolution of 16 keV. The stoichiometry of the films was calculated with the program SIMNRA40 by using the stopping powers of the program SRIM.41 XPS was performed to investigate the chemical composition of the sample surface (Quantera II, Physical Electronics, Chanhassen, MN, USA) applying a monochromatic Al Kα X-ray source (1486.6 eV) operated at a pass energy of 26 eV and a step size of 0.025 eV for elements of interest and a 224 eV pass energy with a 0.1 eV step size for the survey spectrum. The take-off angle was 45°. The binding energy scale was referenced to the C–C signal at 284.8 eV. The quantitative analysis was carried out with CasaXPS. For interpretation, commonly used online databases were used as starting points (http://www.lasurface.com/accueil/index.php, http://xpssimplified.com/elements, accessed April 1, 2015). An Ar-ion beam was used with an energy of 1 keV 2 × 2 for sputtering for 60 s on a spot size of 200 μm × 200 μm to remove ambient impurities. Ultraviolet-visible (UV/Vis) spectroscopy measurements of the samples deposited on quartz substrates were performed using an Agilent Cary 5000 double beam spectrophotometer, subtracting the substrate contribution. Tauc plots based on the UV/Vis data were used to determine the optical band gap.
Acknowledgements
The authors would like to acknowledge the Marie Curie ITN consortium (RAPID) as this project has received funding from the European Union's Seventh Framework Programme for research, technological development and demonstration under grant agreement no. 606889.
Notes and references
-
A. C. Jones and M. L. Hitchman, Chemical vapour deposition. Precursors, processes and applications, Royal Society of Chemistry, Cambridge UK, 2009 Search PubMed.
-
S. Hashmi, Comprehensive Materials Processing, Elsevier Science, 2014 Search PubMed.
-
(a) P. Marchand and C. J. Carmalt, Coord. Chem. Rev., 2013, 257, 3202–3221 CrossRef CAS;
(b) P. Marchand, I. A. Hassan, I. P. Parkin and C. J. Carmalt, Dalton Trans., 2013, 42, 9406–9422 RSC.
-
(a) C. C. Roberts, B. R. Barnett, D. B. Green and J. M. Fritsch, Organometallics, 2012, 31, 4133–4141 CrossRef CAS;
(b) C. Di Iulio, M. Middleton, G. Kociok-Köhn, M. D. Jones and A. L. Johnson, Eur. J. Inorg. Chem., 2013, 1541–1554 CrossRef CAS;
(c) K. A. Gerling, N. M. Rezayee, A. L. Rheingold, D. B. Green and J. M. Fritsch, Dalton Trans., 2014, 43, 16498–16508 RSC.
- D. Bekermann, A. Ludwig, T. Toader, C. Maccato, D. Barreca, A. Gasparotto, C. Bock, A. D. Wieck, U. Kunze, E. Tondello, R. A. Fischer and A. Devi, Chem. Vap. Deposition, 2011, 17, 155–161 CrossRef CAS.
-
(a) D. Bekermann, A. Gasparotto, D. Barreca, L. Bovo, A. Devi, R. A. Fischer, O. I. Lebedev, C. Maccato, E. Tondello and G. van Tendeloo, Cryst. Growth Des., 2010, 10, 2011–2018 CrossRef CAS;
(b) D. Bekermann, A. Gasparotto, D. Barreca, A. Devi, R. A. Fischer, M. Kete, U. Lavrencic Stangar, O. I. Lebedev, C. Maccato, E. Tondello and G. van Tendeloo, ChemPhysChem, 2010, 11, 2337–2340 CrossRef CAS PubMed.
- S. D. Cosham, G. Kociok-Köhn, A. L. Johnson, J. A. Hamilton, M. S. Hill, K. C. Molloy and R. Castaing, Eur. J. Inorg. Chem., 2015, 4362–4372 CrossRef CAS.
- D. Bekermann, D. Rogalla, H.-W. Becker, M. Winter, R. A. Fischer and A. Devi, Eur. J. Inorg. Chem., 2010, 1366–1372 CrossRef CAS.
- J. Holmes, K. Johnson, B. Zhang, H. E. Katz and J. S. Matthews, Appl. Organomet. Chem., 2012, 26, 267–272 CrossRef CAS.
- S. Schulz, R. Schäper, D. Bläser and C. Wölper, Z. Anorg. Allg. Chem., 2012, 638, 2102–2105 CrossRef CAS.
-
(a) M. Tammenmaa, T. Koskinen, L. Hiltunen, L. Niinistö and M. Leskelä, Thin Solid Films, 1985, 124, 125–128 CrossRef CAS;
(b) K. Kobayashi and S. Okudaira, Chem. Lett., 1997, 26, 511–512 CrossRef;
(c) A. Wójcik, M. Godlewski, E. Guziewicz, R. Minikayev and W. Paszkowicz, J. Cryst. Growth, 2008, 310, 284–289 CrossRef.
- K. Kopalko, A. Wójcik, M. Godlewski, E. Łusakowska, W. Paszkowicz, J. Z. Domagała, M. M. Godlewski, A. Szczerbakow, K. Świątek and K. Dybko, Phys. Status Solidi C, 2005, 2, 1125–1130 CrossRef CAS.
-
(a) E. Guziewicz, M. Godlewski, T. Krajewski, Ł. Wachnicki, A. Szczepanik, K. Kopalko, A. Wójcik-Głodowska, E. Przeździecka, W. Paszkowicz, E. Łusakowska, P. Kruszewski, N. Huby, G. Tallarida and S. Ferrari, J. Appl. Phys., 2009, 105, 122413 CrossRef;
(b) V. Lujala, J. Skarp, M. Tammenmaa and T. Suntola, Appl. Surf. Sci., 1994, 82, 34–40 CrossRef;
(c) T. Krajewski, E. Guziewicz, M. Godlewski, L. Wachnicki, I. A. Kowalik, A. Wojcik-Glodowska, M. Lukasiewicz, K. Kopalko, V. Osinniy and M. Guziewicz, Microelectron. J., 2009, 40, 293–295 CrossRef CAS;
(d) P. C. Rowlette, C. G. Allen, O. B. Bromley, A. E. Dubetz and C. A. Wolden, Chem. Vap. Deposition, 2009, 15, 15–20 CrossRef CAS.
-
(a) A. Yamada, B. Sang and M. Konagai, Appl. Surf. Sci., 1997, 112, 216–222 CrossRef CAS;
(b) M. A. Thomas and J. B. Cui, ACS Appl. Mater. Interfaces, 2012, 4, 3122–3128 CrossRef CAS PubMed;
(c) Z. Baji, Z. Lábadi, Z. E. Horváth, G. Molnár, J. Volk, I. Bársony and P. Barna, Cryst. Growth Des., 2012, 12, 5615–5620 CrossRef CAS.
-
(a) K. Tapily, D. Gu, H. Baumgart, G. Namkoong, D. Stegall and A. A. Elmustafa, Semicond. Sci. Technol., 2011, 26, 115005 CrossRef;
(b) T. Tynell and M. Karppinen, Semicond. Sci. Technol., 2014, 29, 43001 CrossRef;
(c) E. Guziewicz, M. Godlewski, T. A. Krajewski, Ł. Wachnicki, G. Łuka, J. Z. Domagała, W. Paszkowicz, B. J. Kowalski, B. S. Witkowski, A. Dużyńska and A. Suchocki, Phys. Status Solidi B, 2010, 247, 1611–1615 CrossRef CAS;
(d) E. Guziewicz, I. A. Kowalik, M. Godlewski, K. Kopalko, V. Osinniy, A. Wójcik, S. Yatsunenko, E. Łusakowska, W. Paszkowicz and M. Guziewicz, J. Appl. Phys., 2008, 103, 33515 CrossRef.
- I. Stassen, D. D. Vos and R. Ameloot, Chem. – Eur. J., 2016, 22, 14452–14460 CrossRef CAS PubMed.
- K. Khaletskaya, S. Turner, M. Tu, S. Wannapaiboon, A. Schneemann, R. Meyer, A. Ludwig, G. van Tendeloo and R. A. Fischer, Adv. Funct. Mater., 2014, 24, 4804–4811 CrossRef CAS.
-
(a) Q. Peng, B. Gong, R. M. VanGundy and G. N. Parsons, Chem. Mater., 2009, 21, 820–830 CrossRef CAS;
(b) J. Liu, B. Yoon, E. Kuhlmann, M. Tian, J. Zhu, S. M. George, Y.-C. Lee and R. Yang, Nano Lett., 2013, 13, 5594–5599 CrossRef CAS PubMed.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- B. Yoon, J. L. O'Patchen, D. Seghete, A. S. Cavanagh and S. M. George, Chem. Vap. Deposition, 2009, 15, 112–121 CrossRef CAS.
- K. Gregorczyk and M. Knez, Prog. Mater. Sci., 2016, 75, 1–37 CrossRef CAS.
- I. Stassen, M. Styles, G. Grenci, H. van Gorp, W. Vanderlinden, S. D. Feyter, P. Falcaro, D. D. Vos, P. Vereecken and R. Ameloot, Nat. Mater., 2016, 15, 304–310 CrossRef CAS PubMed.
- F. Epifano, S. Genovese and M. Curini, Tetrahedron Lett., 2007, 48, 2717–2720 CrossRef CAS.
- R. Laubenstein, M.-D. Serb, U. Englert, G. Raabe, T. Braun and B. Braun, Chem. Commun., 2016, 52, 1214–1217 RSC.
- S. E. Koponen, P. G. Gordon and S. T. Barry, Polyhedron, 2016, 108, 59–66 CrossRef CAS.
- A. Devi, Coord. Chem. Rev., 2013, 257, 3332–3384 CrossRef CAS.
- S. C. Dixon, D. O. Scanlon, C. J. Carmalt and I. P. Parkin, J. Mater. Chem. C, 2016, 4, 6946–6961 RSC.
-
(a) N. Y. Yuan, S. Y. Wang, C. B. Tan, X. Q. Wang, G. G. Chen and J. N. Ding, J. Cryst. Growth, 2013, 366, 43–46 CrossRef CAS;
(b) E. B. Yousfi, J. Fouache and D. Lincot, Appl. Surf. Sci., 2000, 153, 223–234 CrossRef CAS;
(c) S. Jeon, S. Bang, S. Lee, S. Kwon, W. Jeong, H. Jeon, H. J. Chang and H.-H. Park, J. Electrochem. Soc., 2008, 155, H738–H743 CrossRef CAS;
(d) H. Makino, A. Miyake, T. Yamada, N. Yamamoto and T. Yamamoto, Thin Solid Films, 2009, 517, 3138–3142 CrossRef CAS.
- S.-Y. Pung, K.-L. Choy, X. Hou and C. Shan, Nanotechnology, 2008, 19, 435609 CrossRef PubMed.
- E. Guziewicz, M. Godlewski, L. Wachnicki, T. A. Krajewski, G. Luka, S. Gieraltowska, R. Jakiela, A. Stonert, W. Lisowski, M. Krawczyk, J. W. Sobczak and A. Jablonski, Semicond. Sci. Technol., 2012, 27, 74011 CrossRef.
-
(a) E. Janocha and C. Pettenkofer, Appl. Surf. Sci., 2011, 257, 10031–10035 CrossRef CAS;
(b) R. Al-Gaashani, S. Radiman, A. R. Daud, N. Tabet and Y. Al-Douri, Ceram. Int., 2013, 39, 2283–2292 CrossRef CAS.
- F. Säuberlich, J. Fritsche, R. Hunger and A. Klein, Thin Solid Films, 2003, 431–432, 378–381 CrossRef.
- S. B. Amor, M. Jacquet, P. Fioux and M. Nardin, Appl. Surf. Sci., 2009, 255, 5052–5061 CrossRef.
- N. P. Dasgupta, S. Neubert, W. Lee, O. Trejo, J.-R. Lee and F. B. Prinz, Chem. Mater., 2010, 22, 4769–4775 CrossRef CAS.
-
(a) A. Abou Chaaya, R. Viter, M. Bechelany, Z. Alute, D. Erts, A. Zalesskaya, K. Kovalevskis, V. Rouessac, V. Smyntyna, P. Miele and J. Bachmann, Beilstein J. Nanotechnol., 2013, 4, 690–698 CrossRef PubMed;
(b) J. Iqbal, A. Jilani, P. M. Ziaul Hassan, S. Rafique, R. Jafer and A. A. Alghamdi, J. King Saud Univ., Sci., 2016, 28(4), 347–354 CrossRef.
- B. D. Viezbicke, S. Patel, B. E. Davis and D. P. Birnie, Phys. Status Solidi B, 2015, 252, 1700–1710 CrossRef CAS.
-
Agilent Technologies, CrysAlisPro Software, Agilent Technologies UK Ltd, Oxford, UK, 1st edn, 2011 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
-
M. Mayer, SIMNRA User's Guide, Max-Planck-Institut für PLasmaphysik, Garching, Germany, 1997 Search PubMed.
-
Treatise on Heavy-Ion Science, ed. J. F. Ziegler and J. P. Biersack, Springer US, Boston, MA, 1985 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional precursor and thin film analysis. CCDC 1502611–1502614. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt03571j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence