
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron complexes of a bidentate picolyl-NHC ligand: synthesis, structure and reactivity†
Qiuming
Liang
 ,
Trevor
Janes
,
Xhoana
Gjergji
and
Datong
Song
*
,
Trevor
Janes
,
Xhoana
Gjergji
and
Datong
Song
*
Davenport Chemical Research Laboratories, Department of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario M5S 3H6, Canada. E-mail: dsong@chem.utoronto.ca
First published on 4th August 2016
Abstract
The synthesis, structure and reactivity of bidentate picolyl N-heterocyclic carbene (NHC) iron compounds were studied. Compounds [FeBr(HL)2]Br (1), [FeBr(HL)(HMDS)] (2) and [FeBr2(HL)] (3) (HL = 1-mesityl-3-(pyridin-2-ylmethyl)imidazol-1-ylidene, HMDS = hexamethyldisilazide) were prepared from H2LBr with suitable amounts of Fe(HMDS)2 or in situ prepared [Fe(HMDS)Br]. The deprotonation of 1 with 2 eq. of LiHMDS gave [FeL2] (4), featuring dearomatized pyridine moieties with exocyclic C–C double bonds. The protonation of 4 with 2 eq. of PPh3·HBr results in the formation of 1. Attempted deprotonation of 3 using benzyl Grignard as the base resulted in transmetalation products [FeBnBr(HL)] (5) and [FeBn2(HL)] (6). Exposure of 6 to CO resulted in the formation of diamagnetic compound [Fe(CO)3(HL)] (7) and dibenzyl ketone. Prolonged exposure of 7 to CO with heating induces pyridine dissociation, affording [Fe(CO)4(HL-κC)] (8). Treatment of compound 6 with an equimolar amount of p-methoxybenzyl bromide yielded homo- and cross-coupling products.
Introduction
The use of actor ligands in metal complexes, where the ligands can participate in chemical transformation directly, has garnered increasing attention recently as the participation of actor ligands has enabled fascinating reactivity patterns in both stoichiometric and catalytic fashions.1,2 The participation of actor ligands in chemical reactions can be through ligand-based reactivity3,4 or metal–ligand cooperation.5 Depending on the relative positions of the metal centre and the ligand reactive site, metal–ligand cooperation can be proximal2,5–7 or distal.2 Milstein and co-workers have demonstrated an interesting mode of distal metal–ligand cooperation involving dearomatization–rearomatization of pyridine/acridine based tridentate ligands and applied such reactivity to many stoichiometric and catalytic processes.8,9 Our group and others have reported CNN-pincer complexes toward catalytic hydrogenation.10–13 Through deuterium scrambling experiments, we have shown that the dearomatization–rearomatization of the pyridine moiety of the (NHC)NN-ligand can involve the deprotonation–reprotonation of the methylene groups on both the amine and NHC arms, although the intermediate with deprotonated amine arm could not be observed directly.10 Recently, Chirik and co-workers have reported the (CNC)Fe(N2)2 and (CNC)CoR complexes and their high catalytic hydrogenation activity of hindered, unfunctionalized alkenes.14,15 The dearomatization of the pyridine moiety of the pincer ligand from the migration of cobalt-hydride or cobalt-alkyl to the 4-position of pyridine ring is identified as a catalyst deactivation pathway.15In contrast to the pincer complexes mentioned above, complexes of bidentate picolyl-containing ligands rarely show parallel reactivity where the removal of pyridylic proton dearomatizes the pyridine ring.16–19 Waterman and co-workers have recently reported the Zn(II) complexes of bidentate picolyl-phosphine ligands, where the pyridylic protons were removed causing the dearomatization of the pyridine rings prior to coordination to Zn(II).19 To the best of our knowledge, complexes of the analogous deprotonated/dearomatized bidentate picolyl-NHC ligands have not been reported to date. The charge neutral bidentate picolyl-NHC ligands have been incorporated in several transition metal complexes, most of which involve noble metals.20 Despite the increasing interest in base metal complexes, only a few examples with bidentate picoyl-NHC ligands have been reported.21–23 Danopoulos et al. have synthesized the mono and bis(picolyl-NHC) nickel dibromide complexes by transferring the ligands from the corresponding silver complexes.21 Similar bis(picolyl-NHC) nickel dichloride complexes have been synthesized by Jin et al. and applied to olefin polymerization.22 In the case of iron, only the piano-stool iron carbonyl complexes [Fe(CO)1–2(Cp)(picolyl-NHC)X] (X = I, BF4) have been reported.23 In fact, nitrogen/oxygen donor substituted NHC bidentate ligands have received less attention compared to the extensive study on NHC iron complexes.24–26 The first examples with anionic aryloxo-functionalized NHCs, were reported by Shen et al.27,28 Lavoie and co-workers reported the complexes with neutral imine-functionalized NHCs.29,30 The group of Hahn and Chen reported the octahedral complexes with pyridinyl31 and pyrimidinyl32,33 substituted NHC respectively. Herein, we report the syntheses and structures of a series of iron complexes of a bidentate picolyl-NHC ligand, along with their reactivity including the interconversions, transmetalation, C–C bond formations, and ligand deprotonation accompanied by the dearomatization of the pyridine moiety.
Results and discussion
Synthesis and structure of 1–3 bearing neutral picolyl-NHC ligand
Compounds [FeBr(HL)2]Br (1) and [FeBr(HL)(HMDS)] (2) can be synthesized from the reaction of H2LBr with 0.5 and 1 eq. of Fe(HMDS)2, respectively (Scheme 1) in excellent yields, employing the synthetic protocol by Danopoulos.34 The paramagnetic compound 1 is insoluble in THF, diethyl ether, and hydrocarbons, and unstable in dichloromethane and chloroform. Compound 1 crystallizes in the monoclinic space group P21/n along with THF solvent molecules. As shown in Fig. 1, the Fe(II) centre adopts a distorted trigonal bipyramidal geometry with two N donor atoms from two HL ligands occupying the apical positions, and one bromide and two carbon donors occupying the equatorial positions, while the other bromide is outer-sphere to balance the charge. The N1–Fe1–N4 angle is 167.0(1)° while sum of the three bond angles within the equatorial plane is 360.1(2)°. The average Fe–CNHC and Fe–Npyridine bond lengths are 2.115(6) Å and 2.302(4) Å, respectively, similar to those of analogous complexes.24,35–45 The six-membered chelate rings adopt boat conformations. The average bite angle of HL in 1 is 82.8(2)°. Compound 1 is structurally and magnetically distinct from the bis(picolyl-NHC) nickel dihalide analogues,21,22 where the nickel complexes are diamagnetic, square planar at nickel centres with both halides outside the coordination sphere.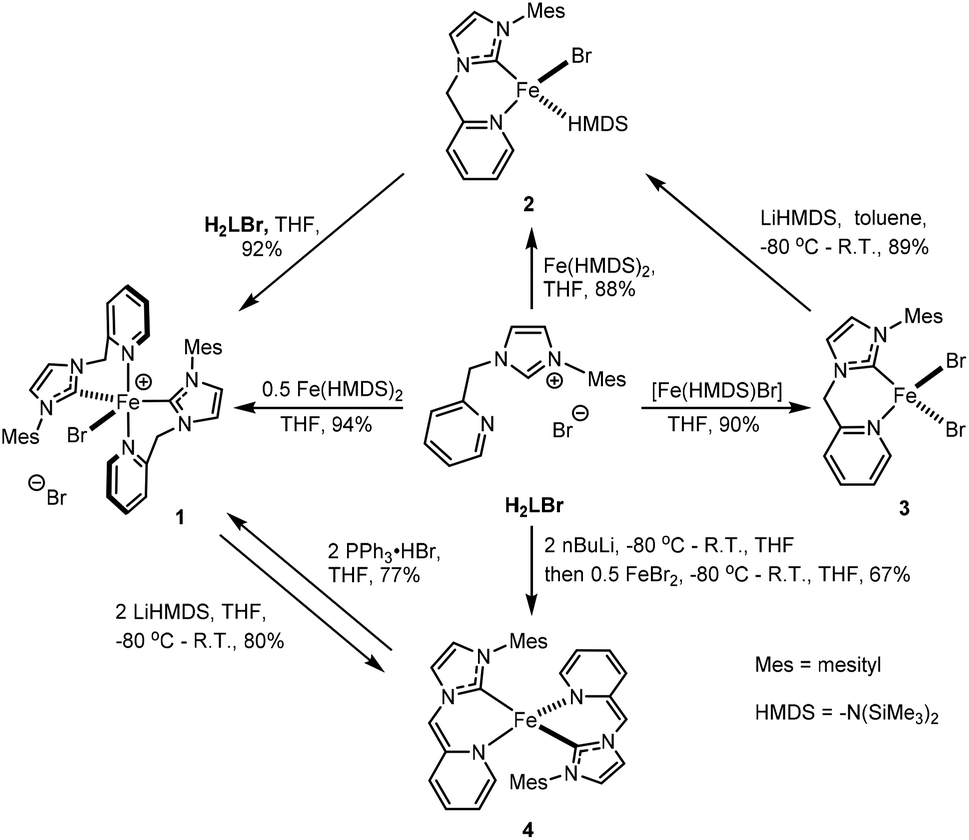 | ||
| Scheme 1 Syntheses of picolyl-NHC iron(II) complexes. | ||
 | ||
| Fig. 1 X-ray structures of 1 (top) and 2 (bottom). Ellipsoids are shown at 50% probability. Only one enantiomer of 2 is shown. Selected bond lengths (Å) and angles (°) for 1: Fe1–C1 2.110(5), Fe1–C19 2.120(5), Fe1–N1 2.292(4), Fe1–N4 2.312(4), Fe1–Br1 2.4114(9), C1–Fe1–C19 119.0(2), C1–Fe1–N1 83.3(2), C19–Fe1–N1 90.1(2), C1–Fe1–N4 91.0(2), C19–Fe1–N4 82.4(2), N1–Fe1–N4 167.0(1), C1–Fe1–Br1 119.1(1), C19–Fe1–Br1 122.0(1), N1–Fe1–Br1 96.8(1), N4–Fe1–Br1 96.2(1); for 2: Fe1–N4 1.943(6), Fe1–C1 2.105(8), Fe1–N3 2.146(6), Fe1–Br1 2.457(1), N4–Fe1–C1 130.5(3), N4–Fe1–N3 107.1(3), C1–Fe1–N3 88.0(3), N4–Fe1–Br1 120.3(2), C1–Fe1–Br1 102.9(2), N3–Fe1–Br1 98.2(2). | ||
The paramagnetic compound 2 is soluble in THF, toluene, and benzene, and slightly soluble in diethyl ether. In solution, 2 slowly decomposes at room temperature into intractable products. When stored in the solid state at −35 °C, 2 is stable for weeks, i.e., without noticeable changes in the appearance of its 1H NMR spectrum. Compound 2 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with a pair of enantiomers in the asymmetric unit. As shown in Fig. 1, the Fe(II) centre adopts a distorted tetrahedral coordination geometry with the nitrogen and carbon donor atoms of HL, the nitrogen donor atom of HMDS, and a bromide ligand occupying the four coordination sites. The average Fe–CNHC and Fe–NHMDS bond lengths of the two enantiomers are 2.095(8) and 1.945(8) Å, respectively, comparable to those of the analogous literature complexes.45,46 The average bite angle of the HL ligand in 2 is 88.1(3)°, wider than that in 1. The largest peak in the 1H NMR spectrum of 2 in C6D6 at 13.92 ppm has been tentatively assigned to the protons of the HMDS group. The reaction of H2LBr with an equimolar amount of 2 in THF cleanly affords 1 in nearly quantitative yield (Scheme 1).
with a pair of enantiomers in the asymmetric unit. As shown in Fig. 1, the Fe(II) centre adopts a distorted tetrahedral coordination geometry with the nitrogen and carbon donor atoms of HL, the nitrogen donor atom of HMDS, and a bromide ligand occupying the four coordination sites. The average Fe–CNHC and Fe–NHMDS bond lengths of the two enantiomers are 2.095(8) and 1.945(8) Å, respectively, comparable to those of the analogous literature complexes.45,46 The average bite angle of the HL ligand in 2 is 88.1(3)°, wider than that in 1. The largest peak in the 1H NMR spectrum of 2 in C6D6 at 13.92 ppm has been tentatively assigned to the protons of the HMDS group. The reaction of H2LBr with an equimolar amount of 2 in THF cleanly affords 1 in nearly quantitative yield (Scheme 1).
Using the protocol by Byers,47 adding in situ prepared [FeBr(HMDS)] (premixing 0.5 eq. of anhydrous FeBr2 and 0.5 eq. of Fe(HMDS)2 in THF) to 1 eq. of H2LBr yields [FeBr2(HL)] (3) in an excellent yield (Scheme 1). The paramagnetic compound 3 is sparingly soluble in diethyl ether and hydrocarbons, slightly soluble in THF, and soluble and stable in dichloromethane and chloroform. Compound 3 crystallizes in the monoclinic space group P21/n. As shown in Fig. 2, the distorted tetrahedral geometry adopted by the Fe(II) centre is similar to those of the methylene-bridged biscarbene iron(II) dihalide complexes.48–50 The bite angle of HL in 3 (89.1(1)°) is similar to that in 2. The Fe–CNHC (Fe1–C1 2.065(3) Å) and Fe–Npyridine (Fe1–N3 2.157(3) Å) bond lengths are shorter than those in 1 and more similar to those in 2.
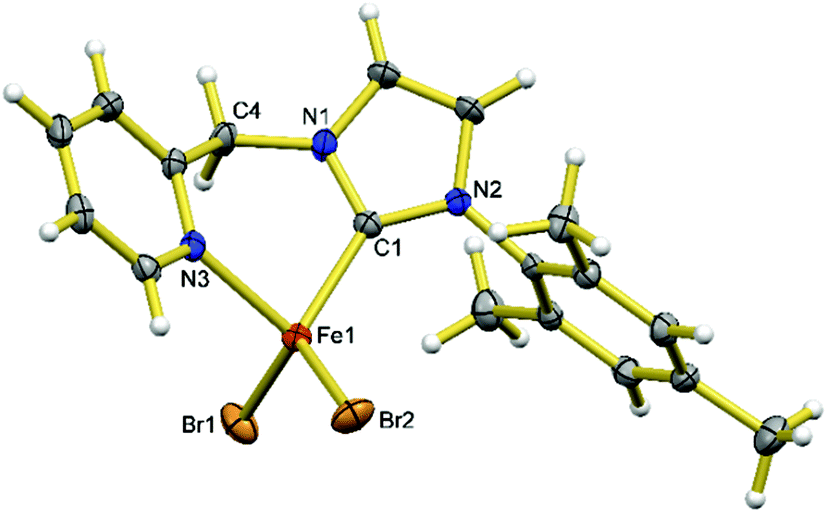 | ||
| Fig. 2 X-ray structures of 3. Ellipsoids are shown at 50% probability. Selected bond lengths (Å) and angles (°): Fe1–C1 2.065(3), Fe1–N3 2.157(3), Fe1–Br2 2.3984(6), Fe1–Br1 2.4004(6), C1–Fe1–N3 89.1(1), C1–Fe1–Br2 118.5(1), N3–Fe1–Br2 103.96(7), C1–Fe1–Br1 109.5(1), N3–Fe1–Br1 111.01(8), Br2–Fe1–Br1 119.85(2). | ||
Synthesis and structure of 4
Complex 3 possesses the charge neutral bidentate HL ligand and two bromide ligands on a four-coordinate Fe(II) centre. It is conceivable that the deprotonation of the CH2 group of HL ligand of 3 could potentially generate an L− ligand with a dearomatized pyridine ring in the form of [FeBrL] with a three-coordinate Fe(II) centre or [FeBrL]2 with four-coordinate Fe(II) centres and two bridging bromides. However, the deprotonation of 3 using 1 eq. of base is often competing with the substitution of the bromide ligand by the base and is also complicated by the rapid ligand redistribution of the deprotonation product. For example, the treatment of 3 with 1 eq. of LiHMDS in toluene at low temperatures cleanly yields 2 (Scheme 1), the ligand substitution product. If this reaction is carried out in diethyl ether or THF, the mixture of 2 and 4 is often obtained under various conditions (i.e., different temperatures, concentrations, and stoichiometries), where the presence of 4 can be confirmed through 1H NMR experiment by comparing with an independently synthesized sample. Compound 4 can be cleanly obtained from the reaction of 2 eq. of LiHMDS with 1 eq. of 1 in THF; although the structure of 4 could not be revealed with NMR experiments due to its paramagnetic nature, the stoichiometry and cleanness of this reaction suggest a formula of [FeL2]. Alternatively, compound 4 can be prepared from the reaction of 1 eq. of in situ generated L− (from 2 eq. of nBuLi and 1 eq. of H2LBr) with 0.5 eq. of FeBr2. The product of the latter method allows for the growth of X-ray quality single crystals and in turn, the structural determination by X-ray crystallography, which confirms the formula of 4 as [FeL2]. Presumably the formation of 4 from the deprotonation of 3 goes through a three-coordinate [FeLBr] intermediate, which undergoes rapid ligand redistribution to form 4 and FeBr2. Using other bases such as alkoxides, LDA, group 1 hydrides, and CH3Li to deprotonate 3 leads to complicate mixtures of products that contain 4. To test the reversibility of dearomatization/aromatization, 4 was treated with 2 eq. of PPh3·HBr, which resulted formation of 1 in good yields.Compound 4 is soluble in THF and toluene, and slightly soluble in diethyl ether, and reacts with dichloromethane and chloroform. It is extremely reactive toward air and moisture, but thermally robust, i.e., no noticeable change in the 1H NMR spectrum after heating a C6D6 solution sealed under N2 at 60 °C for several days. As shown in Fig. 3, the Fe(II) centre in 4 adopts a distorted tetrahedral geometry with two N,C-chelates completing the coordination sphere. The Fe–N (Fe1–N1 2.001(2) Å and Fe1–N4 2.011(3) Å) bonds are much shorter than those in complexes 1–3, while the Fe–C (Fe1–C19 2.024(3) Å and Fe1–C1 2.033(2) Å) bonds are shorter than those in 1–3 to a lesser extent. The C–N bonds in the C5N rings are elongated compared to those in 1–3 and the C–C bonds of the C5N rings display obvious alternating long–short pattern. The C–C bonds exocyclic to the C5N rings (C22–C23 1.368(6) Å and C4–C5 1.371(4) Å) display bond lengths typical for C–C double bonds. The C–N bonds exocyclic to the C3N2 rings (C22–N5 1.429(5) Å and C4–N2 1.412(3) Å) are slightly shortened compared to those in compound 1, showing the partial delocalization of the π electrons in the six-membered chelate ring. However, the shortening of the C–N bonds is much less significant compared to that of the C–C bonds, suggesting that the dominant resonance form has a C–C double bond and C–N single bond between the two rings of the L− ligand in 4. Each chelate ligand is nearly planar with the Fe(II) centre sitting outside the planes by ∼0.5 Å. All these structural features suggest that the desired L− ligand has been obtained, featuring the dearomatized pyridine ring and an exocyclic C–C double bond, analogous to the related CNN pincer ligand10 and picolyl-phosphine bidentate ligand.16–19 The mesityl ring of one L− ligand in 4 is nearly parallel with the C5N ring of the other L− ligand (dihedral angles: 2.75 and 3.96°) with the shortest contact distance of ∼3.6 Å, suggesting weak π–π stacking interactions in between.
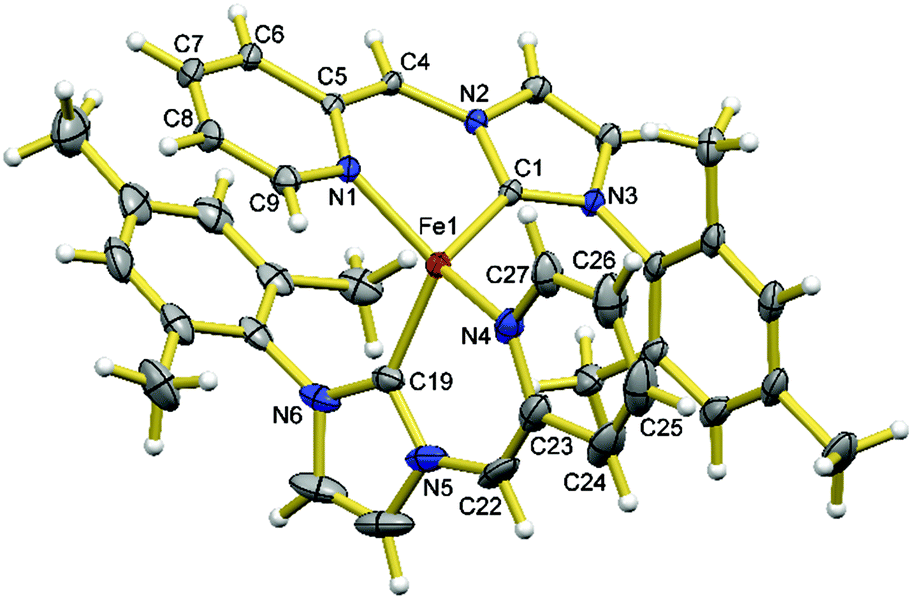 | ||
| Fig. 3 Molecular structures of 4. Ellipsoids are shown at 30% probability. Selected bond lengths (Å) and angles (°): Fe1–N1 2.001(2), Fe1–N4 2.011(3), Fe1–C19 2.024(3), Fe1–C1 2.033(2), C4–C5 1.372(4), C5–C6 1.448(4), C6–C7 1.344(4), C7–C8 1.411(4), C8–C9 1.353(4), N1–C9 1.366(3), N1–C5 1.398(3), C22–C23 1.368(5), C23–C24 1.487(5), C24–C25 1.348(7), C25–C26 1.388(7), C26–C27 1.348(5), N4–C27 1.372(4), N4–C23 1.377(4), N1–Fe1–N4 129.4(1), N1–Fe1–C19 113.6(1), N4–Fe1–C19 93.3(1), N1–Fe1–C1 92.23(9), N4–Fe1–C1 114.7(1), C19–Fe1–C1 115.2(1). | ||
Reaction of 3 with Grignard reagent
When 1.2 eq. of benzyl Grignard reagent is used to deprotonate 3 in THF/dioxane, the transmetalation product [FeBnBr(HL)] (5) can be obtained in good yields (Scheme 2). Compound 5 is soluble in THF, and slightly soluble in diethyl ether and toluene. The reaction of 3 with 2.4 eq. of benzyl Grignard reagent in diethyl ether/dioxane gives the double-transmetalation product [FeBn2(HL)] (6) in good yields. Compound 6 is soluble in THF, toluene and diethyl ether. The molecular structures of 5 and 6 have been confirmed crystallographically (Fig. 4). Each Fe(II) centre adopts a distorted tetrahedral geometry displaying typical bond lengths.42,51,52 The bite angle of the HL ligand in 5 (88.3(2)°) is similar to those of 2 and 3, while the bite angle of HL in 6 (86.05(7)°) is slightly smaller. Both 5 and 6 are thermally unstable and decompose in solution into intractable products at room temperature over a few days.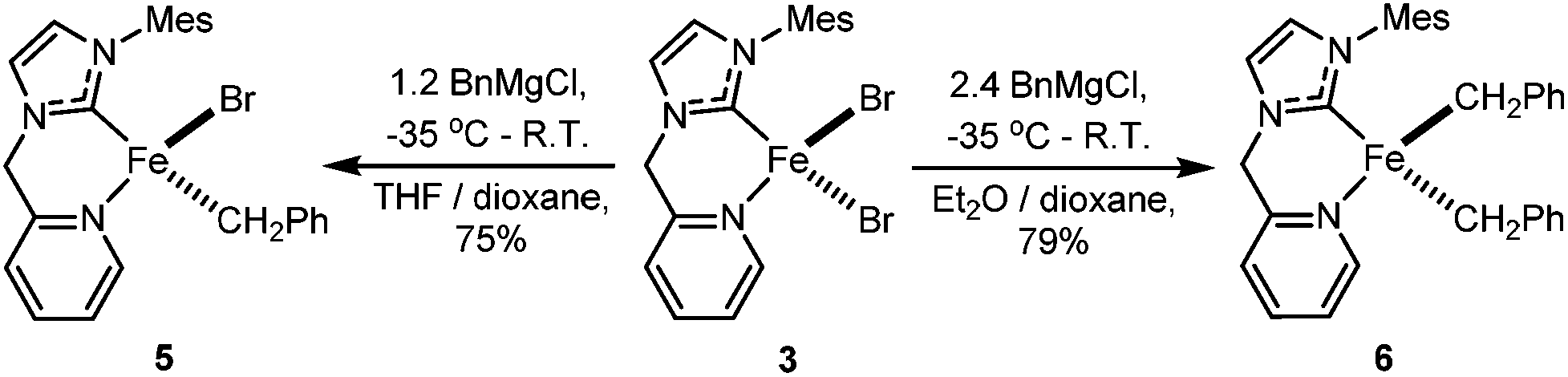 | ||
| Scheme 2 Reactions of 3 with benzyl Grignard reagent. | ||
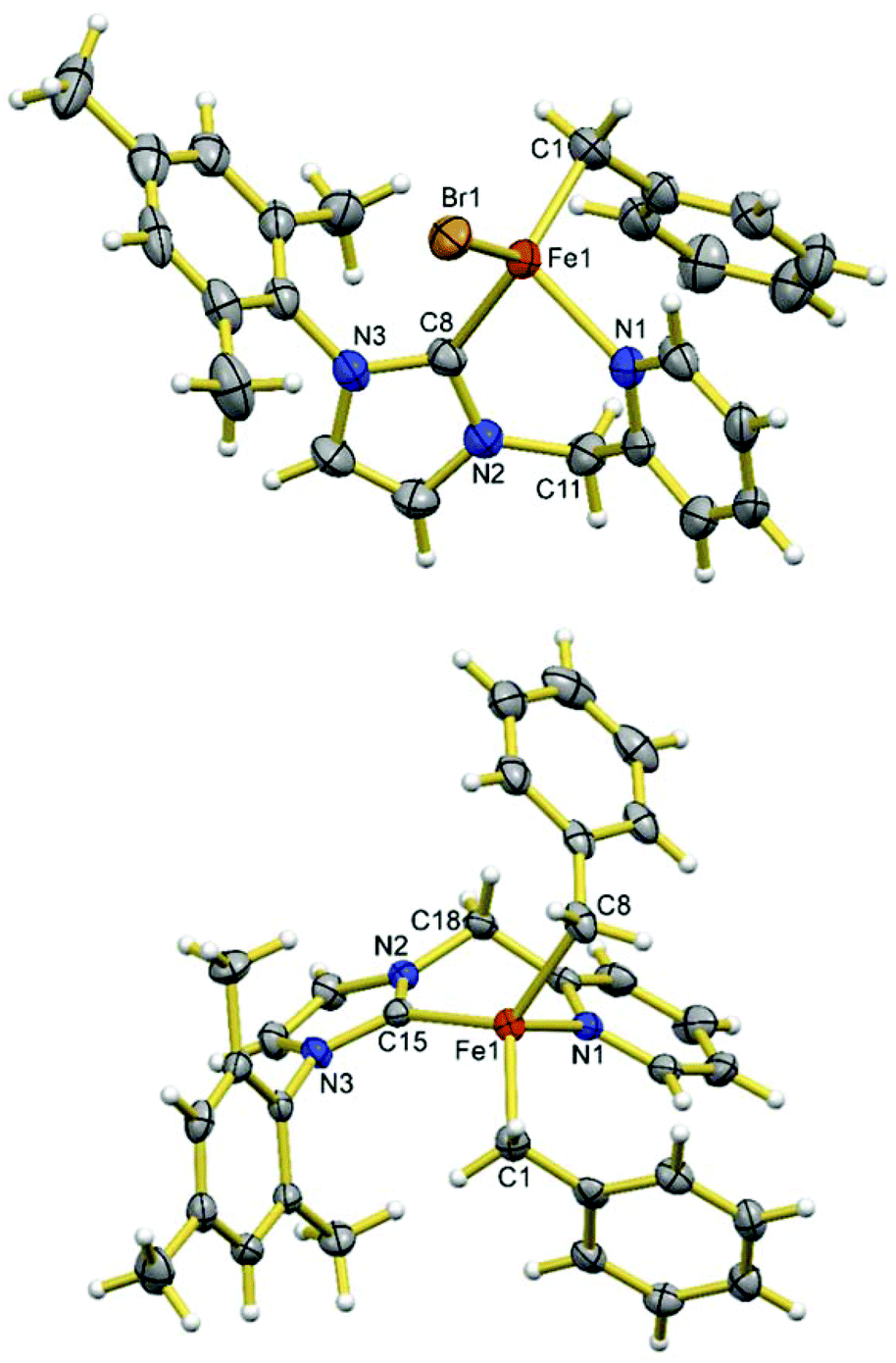 | ||
| Fig. 4 X-ray structures of complexes 5 (top) and 6 (bottom). Ellipsoids are shown at 50% probability. Selected bond lengths (Å) and angles (°) for 5: Fe1–C8 2.060(5), Fe1–C1 2.061(5), Fe1–N1 2.174(4), Fe1–Br1 2.451(1), C8–Fe1–C1 121.9(2), C8–Fe1–N1 88.3(2), C1–Fe1–N1 107.7(2), C8–Fe1–Br1 108.3(1), C1–Fe1–Br1 120.9(2), N1–Fe1–Br1 102.6(1); for 6: Fe1–C15 2.087(2), Fe1–C8 2.090(2), Fe1–C1 2.115(2), Fe1–N1 2.210(2), C15–Fe1–C8 126.81(9), C15–Fe1–C1 114.40(8), C8–Fe1–C1 112.16(9), C15–Fe1–N1 86.05(7), C8–Fe1–N1 95.42(8), C1–Fe1–N1 116.38(8). | ||
Since complex 6 has Fe-bound two benzyl groups as the built-in base, adding π-acceptor ligands to the metal centre could not only force one of the benzyl ligands closer to the to-be-deprotonated CH2 group of the HL ligand, but also increase the acidity of the CH2 group, facilitating the deprotonation. To test this possibility, a toluene solution of 6 was exposed to a CO atmosphere at ambient temperature, which resulted in an immediate colour change from yellow-brown to red-purple and the clean formation of dibenzyl ketone and a diamagnetic compound, 7 (Scheme 3); longer reaction time caused further colour change to yellow, accompanied by the conversion of 7 to 8. At ambient temperature, the 13C NMR spectrum of 7 in CD2Cl2 displays only one carbonyl peak at 222.53 ppm, while the IR spectrum shows three CO stretches at 1953, 1868 and 1832 cm−1, respectively. The singlet at 4.98 ppm with an integration of two protons in its 1H NMR spectrum is diagnostic for the methylene group of the HL ligand. Compared to those of 7, the carbonyl signal of 8 is upfield shifted to 215.94 ppm in its 13C NMR spectrum, while the proton signal of the CH2 group of the HL ligand is downfield shifted to 5.78 ppm in its 1H NMR spectrum. The IR spectrum of 8 in THF has four CO stretches at 2035, 1946, 1924, and 1906 cm−1, respectively.
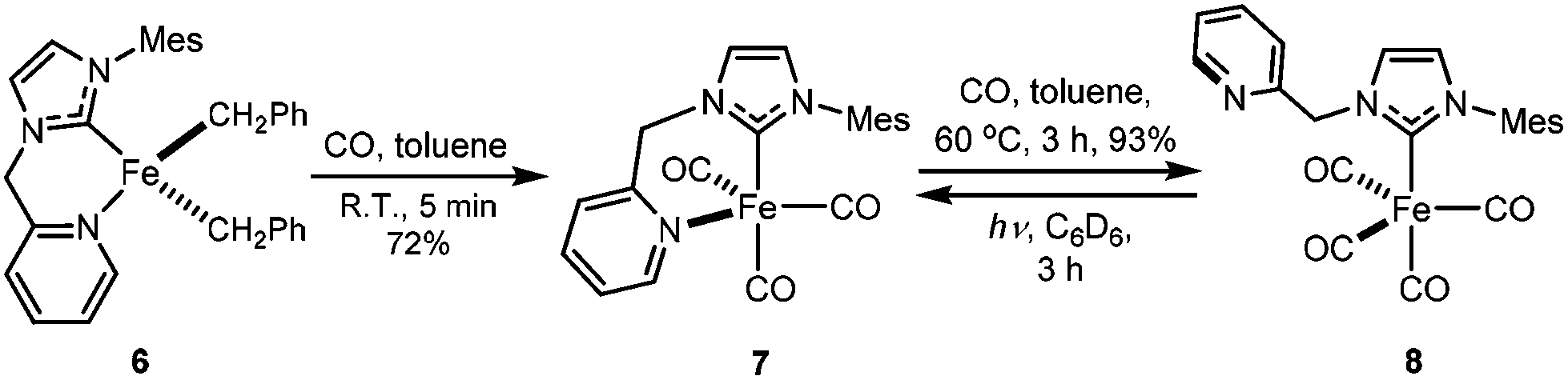 | ||
| Scheme 3 Reaction of 6 with CO. | ||
X-ray crystallography reveals that compounds 7 and 8 have formula of [Fe(CO)3(HL)] and [Fe(CO)4(HL-κC)], respectively (Fig. 5). The Fe(0) centre in 7 adopts a distorted trigonal bipyramidal coordination geometry with the carbene donor of HL and a carbonyl ligand occupying the two axial positions and the pyridine nitrogen donor and the other two carbonyl ligands occupying the three equatorial positions. The angle between two axial bonds (C4–Fe1–C2) is 176.7(1)° and the sum of the three bond angles within equatorial plane is 359.9(1)°. Compound 8 is a ligand substitution product from 7, where the pyridine moiety is replaced by an additional carbonyl ligand, leaving the pyridine donor of HL dangling. The coordination sphere of the Fe(0) centre of 8 is similar to those of [Fe(CO)4(NHC)]53–56 and [Fe(CBA)(CO)4] (CBA = cyclic bent allene)57 compounds where the NHC occupies one of the axial positions. Instead of triggering the desired deprotonation, the addition of CO to compound 6 appears to induce sequential 1,1-insertion and reductive elimination to form 7/8 and dibenzyl ketone. Similar processes have been reported in the literature.57–60 Deng and co-workers have reported the isolation of (Mes)2CO from the reaction of [Fe(Me2IPr)(Mes)2] (Me2IPr = 1,3-diisopropyl-4,5-dimethylimidazolin-2-ylidene) with CO where the presumable iron containing product [Fe(CO)4(Me2IPr)] could not be isolated.58 Unlike the presumable [Fe(CO)4(Me2IPr)]58 and the analogue methylene bridged biscarbene iron tricarbonyl compound [Fe(CO)3(DippC)2CH2] ((DippC)2CH2 = bis(N-Dipp-imidazole-2-ylidene methane)),50 compound 7 is readily isolable and stable under a dinitrogen atmosphere.
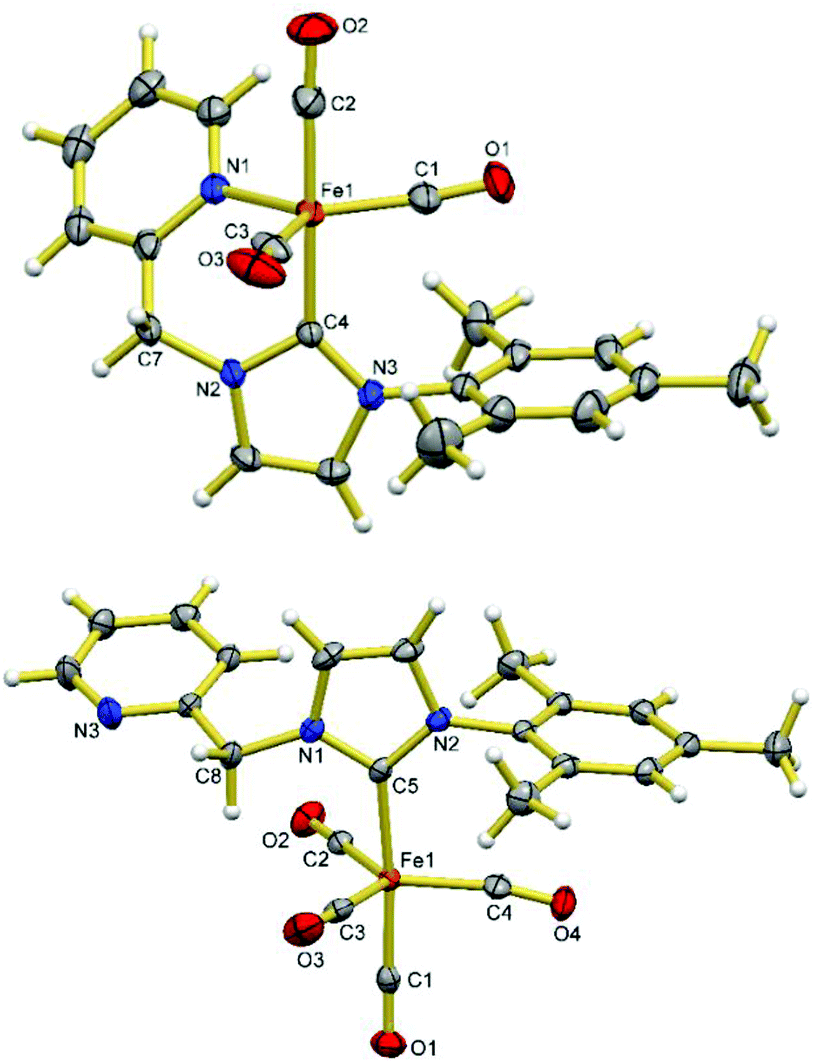 | ||
| Fig. 5 Molecular structure of complexes 7 (top) and 8 (bottom). Ellipsoids are shown at 50% probability level. Selected bond lengths (Å) and angles (°) for 7: Fe1–C1 1.751(2), Fe1–C3 1.753(3), Fe1–C2 1.766(2), Fe1–C4 1.957(2), Fe1–N1 2.074(2), O1–C1 1.167(3), O2–C2 1.157(3), O3–C3 1.157(3), C1–Fe1–C3 115.1(1), C1–Fe1–C2 85.1(1), C3–Fe1–C2 91.8(1), C1–Fe1–C4 95.72(9), C3–Fe1–C4 90.8(1), C2–Fe1–C4 176.7(1), C1–Fe1–N1 129.2(1), C3–Fe1–N1 115.6(1), C2–Fe1–N1 91.82(9), C4–Fe1–N1 85.21(8); for 8: Fe1–C3 1.784(2), Fe1–C1 1.785(2), Fe1–C2 1.794(2), Fe1–C4 1.822(2), Fe1–C5 1.998(2), O1–C1 1.147(2), O2–C2 1.150(2), O3–C3 1.154(2), O4–C4 1.144(2), C3–Fe1–C1 90.61(9), C3–Fe1–C2 123.85(9), C1–Fe1–C2 91.81(9), C3–Fe1–C4 118.58(9), C1–Fe1–C4 86.72(8), C2–Fe1–C4 117.57(9), C3–Fe1–C5 85.02(8), C1–Fe1–C5 174.72(8), C2–Fe1–C5 88.18(8), C4–Fe1–C5 97.97(8). | ||
Compound 8 can also be prepared by heating a toluene solution of 7 at 60 °C under CO atmosphere for three hours (Scheme 3). Compound 8 is light-sensitive pale yellow crystals. The pale yellow solution of 8 slowly turns red under a dinitrogen atmosphere and ambient light at room temperature, forming 7; this reaction is slow even at elevated temperatures. However, the irradiation of a solution of 8 in C6D6 under a UV lamp results in the complete conversion to 7 within three hours at ambient temperature. Similar interconversion between [Fe(CO)1–2(picolyl-NHC)X] (X = I, BF4) compounds have been reported, but the resulting compounds with dangling pyridine moiety could not be cleanly isolated.23
Although we were unable to trigger the deprotonation of HL ligand in 6 by the benzyl ligands on the Fe centre, compound 6 is still intriguing due to its relevance to the proposed key intermediate [Fe(NHC)(R)2] (where R is a hydrocarbyl group) in NHC-Fe-catalyzed Kumada-type coupling reactions.42,61,62 It has been proposed that [Fe(NHC)(R)2] undergoes halogen atom abstraction from the electrophile R′–X to generate a radical (R′˙) and the Fe(III) species [Fe(NHC)(R)2X]; the dissociated R′˙ can then rebound to generate the cross-coupling product R′–R (major) or self-couple to form R′–R′ (minor).42 As shown in Scheme 4, the reaction of 6 with 1 eq. of p-methoxybenzyl bromide results in the formation of the cross-coupling product II in 50% yield and the homo-coupling products I and III in 35% and 28% yield, respectively. The homo-coupling product I might originate from reductive elimination, while the homo-coupling product III might arise from the self-coupling of p-methoxybenzyl radicals.
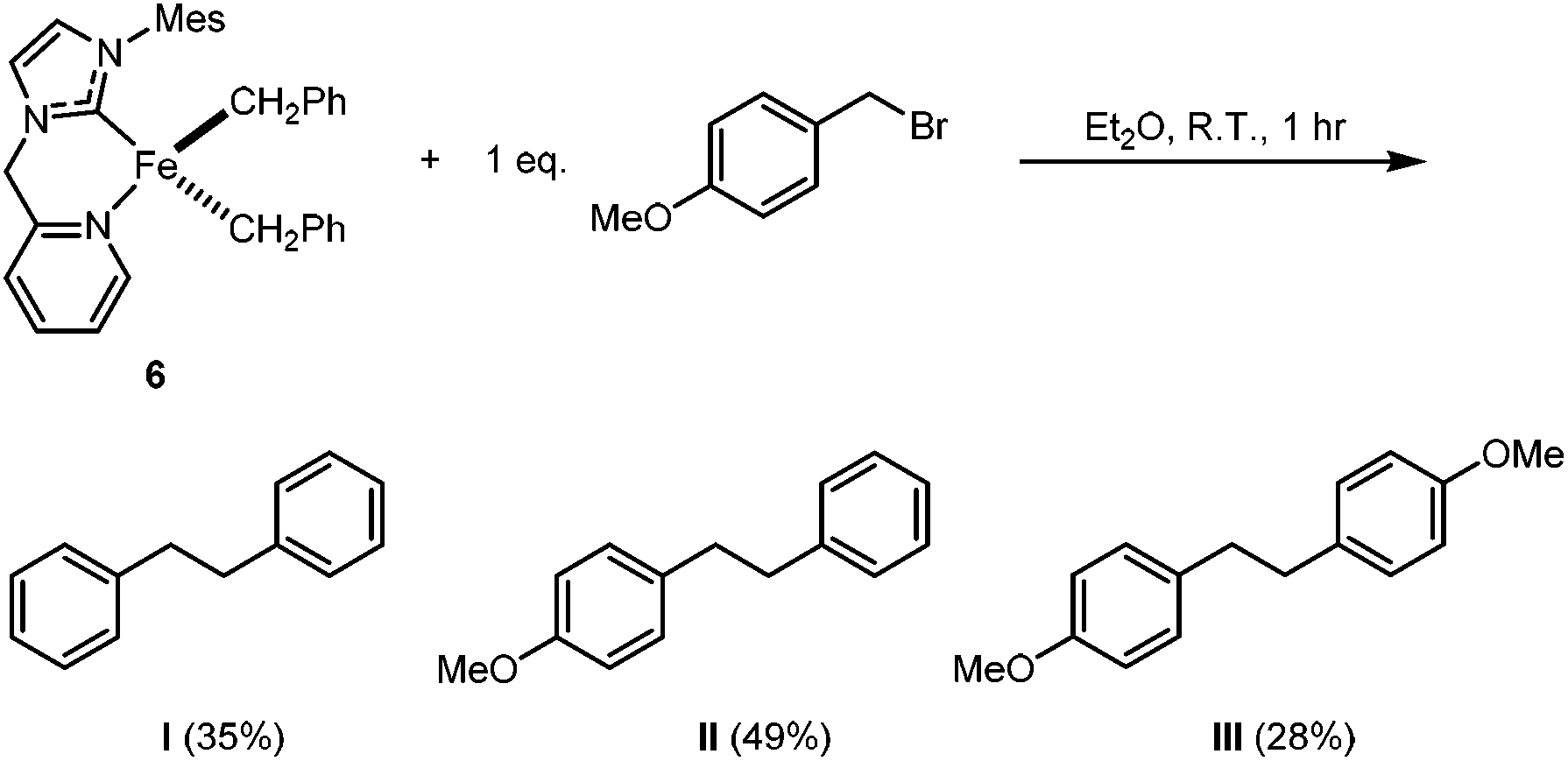 | ||
| Scheme 4 Reaction of 6 with an equimolar amount of p-methoxybenzyl bromide. The percentage yields given in parentheses are the averages of two parallel runs (see ESI†). | ||
Conclusions
In summary, we have synthesized 1–3 from the reactions of H2LBr and appropriate amounts of Fe(HMDS)2 and the in situ prepared [Fe(HMDS)Br], respectively. While the direct deprotonation of the charge neutral HL ligand in 2 and 3 with 1 eq. of base has been unsuccessful, the deprotonation of 1 with 2 eq. of LiHMDS has successfully furnished 4 where the iron(II) centre is coordinated to two L− ligands featuring the dearomatized pyridine moieties and exocyclic C–C double bonds. Alternatively, 4 can be synthesized from 1 eq. of the in situ generated L− ligand and 0.5 eq. of FeBr2. Treating 4 with 2 eq. of PPh3·HBr affords protonated compound 1. Attempts to deprotonate compound 3 using benzyl Grignard as the base cleanly afford the transmetalation products 5 and 6. The dibenzyl complex 6 reacts rapidly with CO affording 7 and dibenzyl ketone. The prolonged exposure of 7 to an atmosphere of CO causes the substitution of the pyridine N-donor by a CO ligand, leading to the formation of 8 where the pyridine moiety of the HL ligand is dangling. Compound 7 can be reformed by irradiating 8 under UV light. Compound 6 is reactive toward p-methoxybenzyl bromide affording homo- and cross-coupling products. The deprotonation of complexes 2 and 3 and their bulkier analogues and the reactivity of 4 toward various small molecules are under investigation in our laboratory.Experimental section
General remarks
All reactions were performed in a nitrogen glovebox or using the standard Schlenk techniques. Glassware was dried in a 180 °C oven overnight. Solvents were dried by a Grubbs-type solvent purification system manufactured by Innovative Technology (dichloromethane, diethyl ether, hexanes, pentane and toluene) or dried by refluxing and distilling over sodium benzophenone ketyl (benzene, dioxane, DME, THF, and C6D6) under dinitrogen. CDCl3 was distilled over P2O5 and CD2Cl2 was distilled over CaH2, degassed through three consecutive freeze–pump–thaw cycles. All solvents were stored over 3 Å molecular sieves prior to use. All NMR spectra were recorded on an Agilent DD2 600 MHz spectrometer at 25 °C. Chemical shifts are referenced to the solvent signals. IR spectra were recorded on a Bruker ALPHA spectrometer equipped with an ATR sampling unit. Elemental analyses were carried out by ANALEST at the University of Toronto. GC/MS analyses were performed on an Agilent 7890A GC System and 5975C inert XL MSD with Triple-Axis Detector. Anhydrous FeBr2 and benzylmagnesium bromide solution were purchased from Strem Chemicals Inc. and Acros Organics, respectively. Lithium hexamethyldisilazide and n-butyl lithium were purchased from Sigma Aldrich. Grade 2.5 carbon monoxide was purchased from Linde. H2LBr,63 Fe(HMDS)2,64 PPh3·HBr65 and p-methoxybenzyl bromide66 were prepared according to literature procedures.[FeBr(HL)2]Br, 1
To the mixture of H2LBr (179.1 mg, 0.50 mmol) and Fe(HMDS)2 (103.7 mg, 0.28 mmol) was added 3 mL of THF. The resulting yellow-orange suspension was stirred overnight at room temperature. The precipitate was collected by vacuum filtration, washed with THF (3 × 2 mL) and pentane (3 × 2 mL), dried under vacuum to afford 1 as a yellow solid (180.6 mg, 94%). X-ray quality crystals were obtained from the reaction of H2LBr and Fe(HMDS)2 in THF with no stirring at room temperature. No NMR spectrum was recorded due to the low solubility and instability in various NMR solvents. Anal. Calcd for C36H38N6FeBr2: C, 56.13; H, 4.97; N, 10.91. Found: C, 55.86; H, 4.92; N, 10.45.[FeBr(HL)(HMDS)], 2
To the mixture of H2LBr (179.2 mg, 0.50 mmol) and Fe(HMDS)2 (188.5 mg, 0.50 mmol) was added 3 mL of THF. The reaction mixture turned red-orange immediately and slowly became homogenous. The mixture was stirred for 3 h at room temperature before all volatiles were removed under vacuum. The oily residue was dissolved in THF and filtered through Celite. The filtrate was concentrated to ∼1 mL, top-layered with 5 mL of pentane, and stored in a −35 °C freezer overnight. The resulting precipitate was collected by filtration, washed with pentane (3 × 2 mL) and dried under vacuum to afford 2 as a yellow solid (252.7 mg, 88%). X-ray quality crystals were obtained by cooling its toluene/hexanes solution at −35 °C. 1H NMR (C6D6): δ 94.53 (1H), 47.96 (1H), 44.96 (1H), 38.70 (1H), 23.48 (1H), 13.92 (18H), 8.31 (1H), 2.13 (2H), 0.47 (3H), −3.28 (2H). Not all proton signals were observed. Anal. Calcd for C24H37N4Si2FeBr: C, 50.26; H, 6.50; N, 9.77. Found: C, 50.32; H, 6.35; N, 9.56.[FeBr2(HL)], 3
To the mixture of anhydrous FeBr2 (107.5 mg, 0.50 mmol) and Fe(HMDS)2 (188.5 mg, 0.50 mmol) was added 3 mL of THF. The mixture was stirred at room temperature for 2 h whereupon all solids were dissolved. The resulting solution was added to H2LBr (358.3 mg, 1.00 mmol) and stirred at room temperature overnight. After the removal of all volatiles, the solid residue was extracted into dichloromethane and filtered through Celite. The filtrate was concentrated to dryness under reduced pressure to give 3 as a yellow solid (445.7 mg, 90%). This compound can be further purified if necessary by top-layering a dichloromethane solution with pentane at −35 °C overnight. X-ray quality crystals were obtained by vapour diffusion of hexanes into a DME solution at −35 °C. 1H NMR (CDCl3): δ 98.88 (1H), 58.12 (1H), 50.09 (1H), 33.03 (1H), 18.00 (3H), 4.42 (2H), 2.56 (4H), −7.16 (6H). Anal. Calcd for C18H19N3FeBr2: C, 43.85; H, 3.88; N, 8.52. Found: C, 43.58; H, 3.91; N, 8.33.[FeL2], 4
To a suspension of H2LBr (114.6 mg, 0.32 mmol, in 3 mL of THF) was slowly added nBuLi (1.6 M in hexanes, 0.40 mL, 0.64 mmol) at −80 °C. The resulting mixture was allowed to warm to room temperature slowly and further stirred for 5 h. The resulting dark orange solution was cooled to −80 °C and slowly added into a pre-cooled (−80 °C) slurry of anhydrous FeBr2 (41.3 mg, 0.19 mmol, in 2 mL of THF). The mixture was allowed to warm to room temperature slowly, stirred overnight, and concentrated under reduced pressure. The dark oily residue was extracted into toluene, filtered through Celite and concentrated under reduced pressure. The residue was then extracted into benzene and filtered through Celite. The filtrate was lyophilized to afford 4 as a brown solid (65.2 mg, 67%). Crystals suitable for X-ray crystallography were obtained by cooling a concentrated THF solution to −35 °C. 1H NMR (C6D6): δ 83.64 (2H), 63.73 (2H), 21.94 (4H), 4.05 (2H), 1.36 (8H), 0.39 (2H), −7.13 (6H), −15.23 (6H), −54.79 (2H), −121.59 (2H). Satisfactory elemental analysis result could not be obtained due to the extreme sensitivity toward air and moisture. Best result: Anal. Calcd for C36H36N6Fe: C, 71.05; H, 5.96; N, 13.81. Found: C, 68.76; H, 5.84; N, 13.44. μeff (Evans67,68) = 5.1μB.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume), filtered through Celite and concentrated to dryness to afford 4 as a brown solid (12.1 mg, 80%).
:1 by volume), filtered through Celite and concentrated to dryness to afford 4 as a brown solid (12.1 mg, 80%).
[FeBnBr(HL)], 5
To a −35 °C suspension of 3 (49.3 mg, 0.1 mmol, in 2 mL of THF and 0.5 mL of dioxane) was slowly added benzylmagnesium chloride solution (90.6 mg, 20 wt% in THF, 0.12 mmol). The resulting red-orange mixture was allowed to warm to room temperature slowly, stirred overnight, and then concentrated to dryness under vacuum. The solid residue was extracted into THF and filtered through Celite. The filtrate was concentrated to ∼2 mL, top-layered with 3 mL of pentane, and stored in a −35 °C freezer overnight to give 5 as an orange crystalline solid. The supernatant was decanted off and the solid was washed with cold diethyl ether (3 × 1 mL) and pentane (3 × 1 mL), dried under vacuum (37.6 mg, 75%). Crystals suitable for X-ray crystallography were obtained by concentrating a THF–toluene (ca. 1/1) solution under vacuum. 1H NMR (C6D6): δ 57.55 (1H), 42.17 (1H), 35.00 (1H), 34.60 (2H), 22.02 (1H), 4.00 (2H), 1.78 (4H), −1.12 (3H), −5.86 (1H), −14.26 (2H), −73.52 (1H), −88.20 (1H). Not all proton signals were observed. Due to the thermal instability of this compound, satisfactory elemental analysis results could not be obtained. Best results: Anal. Calcd for C25H26N3FeBr: C, 59.55; H, 5.20; N, 8.33. Found: C, 58.48; H, 4.97; N, 7.99.[FeBn2(HL)] (6)
To a −35 °C suspension of 3 (246.5 mg, 0.5 mmol, in 5 mL of diethyl ether and 1 mL of dioxane) was slowly added benzylmagnesium chloride solution (905.3 mg, 20 wt% in THF, 1.2 mmol). The resulting dark red-brown mixture was allowed to warm to room temperature slowly, stirred overnight, and concentrated to dryness under reduced pressure. The solid residue was extracted into diethyl ether and filtered through Celite. The filtrate was concentrated to ∼1 mL and top-layered with 2 mL of pentane, and cooled to −35 °C to yield dark red crystals of 6. The supernatant was decanted off and the crystals were washed with cold pentane (3 × 2 mL), and dried under vacuum (204.3 mg, 79%). Crystals suitable for X-ray crystallography were obtained by cooling a concentrated diethyl ether solution at −35 °C. 1H NMR (C6D6): δ 72.31 (1H), 58.27 (1H), 41.52 (1H), 38.73 (1H), 35.91 (4H), 28.24 (1H), 22.58 (2H), 5.12 (2H), 2.76 (3H), −9.00 (6H), −12.98 (1H), −65.25 (3H), −84.41 (2H). Not all proton signals were observed. Due to the thermal instability of this compound, satisfactory elemental analysis results could not be obtained. Best results: Anal. Calcd for C32H33N3Fe: C, 74.56; H, 6.45; N, 8.15. Found: C, 73.63; H, 6.47; N, 8.01.[Fe(CO)3(HL)] (7)
A yellow-brown solution of 6 (51.5 mg, 0.1 mmol, in 5 mL of toluene) was subjected to a freeze–pump–thaw cycle before 1 atm of CO was introduced into the flask, giving a red-purple solution. The solution was allowed to stand for 5 min at room temperature, before another freeze–pump–thaw cycle was done to remove CO from the headspace. After the flask was refilled with dinitrogen gas, the solution was filtered, concentrated to ∼1 mL, top-layered with 5 mL of pentane, and cooled to −35 °C to yield X-ray quality red crystals of 7. The supernatant was decanted off and the crystals were washed with pentane (3 × 5 mL), and dried under vacuum (30.2 mg, 72%). 1H NMR (CD2Cl2) δ 9.05 (ddd, 1H, J = 5.6, 1.5, 0.8 Hz, py-H), 7.66 (td, 1H, J = 7.6, 1.6 Hz, py-H), 7.44 (dt, 1H, J = 7.7, 1.1 Hz, py-H), 7.36 (d, 1H, J = 1.9 Hz, im-H), 7.05 (ddd, 1H, J = 7.3, 5.5, 1.5 Hz, py-H), 6.97 (s, 2H, Mes-o-H), 6.88 (d, 1H, J = 1.9 Hz, im-H), 4.98 (s, 2H, CH2), 2.34 (s, 3H, p-CH3), 1.98 (s, 6H, o-CH3). 13C NMR (CD2Cl2): δ 222.53 (CO), 197.08 (im-C2), 156.92 (py-C), 156.29 (py-C), 139.29 (Mes-C), 136.95 (Mes-C), 136.61 (Mes-C), 136.22 (py-C), 129.10 (Mes-C), 124.17 (py-C), 123.12 (im-C), 122.77 (py-C), 121.83 (im-C), 55.72 (CH2), 21.25 (o-CH3), 17.95 (p-CH3). IR (neat solid):![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (CO) 1953, 1868, 1832 cm−1. Anal. Calcd for C21H19N3O3Fe: C, 60.45; H, 4.59; N, 10.07. Found: C, 60.09; H, 4.51; N, 9.97.
(CO) 1953, 1868, 1832 cm−1. Anal. Calcd for C21H19N3O3Fe: C, 60.45; H, 4.59; N, 10.07. Found: C, 60.09; H, 4.51; N, 9.97.
The supernatant and pentane washes were combined and concentrated under vacuum. The residue was subjected to flash column chromatography with ethyl acetate as eluent. Dibenzyl ketone was isolated as a light yellow oil (16.0 mg, 76%). 1H NMR (CDCl3): δ 7.33–7.30 (m, 4H), 7.28–7.25 (m, 2H), 7.16–7.14 (m, 4H), 3.72 (s, 4H, CH2). 13C NMR (CDCl3): δ 205.77, 134.12, 129.64, 128.86, 127.20, 49.25. MS: m/z calcd for C15H14O+ 210, found 210. NMR data are consistent with literature.69
[Fe(CO)4(HL-κC)] (8)
A red-purple solution of 7 (20.9 mg, 0.05 mmol, in 5 mL of toluene) was subjected to a freeze–pump–thaw cycle, before 1 atm of CO was introduced into the Schlenk bomb. The bomb was then sealed and heated at 60 °C for 3 h. The resulting yellow solution was filtered and concentrated to dryness under vacuum to afford pale yellow crystals of 8 (20.7 mg, 93%). Crystals suitable for X-ray were grown from a concentrated diethyl ether solution at −35 °C. 1H NMR (CD2Cl2): δ 8.62 (ddd, 1H, J = 4.8, 1.9, 1.0 Hz, py-H), 7.72 (td, 1H, J = 7.7, 1.9 Hz, py-H), 7.38 (d, 1H, J = 2.0 Hz, im-H), 7.28 (ddd, 1H, J = 7.7, 4.8, 1.0 Hz, py-H), 7.22 (dt, 1H, J = 7.9, 0.8 Hz, py-H), 7.01 (s, 2H, Mes-o-H), 6.99 (d, 1H, J = 2.0 Hz, im-H), 5.78 (s, 2H, CH2), 2.35 (s, 3H, p-CH3), 2.00 (s, 6H, o-CH3). 13C NMR (CD2Cl2): δ 215.94 (CO), 185.05 (im-C2), 156.24 (py-H), 150.04 (py-C), 140.35 (Mes-C), 137.42 (Mes-C), 137.31 (py-C), 136.71 (Mes-C), 129.43 (Mes-C), 124.44 (im-C), 124.19 (im-C), 123.32 (py-C), 122.73 (py-C), 57.21 (CH2), 21.27 (o-CH3), 17.89 (p-CH3). IR (in THF): (CO) 2037, 1962, 1933, 1916 cm−1. Anal. Calcd for C22H19N3O4Fe: C, 59.35; H, 4.30; N, 9.44. Found: C, 59.50; H, 4.13; N, 9.31.
Reaction of 6 with p-methoxybenzyl bromide
To a solution of 6 (17.7 mg, 34.4 μmol, in 4 mL of diethyl ether), was added p-methoxybenzyl bromide (5 μL, 34.3 μmol) at room temperature. The solution was stirred for 1 h prior and quenched with 1 drop of 1 N HCl and filtered through a pad of anhydrous MgSO4. The filtrate was subjected to GC/MS analysis using hexamethylbenzene as the internal standard (see ESI†).Acknowledgements
We thank NSERC of Canada for funding. We also acknowledge the Canadian Foundation for Innovation Project #19119, and the Ontario Research Fund for funding the CSICOMP NMR lab at the University of Toronto enabling the purchase of several new spectrometers.References
- R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, John Wiley & Sons, Hoboken, New Jersey, 5th edn, 2009 Search PubMed.
- V. T. Annibale and D. Song, RSC Adv., 2013, 3, 11432 RSC.
- V. T. Annibale, D. A. Dalessandro and D. Song, J. Am. Chem. Soc., 2013, 135, 16175 CrossRef CAS PubMed.
- V. T. Annibale and D. Song, Chem. Commun., 2012, 48, 5416 RSC.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236 CrossRef CAS PubMed.
- R. Noyori, M. Yamakawa and S. Hashiguchi, J. Org. Chem., 2001, 66, 7931 CrossRef CAS PubMed.
- B. Zhao, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2013, 52, 4744 CrossRef CAS PubMed.
- C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588 CrossRef CAS PubMed.
- C. Gunanathan and D. Milstein, Top. Organomet. Chem., 2011, 37, 55 CrossRef CAS.
- Y. Sun, C. Koehler, R. Tan, V. T. Annibale and D. Song, Chem. Commun., 2011, 47, 8349 RSC.
- C. del Pozo, M. Iglesias and F. Sánchez, Organometallics, 2011, 30, 2180 CrossRef CAS.
- E. Fogler, E. Balaraman, Y. Ben-David, G. Leitus, L. J. W. Shimon and D. Milstein, Organometallics, 2011, 30, 3826 CrossRef CAS.
- D. Kim, L. Le, M. J. Drance, K. H. Jensen, K. Bogdanovski, T. N. Cervarich, M. G. Barnard, N. J. Pudalov, S. M. M. Knapp and A. R. Chianese, Organometallics, 2016, 35, 982 CrossRef CAS.
- R. P. Yu, J. M. Darmon, J. M. Hoyt, G. W. Margulieux, Z. R. Turner and P. J. Chirik, ACS Catal., 2012, 2, 1760 CrossRef CAS PubMed.
- R. P. Yu, J. M. Darmon, C. Milsmann, G. W. Margulieux, S. C. E. Stieber, S. DeBeer and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 13168 CrossRef CAS PubMed.
- A. Murso and D. Stalke, Dalton Trans., 2004, 2563 RSC.
- I. Objartel, H. Ott and D. Stalke, Z. Anorg. Allg. Chem., 2008, 634, 2373 CrossRef CAS.
- S. Y. de Boer, Y. Gloaguen, J. N. H. Reek, M. Lutzb and J. I. van der Vlugt, Dalton Trans., 2012, 41, 11276 RSC.
- G. I. McGrew, P. A. Khatri, W. E. Geiger, R. A. Kemp and R. Waterman, Chem. Commun., 2015, 51, 15804 RSC.
- Y. Cheng and L. Sun, Chin. J. Org. Chem., 2012, 32, 511 CrossRef CAS.
- S. Winston, N. D. Stylianides, A. A. D. Tulloch, J. A. Wright and A. A. Danopoulos, Polyhedron, 2004, 23, 2813 CrossRef CAS.
- X. Wang, S. Liu and G.-X. Jin, Organometallics, 2004, 23, 6002 CrossRef CAS.
- L. Mercs, G. Labat, A. Neels, A. Ehlers and M. Albrecht, Organometallics, 2006, 25, 5648 CrossRef CAS.
- K. Riener, S. Haslinger, A. Raba, M. P. Hogerl, M. Cokoja, W. W. Herrmann and F. E. Kuhn, Chem. Rev., 2014, 144, 5215 CrossRef and references therein.
- D. Bezier, J.-B. Sortais and C. Darcel, Adv. Synth. Catal., 2013, 355, 19 CrossRef CAS.
- M. J. Ingleson and R. A. Layfield, Chem. Commun., 2012, 48, 3579 RSC.
- Y. Wang, H. Sun, X. Tao, Q. Shen and Y. Zhang, Chin. Sci. Bull., 2007, 52, 3193 CrossRef CAS.
- M. Z. Chen, H. M. Sun, W. F. Li, Z. G. Wang, Q. Shen and Y. Zhang, J. Organomet. Chem., 2006, 691, 2489 CrossRef CAS.
- J. Thagfi and G. G. Lavoie, Organometallics, 2012, 31, 7531 Search PubMed.
- J. Thagfi and G. G. Lavoie, Organometallics, 2012, 31, 2463 CrossRef.
- O. Kaufhold, F. E. Hahn, T. Pape and A. Hepp, J. Organomet. Chem., 2008, 693, 3435 CrossRef CAS.
- Y. Zhang, B. Liu, H. Wu and W. Chen, Chin. Sci. Bull., 2012, 57, 2368 CrossRef CAS.
- B. Liu, Y. Zhang, D. Xu and W. Chen, Chem. Commun., 2011, 47, 2883 RSC.
- A. A. Danopoulos, N. Tsoureas, J. A. Wright and M. E. Light, Organometallics, 2004, 23, 166 CrossRef CAS.
- J. J. Dunsford, D. J. Evans, T. Pugh, S. N. Shah, N. F. Chilton and M. J. Ingleson, Organometallics, 2016, 35, 1098 CrossRef CAS.
- Z. Ouyang, Y. Meng, J. Cheng, J. Xiao, S. Gao and L. Deng, Organometallics, 2016, 35, 1361 CrossRef CAS.
- K. Pal, O. B. Hemming, B. M. Day, T. Pugh, D. J. Evans and R. A. Layfield, Angew. Chem., Int. Ed., 2016, 55, 1690 CrossRef CAS PubMed.
- Z. Ouyang, J. Du, L. Wang, J. L. Kneebone, M. L. Neidig and L. Deng, Inorg. Chem., 2015, 54, 8808 CrossRef CAS PubMed.
- X. Wang, J. Zhang, L. Wang and L. Deng, Organometallics, 2015, 34, 2775 CrossRef CAS.
- Y. Liu, L. Luo, J. Xiao, L. Wang, Y. Song, J. Qu, Y. Luo and L. Deng, Inorg. Chem., 2015, 54, 4752 CrossRef CAS PubMed.
- Y. Liu, L. Wang and L. Deng, Organometallics, 2015, 34, 4401 CrossRef CAS.
- J. A. Przyojski, K. P. Veggeberg, H. D. Arman and Z. J. Tonzetich, ACS Catal., 2015, 5, 5938 CrossRef CAS.
- K. L. Fillman, J. A. Przyojski, M. H. Al-Afyouni, Z. J. Tonzetich and M. L. Neidig, Chem. Sci., 2015, 6, 1178 RSC.
- P. P. Samuel, K. C. Mondal, N. Amin Sk, H. W. Roesky, E. Carl, R. Neufeld, D. Stalke, S. Demeshko, F. Meyer, L. Ungur, L. F. Chibotaru, J. Christian, V. Ramachandran, J. van Tol and N. S. Dalal, J. Am. Chem. Soc., 2014, 136, 11964 CrossRef CAS PubMed.
- W. T. Lee, I. R. Jeon, S. Xu, D. A. Dickie and J. M. Smith, Organometallics, 2014, 33, 5654 CrossRef CAS.
- A. A. Danopoulos, P. Braunstein, N. Stylianides and M. Wesolek, Organometallics, 2011, 30, 6514 CrossRef CAS.
- H. Z. Kaplan, B. Li and J. A. Byers, Organometallics, 2012, 31, 7343 CrossRef CAS.
- S. Zlatogorsky, C. A. Muryn, F. Tuna, D. J. Evans and M. J. Ingleson, Organometallics, 2011, 30, 4974 CrossRef CAS.
- S. Meyer, C. M. Orben, S. Demeshko, S. Dechert and F. Meyer, Organometallics, 2011, 30, 6692 CrossRef CAS.
- S. Zlatogorsky and M. J. Ingleson, Dalton Trans., 2012, 2685 RSC.
- L. Xiang, J. Xiao and L. Deng, Organometallics, 2011, 30, 2018 CrossRef CAS.
- A. A. Danopoulos, P. Braunstein, M. Wesolek, K. Y. Monakhov, P. Rabu and V. Robert, Organometallics, 2012, 31, 4102 CrossRef CAS.
- K. Öfele and C. Kreiter, Chem. Ber., 1972, 105, 529 CrossRef.
- S. Worratz, L. Postigo and B. Royo, Organometallics, 2013, 32, 893 CrossRef.
- H. Li, L. C. M. Castro, J. Zheng, T. Roisnel, V. Dorcet, J. B. Sortais and C. Darcel, Angew. Chem., Int. Ed., 2013, 52, 8045 CrossRef CAS PubMed.
- T. Hashimoto, R. Hoshino, T. Hatanaka, Y. Ohki and K. Tatsumi, Organometallics, 2014, 33, 921 CrossRef CAS.
- C. Pranckevicius and D. W. Stephan, Organometallics, 2013, 32, 2693 CrossRef CAS.
- L. Xiang, J. Xiao and L. Deng, Organometallics, 2011, 30, 2018 CrossRef CAS.
- S. C. Bart, E. J. Hawrelak, A. K. Schmisseur, E. Lobkovsky and P. J. Chirik, Organometallics, 2003, 23, 237 CrossRef.
- A. R. Hermes and G. S. Girolami, Organometallics, 1988, 7, 394 CrossRef CAS.
- C. Cassani, G. Bergonzini and C. J. Wallentin, ACS Catal., 2016, 6, 1640 CrossRef CAS.
- R. B. Bedford, Acc. Chem. Res., 2015, 48, 1485 CrossRef CAS PubMed.
- A. A. D. Tulloch, A. A. Danopoulos, S. Winston, S. Kleinhenz and G. Eastham, J. Chem. Soc., Dalton Trans., 2000, 4499 RSC.
- R. A. Andersen, K. Faegri, J. C. Green, A. Haaland, M. F. Lappert, W.-P. Leung and K. Rypdal, Inorg. Chem., 1988, 27, 1782 CrossRef CAS.
- N. Aoyagi, Y. Furusho and T. Endo, Tetrahedron Lett., 2013, 54, 7031 CrossRef CAS.
- A. K. Ghosh and D. D. Anderson, Org. Lett., 2012, 14, 4730 CrossRef CAS PubMed.
- D. F. Evans, J. Chem. Soc., 1959, 2003 RSC.
- G. J. P. Britovsek, V. C. Gibson, S. K. Spitzmesser, K. P. Tellmann, A. J. P. White and D. J. Williams, J. Chem. Soc., Dalton Trans., 2002, 1159 RSC.
- D. R. Romer, Synthesis, 2011, 2721 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra and X-ray crystallographic experimental details. CCDC 1493390–1493397. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt02792j |
| This journal is © The Royal Society of Chemistry 2016 |