
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRare-earth metal diisopropylamide-catalyzed intramolecular hydroamination†
Tatiana
Spallek
and
Reiner
Anwander
*
Institut für Anorganische Chemie, Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany. E-mail: reiner.anwander@uni-tuebingen.de
First published on 15th September 2016
Abstract
Rare-earth metal diisopropylamide complexes LiLn(NiPr2)4(THF) (Ln = Sc, Y, La), [LiY(NiPr2)4]n, NaLn(NiPr2)4(THF) (Ln = Sc, Y), Sc(NiPr2)3(THF) and Ce(NiPr2)4 were screened as catalysts for the intramolecular hydroamination/cyclization (IHC) of 1-amino-2,2-dimethyl-4-pentene, 1-amino-2,2-diphenyl-4-pentene, and 1-amino-2,2-diphenyl-5-hexene at ambient and moderately increased temperature of 60 °C in C6D6. The lithium ate complexes displayed the most efficient precatalysts with high conversion rates at 60 °C for the phenyl-substituted substrates and Ln = Y and La, affording turnover frequencies Nt as high as 164 h−1. The catalytic activity could be increased by employing THF-free complex [LiY(NiPr2)4]n (Nt = 45.8 h−1 at 26 °C; 34.1 h−1 for LiY(NiPr2)4(THF)). In situ generation of putative LiY(NiPr2)4(THF) from YCl3(THF)3.3 and four equivalents of LiNiPr2 (LDA) in C6D6 generated a catalyst revealing Nt comparable to pre-isolated crystallized LiY(NiPr2)4(THF) but yielding even higher substrate conversion. The IHC reactions were also examined for rare-earth metal bis(trimethylsilyl)amide catalysts Ln[N(SiMe3)2]3 (Ln = Sc, Y, La) as well as for LDA using the same reaction conditions, revealing overall superior activity of the silylamide derivatives but poor performance of LDA compared to the rare-earth metal diisopropylamide complexes LiLn(NiPr2)4(THF). Cyclization of 1-amino-2,2-diphenyl-5-hexene to the 6-membered heterocycle 2-methyl-4,4-diphenylpiperidine by lanthanum derivative LiLa(NiPr2)4(THF) was accompanied by a competitive isomerization reaction affording max. 20% of 1-amino-2,2-diphenyl-4-hexene after 2 h at 60 °C. Crystalline tetravalent Ce(NiPr2)4 showed a better IHC performance than crystalline trivalent Sc(NiPr2)3(THF) as preliminary examined for 1-amino-2,2-diphenyl-4-pentene at 26 °C (Nt = 5.6 and 0.9 h−1, respectively), but cyclization came to a halt after 2 h, probably due to decomposition of the catalyst.
Introduction
Over the past 25 years, the hydroamination of olefins, and in particular the intramolecular hydroamination/cyclization (IHC) of aminoolefins and aminoalkynes, has developed into a most prominent catalytic reaction in (organo)rare-earth metal chemistry.1–5 The IHC affords direct and atom-efficient access to natural products like alkaloids and other functionalized mono- and polyazacycles.1–5 The ground breaking discovery by Marks et al. in 1989 revealed that rare-earth metallocene-based catalysts of the type [(C5Me5)2LnH]2 or (C5Me5)2LnCH(SiMe3)2 convert 2,2-dimethyl-4-penten-1-amine (herein denoted as aminoalkene S1) very efficiently into a pyrrolidine derivative (herein denoted as product P1), with the catalytic activity being metal-size-dependent (maximum turnover frequencies Nt for lanthanum: 125 and 95 h−1 (25 °C), respectively).6 Later on, catalyst design put main emphasis on chelating diamido- and diaryloxo ancillary ligands L2− as well as on readily exchangeable monoanionic amido ligands NR2 (= actor ligand) targeting well-defined discrete complexes LLnIIINR2.4,7–13 Many of these non-metallocene complexes have been accessed via protonolysis reactions – often generated in situ – using silylamide complexes Ln[N(SiMe3)2]3 or Ln[N(SiHMe2)3]3(THF)x.7,8 More recently, the implementation of chiral variants for asymmetric hydroamination has been successful5,9–13 and the feasibility of intermolecular hydroamination has been demonstrated at elevated temperatures.4,14Ln(III) precatalysts of the type L′LnIII(NR2)2,7,9 LnIII(NR2)3,8,15 and LnIII(NR2)4− bearing two, three or four exchangeable monoanionic amido ligands NR2, respectively, did receive much less attention. Lack of a rigid stabilizing ancillary ligand backbone, ligand redistribution, and multiple active sites have to be considered potential drawbacks. Nevertheless, the C2-symmetric bis(oxazolinato) bis(amido) complex [(4R,5S)-Ph2Box]La[N(SiMe3)2]2 exhibited good rates (e.g., S1: Nt = 25 h−1; 23 °C)9 and homoleptic Y[N(SiMe3)2]3 was advertised as a commercially available comparatively inexpensive precatalyst (e.g., S1: Nt = 11.6 h−1; 25 °C).15 Moreover, surface grafting of the latter homoleptic derivative on large-pore mesoporous silica afforded hybrid material Y[N(SiMe3)2]3@SBA-15-LP500 featuring markedly enhanced catalytic activity (e.g., S1: Nt = 36 h−1; 50 °C).16
Referring to the initiating step of the IHC catalytic cycle (= X/amine ligand exchange; Scheme 1) it has been pointed out several times that tuning of the actor ligand can be beneficial for enhancing the reaction rate or turnover frequencies (Nt). For diamidoamine complexes [(2,4,6-Me3C6H2NCH2CH2)2NMe]YX it was found that the least basic actor ligand X = N(SiHMe2)2 gave significantly reduced reaction rates (S1: Nt = 1.3 h−1; 40 °C) compared to X = N(SiMe3)2 (S1: Nt = 8.4 h−1; 25 °C) and an aryl derivative (X = C6H4CH2NMe2-6; S1: Nt = 10 h−1; 25 °C).17 In general, the rate dependence on actor ligands X = N(SiMe3)2versus X = CH(SiMe3)2 was less revealing, but for selected ancillary ligand systems, the higher basicity of the silylalkyl ligand gave higher Nt values. Representative examples feature C1-symmetric ansa-metallocene complexes Me2Si(Me4C5)(C5H3R*)LnX (R* = chiral auxiliary, e.g., (+)neomenthyl; S1: Nt = 21 versus 38 h−1; 25 °C)18 or chiral 3,3′-bis(trisarylsilyl)-substituted binaphtholate complexes R-[{Binol-Si(3,5-xylyl)3}LaX(THF)2] (S1: Nt = ≥60 versus 94 h−1; 22 °C).11 Surprisingly, rare-earth metal alkylamido moieties, e.g., Ln–NiPr2, have barely been considered as actor ligands, despite of their enhanced basicity/exchangeability (e.g., pKa,THF = 22.8 (HN(SiHMe2)2),19 35.7 (HNiPr2)),20 ready availability from LnCl3 (THF)x and alkali metal amides like lithium diisopropyl amide (LDA),21 and inexpensiveness compared to silylamide derivatives.‡22–24 In addition, any released isopropylamine during the initial substrate coordination displays a weak donor ligand/nucleophile. In 2003, Scott et al. employed discrete tBu-3,5-functionalized bisarylamide silylamide complexes (X = N(SiHMe2)2; Ln = Y: S1 full conversion after 14 d at 60 °C) for enantioselective IHC reactions, but emphasized that use of the more basic diisopropylamido ligand (X = NiPr2) – precatalyst generated in situ via protonolysis of Y(NiPr2)3(THF)2 (Scheme 1, box) – gave significantly increased turnovers (Ln = Y: S1 full conversion after 5 d at 35 °C).25 Moreover, the R-N,N′-diisopropyl-1.1′-binaphthyl-2,2′-diamide complex [(R)-C20H12(NiPr)2]Y(NiPr2)(THF){Li(THF)2} (Scheme 1, box), obtained via salt metathesis from the chloro derivative and LDA and used as crude reaction product, proved to be much more reactive than ate complex [Li(THF)4]Y[(R)-C20H12(NiPr)2]2.26 Latter ate complexes have been investigated in detail for IHC,27–30 and the effect of the alkali metal counterion (Li+versus K+)28 and the presence of LiCl29 scrutinized. Given the catalytic activity of such 1.1′-binaphthyl-2,2′-diamide ate complexes it seems like an oversimplification to discuss the IHC inactivity of half-sandwich anilide complexes like [C5H2(SiMe3)3-1,2,4]LaI(NHC6H3iPr2-1,6)py2 solely on the basis of relative pKa values while ruling out any effect of halo ligands.31 In our preceding study we have described the syntheses and structural chemistry of a series of rare-earth metal diisopropylamide complexes.21 Herein we present a comparative study of the catalytic activity of a number of structurally defined rare-earth metal ate complexes, LiLn(NiPr2)4(THF) (Ln = Sc, Y, La) and NaLn(NiPr2)4(THF)2 (Ln = Sc, Y), donor(THF)-free [LiY(NiPr2)4]n, as well as of rare-earth metal silylamides Ln[N(SiMe3)2]3 (Ln = Sc, Y, La) for the IHC of selected terminal aminoalkenes.
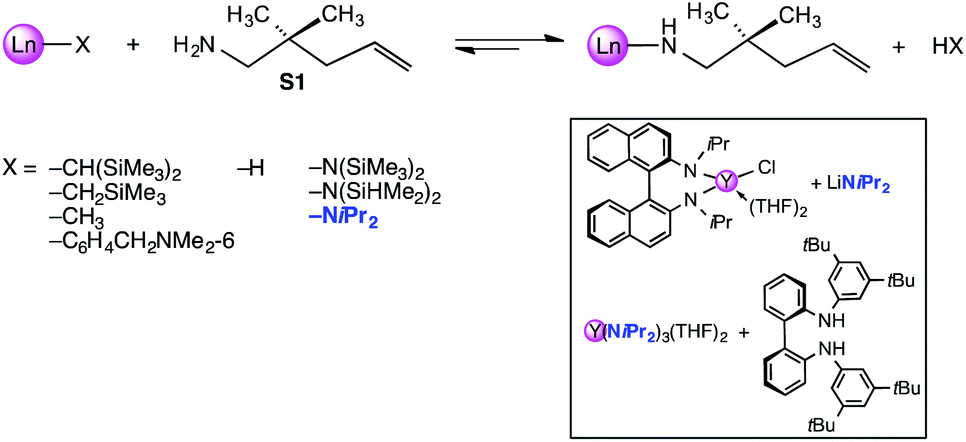 | ||
| Scheme 1 Protonolysis of the precatalyst as initiating step of the IHC catalytic cycle: scope of routinely employed actor ligands X and relevant examples of diisopropylamide complexes. | ||
Results and discussion
IHC activity of bimetallic ate complexes LiLn(NiPr2)4 (THF)
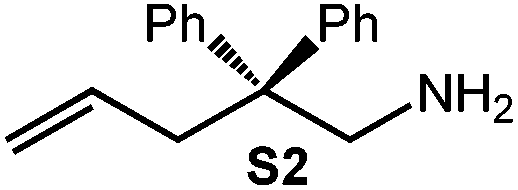
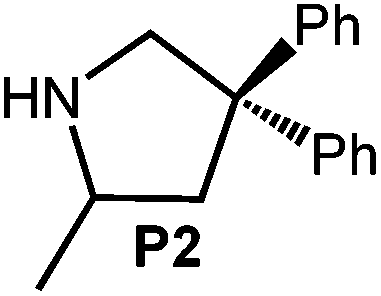
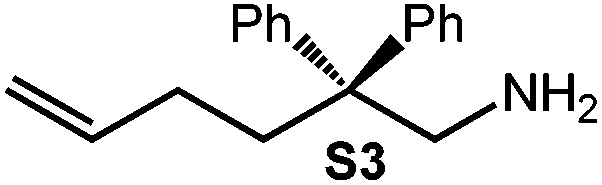
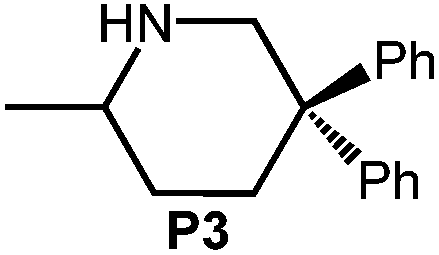
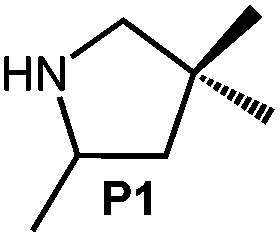
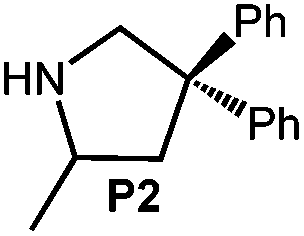
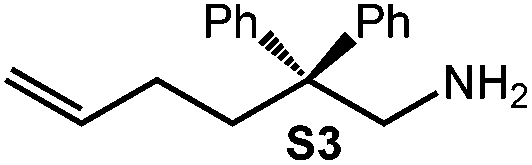
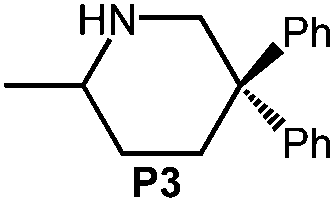
Because of their easy accessibility for the entire rare-earth metal series, we examined in detail the performance of the bimetallic ate complexes LiLn(NiPr2)4(THF) (Ln = Sc (1a), Y (1b), La (1c)) in the hydroamination/cyclization of terminal aminoalkenes (Scheme 2). It has been previously shown that the substrates amino-2,2-dimethyl-4-pentene (S1), 1-amino-2,2-diphenyl-4-pentene (S2), and 1-amino-2,2-diphenyl-5-hexene (S3) give the cyclized products 2,4,4-trimethylpyrrolidine (P1), 2-methyl-4,4-diphenylpyrrolidine (P2), and 2-methyl-5,5-diphenylpiperidine (P3), respectively.3 The scope of substrates, products and well-defined catalysts under study are illustrated in Scheme 2.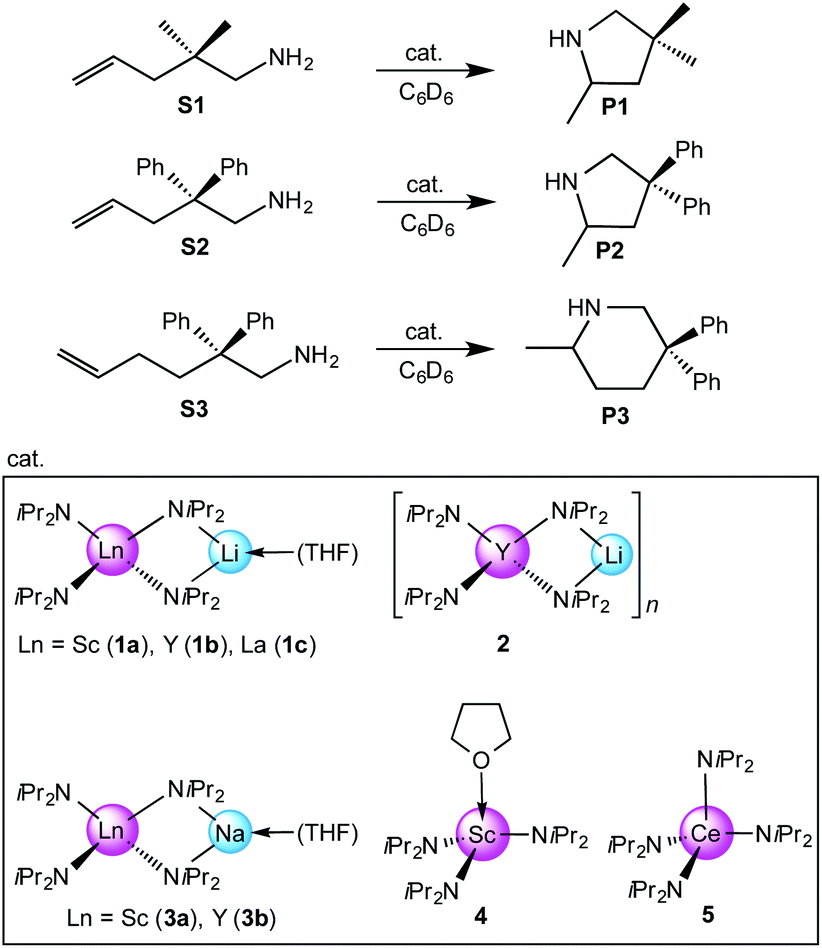 | ||
| Scheme 2 IHC of terminal aminoalkenes 1-amino-2,2-dimethyl-4-pentene (S1), 1-amino-2,2-diphenyl-4-pentene (S2), and 1-amino-2,2-diphenyl-5-hexene (S3) promoted by rare-earth metal diisopropylamide complexes LiLn(NiPr2)4(THF) (Ln = Sc (1a), Y (1b), La (1c)), [LiY(NiPr2)4]n (2), NaLn(NiPr2)4(THF) (Ln = Sc (3a), Y (3b)), Sc(NiPr2)3(THF) (4) and Ce(NiPr2)4 (5). | ||
The catalytic reactions have been monitored by 1H NMR spectroscopy using C6D6 as a solvent, and 2 or 4 mol% of precatalyst at 26 °C or 60 °C (Table 1). For phenyl-substituted aminoalkenes S2 and S3, the color of the reaction mixtures changed from colourless to yellow at the beginning of the catalytic tranformations. The progress of ring closure was determined by the integral ratios of the 1H NMR specific peaks of the substrates and products relative to the signal of free HNiPr2. Protonolysis of the NiPr2 ligands proceeded rapidly after aminoalkene addition to the precatalyst solution and stayed constant during the catalytic runs, as already observed by Marks et al.6b and others for actor ligand N(SiMe3)2.16
| Entry | Aminoalkene | Product | Catalyst | T/°C | t/h | Conv./%b |
N
t![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c/h−1 c/h−1 |
|---|---|---|---|---|---|---|---|
|
a 4 mol% 1 in C6D6.
b All conversion data were derived from 1H NMR spectra referring to the corresponding duration of reaction.
c Initial Ntversus overall Nt (taken at 80% conversion, unless otherwise stated).
d 2 mol% 1b in C6D6.
e Ratio cyclized product P3: isomerized aminoalkene P4 ≈ 4:1.
f Conversion completed after <9 min.
g At 99% conversion.
h Initial Nt = Nt at 80%.
i
N
t at time t given in this table.
|
|||||||
| 1 |
|
|
1a (Sc) | 60 | 4.84 | <1 | <1 |
| 2 | 1b (Y) | 60 | 4.84 | 24 | 1.6/1.2i | ||
| 3d | 1b (Y) | 60 | 9.7 | <1 | <1 | ||
| 4 | 1c (La) | 60 | 4.84 | 47 | 18.1/2.9i | ||
| 5 |
|
|
1a (Sc) | 60 | 1.0 | 43 | 10.6/5.2 |
| 6 | 1a (Sc) | 26 | 4.84 | 26 | 3.3/1.4i | ||
| 7 | 1b (Y) | 60 | 0.15 | >99 | —f/164 | ||
| 8d | 1b (Y) | 60 | 0.6 | >98 | 88/90 | ||
| 9 | 1b (Y) | 26 | 1.5 | >88 | 67.3/34.1 | ||
| 10 | 1c (La) | 60 | 0.15 | >99 | —f/164 | ||
| 11 | 1c (La) | 26 | 0.6 | >99 | 151.4/4g | ||
| 12 |
|
|
1a (Sc) | 60 | 2.27 | >99 | 21.8/14.7 |
| 13 | 1b (Y) | 60 | 0.15 | >99 | —f/164 | ||
| 14d | 1b (Y) | 60 | 0.9 | >99 | 69.0h | ||
| 15 | 1b (Y) | 26 | 2.12 | >99 | 13.6/11.3 | ||
| 16d | 1b (Y) | 26 | 4.84 | 36 | 5.4/4.3i | ||
| 17 | 1c (La) | 60 | 4.84 | >99e | 15.5/9.7 | ||
| 18 | 1c (La) | 26 | 288 | >98e | 0.2/0.1i | ||
Exemplarily, the progress of the catalytic transformation of substrate S3 (entry 14) is shown in Fig. S1† and clearly corroborated by the disappearance of the olefinic signals at around 5–6 ppm. The structures/compositions of the products P1–P4 were assigned by comparison with literature data.32
Generally, complexes LiLn(NiPr2)4(THF) (Ln = Sc (1a), Y (1b), La (1c)) follow the prevailing rules for IHC observed for rare-earth metal catalysts.3 Comparison of entries 1, 5, and 12 (Table 1) shows that the ring closing for the investigated aminoalkenes S1, S2, and S3 catalyzed by the same precatalyst proceeds according to the Thorpe-Ingold-effect.33,34 The rates of cyclization revealed the trends consistent with the Baldwin's guidelines for ring closure,35 meaning that aminoalkene S1 is the least reactive, while S2 is easily converted. Furthermore, as previously observed the catalytic activity increased with increasing ionic radius of the rare-earth metal centre (e.g., Table 1, entries 1, 2, and 4).6 Quantitative conversion of aminoalkenes S2 and S3 with precatalysts 1b (Y; Table 1, entries 7 and 13) and 1c (La; Table 1, entry 10) was observed after 0.15 h at 60 °C with the highest Nt = 164 h−1. Therefore, the cyclization of S2 was additionally monitored at 26 °C. La-catalyst 1c with the largest rare-earth metal centre is the most efficient displaying 99% conversion after 0.6 h at 26 °C (Table 1, entry 11), followed by Y-catalyst 1b (Table 1, entry 9) with 88% conversion after 1.5 h. After ca. 5 h, the respective Sc-catalyst 1a had converted only 26% of S2 (Table 1, entry 6). This latter effect of the rare-earth metal onto the IHC of aminoalkene S2 is depicted in Fig. 1. The experimental conversion/time data plots for all three aminoalkenes and LiY(NiPr2)4(THF) (1b) as the precatalyst are shown in Fig. 2. Significant transformation of substrate S1 occurred only at 60 °C. Surprisingly, La complex 1c did not convert aminoalkene S3 as efficiently as anticipated and the reactions seemed not completed even after several days (Table 1, entries 17 and 18). Careful analysis of the 1H NMR spectroscopic data, however, revealed the co-formation of isomerized byproduct 1-amino-2,2-diphenyl-4-hexene (P4, Fig. 3). The formation of P4 started parallel to that of the main cyclized product P3 and reached ca. 20% conversion (relative to the concentration of the substrate), independent of the reaction temperature (60 °C or 26 °C). The experimental conversion/time data plots using 4 mol% LiLa(NiPr2)4(THF) (1c, Table 1, entry 17) are shown in Fig. 4, corroborating that the competitive isomerization reaction for S3 slowed down markedly the formation of the major cyclized product P3. Such concomitant aminoalkene isomerizations were observed previously in IHC reactions employing alkali metal bases such as NaK alloy or n-BuLi,23,24 and alkaline-earth metal complexes.36 While the use of NaK gave the isomerized aminoalkene as the major product, treatment of amino-2,2-dimethyl-4-pentene (S1) with n-butyllithium in THF led to cyclization.23 Later, Markó et al. treated 1-amino-4-pentene with 16 mol% n-BuLi which led to the cyclization product, but the isomerized alkene formed to different extents.24 A detailed investigation and discussion of the formation of isomerized aminoalkene in the course of IHC was performed by Hill et al. for the cyclization of 1-amino-2,2-dimethyl-5-hexene and 1-amino-2,2-diphenyl-5-hexene (S3) in the presence of the β-diketiminate-supported calcium complex {[ArNC(Me)CHC(Me)NAr]Ca[N(SiMe3)2](THF)}.36a For the S3-reaction, the ca. 10% yield of the isomerized byproduct (which was not observed for the magnesium precatalyst {[ArNC(Me)CHC(Me)NAr]MgMe(THF)}) were ascribed to stereoelectronic effects: formation of a boat-like six-membered ring transition state might lead to an allyl species via intramolecular proton transfer and subsequently to the isomerized product P4 (or regenerated S3) via protonolysis.36a To the best of our knowledge such competitive isomerization/IHC reactions have not been observed yet for rare-earth metal catalysts.37 Any competing effect of the lithium cation might be ruled out on the basis of a VT NMR study since the Li(THF)+ fragment shows approximately the same mobility in the respective yttrium (1b) and lanthanum complexes (1c).21
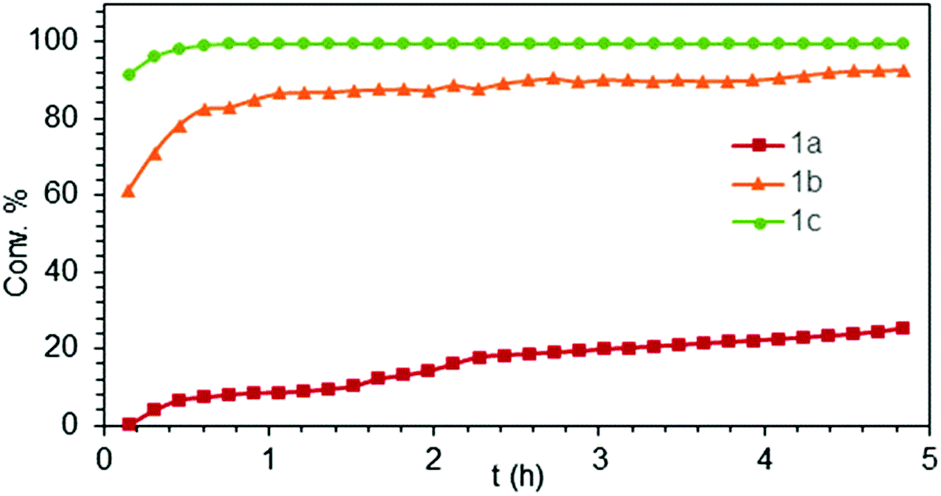 | ||
| Fig. 1 Experimental conversion/time data plots for the IHC of aminoalkene S2 at 26 °C by using 4 mol% LiLn(NiPr2)4(THF) (Ln = Sc (1a, Table 1, entry 6), Y (1b, Table 1, entry 9), La (1c, Table 1, entry 11)) as precatalysts. | ||
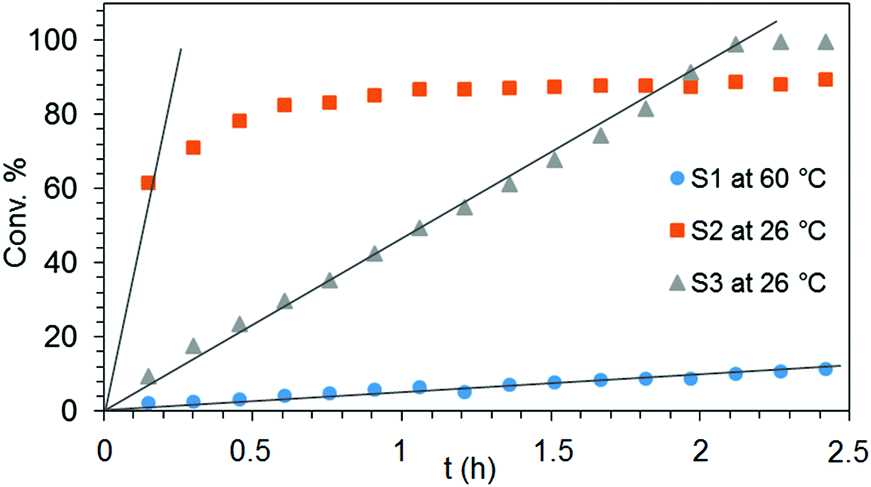 | ||
| Fig. 2 Experimental conversion/time data plots for the IHC of aminoalkenes S1 at 60 °C (Table 1, entry 2), S2 at 26 °C (Table 1, entry 9) and S3 at 26 °C (Table 1, entry 15) by using 4 mol% LiY(NiPr2)4(THF) as precatalyst. | ||
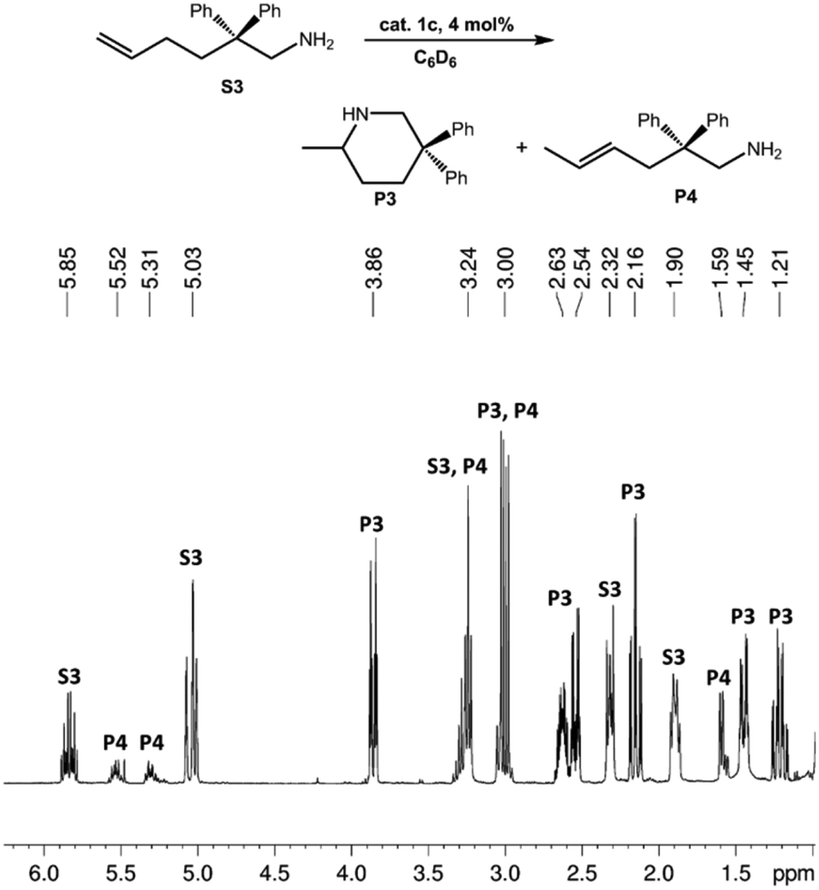 | ||
| Fig. 3 1H NMR spectrum (shown in the region 1.0–6.2 ppm) of the catalytic IHC and isomerization reactions of 1-amino-2,2-diphenyl-5-hexene (S3) to main product 2-methyl-5,5-diphenylpiperidine (P3) and isomerized side product 1-amino-2,2-diphenyl-4-hexene (P4) using La-catalyst 3c (Table 1, entry 17). | ||
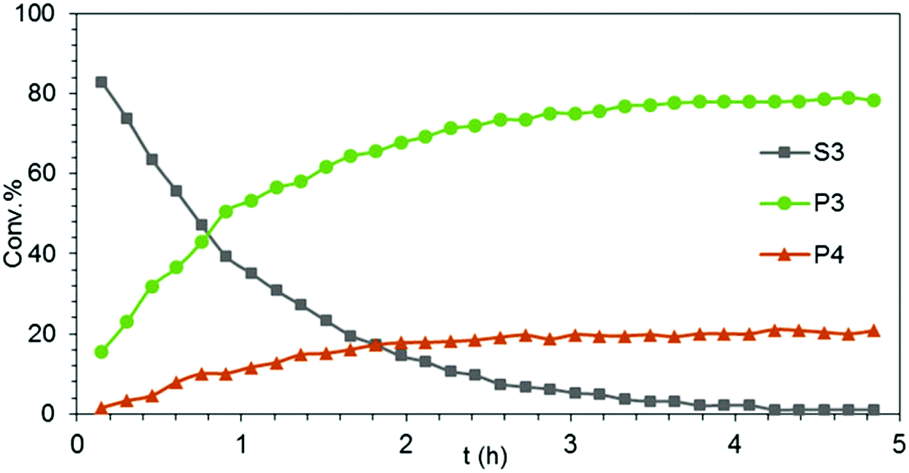 | ||
| Fig. 4 Experimental conversion/time data plots for the transformation of aminoalkene S3 into cyclized main product P3 and isomerized side product P4 using 4 mol% LiLa(NiPr2)4(THF) as precatalyst (Table 1, entry 17). | ||
As in the case of the aforementioned Mg/Ca transformations, the occurrence of the isomerization reaction in the presence of LiLa(NiPr2)4(THF) (1c) can be rationalized on the basis of the large size of La(III). In this sense the scandium (1a) and yttrium (1b) catalysts did not yield any detectable amount of P4.36a Similar to the study by Hill et al. the isomerized byproduct does not undergo subsequent cyclization. Overall, the distinct reactivity of 1c toward S3, which is comparable to that of {[ArNC(Me)CHC(Me)NAr]Ca[N(SiMe3)2](THF)} seems to originate from similarities of the calcium(II) and lanthanum(III) cations e.g., Lewis acidity38 and comparable size of the effective ionic radii (Ca2+ 1.00–1.34 Å and La3+ 1.03–1.36 Å, depending on the coordination number).39 Precatalyst 1c did not promote the isomerization of aminoalkene S1 and S2.
IHC catalytic activity of LDA
Early research on the generation of carbon–nitrogen bonds involved the alkali metal-catalyzed amination of olefins in 195740 and 1985.41 The application of LDA supported by irradiation (150 W tungsten bulb) was described by Trost et al. as a viable protocol in alkaloid synthesis.42 Moreover, Markó et al. reported on the addition of diisopropylamine to the lithiated substrate 1-amino-4-pentene or treatment with either catalytic quantities or with 1 equiv. of LDA to afford the cyclized product 2-methylpyrrolidine.24 Furthermore, mixtures of n-BuLi/HNiPr243 and LiN(SiMe3)2/TMEDA,44 as well as a chiral lithium amides45 were successfully utilized in base-catalyzed IHC reactions. In order to exclude any significant effect of LDA, the separation of which might occur in ate complex (1)/substrate (S) reaction mixtures, we probed the IHC catalytic activity of LDA for the aminoalkenes under study. Putative cyclization of S2, which benefits most from the Thorpe-Ingold effect, did not occur in the presence of 4 mol% LDA even after 96 h at 60 °C (Table 2, entry 1). The formation of a significant amount of cyclized product P2 (ca. 18%) could be observed after 5 h, using 12 mol% LDA at 26 °C (Table 2, entry 2). Further increase of both the reaction temperature (60 °C) and concentration of LDA (21 mol%) led to full conversion of substrate S2 after 4 h (Table 2, entry 3).| Entry | Aminoalkene | Product | Conc./mol% | T/°C | t/h | Conv./%a |
N
tb/h−1 |
|---|---|---|---|---|---|---|---|
| a All conversion data were derived from 1H NMR spectra referring to the corresponding duration of reaction. b Initial Ntversus overall Nt (taken at 80% conversion, unless otherwise stated). c N t at time t given in this table. | |||||||
| 1 |
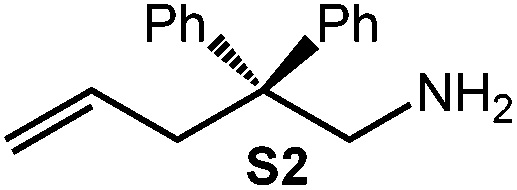
|
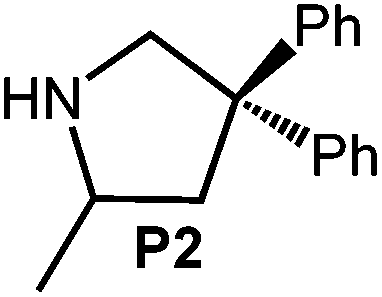
|
4 | 60 | 96 | <1 | <1c |
| 2 | 12 | 26 | 4.84 | 18 | 1.9/0.3c | ||
| 96 | 43 | <0.1 | |||||
| 3 | 21 | 60 | 4 | >99 | 1.2/1.2 | ||
| 4 |
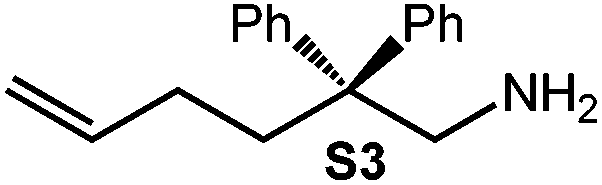
|
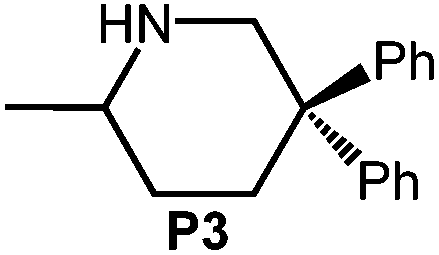
|
4 | 26 | 4.84 | <1 | <1c |
As mentioned in the introduction, the only known rare-earth metal ate complexes, which have been investigated in detail for the catalytic IHC, are bimetallic lanthanide-binaphthylamide complexes of the A and B-type (Chart 1).27–29,46,47 While addition of LiCl to [(R)-C20H12(NR)2]Y(CH2SiMe3)(THF)2 and [(R)-C20H12(NR)2]Y(NiPr2)(THF)2 (R = c-C5H9, SiMe3, SiMe2tBu) mainly produced higher enantioselectivities,29 lithium-ate complexes [Li(THF)4]Ln[(R)-C20H12(NR)2]2 displayed higher catalytic activity than the potassium analogues [K(THF)5]Ln[(R)-C20H12(NR)2]2 (Ln = Y, Yb; R = c-C5H9, CH2tBu).28 It is noteworthy that such bimetallic rare-earth metal binaphthylamide complexes were employed to some extent generated in situ48 and required higher concentrations of the catalysts and longer reaction times28 compared to our ate complexes LiLn(NiPr2)4(THF). Diethylamide ate complexes of type C (Chart 1) were described as efficient catalysts for asymmetric IHC reactions.12a
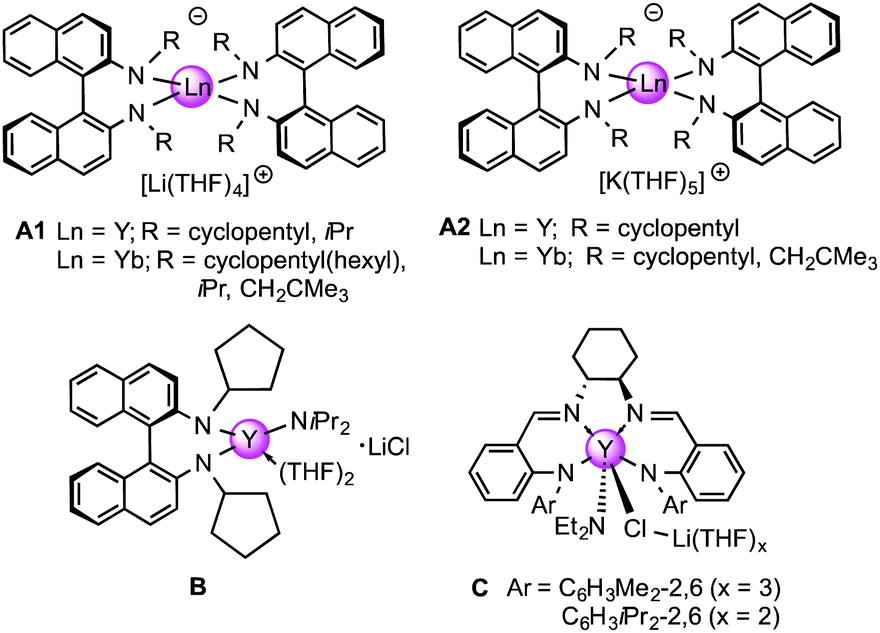 | ||
| Chart 1 Bimetallic rare-earth metal ate complexes previously employed as precatalyts for (asymmetric) IHC reactions. | ||
The Li/Y diisopropylamide catalyst system
The IHC activity of complex LiY(NiPr2)4(THF) (1b) was further elaborated at a concentration of 2 mol% (cf., Table 1). Compared to the 4 mol%-reaction, which gave 24% conversion of S1 after 4.84 h at 60 °C (Table 1, entry 2), use of half of the amount of 1b did not produce any detectable P1 even after 9.7 h (Table 1, entry 3). In contrast, under the same conditions 98% of aminoalkene S2 were converted after 0.6 h (Table 1, entry 8) and, similarly, aminoalkene S3 could be cyclized after 0.9 h (Table 1, entry 14). At 26 °C, cyclization of S3 in the presence of 2 mol% 1b was noted at 36% after 4.84 h (Table 1, entry 16). These results underline the high catalytic activity of the investigated amide complexes, the impact of the Thorpe-Ingold effect, as well as the different rates of the formation of five- and six-membered rings. The experimental conversion/time data plots for the IHC of aminoalkenes S2 and S3 using 2 mol% 1b as the precatalyst are depicted in Fig. 5.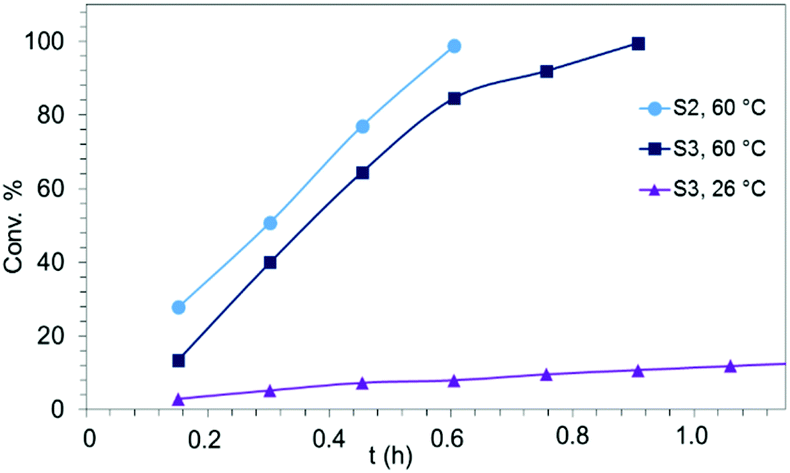 | ||
| Fig. 5 Experimental conversion/time data plots for the IHC of aminoalkenes S2 and S3 using 2 mol% LiY(NiPr2)4(THF) (1b) as precatalyst (Table 1, entries 8, 14, and 16). | ||
In our preceding work on rare-earth metal diisopropylamide complexes, the diversity of molecular compositions, solution behaviours and solid-state structures depending on the ratio MNiPr2/LnCl3 (M = Li, Na) and presence of donor solvent was emphasized.21 In the following experiments we assessed the catalytic performance of (a) [LiY(NiPr2)4]n (2), that is, in the absence of donor solvent THF (Table 3, entry 3), (b) in situ generated bimetallic catalysts using LiNiPr2/LnCl3 ratios of 4 (aiming at 1b; Table 3, entry 4) and 2.5 (aiming at Y(NiPr2)3(THF)x; Table 3, entry 5), (c) in situ generated LiY[N(SiHMe2)2]4(THF) (from YCl3(THF)3.3 + 4 LiN(SiHMe2)2) (Table 3, entry 6), and (d) crystalline NaY(NiPr2)4(THF) (3b) to probe any alkali metal effect, compared to pure crystalline complex 1b (Table 1, entry 9) and LDA (Table 2, entry 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Conc./mol% | t/h | Conv./%a |
N
tb/h−1 |
| a All conversion data were derived from 1H NMR spectra referring to the corresponding duration of reaction. b Initial Ntversus overall Nt (taken at 80% conversion, unless otherwise stated). c cf., Table 1, entry 9. d cf., Table 2, entry 2. e N t at time t given in this table. f The presence of 10 equivalents of HNiPr2 did not affect the catalytic transformation. | |||||
| 1c | LiY(NiPr2)4(THF) (1b) | 4 | 1.5 | >88 | 67.3/34.1 |
| 2d | LDA | 12 | 4.84 | 18 | 1.9/0.3e |
| 3f | [LiY(NiPr2)4]n (2) | 4 | 1.1 | >99 | 60.5/45.8 |
| 4 | In situ 1b (YCl3(THF)3.3 + 4 LDA) | 4 | 1.7 | >99 | 30.6/28.7 |
| 5 | In situ Y(NiPr2)3(THF)2 [YCl3(THF)3.3 + 2.5 LDA] | 4 | 4.84 | 4 | 0.2e |
| 6 | In situ LiY[N(SiHMe2)2]4(THF) [YCl3(THF)3.3 + 4 LiN(SiHMe2)2] | 4 | 192 | 34 | <0.1e |
| 288 | 47 | ||||
| 7 | Sc(NiPr2)3(THF) (4) | 4 | 4.84 | 18 | 4.5/0.9e |
| 8 | Ce(NiPr2)4 (5) | 4 | 2.0 | 44 | 17.7/5.6e |
(a) Indeed, the THF-free polymeric complex [LiY(NiPr2)4]n (2), which is soluble in aliphatics and aromatics, revealed the highest catalytic activity of all isopropylamide complexes under study (Fig. 6). Using 4 mol% of 2 the conversion of S2 was completed after 1.1 h even at ambient temperature (Table 3, entry 3 and Fig. 6). The superior reactivity of 2 compared to the THF-coordinate congener 1b (Table 1, entry 9 and Fig. 6) can be rationalized on the basis of their distinct dynamic behaviours in solution as revealed by VT NMR spectroscopic studies, suggesting distinct Li–N bonding in 2. This might affect the intitial protonolysis reaction (NiPr2/substrate exchange, Scheme 1). Moreover, upon elimination of a bulky NiPr2 ligand the Lewis base THF present in 1b could switch from Li to Y coordination and thus compete with any vacant coordination site of the catalyst, the availability of which is essential for the subsequent insertion/cyclization step of the catalytic cycle. The IHC reaction of S2 catalyzed by 2 was repeated under the same conditions using 5.09 μmol of tetrakis(p-tolyl)silane as an internal standard revealing the same conversion and NMR yield >99% after 1.1 h (initial Nt = 62.4 h−1 and overall Nt = 45.92 h−1 at 80% conversion (Fig. S2†)). The negative effect of THF on IHC reactions was early observed by Marks et al.6
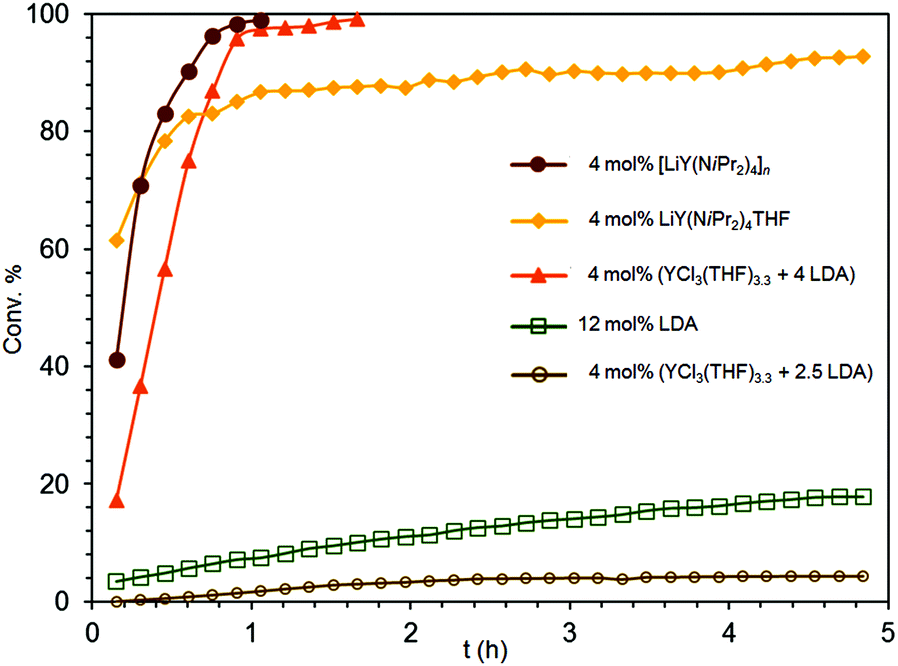 | ||
| Fig. 6 Experimental conversion/time data plots for the IHC of aminoalkene S2 promoted by 4 mol% LiY(NiPr2)4(THF) (1b), 12 mol% LDA, 4 mol% [LiY(NiPr2)4]n (2), mol% YCl3(THF)3.3/4 LDA, and 4 mol% YCl3(THF)3.3/2.5 LDA (cf., Table 3, entries 1–5). | ||
(b) More interestingly, isolation and crystallization of complexes 1b or 2 seem not be a prerequisite for efficiently catalyzing the IHC reaction. Although crystalline complex 1b afforded 78% conversion of S2 after 0.5 h, the reaction decelerated drastically afterwards (Table 1, entry 9 and Fig. 6). The corresponding reaction employing the respective in situ generated catalyst, preformed from YCl3(THF)3.3 and 4 equivalents of LiNiPr2 in C6D6 (preformation time 5 min), was at the beginning slower, but afforded 97% conversion of the substrate already after 1 h (Table 3, entry 4 and Fig. 6). It is noteworthy that for both reactions ca. 80% cyclization was accomplished after 0.75 h (see Fig. 6). In contrast, in situ generated Y(NiPr2)3(THF)x, obtained from YCl3(THF)3.3 and 2.5 equivalents of LiNiPr2 in C6D6,21 displayed only minor catalytic activity (Table 2, entry 5 and Fig. 6). Applying 4 mol% of the catalyst, only 4% conversion of S2 was detected after 3 h followed by a stagnation of the IHC reaction. As importantly for assessing the role of LDA is the fact that pure LDA promotes the IHC of substrate S2 not until a concentration of 12 mol% (Table 2). Moreover, crystallized mixed NiPr2/Cl complexes such as choro-bridged [Ln(NiPr2)2(μ-Cl)(THF)]2 displayed generally lower IHC catalytic reactivity for aminoalkene S2. For example, [Sc(NiPr2)2(μ-Cl)(THF)]2 converted 31% of the substrate after 48 h (not displayed in Table 3), accounting for a smaller catalytic efficiency per metal centre, compared to catalysts 1a and 4 (vide supra).
(c) Yttrium complexes featuring the less basic but similarly sized bis(dimethylsilyl)amido ligand have been employed previously for catalytic IHC reactions.17 For direct comparison, {LiY[N(SiHMe2)2]4(THF)}49 was generated in situ from YCl3(THF)3.3 and four equivalents of LiN(SiHMe2)2) in C6D6 (preformation time 5 minutes) (Table 3, entry 6). Not unexpectedly, cyclization of S2 proceeded very slowly at ambient temperature, affording 34% conversion after 8 d and 47% after another 4 d.
(d) Having synthesized the sodium congeners NaLn(NiPr2)4(THF) (Ln = Sc (3a), Y (3b)) as well,21 we were also interested in elucidating the effect of the alkali metal ion on the IHC reactions (Table S1†). We found that the Y–Na catalyst 3b exhibits markedly lower activity than the Y–Li catalyst 1b, converting 37% of S2 after a reaction time of 3 h at 60 °C and achieving full conversion only after 24 h (Table S1,† entry 2) (1b: complete conversion after 9 minutes) (Table 1, entry 7). The decreased catalytic activity of the Y–Na derivative 3b was further supported by the outcome of the cyclization of substrate S3 (Table S1†), which gave 15% and complete cyclization after 3 h and 48 h at 60 °C, respectively (Table S1,† entry 4), 1b: complete conversion after 9 minutes (Table 1, entry 13). The decreased reactivity of the sodium ate complexes might originate from their changed dynamic behaviour in solution and decreased thermal stability. A VT NMR spectroscopic study revealed that complex 3b is highly fluxional even at ca. −70 °C (1b: above ca. 0 °C), which was attributed to a weaker (ionic) bonding of the Na(THF)+ fragment with the amido ligands compared to the Li(THF)+ fragment.21 The comparatively weaker bonding of the Na(THF)+ fragment might exert a destabilizing effect on the Ln(NiPr2)4− counterion as revealed by accelerated decomposition (formation of HNiPr2; see also ref. 28: Li+versus K+). As observed for complexes LiLn(NiPr2)4(THF) (1), the Sc–Na derivative 3a was far less active than the Y–Na complex 3b. Complex 3a converted only traces of the substrates S2 and S3 after 24 h reaction time and reached full conversion after 3 and 12 d, respectively (Table S1,† entries 1 and 3). Although the catalytic role and influence of the alkali metals in such bimetallic complexes is not clarified until now, their presence and type clearly affects the catalytic performance of amide complexes. Similar observations were made by Schulz et al. when probing bimetallic rare-earth metal binaphthylamide complexes of the type [M(THF)n]Ln[(R)-C20H12(NR)2]2 (M = Li, K) as catalysts for IHC.28
IHC catalytic activity of complexes Sc(NiPr2)3(THF) (4) and Ce(NiPr2)4 (5)
Crystalline alkali-metal free tris(amido) complex Sc(NiPr2)3(THF) (4) can be readily obtained.21 Although it is routinely observed that scandium catalysts are less active than their yttrium counterparts, complex 4 had converted 18% of S2 after 5 h (Table 3, entry 7) thus kind of outperforming in situ formed Y(NiPr2)3(THF)x, which cyclized 4% during the same time period (Table 3, entry 5). The inefficiency of the latter yttrium-reaction, however, might be also a result of the in situ protocol applied for the synthesis of Y(NiPr2)3(THF)x,50 allowing a preformation time of 5 min only. Matching the present results, complex 4 is significantly less active than lithium ate complex LiSc(NiPr2)4(THF) (1a) (Table 1, entry 6). Next, we initially probed the IHC catalytic activity of the alkali-metal-free homoleptic tetravalent complex Ce(NiPr2)4 (5).51 Like complexes LiLn(NiPr2)4(THF) (1), the cerium complex 5 adopts a tetrahedral coordination geometry, but features a more Lewis-acidic rare-earth metal center and shorter Ln–N(amido) bonds as a consequence of the tetravalent oxidation state. Generally, there exist only a few reports on molecular organo-Ce(IV) catalysis.52 Complex 5 achieved 44% conversion of substrate S2 after 2 h, displaying an Nt = 5.6 h−1 (Table 3, entry 8). By then the color of the reaction mixture changed from blue to brown and the cyclization came to an halt, probably due to decomposition of the catalyst.IHC catalytic activity of rare-earth metal diisopropylamide complexes compared to bis(trimethylsilyl)amides Ln[N(SiMe3)2]3
The final part of this study compares the catalytic activities of the investigated rare-earth metal diisopropylamide complexes with those of the ubiquitous rare-earth metal bis(trimethylsilyl)amides Ln[N(SiMe3)2]3 (Ln = Sc (6a), Y (6b), and La (6c)). Complexes 6 were previously found to efficiently catalyze the IHC of aminoolefins and aminoalkynes.8,15 For direct comparison the reactions with precatalysts 6, which were sublimed prior to use, were carried out using exactly the same conditions as for LiLn(NiPr2)4(THF) (Ln = Sc (1a), Y (1b), and La (1c)) described above. The obtained results as listed in Table 4 are compared in the following with entries 1, 2, 4, 5, 7, 10, 12, 13, and 17 of Table 1. The majority of the reactions revealed that the rare-earth metal bis(trimethylsilyl)amide complexes 6 exhibit overall higher cyclization rates independent of the substrate. In the case of substrate S3, an activity increase from 6a (Sc) to La (6c) could be observed as expected (Table 4, entries 7–9) but 6a afforded almost the same Nt as scandium ate complex 1a (Table 1, entry 12). It also occurred for S3, that the yttrium diisopropylamide complex outperformed the silylamide one (1b: Nt = 164 h−1 (Table 1, entry 13); 6b: Nt = 13.9 h−1 (Table 4, entry 8)). Moreover, the competitive isomerization reaction of S3 to give P4 (Fig. 3 and 4) detected for LiLa(NiPr2)4(THF) (1c) could not be observed for La[N(SiMe3)2]3 (6c) which efficiently produced cyclized P3 (Table 4, entry 10). It is noteworthy that unsublimed Y[N(SiMe3)2]3 (6b) gave only traces of cyclized product P3 after 4.84 h at 26 °C (Table 4, entry 9) but full conversion was reached after 4 days. The overall better catalytic performance of the bis(trimethylsilyl)amide derivatives can be rationalized on the basis of less sterically saturated Ln(III) centres (4- versus 3-coordinate) allowing better initial access of the substrate molecules. Although diisopropylamine displays a similar basicity as the substrate molecules (pKa values of protonated amines in H2O: HNiPr2 = 11.05; primary amines = ca. 10; cyclized products pyrrolidine = 11.27 and piperidine = 11.22) the addition of 10 equivalents of HNiPr2 to the reaction system S2/2 did not affect the IHC transformation significantly, arguing against an enhanced coordination capability compared to the silylamine.53,54§| Entry | Aminoalkene | Product | catalyst | T/°C | t/h | Conv./%b |
N
tc/h−1 |
|---|---|---|---|---|---|---|---|
| a 4 mol% 6 in C6D6. b All conversion data were derived from 1H NMR spectra referring to the corresponding duration of reaction. c Initial Ntversus overall Nt (taken at 80% conversion, unless otherwise stated). d Conversion completed after <10 min. e Initial Nt = Nt at 80%. f Unsublimed 6b. | |||||||
| 1 |
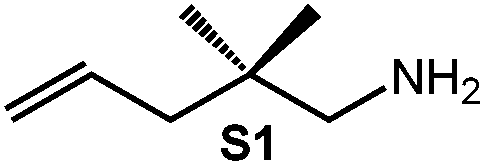
|
|
6a (Sc) | 60 | 4.84 | — | — |
| 2 | 6b (Y) | 60 | 2.0 | >99 | 37.2/17.1 | ||
| 3 | 6c (La) | 60 | 1.0 | >99 | 49.1/42.6 | ||
| 4 |
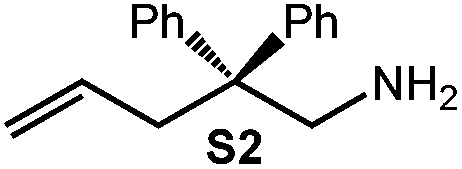
|
|
6a (Sc) | 26 | 0.17 | >99 | —d/149 |
| 5 | 6a (Y) | 26 | 0.17 | >99 | —d/149 | ||
| 6 | 6b (La) | 26 | 0.17 | >99 | —d/149 | ||
| 7 |
|
|
6a (Sc) | 60 | 2.3 | >99 | 64.3/22.5 |
| 8 | 6b (Y) | 60 | 1.5 | >99 | 13.9e | ||
| 9f | 6b (Y) | 26 | 4.84 | traces | — | ||
| 10 | 6b (La) | 60 | 1.1 | >99 | 17.4e | ||
Conclusions
Easily available rare-earth metal diisopropylamide complexes LiLn(NiPr2)4(THF) (Ln = Y, La)21 display high catalytic activity for regio-selective intramolecular hydroamination/cyclization reactions of terminal aminoalkenes depending on the substrate and the rare-earth metal centre at mild temperatures and low catalyst concentrations. The catalysts can be conveniently obtained in situ combining YCl3(THF)3.3 and four equivalents of LiNiPr2 (LDA), briefly before addition of aminoalkene substrate. Although the largest rare-earth metal centre exhibits the highest IHC activity it is prone to side reactions as shown for a competitive isomerization reaction in case of 1-amino-2,2-diphenyl-5-hexene (formation of 1-amino-2,2-diphenyl-4-hexene versus the 6-membered heterocycle 2-methyl-4,4-diphenylpiperidine). The occurrence of such isomerization reactions in tandem with hydroamination was previously reported for alkali and alkaline-earth metal-promoted catalysis but not in the case of rare-earth metal catalysis. Moreover, the present diisopropylamide case study clearly revealed the effect of donor solvent (THF), halo co-ligands and alkali metals on the catalytic performance of the rare-earth metal complex. It is shown that the presence of THF slows down drastically substrate conversion (LiLn(NiPr2)4(THF) versus [LiLn(NiPr2)4]n), most likely by competing with substrate coordination/cyclization at the catalytically active centre. Similarly, the presence of highly mobile AM(THF)+ fragments decelerates the conversion rates (AM = alkali metal; LiLn(NiPr2)4(THF) ≫ NaLn(NiPr2)4(THF)). The presence of chloro ligands seems to counteract an efficient catalytic process as well as shown for heteroleptic complex [Sc(NiPr2)2(μ-Cl)(THF)]2). Compared to the ubiquitous rare-earth metal bis(trimethylsilyl)amides Ln[N(SiMe3)2]3 (Ln = Sc, Y, La), which require sublimation prior to use, the investigated rare-earth metal diisopropylamide complexes show significantly lower catalytic activity. Exceptionally, 4-coordinate complex LiY(NiPr2)4(THF) outperformed three-coordinate Ln[N(SiMe3)2]3 for the IHC of 1-amino-2,2-diphenyl-5-pentene. We assume that the enhanced steric saturation of the rare earth-metal centre in ate complexes AMLn(NiPr2)4(THF)x and enhanced thermal instability compared to Ln[N(SiMe3)2]3 overcompensate the comparatively higher reactivity of the Ln–NiPr2 bond (versus Ln–N(SiMe3)2). Nevertheless, ate complexes AMLn(NiPr2)4(THF)x display an overall higher IHC efficiency than the previously reported rare-earth metal diamidobinaphthyl ate complexes.27–29,47,48 Finally, our preliminary observations that tetravalent cerium complex Ce(NiPr2)4 revealed higher conversion rates than Sc(NiPr2)3(THF) might significantly broaden the scope of cerium(IV) catalysis.Experimental section
General considerations
All operations were performed with rigorous exclusion of air and moisture, using standard Schlenk, high-vacuum, and glovebox techniques (MB Braun MB150B-G; <1 ppm O2, <1 ppm H2O). n-Hexane, n-pentane, and THF were purified by using Grubbs columns (MBraun SPS, Solvent Purification System) and stored in a glovebox. n-Butyllithium (2.5 M in n-hexane) and diisopropylamine (99.95%), isobutyronitrile (99.6%), allyl bromide (99%), lithium aluminium hydride (95%), 4-bromo-1-butene (97%), and tetrakis(p-tolyl)silane (99%) were obtained from Sigma-Aldrich, diphenylacetonitrile (99%) was obtained from ABCR. Benzene-d6 (99.6%) was received from Sigma Aldrich, dried over NaK alloy for a minimum of 48 h, and filtered twice through a filter pipette (Whatman) before use. Chloroform-d was obtained from Eurisotop and used as received. Complexes LiLn(NiPr2)4(THF) (Ln = Sc (1a), Y (1b), La (1c)), [LiY(NiPr2)4]n (2), NaLn(NiPr2)4(THF) (Ln = Sc (3a), Y (3b)) and Sc(NiPr2)3(THF) (4) were synthesized as described previously.21 Complexes Ce(NiPr2)4 (5),51 Ln[N(SiMe3)2]3 (Ln = Sc (6a), Y (6b), La (6b)),55 and LDA56 were synthesized according to literature procedures. Ln[N(SiMe3)2]3 were sublimed twice prior to use. NMR spectra were recorded by using J. Young valve NMR tubes and obtained on a Bruker AVII+400 (1H: 400.11 MHz, 13C: 100.61 MHz), and on a Bruker AVII+500 (1H: 500.13 MHz) spectrometer. 1H and 13C signals are referenced to internal solvent resonances and reported in parts per million relative to TMS. All NMR spectra of the cyclized products were obtained by in situ NMR scale catalytic hydroamination/cyclization reactions of the corresponding aminoalkene with the appropriate precatalyst under the reported conditions.Representative IHC procedure
In a glovebox, the desired amount of precatalyst or appropriate mixture of the in situ generated catalyst were introduced in a J. Young NMR tube and 0.4 mL benzene-d6 was added. The mixture was frozen at −35 °C and a solution of the aminoalkene (0.28 mmol) in 0.3 mL benzene-d6 was injected onto the solid mixture (= t0). The tube was sealed and frozen again. Then, the tube was removed from the glovebox, loaded immediately into the spectrometer and the progress of the reaction monitored by 1H NMR spectroscopy at the desired temperature (26 °C or 60 °C). The kinetics for rare-earth metal diisopropylamide complexes 1–5 and Ln[N(SiMe3)2]3 (6) as catalysts were measured by integrating the intrinsic signals of the educt (H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH) and product (H-2), respectively. The signals of hexamethyldisilazane or diisopropylamine which were formed by protonolysis of the appropriate precatalysts at the beginning of the reactions and stayed unchanged during the catalytic runs were chosen as internal standards. The cyclized amines P1, P2 and P3 as well as the isomerized aminoalkene P4 were identified by 1H NMR spectroscopy (NMR resonances included in the ESI†), the chemical shifts comparable to those described in literature.36,57
CH) and product (H-2), respectively. The signals of hexamethyldisilazane or diisopropylamine which were formed by protonolysis of the appropriate precatalysts at the beginning of the reactions and stayed unchanged during the catalytic runs were chosen as internal standards. The cyclized amines P1, P2 and P3 as well as the isomerized aminoalkene P4 were identified by 1H NMR spectroscopy (NMR resonances included in the ESI†), the chemical shifts comparable to those described in literature.36,57
Acknowledgements
We thank the German Science Foundation (Grant AN 238/16-1) for funding and D. Schneider for a sample of Ce(NiPr2)4.Notes and references
- T. E. Müller and M. Beller, Chem. Rev., 1998, 98, 675 CrossRef.
- S. Hong and T. J. Marks, Acc. Chem. Res., 2004, 37, 673 CrossRef CAS PubMed.
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef PubMed.
- (a) A. L. Reznichenko and K. C. Hultzsch, Struct. Bonding, 2010, 137, 1 CrossRef CAS; (b) A. L. Reznichenko and K. C. Hultzsch, Hydroamination of Alkenes, in Organic Reactions, Wiley, Hoboken, NJ, 2016, pp. 1–554 Search PubMed.
- (a) P. W. Roesky and T. E. Müller, Angew. Chem., Int. Ed., 2003, 42, 2708 CrossRef CAS PubMed; (b) J. Hannedouche and E. Schulz, Chem. – Eur. J., 2013, 19, 4972 CrossRef CAS PubMed.
- (a) M. R. Gagne and T. J. Marks, J. Am. Chem. Soc., 1989, 111, 4108 CrossRef CAS; (b) M. R. Gagne, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1992, 114, 275 CrossRef CAS.
- (a) M. R. Bürgstein, H. Berberich and P. W. Roesky, Organometallics, 1998, 17, 1452 CrossRef; (b) M. Rastätter, A. Zulys and P. W. Roesky, Chem. Commun., 2006, 874 RSC; (c) M. Rastätter, A. Zulys and P. W. Roesky, Chem. – Eur. J., 2007, 13, 3606 CrossRef PubMed; (d) H. F. Yuen and T. J. Marks, Organometallics, 2008, 27, 155 CrossRef CAS; (e) L. J. E. Stanlake and L. L. Schafer, Organometallics, 2009, 28, 3990 CrossRef CAS; (f) A. Otero, A.- Lara-Sánchez, J. A. Castro-Osma, I. Márquez-Segovia, C. Alonso-Moreno, J. Fernámdez-Baeza, L. F. Sánchez-Barba and A. M. Rodriguez, New J. Chem., 2015, 39, 7672 RSC.
- (a) Y. K. Kim and T. Livinghouse, Angew. Chem., Int. Ed., 2002, 41, 3645 CrossRef CAS; (b) Y. K. Kim, T. Livinghouse and Y. Horino, J. Am. Chem. Soc., 2003, 125, 9560 CrossRef CAS PubMed; (c) Y. K. Kim, T. Livinghouse and J. E. Bercaw, Tetrahedron Lett., 2001, 42, 2933 CrossRef CAS; (d) J. Y. Kim and T. Livinghouse, Org. lett., 2005, 7, 4391 CrossRef CAS PubMed; (e) C. Quinet, A. Ates and I. E. Markó, Tetrahedron Lett., 2008, 49, 5032 CrossRef CAS; (f) J. Y. Kim and T. Livinghouse, Org. lett., 2005, 7, 1737 CrossRef CAS PubMed; (g) X. Yu and T. J. Marks, Organometallics, 2007, 26, 365 CrossRef CAS; (h) T. Jiang and T. Livinghouse, Org. Lett., 2010, 12, 4271 CrossRef CAS PubMed.
- S. Hong, S. Tian, M. V. Metz and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 14768 CrossRef CAS PubMed.
- P. N. O'Shaughnessy, K. M. Gillespie, P. D. Knight, I. J. Munslow and P. Scott, Dalton Trans., 2004, 2251 RSC.
- D. V. Gribkov, K. C. Hultzsch and F. Hampel, J. Am. Chem. Soc., 2006, 128, 3748 CrossRef CAS PubMed.
- (a) Y. Zhang, W. Yao, H. Li and Y. Mu, Organometallics, 2012, 31, 4670 CrossRef CAS; (b) Z. Chai, D. Hua, K. Li, J. Chu and G. Yang, Chem. Commun., 2014, 50, 177 RSC and references therein.
- A. L. Reznichenko, A. J. Nawara-Hultzsch and K. C. Hultzsch, Top. Curr. Chem., 2014, 343, 191–260 CrossRef CAS PubMed.
- (a) A. L. Reznichenko, H. N. Nguyen and K. C. Hultzsch, Angew. Chem., Int. Ed., 2010, 49, 8984 CrossRef CAS PubMed; (b) A. L. Reznichenko and K. C. Hultzsch, Organometallics, 2013, 32, 1394 CrossRef CAS.
- M. R. Bürgstein, H. Berberich and P. W. Roesky, Chem. – Eur. J., 2001, 7, 3078 CrossRef.
- E. L. Roux, Y. Liang, M. P. Storz and R. Anwander, J. Am. Chem. Soc., 2010, 132, 16368 CrossRef PubMed.
- K. C. Hultzsch, F. Hampel and T. Wagner, Organometallics, 2004, 23, 2601 CrossRef CAS.
- (a) M. A. Giardello, V. P. Conticello, L. Brard, M. Sabat, A. L. Rheingold, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1994, 116, 10212 CrossRef CAS; (b) M. A. Giardello, V. P. Conticello, L. Brard, M. R. Gagne and T. J. Marks, J. Am. Chem. Soc., 1994, 116, 10241 CrossRef CAS.
- J. Eppinger, M. Spiegler, W. Hieringer, W. A. Herrmann and R. Anwander, J. Am. Chem. Soc., 2000, 122, 3080 CrossRef CAS.
- R. R. Fraser, M. Bresse and T. S. Mansour, J. Chem. Soc., Chem. Commun., 1983, 620 RSC.
- T. Spallek, O. Heß, M. Meermann-Zimmermann, C. Meermann, M. G. Klimpel, F. Estler, D. Schneider, W. Scherer, M. Tafipolsky, K. W. Törnroos, C. Maichle-Mössmer, P. Sirsch and R. Anwander, Dalton Trans., 2016, 45, 13750 RSC.
- L. Ackermann, L. T. Kaspar and A. Althammer, Org. Biomol. Chem., 2007, 5, 1975 CAS.
- A. Ates and C. Quinet, Eur. J. Org. Chem., 2003, 1623 CrossRef CAS.
- C. Quinet, P. Jourdain, C. Hermans, A. Ates, I. Lucas and I. E. Markó, Tetrahedron, 2008, 64, 1077 CrossRef CAS.
- P. N. O'Shaughnessy and P. Scott, Tetrahedron: Asymmetry, 2003, 14, 1979 CrossRef.
- D. Riegert, J. Collin, J.-C. Daran, T. Fillebeen, E. Schulz, D. Lyubov, G. Fukin and A. Trifonov, Eur. J. Inorg. Chem., 2007, 2007, 1159 CrossRef.
- (a) J. Collin, J. C. Daran, E. Schulz and A. Trifonov, Chem. Commun., 2003, 3048 RSC; (b) J. Collin, J. C. Daran, O. Jacquet, E. Schulz and A. Trifonov, Chem. – Eur. J., 2005, 11, 3455 CrossRef CAS PubMed; (c) D. Riegert, J. Collin, A. Meddour, E. Schulz and A. Trifonov, J. Org. Chem., 2006, 71, 2514 CrossRef CAS PubMed; (d) I. Aillaud, K. Wright, J. Collin, E. Schulz and J.-P. Mazaleyrat, Tetrahedron: Asymmetry, 2008, 19, 82 CrossRef CAS.
- I. Aillaud, J. Collin, C. Duhayon, R. Guillot, D. Lyubov, E. Schulz and A. Trifonov, Chem. – Eur. J., 2008, 14, 2189 CrossRef CAS PubMed.
- Y. Chapurina, R. Guillot, D. Lyubov, A. Trifonov, J. Hannedouche and E. Schulz, Dalton Trans., 2013, 42, 507 RSC.
- K2Ca anilide ate complexes catalyze the intermolecular hydroamination of butadiynes: (a) C. Glock, H. Görls and M. Westerhausen, Chem. Commun., 2012, 48, 7094 RSC; (b) F. M. Younis, S. Krieck, H. Görls and M. Westerhausen, Organometallics, 2015, 34, 3577 CrossRef CAS and references therein.
- G. R. Giesbrecht, G. E. Collis, J. C. Gordon, D. L. Clark, B. L. Scott and N. J. Hardman, J. Organomet. Chem., 2004, 689, 2177 CrossRef CAS.
- M. Arrowsmith, M. S. Hill and G. Kociok-Köhn, Organometallics, 2014, 33, 206 CrossRef CAS.
- R. M. Beesley, C. K. Ingold and J. F. Thorpe, J. Chem. Soc., Dalton Trans., 1915, 107, 1080 RSC.
- M. E. Jung and G. Piizi, Chem. Rev., 2005, 105, 1735 CrossRef CAS PubMed.
- J. E. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 734 RSC.
- (a) M. R. Crimmin, M. Arrowsmith, A. G. M. Barrett, I. J. Casely, M. S. Hill and P. A. Procopiou, J. Am. Chem. Soc., 2009, 131, 9670 CrossRef CAS PubMed; (b) J. Jenter, R. Köppe and P. W. Roesky, Organometallics, 2011, 30, 1404 CrossRef CAS.
- For representative examples of α-olefin isomerization at Ln(III) centres, see: (a) E. Bubel, B. J. Burger and J. E. Bercaw, J. Am. Chem. Soc., 1988, 110, 976 CrossRef; (b) C. Qian, Y. Ge, D. Deng, Y. Gu and C. Zhang, J. Organomet. Chem., 1988, 344, 175 CrossRef CAS.
- S. Harder, Chem. Rev., 2010, 110, 3852 CrossRef CAS PubMed.
- R. Shannon, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1976, 32, 751 CrossRef.
- R. D. Closson, J. P. Napolitano, G. G. Ecke and A. J. Kolka, J. Org. Chem., 1957, 22, 646 CrossRef CAS.
- G. P. Pez and J. E. Galle, Pure Appl. Chem., 1985, 57, 1917 CrossRef CAS.
- (a) B. M. Trost and W. Tang, J. Am. Chem. Soc., 2002, 124, 14542 CrossRef CAS PubMed; (b) B. M. Trost and W. Tang, J. Am. Chem. Soc., 2003, 125, 8744 CrossRef CAS PubMed.
- (a) T. Ogata, A. Ujihara, S. Tsuchida, T. Shimizu, A. Kaneshige and K. Tomioka, Tetrahedron Lett., 2007, 48, 6648 CrossRef CAS; (b) S. Tsuchida, A. Kaneshige, T. Ogata, H. Baba, Y. Yamamoto and K. Tomioka, Org. Lett., 2008, 10, 3635 CrossRef CAS PubMed.
- P. Horrillo-Martínez, K. C. Hultzsch, A. Gil and V. Branchadell, Eur. J. Org. Chem., 2007, 3311 CrossRef.
- P. H. Martinez, K. C. Hultzsch and F. Hampel, Chem. Commun., 2006, 2221 RSC.
- I. Aillaud, D. Lyubov, J. Collin, R. Guillot, J. Hannedouche, E. Schulz and A. Trifonov, Organometallics, 2008, 27, 5929 CrossRef CAS.
- Y. Chapurina, J. Hannedouche, J. Collin, R. Guillot, E. Schulz and A. Trifonov, Chem. Commun., 2010, 46, 6918 RSC.
- Y. Chapurina, H. Ibrahim, R. Guillot, E. Kolodziej, J. Collin, A. Trifonov, E. Schulz and J. Hannedouche, J. Org. Chem., 2011, 76, 10163 CrossRef CAS PubMed.
- C. Meermann, G. Gerstberger, M. Spiegler, K. W. Törnroos and R. Anwander, Eur. J. Inorg. Chem., 2008, 2014 CrossRef CAS.
- F. Estler, G. Eickerling, E. Herdtweck and R. Anwander, Organometallics, 2003, 22, 1212 CrossRef CAS.
- D. Schneider, T. Spallek, C. Maichle-Mössmer, K. W. Törnroos and R. Anwander, Chem. Commun., 2014, 50, 14763 RSC.
- For examples of Ce(IV)-promoted homogeneous catalysis, see: (a) I. E. Markó, A. Ates, A. Gautier, B. Leroy, J.-M. Plancher, Y. Quesnel and J.-C. Vanherck, Angew. Chem., Int. Ed., 1999, 38, 3207 CrossRef; (b) K. K. Laali, M. Herbert, B. Cushnyr, A. Bhatt and D. Terrano, J. Chem. Soc., Perkin Trans. 1, 2001, 578 RSC; (c) N. Nomura, A. Tiara, A. Nakase, T. Tomioka and M. Okada, Tetrahedron, 2007, 63, 8478 CrossRef CAS; (d) E. M. Broderick and P. L. Diaconescu, Inorg. Chem., 2009, 48, 4701 CrossRef CAS PubMed; (e) A. Sauer, J.-C. Buffet, T. P. Spaniol, H. Nagae, K. Maschima and J. Okuda, ChemCatChem, 2013, 5, 1088 CrossRef CAS; (f) H. Yin, P. J. Carrol, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 5984 CrossRef CAS PubMed.
- H. K. Hall, J. Am. Chem. Soc., 1957, 79, 5441 CrossRef CAS.
- F. Lauterwasser, P. G. Hayes, S. Bräse, W. E. Piers and L. L. Schafer, Organometallics, 2004, 23, 2234 CrossRef CAS.
- (a) E. C. Alyea, D. C. Bradley and R. G. Copperthwaite, J. Chem. Soc., Dalton Trans., 1972, 1580 RSC; (b) D. C. Bradley, J. S. Ghotra and F. A. Hart, J. Chem. Soc., Chem. Commun., 1972, 349 RSC; (c) D. C. Bradley, J. S. Ghotra and F. A. Hart, J. Chem. Soc., Dalton Trans., 1973, 1021 RSC.
- W. Bauer and D. Seebach, Helv. Chim. Acta, 1984, 67, 1972 CrossRef CAS.
- M. Arrowsmith, M. R. Crimmin, A. G. M. Barrett, M. S. Hill, G. Kociok-Köhn and P. A. Procopiou, Organometallics, 2011, 30, 1493 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of experimental procedures, NMR and catalytic data. See DOI: 10.1039/c6dt03045a |
| ‡ It has been shown that LDA22 or treatment of lithiated aminoalkene with diisopropylamine23,24 can efficiently catalyze IHC of non-actived olefins. |
| § Stoichiometric reactions, similarly examined by Piers et al.,54 performed overnight at 26 °C and involving mixtures Y[N(SiMe3)2]3/3 S3, LiSc(NiPr2)4(THF)/4 S2 and LiNiPr2/S2(S3) led to the complete disappearance of the olefinic and NH protons of the substrates, suggesting conversion to the cyclized product which remains coordinated to the metal centre. Unfortunately, several attempts to obtain single crystals of the ligand exchanged products were unsuccessful. |
| This journal is © The Royal Society of Chemistry 2016 |