DOI:
10.1039/C6DT02709A
(Paper)
Dalton Trans., 2016,
45, 14265-14276
Copper(I) clusters with bulky dithiocarboxylate, thiolate, and selenolate ligands†
Received
8th July 2016
, Accepted 28th July 2016
First published on 18th August 2016
Abstract
The coordination chemistry of copper has interest due to its use in biological systems as well as for photochemical and medicinal properties. We report the coordination chemistry of copper(I) complexes using terphenyl-based dithiocarboxylate, thiolate, and selenolate ligands. The number of metal ions of the resulting complexes can be tuned by varying the steric properties of the terphenyl ligands, changing the starting material, as well as adding PEt3. In addition, the steric crowding of the terphenyl ligand leads to varying reactivity. For example, while the reaction of carbon disulfide with [Cu(2,6-(Ph)2C6H3)]2 results in an insertion into the copper–carbon bond, no reaction occurs with [Cu(2,4,6-(Mes)2C6H3)], Mes = 2,4,6-Me3C6H2, or [Et3PCu(2,4,6-(Mes)2C6H2)2C6H3]. The synthesis and characterization of new copper(I) complexes using NMR and IR spectroscopy, as well as X-ray crystallography is described.
Introduction
Copper has attracted attention due to its photochemical,1 catalytic,2,3 and anti-microbial properties,4 as well as cytotoxicity.5 In nature, copper mainly occurs in the +1 and +2 oxidation states, and it is well known that copper(I) prefers to coordinate to soft donor ligands. Thus, the coordination chemistry of copper in nature is mainly dominated by amino acids such as histidine, cysteine, and methionine or ligands containing soft donor atoms such as sulfur.6 A variety of coordination numbers exist, ranging from two to five, and are predominantly linear, trigonal planar or tetrahedral geometries are observed. Copper-transporting proteins, such as eukaryote Atx1, are flexible in nature and feature copper in coordination numbers of two or three,7 while CusA a protein that is essential in bacteria for copper resistance,8 was found to contain one Cu(I) in a trigonal planar geometry, Fig. 1.
 |
| | Fig. 1 Active site of CusA is shown. | |
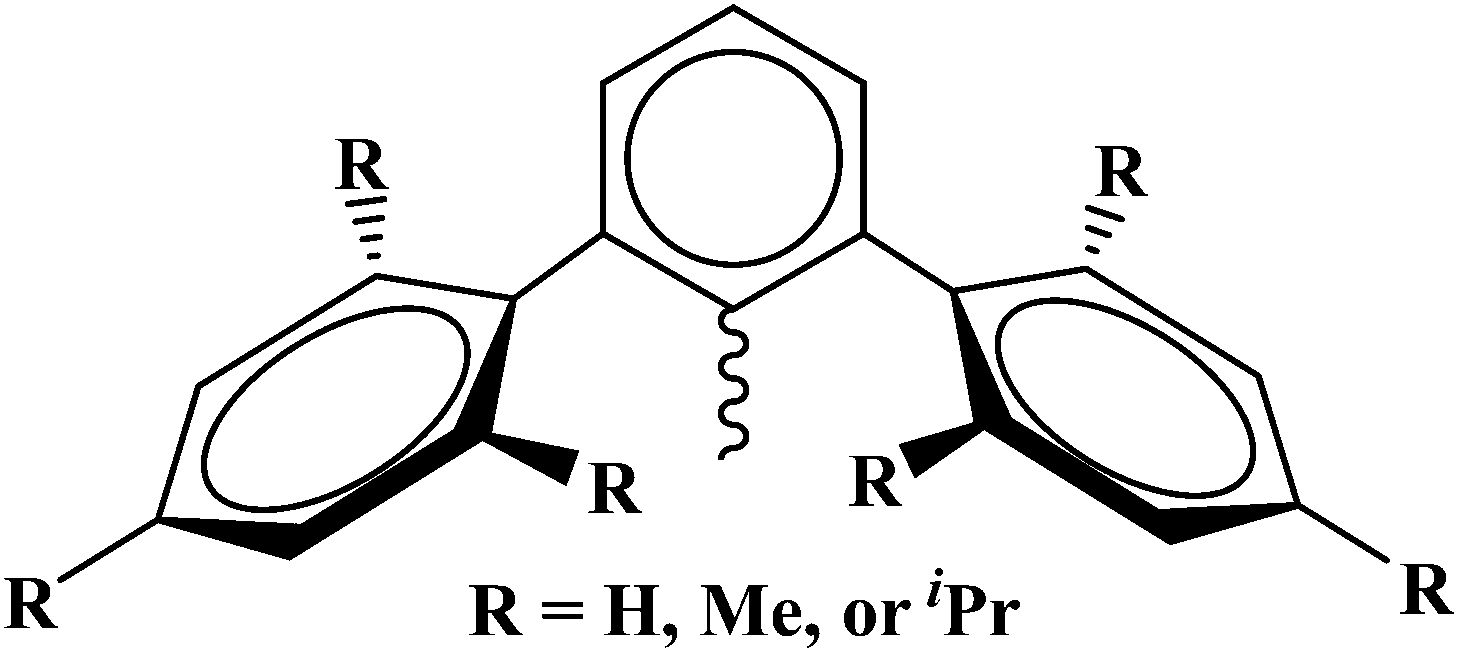
The enforcement of low coordination numbers can be achieved by having a ligand with large steric demand. The terphenyl-based ligand frameworks, Fig. 2, can provide such steric properties. Several derivatives of terphenyl ligands with nearly every main group element have been synthesized.9 Our interest was to use dithiocarboxylate, thiolate, and selenolate derivatives to produce copper(I) complexes to model the active site of copper-containing enzymes with linear, trigonal planar, or tetrahedral coordination geometries. We have previously used amidinate ligands to examine the dinuclear copper CuA site10 and used the insertion of carbon disulfide into copper–nitrogen bonds to produce polymetallic clusters and control the number of metal ions using the steric properties of the amidinate ligand. Herein, we report the synthesis and characterization of a series of copper complexes with coordination numbers of two, three, and four with linear, trigonal planar and tetrahedral geometries using sulfur and selenium-based terphenyl ligands.
 |
| | Fig. 2 General drawing of the terphenyl ligands used in this manuscript. | |
Results and discussion
Dithiocarboxylate complexes
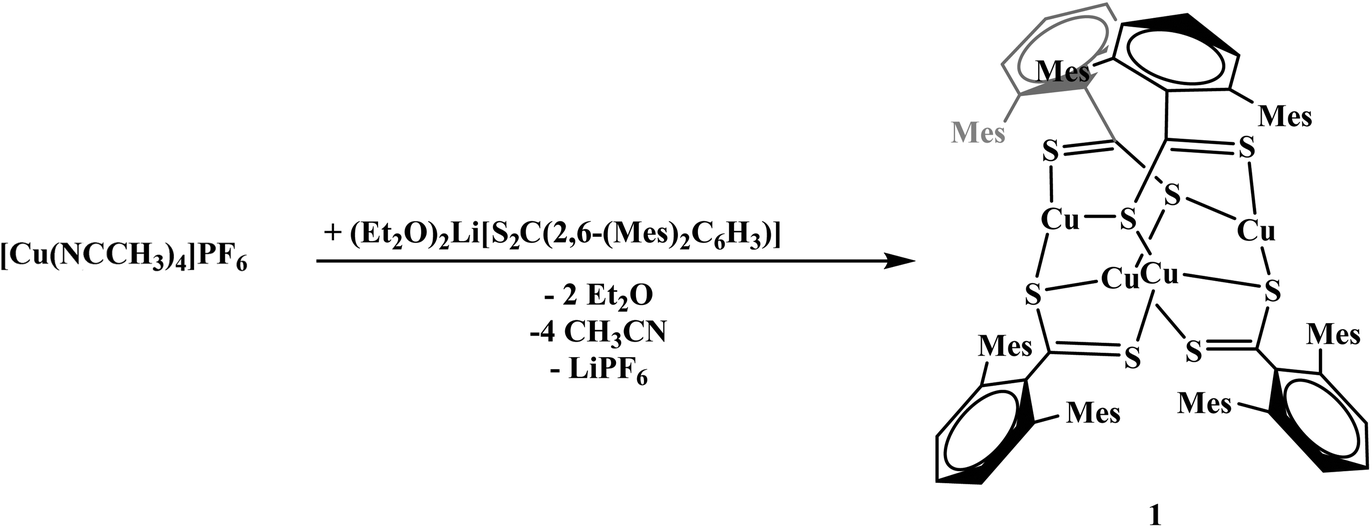
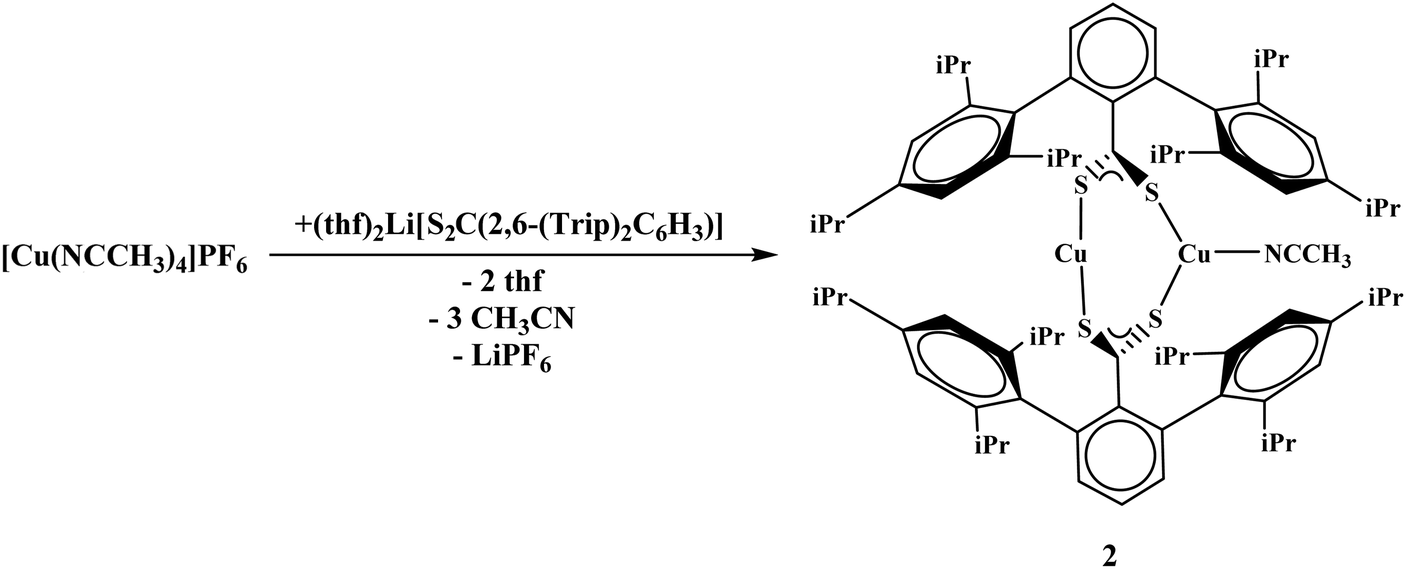
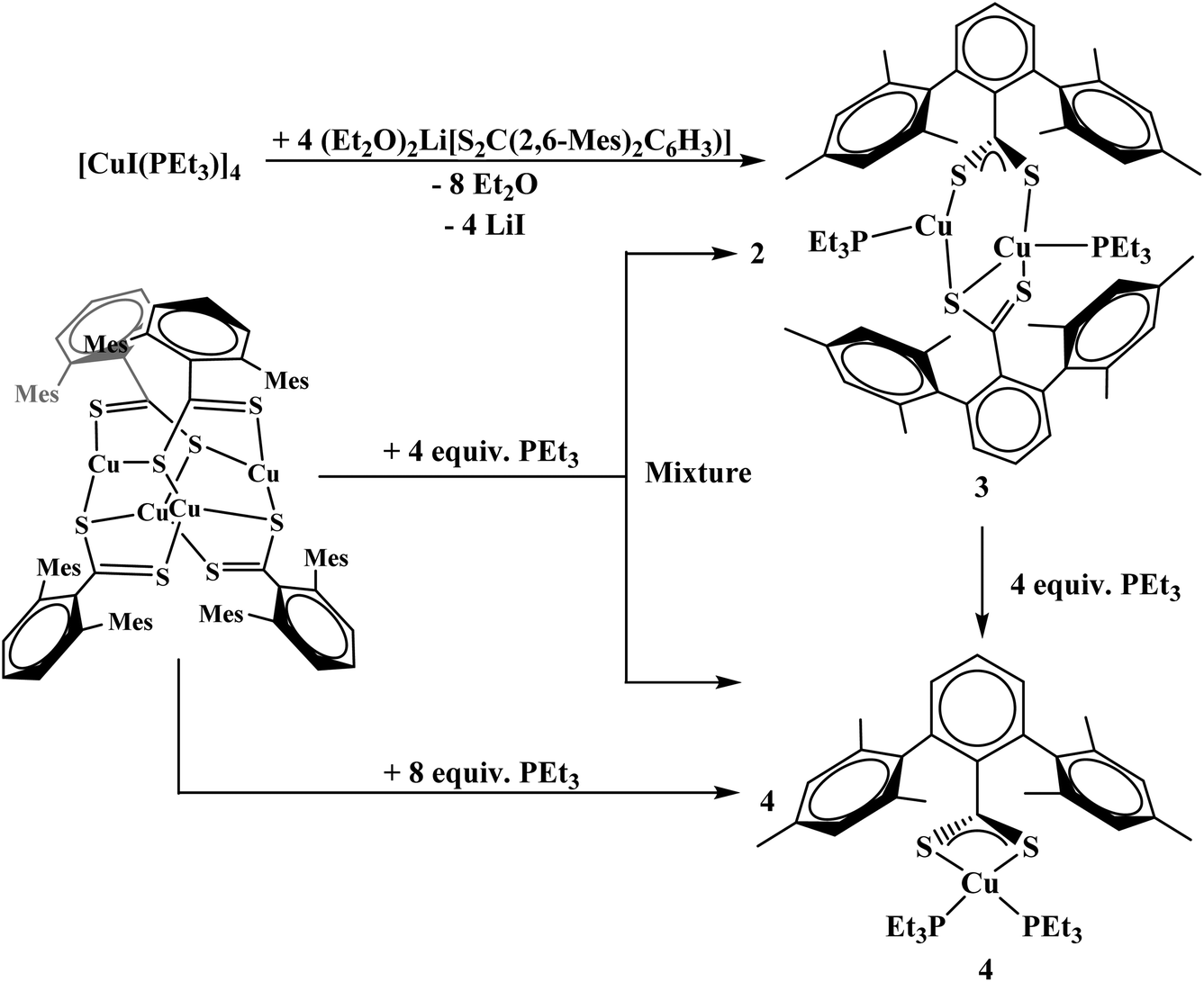
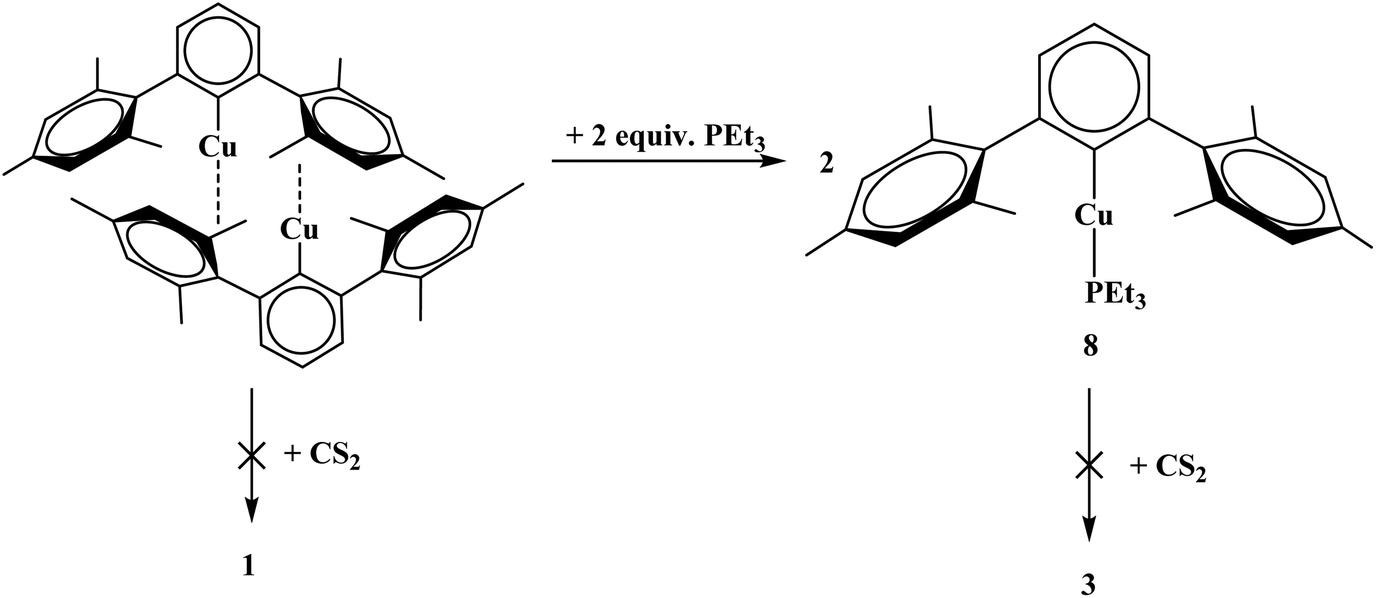
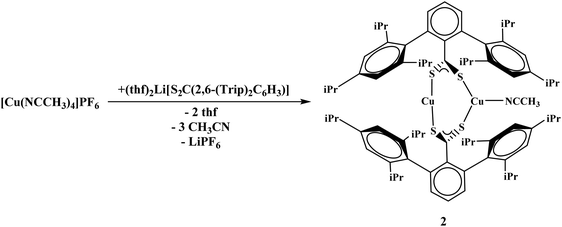
To generate the dithiocarboxylate copper complexes, (Et2O)2Li[S2C(2,6-(Mes)2C6H3)] (Mes = 2,4,6-Me3C6H2)11 and (thf)2Li[S2C(2,6-(Trip)2C6H3)] (Trip = 2,4,6-iPr3C6H2)11 were reacted with [Cu(NCCH3)4]PF6. The reaction of (Et2O)2Li[S2C(2,6-(Mes)2C6H3)] with [Cu(NCCH3)4]PF6 results in an immediate color change from orange to dark brown. After workup, the product, Cu4[S2C(2,6-Mes)2C6H3]4, 1, could be isolated as a brown solid in moderate yield, eqn (1). The reaction of (thf)2Li[S2C(2,6-(Trip)2C6H3)] with [Cu(NCCH3)4]PF6 also resulted in an immediate color change to dark brown, eqn (2), and Cu2[S2C(2,6-Trip)2C6H3]2(NCCH3), 2, was recovered as a brown solid. Unfortunately, 2 could not be obtained in acceptable purity for further characterization and we note that if a non-coordinating solvent such as toluene is used for recrystallization, a different product is obtained, but we have been unable to identify the resulting compound.| | 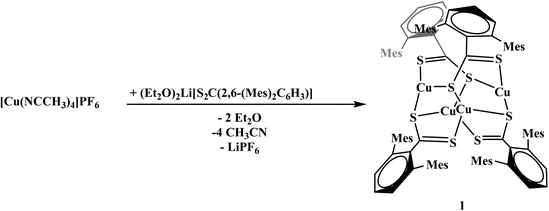 | (1) |
| |  | (2) |
The reaction of complex 1 with four equivalents of triethylphosphine (PEt3) yields two products: Cu2[S2C(2,6-Mes)2C6H3]2(PEt3)2, 3, and Cu[S2C(2,6-Mes)2C6H3](PEt3)2, 4, Scheme 1. During the course of the reaction of 1 with PEt3, a color change from brown to orange could be observed. Complex 4 could also be synthesized from adding PEt3 to 3. To generate complex 3 with an acceptable purity, PEt3 was preinstalled on the copper center by using [CuI(PEt3)]4, Scheme 1.
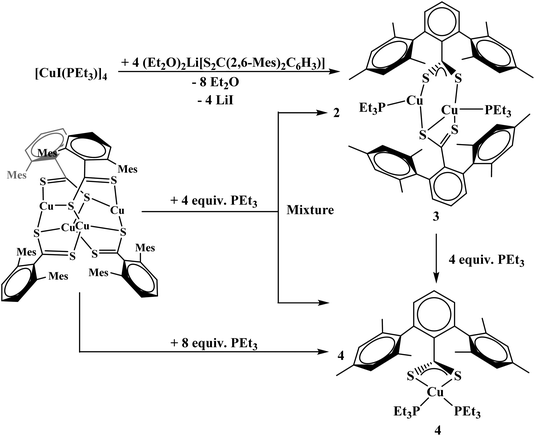 |
| | Scheme 1 Synthesis of 3 and 4. | |
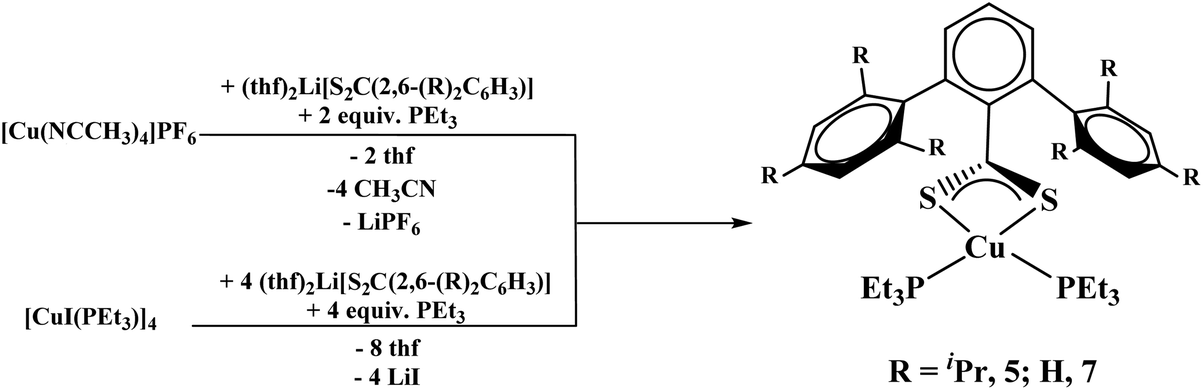
The reaction of (thf)2Li[S2C(2,6-(Trip)2C6H3)] with [Cu(NCCH3)4]PF6 in the presence of two equivalents of PEt3 or [CuI(PEt3)]4 in the presence of one equivalent of PEt3 both yielded Cu[S2C(2,6-Trip)2C6H3](PEt3)2, 5, in good yield, eqn (3).
| | 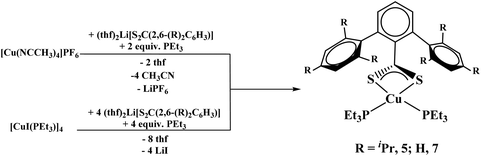 | (3) |
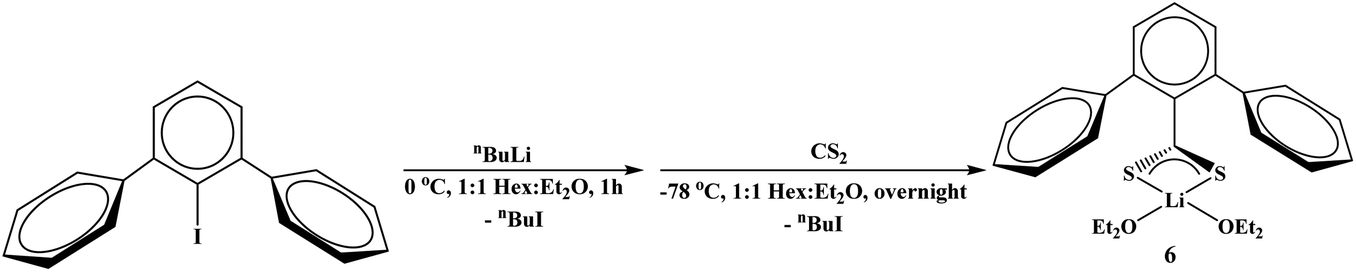
Finally, we wanted to examine an unsubstituted terphenyl ligand, which has not been examined as thoroughly as the mesityl and triisopropyl-derivatives. The reaction of 2,6-diphenyliodobenzene with nBuLi followed by addition of CS2 at −78 °C produced the dithiocarboxylate complex, (Et2O)2Li[S2C(2,6-Ph)2C6H3], 6, eqn (4). When 6 is reacted with [Cu(NCCH3)4]PF6 in the presence of two equivalents of PEt3, or [CuI(PEt3)]4 in the presence of one equivalent of PEt3, both yielded 8 in good yield (>75%), eqn (3).
| | 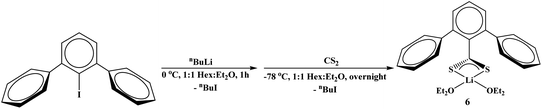 | (4) |
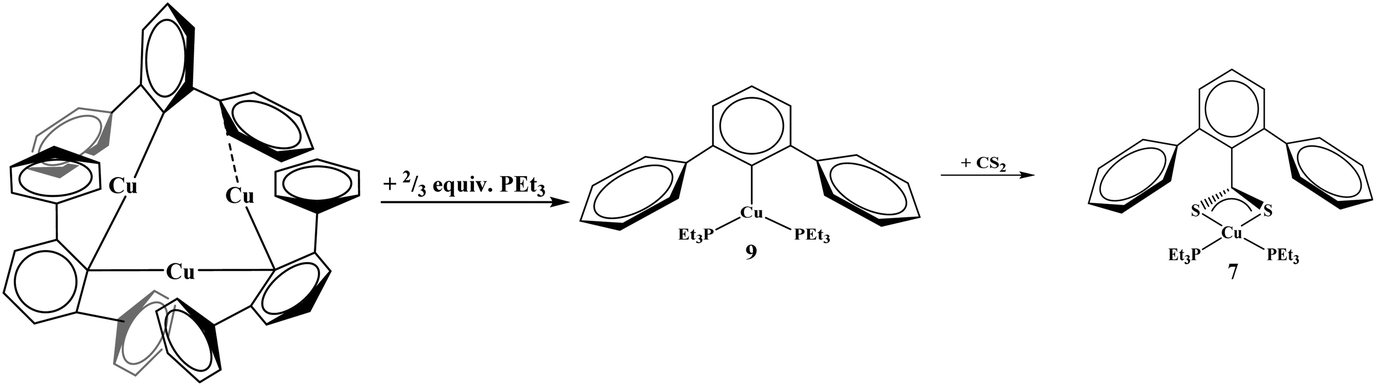
Given that complexes 1–5 and 7 could be made from metathesis with dithiocarboxylate salts, we investigated the possibility of producing the identical compounds by insertion of CS2 into the copper–aryl bond of the corresponding terphenyl derivative. Complex 8 is made from Cu2[(2,6-(Mes)2C6H3)]2 with PEt3. However, the reaction of Cu2[(2,6-(Mes)2C6H3)]2 or Cu[2,6-(Mes)2C6H3](PEt3), 8, with CS2 to generate copper–sulfur containing complexes did not yield the desirable product, Scheme 2. In fact, no reaction was obtained with either 8 or Cu2[(2,6-(Mes)2C6H3)]2 when reacted with CS2. The steric bulk of the terphenyl ligand is believed to be the main contributor to the inertness of the complex. When two equivalents of PEt3 are added to Cu3[(2,6-(Ph)2C6H3)]3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 (Ph = phenyl) in hexanes, Cu[2,6-(Ph)2C6H3](PEt3)2, 9, is produced, eqn (5). However, when 9 is reacted with CS2, 7 was isolated, eqn (5). This demonstrates the potential for enhanced reactivity with the unsubstituted terphenyl ligand over their more sterically encumbering counterparts.
12 (Ph = phenyl) in hexanes, Cu[2,6-(Ph)2C6H3](PEt3)2, 9, is produced, eqn (5). However, when 9 is reacted with CS2, 7 was isolated, eqn (5). This demonstrates the potential for enhanced reactivity with the unsubstituted terphenyl ligand over their more sterically encumbering counterparts.
| | 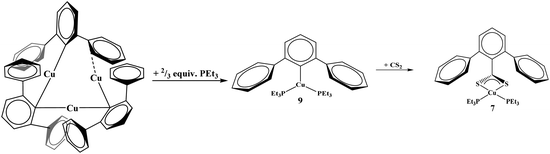 | (5) |
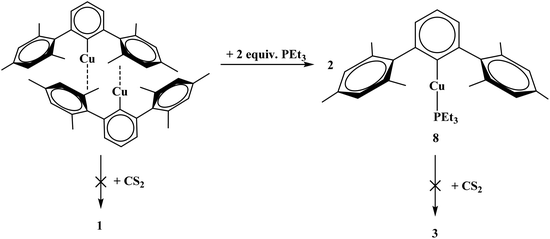 |
| | Scheme 2 Synthesis of 8 from Cu2[(2,6-(Mes)2C6H3)]2 and reactivity toward carbon disulfide (CS2) towards both complexes. | |
Chalcogenide complexes
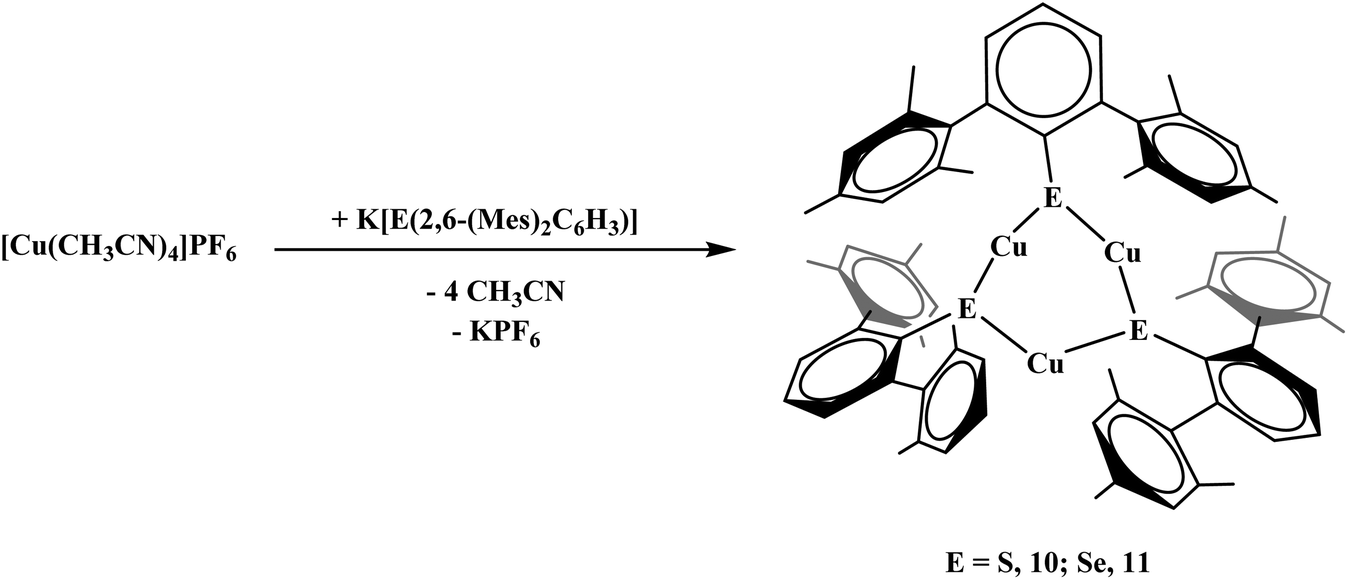
To examine differences in dithiocarboxylate versus chalcogenide ligands, the thiolate and selenolate salts were synthesized. The reaction of [Cu(NCCH3)4]PF6 and K[S(2,6-(Mes)2C6H3)] proceeded with no color change and afforded Cu3[S(2,6-(Mes)2C6H3)]3, 10, eqn (6). The product could be isolated in nearly quantitative yield (>98%). The reaction of [Cu(NCCH3)4]PF6 and K[Se(2,6-(Mes)2C6H3)] was carried out in a similar fashion to 10 yielding Cu3[Se(2,6-(Mes)2C6H3)]3, 11, eqn (6).| | 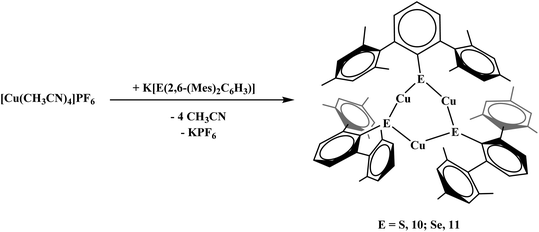 | (6) |
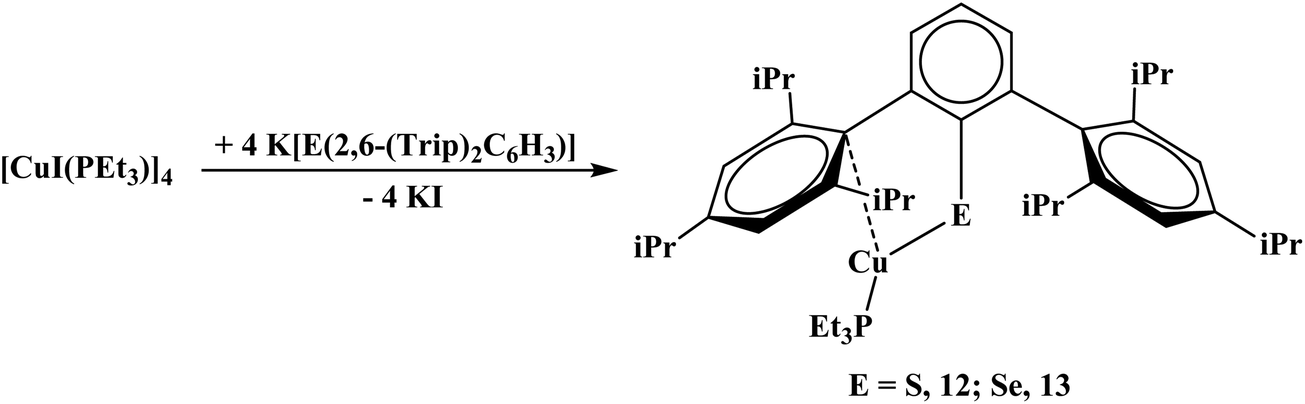
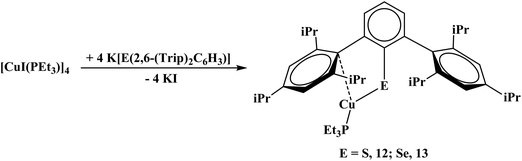
With the triisopropyl derivatives, the reaction of K[E(2,6-(Trip)2C6H3)] with [CuI(PEt3)]4 yielded mononuclear complexes, Cu[E(2,6-(Trip)2C6H3)](PEt3), E = S, 12; Se, 13, eqn (7). Without PEt3, no product could be readily isolated. Interestingly, all of the chalcogenide complexes, 10–13, are colorless while the dithiocarboxylate complexes are brown or orange. Reactivity with the unsubstituted terphenyl chalcogenide salts was attempted but yielded multiple products based on the 1H NMR spectrum which could not be readily discriminated.
| |  | (7) |
All complexes were characterized using 1H and 13C NMR spectroscopy and, when appropriate, 31P and 77Se NMR spectroscopy, Table 1. The 77Se NMR resonance for Cu3[Se(2,6-(Mes)2C6H3)]3, 11, was observed at −96.2 ppm while 13 showed a resonance at 171.6 ppm. This indicates that the donation from the selenium atom is far greater in 13 with a more downfield chemical shift.
Table 1 Select 1H, 13C, and 31P NMR resonances (in ppm) for complexes 1, 3–13
| Compound |
13C NMR CS2 |
1H NMR |
13P NMR |
|
o-Mes |
p-Mes |
o-CH(CH3)2 |
p-CH(CH3)2 |
o-CH(CH3)2 |
p-CH(CH3)2 |
| Cu4[S2C(2,6-(Mes)2C6H3)]4, 1 |
229.2 |
2.21 |
2.16 |
|
|
|
|
|
| Cu2[S2C(2,6-(Mes)2C6H3)]2(PEt3)2, 3 |
246.8 |
2.43 |
2.29 |
|
|
|
|
−8.97 |
| Cu[S2C(2,6-(Mes)2C6H3)](PEt3)2, 4 |
251.7 |
2.54 |
2.26 |
|
|
|
|
−7.01 |
| Cu[S2C(2,6-(Trip)2C6H3)](PEt3)2, 5 |
251.4 |
|
|
1.21, 1.59 |
1.34 |
3.44 |
2.92 |
−7.61 |
| (Et2O)2Li[S2C(2,6-(Ph)2C6H3)], 6 |
251.4 |
|
|
|
|
|
|
|
| Cu[S2C(2,6-(Ph)2C6H3)](PEt3)2, 7 |
251.9 |
|
|
|
|
|
|
−7.73 |
| Cu[(2,6-(Mes)2C6H3)](PEt3), 8 |
|
2.40 |
2.29 |
|
|
|
|
−2.87 |
| Cu[(2,6-(Ph)2C6H3)](PEt3)2, 9 |
|
|
|
|
|
|
|
−14.49 |
| Cu3[S(2,6-(Mes)2C6H3)]3, 10 |
|
2.06 |
2.24 |
|
|
|
|
|
| Cu3[Se(2,6-(Mes)2C6H3)]3, 11 |
|
2.07 |
2.26 |
|
|
|
|
|
| Cu[S(2,6-(Trip)2C6H3)](PEt3), 12 |
|
|
|
1.27, 1.60 |
1.23 |
3.21 |
2.80 |
−9.01 |
| Cu[Se(2,6-(Trip)2C6H3)](PEt3), 13 |
|
|
|
1.25, 1.62 |
1.24 |
3.18 |
2.81 |
−9.51 |
X-ray crystallographic analysis
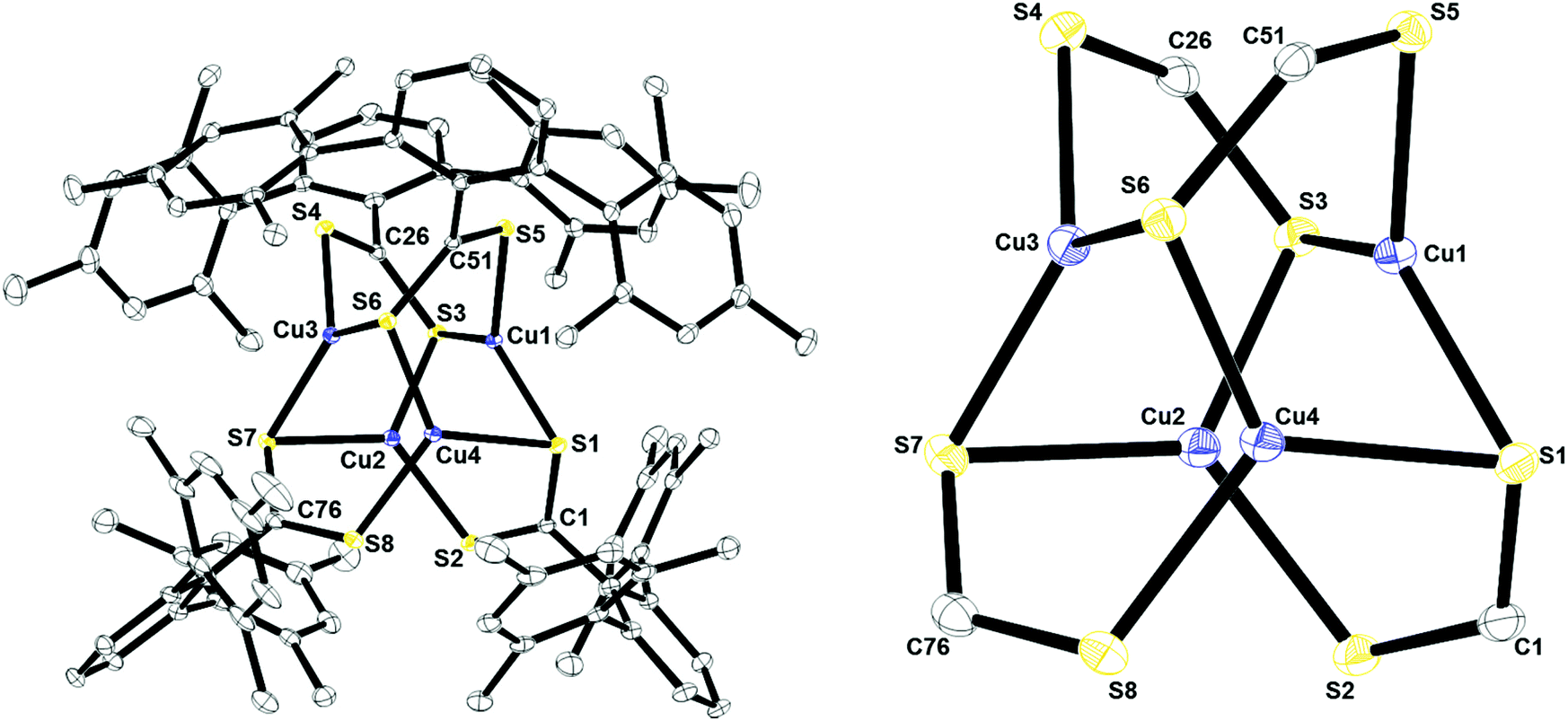
X-ray structural analysis was used to determine the molecular structure of Cu4[S2C(2,6-Mes2C6H3)]4, 1, shown in Fig. 3. All four ligands in the complex are μ3:η3 featuring one bridging sulfur and one sulfur which is directly coordinated to one copper atom. The geometry around each copper center could be described as pseudo-trigonal. This is similar to the geometry and coordination environment for copper in the active site of CusA (see Fig. 1). Complex 1 also features two different types of C–S bonds. C(1)–S(1), C(26)–S(3), C(51)–S(6), and C(76)–S(7) all have relatively similar bond distances, 1.7313(18), 1.7404(18), 1.73820(4), and 1.739(2) Å, respectively. The average bond distance for all four bonds is 1.737 Å which resembles that of a C–S single bond.13 It should also be noted that all four sulfurs are bound to two copper atoms each in a bridging manner. On the other hand, C(1)–S(2), C(26)–S(4), C(51)–S(5), and C(76)–S(8) all have slightly shorter bond lengths of 1.6618(19), 1.6580(19), 1.6575(19), 1.660(2) Å, respectively, which resembles a C–S double bond.13 This indicates that the anionic charge on the ligand is localized. Interestingly, 1 contains a striking feature in its quaternary carbon resonance on the thiocarboxylate group (229.2 ppm), which was significantly shifted downfield. This localization may be the reason for the observed shift since this is the only compound where localization is observed. The Cu–Cu interactions of 2.6–2.86 Å as well as the Cu–S bond lengths between 2.25–2.30 Å are similar to those observed in other tetranuclear dithiocarboxylate copper(I) clusters.14,15
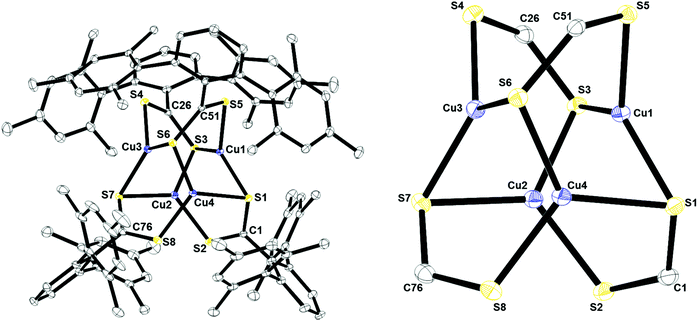 |
| | Fig. 3 Thermal ellipsoid plot of 1 shown at the 50% probability level. All hydrogen atoms and residual solvent molecule are omitted for clarity. The Cu4S8 core is shown on the right. Selected bond lengths: C1–S1: 1.7313(18) Å; C1–S2: 1.6618(19) Å; C26–S3: 1.7404(18) Å; C26–S4: 1.6580(19); C51–S5: 1.6575(19) Å; C51–S6: 1.73820(4) Å; C76–S7: 1.739(2) Å; C76–S8: 1.660(2) Å; Cu1–S5: 2.2500(5) Å; Cu1–S1: 2.2906(5) Å; Cu1–S3: 2.2907(5) Å; Cu1–Cu2: 2.6029(4) Å; Cu1–Cu4: 2.6113(4) Å; Cu1–Cu3: 2.8575(4) Å. | |
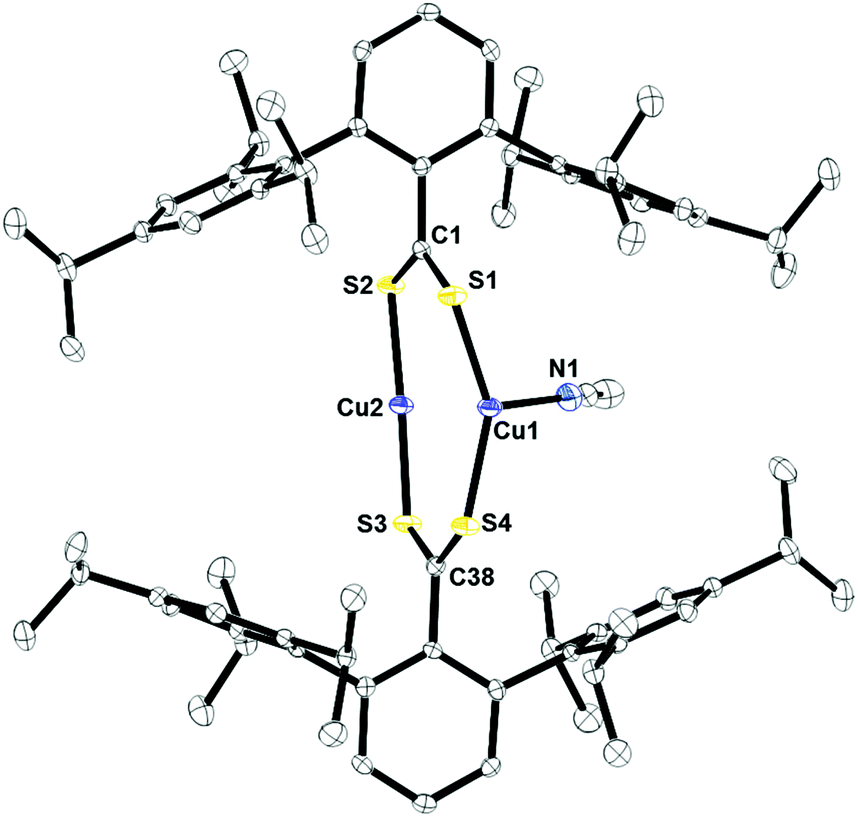
Fig. 4 shows the crystal structure of 2 obtained from acetonitrile by slow evaporation. The structure features a dinuclear species with a μ2:η2 binding mode of the two dithiocarboxylate ligands. One copper, Cu2, is two-coordinate and has a near linear S2–Cu2–S3 bond angle of 169.01(3)°. The other copper ion, Cu1, is three-coordinate in a pseudo-trigonal geometry. The Cu1–S1 and Cu1–S4 bond distances of 2.2277(5) and 2.2205(5) Å, respectively, are slightly longer than Cu2–S2 and Cu2–S3 of 2.1543(5) and 2.1579(5) Å. This is due to the electron density siphoned by the nitrogen atom of acetonitrile bound to Cu1, which leads to longer copper–sulfur bond distances. The Cu–N(acetonitrile) bond distance is 2.004(2) Å.
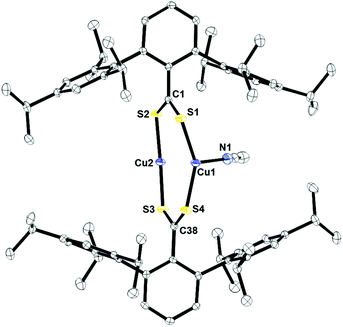 |
| | Fig. 4 Thermal ellipsoid plot of 2 shown at the 50% probability plot. All hydrogen atoms are omitted for clarity. Selected bond lengths: C1–S1: 1.6753(19) Å; C1–S2: 1.6912(19) Å; C38–S3: 1.6958(19) Å; C38–S4: 1.6677(19) Å; Cu1–S1: 2.2277(5) Å; Cu1–S4: 2.2205(5) Å; Cu2–S2: 2.1543(5) Å; Cu2–S3: 2.1579(5) Å; Cu1–N1: 2.004(2) Å. | |
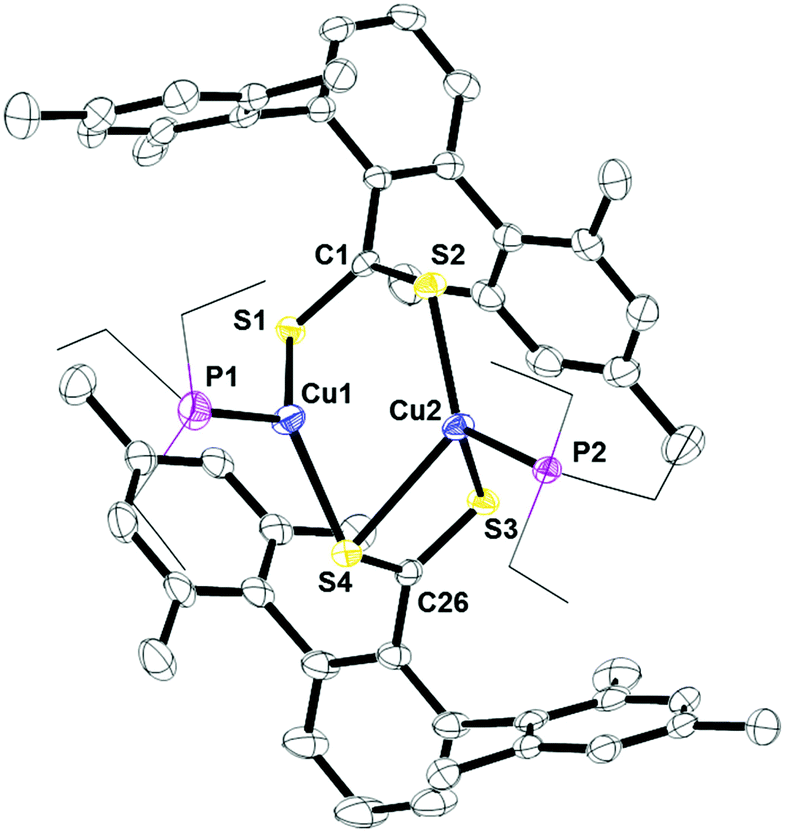
The structures of 3 and 4 were determined by X-ray crystallography and revealed dinuclear and mononuclear complexes, respectively. In the solid-state, 3 forms a bimetallic core where the two ligands feature two different binding modes μ2:η3 and μ2:η2, Fig. 5. Within the μ2:η2 unit, the two C–S bonds are essentially the same, indicating that the anionic charge of the ligand is delocalized along the S–C–S unit. The μ2:η3 unit features two different C–S bonds. The longer C–S bond in C26–S4 resembles that of a single bond while C26–S3 is close to a double bond. Similar to 1, the sulfur with the longer C–S bond distance is acting as a bridge between two copper centers. Since only one 13C NMR resonance is found for the quaternary carbon, the two S–C–S units are the same on the NMR time scale. This type of bonding has been observed in dinuclear copper(I) complexes previously.16
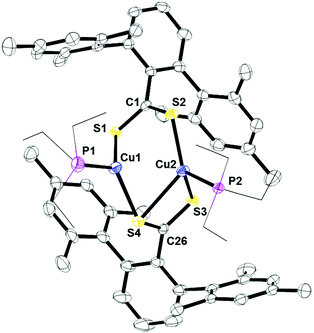 |
| | Fig. 5 Thermal ellipsoid plot of 3 shown at the 50% probability level. All hydrogen atoms, carbon atoms on PEt3, and residue solvent molecule are omitted for clarity. Selected bond lengths: C1–S1: 1.6720(3) Å; C1–S2: 1.6720(3) Å; Cu1–S1: 2.2702(4) Å; Cu2–S2: 2.2440(3) Å; C26–S3: 1.6648(2) Å; C26–S4: 1.7107(3) Å; Cu1–S4: 2.2635(3) Å; Cu2–S4: 2.6307(4) Å; Cu2–S3: 2.3949(7) Å; Cu1–P1: 2.2201(8) Å; Cu2–P2: 2.2243(7) Å. | |
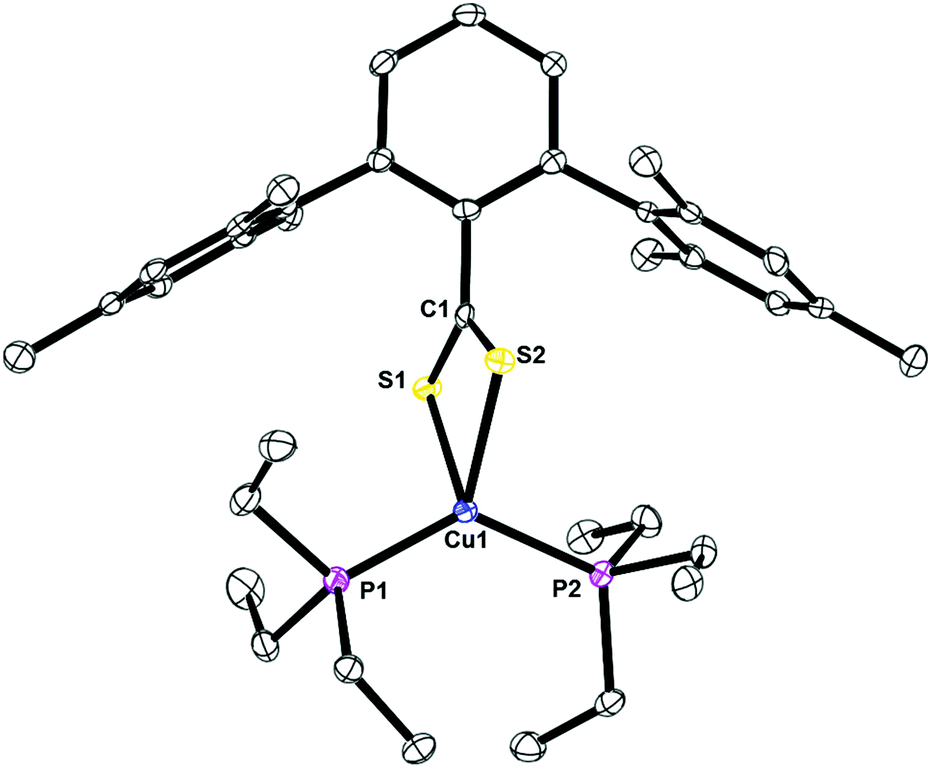
The structures of 4, Fig. 6, 5, Fig. S1,† and 7, Fig. S2,† are mononuclear copper(I) complexes with pseudo-tetrahedral geometry with two sulfur and two phosphorus atoms coordinated. The data collection for 5 was done at 298 K due to a phase transition at low temperature. In all three compounds, the C–S bond lengths are nearly identical, which indicate that the anionic charge of the ligand is delocalized. The copper–sulfur and –phosphorus bond distances and angles are similar to others reported.
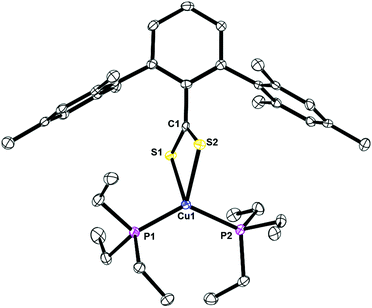 |
| | Fig. 6 Thermal ellipsoid plot of 4 shown at the 50% probability level. All hydrogen atoms and residue solvent molecule are omitted for clarity. Selected bond lengths: C1–S1: 1.6795(2) Å; C1–S2: 1.6851(4) Å; Cu1–P1: 2.2175(3) Å; Cu1–P2: 2.2360(3) Å; P1–Cu1–P2: 130.71(4)°; S1–Cu1–P1: 112.35(3)°; S2–Cu1–P1: 112.63(4)°; S1–Cu1–S2: 73.60(3)°. | |
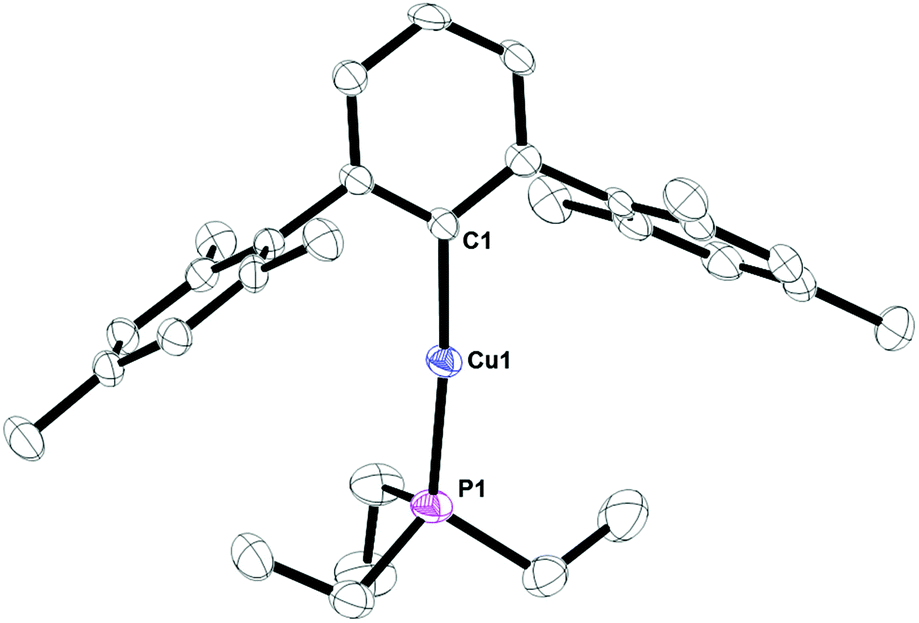
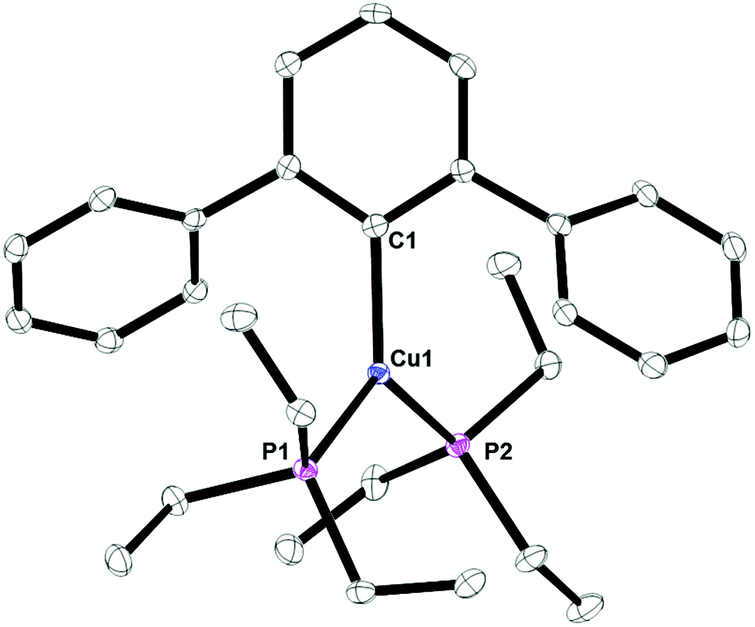
Complex 8, Fig. 7, contains a two-coordinate copper center with a C1–Cu1–P1 bond angle of 168.380(3)°. Cu2[2,6-(Mes)2C6H3]2 has been reported with a Cu1–C1 bond distance of 1.927(5) Å.12 This is nearly identical to the 1.9282(4) Å bond length in 8. In addition, the analog of 8 with PPh3 is known and it contains C1–Cu1–P1 bond angle of 168.82(8)° and Cu1–C1 bond distance of 1.922(3) Å.17 Complex 9, Fig. 8, is a three-coordinate trigonal planar copper compound with one Cu–C bond of 1.9720(2) Å and all C–Cu–P bond angles of 120°.
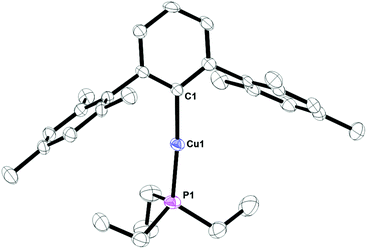 |
| | Fig. 7 Thermal ellipsoid plot of 8 shown at the 50% probability level. All hydrogen atoms and residue solvent molecule are omitted for clarity. Selected bond lengths and angles: C1–Cu1: 1.9282(4) Å; P1–Cu1: 2.2003(4) Å; C1–Cu1–P1: 168.380(3)°. | |
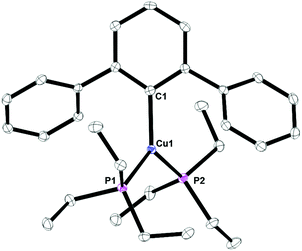 |
| | Fig. 8 Thermal ellipsoid plot of 9 shown at the 50% probability level. All hydrogen atoms are omitted for clarity. Selected bond lengths and angles: C1–Cu1: 1.9790(2) Å; Cu1–P1: 2.2495(4) Å; Cu1–P2: 2.558(2) Å; P1–Cu1–P2: 119.526(6)°; C1–Cu1–P2: 119.790(11)°; C1–Cu1–P1: 120.682(6)°. | |
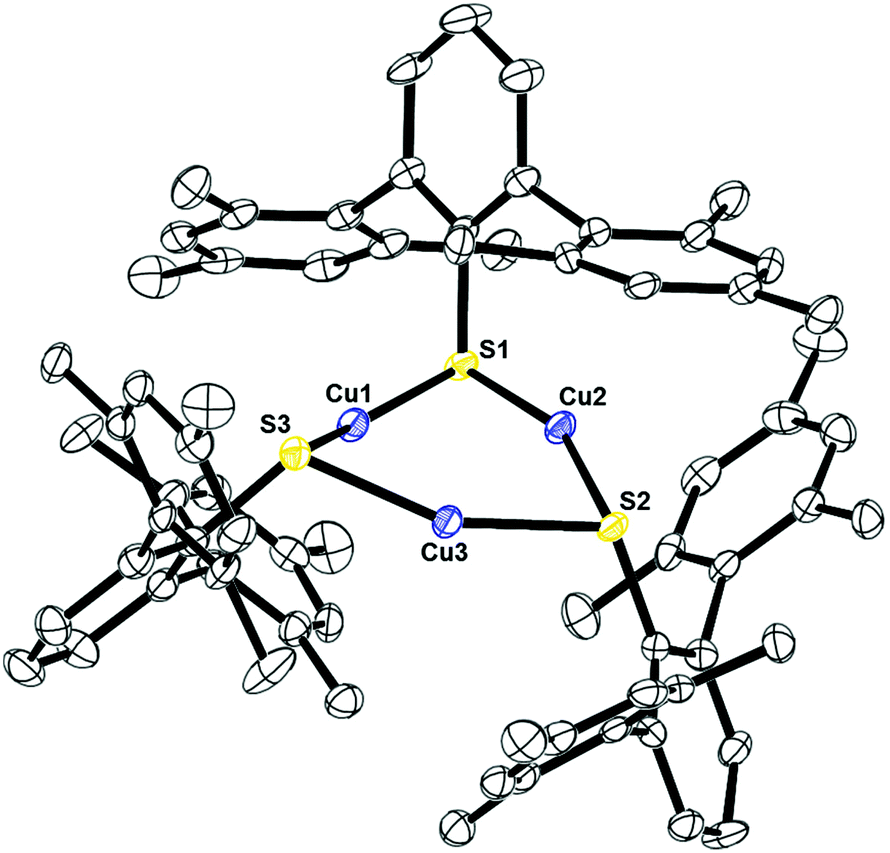
Complexes 10, Fig. 9, and 11, Fig. S3,† are structural analogs with three copper(I) centers and three mesityl-terphenyl ligands. The geometry around each copper is angular with E–Cu–E (E = S, Se) bond angles between 155–160°. Complex 11 is structurally similar to the previously reported [iPr3PCuSPh]318 and (μ-SPh)3Cu3(PPh3)4.19 Few copper–selenium clusters have been reported, but the Cu–Se bond distances in 11 of 2.32 Å are comparable to 2.370(2) to 2.412(2) Å in {Cu[Se(2,4,6-iPr3C6H2)]}6.20
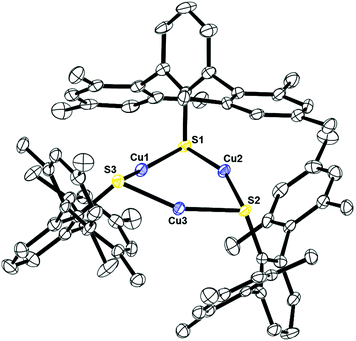 |
| | Fig. 9 Thermal ellipsoid plot of 10 shown at the 50% probability level. All hydrogen atoms are omitted for clarity. Selected bond lengths and angles: Cu1–S1: 2.21032(10) Å; Cu2–S1: 2.21033(14) Å; Cu2–S2: 2.22210(13) Å; Cu3–S2: 2.19521(10) Å; Cu3–S3: 2.20693(11) Å, Cu1–S3: 2.20536(11) Å; Cu1–Cu2: 2.92243(16) Å; Cu1–Cu3: 2.85052 Å; Cu3–Cu2: 2.87843(14) Å; S1–Cu1–S3: 155.1371(17)°; S2–Cu3–S3: 157.8052(15)°; S1–Cu2–S2: 159.3547(14)°. | |
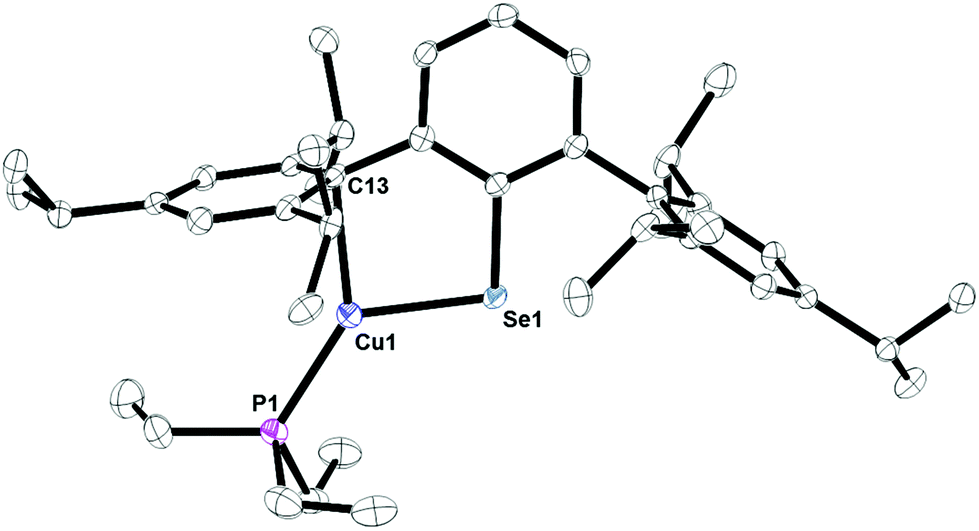
Complexes 12, Fig. S4,† and 13, Fig. 10, are three-coordinate complexes which feature the copper center coordinated through a chalcogenide, phosphorus, and an ipso-carbon interaction. There is no evidence for the ipso-carbon interaction in solution. The Cu1–S1 bond distance of 2.1734(10) Å is close to the 2.1470(8) Å distance in the 3,4,7,8-tetramethyl-1,10-phenanthroline (tmphen) 2,6-diphenyl-thiophenolatocopper(I) complex21 as well as 2.194(1) Å in (Trip)CuPPh3.15 The C1–S1–Cu1 bond angle of 103.53(7)° is comparable to the 102.99(13)° to the corresponding C–S–Cu angle in (Trip)CuPPh3, but larger than the 98.7(1)° in (tmphen)Cu[S(2,4,6-Me3C6H2)], tmphen = 3,4,7,8-tetramethyl-1,10-phenanthroline.21 The C1–Se1–Cu1 angle in 13 is larger, 97.56(6)°, due to the larger size of selenium versus sulfur. The ipso-carbon interactions, Cu1–C13, in 12 and 13 of 2.378(3) and 2.28020(12) Å are similar to those seen in [Cu(2,6-(Mes)2C6H3)]2 of 2.295(4) and 2.123(4) Å or 2.298(2) and 2.271(2) Å in [Cu(C6F5)]4(η2-toluene)2.22
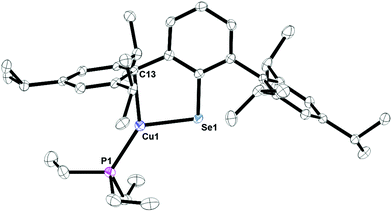 |
| | Fig. 10 Thermal ellipsoid plot of 13 shown at the 50% probability level. All hydrogen atoms are omitted for clarity. Selected bond length Cu1–Se1: 2.30182(12) Å, Cu1–C13: 2.28020(12) Å; Cu1–P1: 2.20287(13) Å; Se1–Cu1–C13: 89.276(4)°; C1–Se1–Cu1: 97.56(6)°. | |
Conclusion
We have synthesized twelve new copper(I) complexes with chalcogen-substituted terphenyl-based ligands to produce mono-, di-, tri-, and tetranuclear complexes. This afforded complexes that structurally mimic the active site of several copper-containing proteins such as Atx1 and CusA. The steric properties, in tandem with varying equivalents of PEt3, forced copper into geometries of near-linear, angular, pseudo-trigonal, and pseudo-tetrahedral. While these geometries are not unusual for copper(I) metal centers, the manipulation of the coordination environment to achieve different geometries is novel and demonstrates the necessity for the large protein manifolds to force the metal centers in alignment for proper function.
In addition, we have synthesized a new lithium dithiocarboxylate terphenyl complex. When the unsubstituted terphenyl ligand was employed, carbon disulfide insertion into the copper–carbon bond was observed, while this was not the case with the mesityl and triisopropyl-derivatives. The chemistry of the terphenyl ligand has been largely unexplored compared to the mesityl and triisopropyl-derivatives and the coordination chemistry and reactivity of this species is under further investigation.
Experimental
General considerations
All syntheses were carried out under a dry N2 atmosphere using standard glovebox and Schlenk line techniques. Organic solvents were obtained from Acros and Sigma Aldrich and dried by standard procedures. Potassium tert-butoxide (Fisher), [Cu(NCCH3)4]PF6 and 10 wt% triethylphosphine (PEt3) in hexane (Strem), potassium bis(trimethylsilyl)amide, carbon disulfide, and 2.5 M n-butyllithium (Aldrich) were purchased. Cu2[(2,6-(Mes)2C6H3)]2,12 (Et2O)2Li[S2C(2,6-(Mes)2C6H3)],11 (thf)2Li[S2C(2,6-(Trip)2C6H3)],11 HS(2,6-(Mes)2C6H3),23 HSe(2,6-(Mes)2C6H3),24 [CuI(PEt3)]4,25 K[S(2,6-(Trip)2C6H3)],23 K[Se(2,6-(Trip)2C6H3)],26 2,6-(Ph)2C6H3I (Ph = C6H5),27 and Cu3[(2,6-(Ph)2C6H3)]312 were prepared as previously described. K[Se(2,6-(Mes)2C6H3)] and K[S(2,6-(Mes)2C6H3)] were made from the following procedure: K[S(2,6-(Mes)2C6H3)] was generated from HS(2,6-(Mes)2C6H3) and potassium tert-butoxide. The reaction was done in thf at room temperature for 1 hour, followed by removal of solvent in vacuo to yield solid K[S(2,6-(Mes)2C6H3)]. K[Se(2,6-(Mes)2C6H3)] was generated from HSe(2,6-(Mes)2C6H3) and potassium bis(trimethylsilyl)amide in toluene. Solid K[Se(2,6-(Mes)2C6H3)] was separated from solution by centrifugation and dried in vacuo. Acetonitrile-d3 and benzene-d6 (Cambridge Isotope Laboratories) were dried over molecular sieves and degassed with three freeze–evacuate–thaw cycles. All 1H and 13C{1H} NMR data were obtained from 500 MHz and 600 MHz DRX Bruker spectrometer. All 77Se and 7Li NMR data were obtained from 300 MHz DRX Bruker spectrometer. All 31P NMR data were obtained from 250 MHz and 300 MHz DRX Bruker spectrometer. 1H NMR shifts given were referenced internally to the residual solvent peaks at δ 7.16 ppm for C6D5H and 7.26 ppm for CHCl3. All 31P NMR shifts given were referenced externally to 85% H3PO4 at 0 ppm. All 77Se NMR shifts given were referenced externally to diphenyldiselenide in CDCl3 at 463 ppm. All 7Li NMR shifts given were referenced externally to 1 M solution of LiCl in D2O at 0 ppm. Infrared spectra were recorded as KBr pellets on Perkin-Elmer Spectrum One FT-IR spectrometer. UV-Vis spectra were recorded on a Varian CARY 100 Bio spectrophotometer. Elemental analyses were performed by Atlantic Microlabs, Inc. (Norcross, GA).
Synthesis of Cu4[S2C(2,6-(Mes)2C6H3)]4, 1.
In a 20 mL scintillation vial, 2.5 mL of thf was added to [Cu(NCCH3)4]PF6 (68 mg, 0.18 mmol) followed by 7.5 mL of (Et2O)2Li[S2C(2,6-(Mes)2C6H3)] (100 mg, 0.18 mmol) in thf to yield a dark brown colored solution. The mixture was stirred at room temperature overnight after which the solvent was removed in vacuo. The brown solid was extracted with toluene then filtered through Celite and the solvent was removed in vacuo to yield a brown powder (60 mg, 72%). X-ray quality crystals were obtained from a concentrated thf solution at −24 °C. 1H NMR (600 MHz, C6D6): δ 2.16 (s, 24H, p-Mes), 2.21 (s, 48H, o-Mes), 6.83 (s, 16H, m-Mes), 6.89 (d, 8H, 3JH–H = 7.2 Hz, m-C6H3), 7.07 (t, 4H, 3JH–H = 7.2 Hz, p-C6H3). 13C{1H} NMR (C6D6): δ 21.4, 21.5, 128.3, 129.3, 129.6, 136.5, 137.1, 137.2, 137.9, 145.5, 229.2. IR (KBr, cm−1): 2956 (m), 2917 (s), 2855 (w), 1611 (w), 1567 (w), 1449 (s), 1376 (w), 1263 (w), 1099 (s), 1018 (vs), 885 (w), 849 (s), 806 (w), 758 (w), 737 (w), 580 (w), 470 (w). Anal. Calcd for C104H110OCu4S8: C, 66.21; H, 5.88. Found: C, 66.73; H, 5.87.
Synthesis of Cu2[S2C(2,6-(Trip)2C6H3)]2(NCCH3), 2.
The synthesis was carried out in a similar manner to Cu4[S2C(2,6-(Mes)2C6H3)]4 with [Cu(NCCH3)4]PF6 (65 mg, 0.17 mmol) and (thf)2Li[S2C(2,6-(Trip)2C6H3)] (100 mg, 0.14 mmol) to yield an orange solid. The crude product was dissolved in acetonitrile, filtered through Celite, and allowed to evaporate at room temperature outside of the glovebox to yield a small amount of Cu2[S2C(2,6-(Trip)2C6H3)]2(NCCH3) crystals.
Synthesis of Cu2[S2C(2,6-(Mes)2C6H3)]2(PEt3)2, 3.
In a 20 mL scintillation vial, 5 mL of thf was added to of (Et2O)2Li[S2C(2,6-(Mes)2C6H3)] (100 mg, 0.18 mmol) followed by 5 mL of [CuI(PEt3)]4 (57 mg, 0.046 mmol) in thf to yield a deep red solution. The mixture was stirred at room temperature overnight after which the solvent was removed in vacuo. The deep red solid was extracted with hexanes the filtered through Celite and the solvent was removed in vacuo to yield deep red powder (101 mg, 96%). X-ray quality crystals were obtained from a concentrated hexanes solution at −24 °C. 1H NMR (500 MHz, C6D6): 0.83–0.9 (m, 18H, P(CH2CH3)3), 1.02–1.05 (m, 12H, P(CH2CH3)3), 2.29 (s, 12H, p-Mes), 2.43 (s, 24H, o-Mes), 6.85 (s, 8H, m-Mes), 6.92 (d, 4H, 3JH–H = 7.5 Hz, m-C6H3), 7.11 (t, 2H, 3JH–H = 7.5 Hz, p-C6H3). 13C{1H} NMR (C6D6): 8.7, 17.1 (d, 1JC–P = 15 Hz), 21.5, 22.2, 127.5, 128.4, 129.3, 135.8, 137.1, 137.8, 139.5, 149.8, 246.8. 31P{1H} NMR (C6D6, 300 K): δ −8.97. IR (KBr, cm−1): 2962 (vs), 2913 (vs), 2877 (s), 2729 (w), 1610 (w), 1570 (w), 1453 (s), 1376 (m), 1260 (w), 1220 (w), 1180 (w), 1158 (w), 1095 (w), 1019 (vs), 907 (m), 848 (m), 805 (w), 753 (m), 694 (w), 622 (w), 581 (w). Anal. Calcd for C62H80Cu2P2S4: C, 65.17; H, 7.06. Found: C, 65.26; H, 7.33.
Synthesis of Cu[S2C(2,6-(Mes)2C6H3)](PEt3)2, 4.
In a 20 mL scintillation vial, 5 mL of toluene was added to [CuI(PEt3)]4 (57 mg, 0.046 mmol) followed by 10 wt% PEt3 in hexane (217 mg, 0.18 mmol). The solution was then added to a suspension of (Et2O)2Li[S2C(2,6-(Mes)2C6H3)] (100 mg, 0.18 mmol) in toluene. The mixture was stirred at room temperature overnight after which the solution was filtered through Celite and the solvent was removed in vacuo to yield orange powder (61 mg, 48%). X-ray quality crystals were obtained from a concentrated toluene solution at −24 °C. 1H NMR (600 MHz, C6D6): δ 0.81–0.86 (m, 18H, P(CH2CH3)3), 1.07–1.11 (m, 12H, P(CH2CH3)3), 2.26 (s, 6H, p-Mes), 2.54 (s, 12H, o-Mes), 6.88 (s, 4H, m-Mes), 6.98 (d, 2H, 3JH–H = 7.2 Hz, m-C6H3), 7.15 (t, 1H, 3JH–H = 7.2 Hz, p-C6H3). 13C{1H} NMR (C6D6): 8.6, 17.2 (t, 1JC–P = 8.4 Hz), 21.4, 22.2, 127.1, 127.9, 129.1 135.5, 137.4, 137.7, 139.8, 150.8, 251.7. 31P{1H} NMR (C6D6, 300 K): δ −7.01. IR (KBr, cm−1): 3035 (m), 3005 (m), 2957 (vs), 2911 (vs), 2877 (vs), 2727 (w), 1607 (w), 1568 (w), 1483 (w), 1452 (s), 1411 (m), 1374 (s), 1247 (w), 1219 (w), 1180 (w), 1096 (w), 1013 (br-vs), 923 (m), 854 (m), 804 (w), 773 (vs), 749 (vs), 703 (m), 675 (m), 636 (m), 582 (w), 526 (w). Anal. Calcd for C37H55Cu1P2S2: C, 64.46; H, 8.04. Found: C, 64.70; H, 7.92.
Synthesis of Cu[S2C(2,6-(Trip)2C6H3)](PEt3)2, 5.
The synthesis was carried out in a similar manner to Cu[S2C(2,6-(Mes)2C6H3)](PEt3)2 with (thf)2Li[S2C(2,6-(Trip)2C6H3)] (100 mg, 0.14 mmol), [CuI(PEt3)]4 (43 mg, 0.04 mmol), and 10 wt% PEt3 in hexane (166.5 mg, 0.14 mmol) to yield an orange powder of Cu[S2C(2,6-(Trip)2C6H3)](PEt3)2 (53 mg, 44%). X-ray quality crystals were obtained from a concentrated hexanes solution at −24 °C. 1H NMR (600 MHz, C6D6): δ 0.8–0.9 (m, 18H, P(CH2CH3)3), 1.06–1.12 (m, 12H, P(CH2CH3)3), 1.21 (d, 12H, 3JH–H = 7.2 Hz, o-CH(CH3)2), 1.34 (d, 12H, 3JH–H = 7.2 Hz, p-CH(CH3)2), 1.59 (d, 12H, 3JH–H = 7.2 Hz, o-CH(CH3)2), 2.92 (sept, 2H, 3JH–H = 7.2 Hz, p-CH(CH3)2), 3.44 (sept, 4H, 3JH–H = 7.2 Hz, o-CH(CH3)2), 7.08 (t, 1H, 3JH–H = 7.5 Hz, p-C6H3), 7.19 (d, 2H, 3JH–H = 7.5 Hz, m-C6H3), 7.21 (s, 4H, m-Trip). 13C{1H} NMR (C6D6): δ 8.7, 17.3 (dd, 1JC–P = 8.6 Hz, 2JC–P = 8.4 Hz), 23.4, 23.5, 26.6, 31.6, 34.7, 120.4, 124.6, 130.8, 135.8, 137.6, 147.3, 148.1, 150.1, 251.4. 31P{1H} NMR (C6D6, 300 K): δ −7.61. IR (KBr, cm−1): 3050 (m), 2929 (vs), 2962 (vs), 2869 (vs), 1604 (m), 1566 (w), 1458 (s), 1419 (m), 1479 (m), 1360 (m), 1316 (w), 1258 (w), 1166 (w), 1101 (m), 1026 (s), 931 (w), 874 (m), 802 (w), 764 (s), 712 (w), 651 (w), 627 (w), 464 (w), 432 (w), 408 (w). Anal. Calcd for C49H79Cu1P2S2: C, 68.61; H, 9.28. Found: C, 68.32; H, 9.30.
Synthesis of (Et2O)2Li[S2C(2,6-(Ph)2C6H3)], 6.
In a Schlenk flask, 20 mL of n-hexane and 20 mL of diethylether was added to 2,6-(Ph)2C6H3I (1.0 g, 2.8 mmol). On the Schlenk line, the flask was cooled to 0 °C in an ice bath. 2.5 M n-butyllithium (1.2 mL, 3 mmol) was added via syringe. Formation of white precipitate was observed immediately. The flask was allowed to stir at 0 °C for an hour. Ice bath was removed and replaced with dry ice/acetone bath. Once the solution reached −70 °C, carbon disulfide (0.2 mL, 3.3 mmol) was added via syringe. The flask was then allowed to stir in dry ice/acetone bath overnight. The resulting orange suspension in light orange liquid was dried under vacuum and brought into N2 glovebox. The residue was partially dissolved in a 1:1 ratio of n-hexane and diethyl ether and transferred a 20 mL scintillation vial. The content in the scintillation vial was concentrated and cooled to −24 °C overnight. The product was isolated as an orange powder which was with n-hexane and dried under vacuum (859 mg, 66% yield). X-ray quality crystals were obtained from concentrated a n-hexane/diethyl ether solution at −24 °C. 1H NMR (600 MHz, C6D6): δ 0.92 (t, 12H, O(CH2CH3)2), 3.12 (q, 8H, O(CH2CH3)2), 7.10 (t, 1H, 3JH–H = 7.8 Hz, p-C6H3), 7.10–7.13 (m, 2H, p-Ph), 7.17–7.20 (m, 4H, m-Ph), 7.24 (d, 2H, 3JH–H = 7.8 Hz, m-C6H3), 7.94–7.95 (m, 4H, o-Ph). 13C{1H} NMR (C6D6): δ 15.0, 65.9, 125.9, 126.3, 127.5, 130.0, 130.5, 136.7, 143.6, 154.9, 259.9. 7Li{1H} NMR (C6D6, 297 K): −0.17. IR (KBr, cm−1): 3054 (m), 3027 (w), 2974 (s), 2931 (m), 2884 (m), 1597 (w), 1494 (m), 1439 (m), 1385 (m), 1221 (w), 1178 (w), 1154 (w), 1091 (s), 1065 (s), 1021 (vs), 999 (s), 967 (w), 914 (m), 776 (s), 759 (vs), 698 (vs), 613 (m), 530 (w). Anal. Calcd for C27H33Li1O1; C, 70.40; H, 7.22. Found: C, 70.42; H, 7.09.
Synthesis of Cu[S2C(2,6-(Ph)2C6H3)](PEt3)2, 7.
The synthesis was carried out in a similar manner to Cu[S2C(2,6-(Mes)2C6H3)](PEt3)2 with (Et2O)2Li[S2C(2,6-(Ph)2C6H3)] (100 mg, 0.22 mmol), [CuI(PEt3)]4 (67 mg, 0.05 mmol), and 10 wt% PEt3 in hexane (257 mg, 0.22 mmol) to yield an orange powder of Cu[S2C(2,6-(Ph)2C6H3)](PEt3)2 (66 mg, 50%). X-ray quality crystals were obtained from a concentrated n-hexane solution at −24 °C. 1H NMR (600 MHz, C6D6): δ 0.83 (s, 18H, P(CH2CH3)3), 1.12 (q, 12H, P(CH2CH3)3), 7.06 (t, 1H, 3JH–H = 7.2 Hz, p-C6H3), 7.07–7.17 (m, 2H, p-Ph), 7.17 (d, 2H, 3JH–H = 7.2 Hz, m-C6H3), 7.20–7.22 (m, 4H, m-Ph), 7.91–7.92 (m, 4H, o-Ph). 13C{1H} NMR (C6D6): δ 8.5, 17.2, 125.9, 126.3, 127.4, 130.1, 130.9, 138.4, 143.8, 151.6, 251.9. 31P{1H} NMR (C6D6, 300 K): −7.73. IR (KBr, cm−1): 3053 (w), 3025 (w), 2960 (s), 2978 (s), 2901 (s), 2872 (s), 2820 (w), 1598 (w), 1572 (w), 1492 (w), 1451 (s), 1416 (m), 1375 (m), 1251 (w), 1232(w), 1178 (w), 1153 (w), 1094 (w), 1024 (vs), 926 (m), 839 (w), 802 (w), 757 (vs), 694 (vs), 615 (m), 532 (m). Anal. Calcd for C31H43Cu1S2P2; C, 61.51; H, 7.16. Found: C, 61.07; H, 7.10.
Synthesis of Cu[(2,6-(Mes)2C6H3)](PEt3), 8.
In a 20 mL scintillation vial, 5 mL of hexanes was added to Cu2[(2,6-(Mes)2C6H3)]2 (43 mg, 0.057 mmol) followed by 0.1 mL of 10 wt% PEt3 in hexane. The mixture was stirred at room temperature overnight after which the solution was filtered through Celite and the solvent was removed in vacuo to yield a white powder (44 mg, 78%). X-ray quality crystals were obtained from a concentrated hexanes solution at −24 °C. 1H NMR (600 MHz, C6D6): δ 0.48 (dt, 9H, 3JH–P = 16.8 Hz, 3JH–H = 7.8 Hz, P(CH2CH3)3), 0.61–0.66 (m, 6H, P(CH2CH3)3), 2.29 (s, 6H, p-Mes), 2.40 (s, 12H, o-Mes), 6.93 (s, 4H, m-Mes), 7.22 (d, 2H, 3JH–H = 7.8 Hz, m-C6H3), 7.42 (t, 1H, 3JH–H = 7.8 Hz, p-C6H3). 13C{1H} NMR (C6D6, 298 K): 8.7 (d, 2JC–P = 2.4 Hz), 16.3 (d, 1JC–P = 21.9 Hz), 21.2, 22.1, 124.3, 125.7, 128.0, 134.2, 136.2, 147.4, 151.0, 169.3. 31P{1H} NMR (C6D6, 300 K): δ −2.87. IR (KBr, cm−1): 3027 (m), 2963 (vs), 2933 (vs), 2909 (vs), 2725 (w), 1607 (m), 1544 (m), 1446 (s), 1374 (m), 1233 (m), 1163 (w), 1077 (m), 1034 (s), 850 (m), 796 (m), 762 (m), 729 (m), 579 (w), 548 (w). Anal. Calcd for C30H40Cu1P2: C, 72.77; H, 8.14. Found: C, 72.10; H, 8.22.
Synthesis of Cu[(2,6-(Ph)2C6H3)](PEt3)2, 9.
The synthesis was carried out in a similar manner to Cu[(2,6-(Mes)2C6H3)](PEt3) with Cu3[(2,6-(Ph)2C6H3)]3 (77 mg, 0.09 mmol) and 10 wt% PEt3 in hexane (313 mg, 0.26 mmol) to yield a light yellow powder of Cu[(2,6-(Ph)2C6H3)](PEt3)2 (82 mg, 59%). X-ray quality crystals were obtained from a concentrated n-hexane solution at −24 °C. 1H NMR (600 MHz, C6D6): δ 0.74 (dt, 18H, P(CH2CH3)3, 3JH–P = 15 Hz, 3JH–H = 7.8 Hz), 0.99–1.02 (m, 12H, P(CH2CH3)3), 7.05–7.20 (m, 2H, p-Ph), 7.29–7.31 (m, 4H, m-Ph), 7.42 (t, 1H, 3JH–H = 7.8 Hz, p-C6H3), 7.72 (d, 2H, 3JH–H = 7.8 Hz, m-C6H3), 7.96–7.97 (m, 4H, o-Ph). 13C{1H} NMR (C6D6): 8.5 (d, 2JC–P = 4.5 Hz), 17.4 (d, 2JC–P = 10.5 Hz), 124.6, 125.0, 125.1, 128.4, 128.5, 150.9, 152.3, 177.4. 31P{1H} NMR (C6D6, 297 K): −14.49. IR (KBr, cm−1): 3073 (m), 3053 (m), 3018 (m), 2962 (s), 2932 (s), 2898 (s), 2873 (s), 2813 (w), 2726 (w), 1593 (m), 1573 (w), 1559 (w), 1490 (m), 1457 (s), 1442 (m), 1430 (m), 1412 (s), 1376 (m), 1285 (w), 1276 (w), 1236 (m), 1154 (w), 1089 (w), 1068 (w), 1026 (s), 903 (w), 798 (w), 751 (vs), 721 (s), 698 (vs), 613 (m), 554 (m), 500 (w), 419 (w). Anal. Calcd for C30H43Cu1P2; C, 68.09; H, 8.19. Found: C, 67.96; H, 8.04.
Synthesis of Cu3[S(2,6-(Mes)2C6H3)]3, 10.
In a 20 mL scintillation vial, 5 mL of thf was added to [Cu(NCCH3)4]PF6 (81 mg, 0.22 mmol) followed by 5 mL of K[S(2,6-(Mes)2C6H3)] (84 mg, 0.22 mmol) in thf to yield a colorless solution. The mixture was stirred at room temperature overnight after which the solvent was removed in vacuo. The white solid was extracted with toluene then filtered through Celite and the solvent was removed in vacuo to yield white powder in nearly quantitative yield. X-ray quality crystals were obtained from a concentrated toluene/acetonitrile solution at room temperature. 1H NMR (500 MHz, C6D6): δ 2.06 (s, 36H, o-Mes), 2.24 (s, 18H, p-Mes), 6.74 (d, 6H, 3JH–H = 7.5 Hz, m-C6H3), 6.82 (s, 12H, m-Mes), 6.91 (t, 3H, 3JH–H = 7.5 Hz, p-C6H3). 13C{1H} NMR (C6D6): δ 21.3, 21.7, 125.9, 129.1, 129.7, 135.3, 135.9, 136.7, 139.8, 144.4. IR (KBr, cm−1): 3031 (m), 2968 (s), 2913 (vs), 2853 (s), 2727 (w), 1609 (s), 1569 (m), 1481 (m), 1445 (vs), 1388 (s), 1375 (s), 1113 (w), 1090 (w), 1036 (s), 850 (vs), 798 (s), 771 (w), 741 (vs), 589 (m), 547 (w), 465 (w). Anal. Calcd for C72H75Cu3S3: C, 70.47; H, 6.16. Found: C, 70.42; H, 6.10.
Synthesis of Cu3[Se(2,6-(Mes)2C6H3)]3, 11.
The synthesis was carried out in a similar manner to Cu3[S(2,6-(2,4,6-Me3C6H3))]3 with K[Se(2,6-(Mes)2C6H3)] (100 mg, 0.25 mmol) and [Cu(NCCH3)4]PF6 (95 mg, 0.25 mmol) to yield an white solid of Cu3[Se(2,6-(2,4,6-Me3C6H3))]3 (50 mg, 43%). X-ray quality crystals were obtained from concentrated toluene/acetonitrile solution at room temperature. 1H NMR (600 MHz, C6D6): δ 2.07 (s, 36H, o-Mes), 2.26 (s, 18H, p-Mes), 6.71 (d, 6H, 3JH–H = 7.5 Hz, m-C6H3), 6.80 (s, 12H, m-Mes), 6.94 (t, 3H, 3JH–H = 7.5 Hz, p-C6H3). 1H NMR (600 MHz, CDCl3): δ 1.88 (s, 36H, o-Mes), 2.26 (s, 18H, p-Mes), 6.76 (d, 6H, 3JH–H = 7.8 Hz, m-C6H3), 6.82 (s, 12H, m-Mes), 7.12 (t, 3H, 3JH–H = 7.8 Hz, p-C6H3). 13C{1H} NMR (CDCl3): δ 21.3, 21.4, 126.4, 128.5, 129.2, 130.9, 135.2, 136.7, 140.7, 145.4. 77Se{1H} NMR (thf, 297 K): −96.2. IR (KBr, cm−1): 2912 (vs), 2854 (s), 2726 (w), 1605 (m), 1565 (m), 1443 (vs), 1379 (s), 1271 (w), 1228 (w), 1172 (w), 1093 (w), 1024 (s), 848 (s), 797 (s), 735 (s), 576 (m). Anal. Calcd for C72H75Cu3Se3: C, 63.22; H, 5.53. Found: C, 63.11; H, 5.64.
Synthesis of Cu[S(2,6-(Trip)2C6H3)](PEt3), 12.
In a 20 mL scintillation vial, 5 mL of thf was added to [CuI(PEt3)]4 (56 mg, 0.045 mmol) followed by 5 ml of K[S(2,6-(Trip)2C6H3)] (100 mg, 0.18 mmol). The mixture was stirred at room temperature overnight yielding a cloudy white solution after which the solvent was removed in vacuo. The white solid was extracted with hexane then filtered through Celite and the solvent was removed in vacuo to yield white powder (92 mg, 73%). X-ray quality crystals were obtained from a concentrated hexanes solution at −24 °C. 1H NMR (600 MHz, C6D6): δ 0.58 (dt, 9H, 3JH–P = 16.2 Hz, 3JH–H = 7.2 Hz, P(CH2CH3)3), 0.78–0.83 (m, 6H, P(CH2CH3)3), 1.23 (d, 12H, 3JH–H = 6.6 Hz, p-CH(CH3)2), 1.27 (d, 12H, 3JH–H = 6.6 Hz, o-CH(CH3)2), 1.60 (d, 12H, 3JH–H = 7.2 Hz, o-CH(CH3)2), 2.80 (sept, 1H, 3JH–H = 7.2 Hz, p-CH(CH3)2), 3.21 (sept, 2H, 3JH–H = 6.6 Hz, o-CH(CH3)2), 6.94 (t, 1H, 3JH–H = 7.5 Hz, p-C6H3), 7.01 (d, 2H, 3JH–H = 7.5 Hz, m-C6H3), 7.31 (s, 4H, m-Trip). 13C{1H} NMR (C6D6): δ 8.0, 15.6 (d, 1JC–P = 21 Hz), 24.1, 24.9, 25.0, 31.3, 34.3, 120.3, 122.4, 128.7, 139.2, 141.2, 146.2, 146.6, 153.6. 31P{1H} NMR (C6D6, 297 K): δ −9.01. IR (KBr, cm−1): 3040 (w), 2960 (vs), 2929 (vs), 2867 (s), 2750 (w), 1599 (w), 1565 (w), 1459 (s), 1413 (m), 1383 (s), 1359 (m), 1314 (m), 1254 (w), 1167 (w), 1107 (w), 1075 (w), 1044 (m), 1008 (w), 940 (w), 874 (m), 791 (w), 767 (m), 736 (m), 694 (w), 648 (w), 627 (w), 521 (w). Anal. Calcd for C42H64Cu1P1S1: C, 72.53; H, 9.27. Found: C, 72.32; H, 9.47.
Synthesis of Cu[Se(2,6-(Trip)2C6H3)](PEt3), 13.
The synthesis was carried out in a similar manner to Cu[S(2,6-(Trip)2C6H3)](PEt3) with K[Se(2,6-(Trip)2C6H3)] (93 mg, 0.17 mmol) and [CuI(PEt3)]4 (51.9 mg, 0.04 mmol) to yield a white solid of Cu[Se(2,6-(Trip)2C6H3)](PEt3) (71 mg, 42%). X-ray quality crystals were obtained from a concentrated hexanes solution at −24 °C. 1H NMR (600 MHz, C6D6): δ 0.61 (br-s, 9H, P(CH2CH3)3), 0.84 (br-s, 6H, P(CH2CH3)3), 1.24 (d, 12H, 3JH–H = 6.9 Hz, p-CH(CH3)2), 1.25 (d, 12H, 3JH–H = 6.9 Hz, o-CH(CH3)2), 1.62 (d, 12H, 3JH–H = 6.9 Hz, o-CH(CH3)2), 2.81 (sept, 1H, 3JH–H = 6.9 Hz, p-CH(CH3)2), 3.18 (sept, 2H, 3JH–H = 6.9 Hz, o-CH(CH3)2), 6.95 (d, 2H, 3JH–H = 7.8 Hz, m-C6H3), 7.00 (t, 1H, 3JH–H = 7.8 Hz, p-C6H3), 7.32 (s, 4H, m-Trip). 13C{1H} NMR (C6D6): δ 8.02, 15.7 (d, 2JC–P = 19.1 Hz), 24.1, 25.0, 25.1, 31.4, 34.3, 121.8, 122.6, 128.3, 140.2, 143.6, 145.7, 146.5, 147.7. 31P{1H} NMR (C6D6, 300 K): δ −9.51. 77Se{1H} NMR (C6D6, 300 K): δ 171.6 (d, 2JSe–P = 49 Hz). IR (KBr, cm−1): 3042 (w), 2958 (vs), 2929 (vs), 2866 (vs), 2748 (w), 1604 (w), 1594 (w), 1565 (m), 1536 (w), 1459 (s), 1412 (m), 1383 (vs), 1360 (s), 1336 (w), 1314 (m), 1255 (m), 1187 (w), 1167 (w), 1151 (w), 1106 (w), 1079 (w), 1068 (w), 1035 (s), 955 (w), 939 (m), 921 (w), 873 (s), 791 (m), 767 (s), 735 (s), 716 (m), 694 (w), 648 (w), 627 (w), 587 (w), 506 (w), 430 (w). Anal. Calcd for C42H64Cu1P1Se1: C, 67.95; H, 8.69. Found: C, 67.65; H, 8.75.
Crystal structure determination and refinement
The selected single crystal of 1–13 was mounted on a nylon cryoloop using viscous hydrocarbon oil. X-ray data collection was performed at 100(2) K except for 5 which was conducted at room temperature due to a phase transition. The X-ray data were collected on a Bruker CCD diffractometer with monochromated Mo-Kα radiation (λ = 0.71073 Å). The data collection and processing utilized Bruker Apex2 suite of programs.28 The structures were solved using direct methods and refined by full-matrix least-squares methods on F2 using Bruker SHELX-97 program.29 All non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were placed at calculated positions and included in the refinement using a riding model. Thermal ellipsoid plots were prepared by using Olex230 with 50% of probability displacements for non-hydrogen atoms. Crystal data and details for data collection for complexes 1–5, 6–11, and 12–13 are provided in Tables 2, 3, and 4, respectively.
Table 2 X-ray crystallographic data shown for complexes 1–5
| |
1
|
2
|
3
|
4
|
5
|
| CCDC deposit number |
1491737
|
1491738
|
1491739
|
1491740
|
1491741
|
| Empirical formula |
C100H100Cu4S8·C4H10O |
Cu76H101Cu2NS4·C2H3N |
C62H80Cu2P2S4·C3H7 |
C37H55CuP2S2 |
C49H79CuP2S2 |
| Formula weight (g mol−1) |
1886.55 |
1324.95 |
1185.60 |
689.41 |
857.72 |
| Crystal habit, color |
Plate, red |
Prism, orange |
Plate, red |
Prism, orange |
Prism, red |
| Temperature (K) |
100(2) |
100(2) |
100(2) |
100(2) |
297(2) |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Pc
|
P |
P21 |
P21/n |
| Crystal system |
Triclinic |
Monoclinic |
Triclinic |
Orthorhombic |
Monoclinic |
| Volume (Å3) |
4634.03(18) |
3619.9(4) |
3162.1(10) |
3628.1(11) |
5258.88(19) |
|
a (Å) |
13.6538(3) |
12.9612(9) |
11.944(2) |
11.887(2) |
13.6652(3) |
|
b (Å) |
15.0404(3) |
17.4811(12) |
13.738(3) |
11.899(2) |
26.0431(5) |
|
c (Å) |
24.7778(6) |
16.5758(11) |
20.459(4) |
25.649(5) |
14.8022(3) |
|
α (°) |
87.0640(10) |
90 |
84.180(2) |
90 |
90 |
|
β (°) |
82.3660(10) |
105.4550(10) |
88.550(2) |
90 |
93.3470(10) |
|
γ (°) |
66.7600(10) |
90 |
71.227(2) |
90 |
90 |
|
Z
|
2 |
2 |
2 |
4 |
4 |
| Calculated density (Mg m−3) |
1.352 |
1.216 |
1.245 |
1.262 |
1.083 |
| Absorption coefficient (mm−1) |
3.080 |
0.745 |
0.892 |
0.830 |
2.101 |
| Final R indices [I > 2σ(I)] |
R = 0.0287 |
R = 0.0300 |
R = 0.0401 |
R = 0.0245 |
R = 0.0754 |
|
R
w = 0.0697 |
R
w = 0.0788 |
R
w = 0.1027 |
R
w = 0.0574 |
R
w = 2458 |
Table 3 X-ray crystallographic data shown for complexes 6–11
| |
6
|
7
|
8
|
9
|
10
|
11
|
| CCDC deposit number |
1491742
|
1491743
|
1491744
|
1491745
|
1491746
|
1491747
|
| Empirical formula |
C27H33LiO2S2 |
C62H86Cu2P4S4 |
C30H40CuP |
C30H43CuP2 |
C72H75Cu3S3 |
C72H75Cu3Se3 |
| Formula weight (g mol−1) |
460.59 |
1210.50 |
495.13 |
529.12 |
1227.12 |
1367.82 |
| Crystal habit, color |
Prism, yellow |
Prism, red |
Prism, colorless |
Prism, colorless |
Prism, colorless |
Plate, colorless |
| Temperature (K) |
100(2) |
100(2) |
100(2) |
100(2) |
100(2) |
100(2) |
| Space group |
P21/c |
P21/c |
P21/n |
Cc
|
P |
P |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
Triclinic |
Triclinic |
| Volume (Å3) |
2582.2(6) |
6322.2(7) |
2710.1(10) |
2813.3(9) |
2988.2(3) |
3027.1(15) |
|
a (Å) |
8.8758(12) |
17.1313(11) |
11.938(3) |
15.958(3) |
13.0283(6) |
13.183(4) |
|
b (Å) |
22.935(3) |
14.1534(9) |
11.356(2) |
10.5272(19) |
14.0454(7) |
14.033(4) |
|
c (Å) |
12.6872(17) |
26.5086(16) |
20.088(4) |
17.379(3) |
18.6504(9) |
18.660(5) |
|
α (°) |
90 |
90 |
90 |
90 |
85.028(4) |
85.712(3) |
|
β (°) |
91.084(2) |
100.3840(10) |
95.631(3) |
105.505(2) |
74.327(4) |
73.509(3) |
|
γ (°) |
90 |
90 |
90 |
90 |
65.477(3) |
66.271(3) |
|
Z
|
4 |
4 |
4 |
4 |
2 |
2 |
| Calculated density (Mg m−3) |
1.185 |
1.272 |
1.214 |
1.249 |
1.364 |
1.501 |
| Absorption coefficient (mm−1) |
0.227 |
0.942 |
0.880 |
0.906 |
1.205 |
2.890 |
| Final R indices [I > 2σ(I)] |
R = 0.0434 |
R = 0.0340 |
R = 0.0425 |
R = 0.0235 |
R = 0.0681 |
R = 0.0339 |
|
R
w = 0.0967 |
R
w = 0.0736 |
R
w = 0.1153 |
R
w = 0.0509 |
R
w = 0.1791 |
R
w = 0.0719 |
Table 4 X-ray crystallographic data shown for complexes 12–13
| |
12
|
13
|
| CCDC deposit number |
1491748
|
1491749
|
| Empirical formula |
C42H64CuPS |
Cu42H64CuPSe |
| Formula weight (g mol−1) |
695.50 |
742.40 |
| Crystal habit, color |
Prism, colorless |
Prism, colorless |
| Temperature (K) |
100(2) |
100(2) |
| Space group |
P21/c |
P21/c |
| Crystal system |
Monoclinic |
Monoclinic |
| Volume (Å3) |
4002.0(5) |
3980.3(5) |
|
a (Å) |
12.9452(8) |
12.948(10) |
|
b (Å) |
18.3134(12) |
17.9243(13) |
|
c (Å) |
17.8762(12) |
18.0827(14) |
|
α (°) |
90 |
90 |
|
β (°) |
109.2060(10) |
109.0860(10) |
|
γ (°) |
90 |
90 |
|
Z
|
4 |
4 |
| Calculated density (Mg m−3) |
1.154 |
1.239 |
| Absorption coefficient (mm−1) |
0.664 |
1.530 |
| Final R indices [I > 2σ(I)] |
R
1 = 0.0400, wR2 = 0.1003 |
R
1 = 0.0287, wR2 = 0.0722 |
Acknowledgements
We gratefully acknowledge the University of Missouri College of Arts & Science Alumni Faculty Incentive Grant and PRIME fund for support of this work.
Notes and references
- P. C. Ford, E. Cariati and J. Bourassa, Chem. Rev., 1999, 99, 3625–3648 CrossRef CAS PubMed.
- C. Chen, Z. Weng and J. F. Hartwig, Organometallics, 2012, 31, 8031–8037 CrossRef CAS PubMed.
- P. S. Fier, J. Luo and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 2552–2559 CrossRef CAS PubMed.
- J. Rogers, A. B. Dowsett, P. J. Dennis, J. V. Lee and C. W. Keevil, Appl. Environ. Microbiol., 1994, 60, 1585–1592 CAS.
- J. D. Ranford, P. J. Sadler and D. A. Tocher, J. Chem. Soc., Dalton Trans., 1993, 3393–3399 RSC.
- R. A. Festa and D. J. Thiele, Curr. Biol., 2011, 21, R877–R883 CrossRef CAS PubMed.
- R. A. Pufahl, C. P. Singer, K. L. Peariso, S.-J. Lin, P. J. Schmidt, C. J. Fahrni, V. C. Culotta, J. E. Penner-Hahn and T. V. O'Halloran, Science, 1997, 278, 853–856 CrossRef CAS PubMed.
- F. Long, C.-C. Su, M. T. Zimmermann, S. E. Boyken, K. R. Rajashankar, R. L. Jernigan and E. W. Yu, Nature, 2010, 467, 484–488 CrossRef CAS PubMed.
- R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed.
- A. C. Lane, C. L. Barnes, W. E. Antholine, D. Wang, A. T. Fiedler and J. R. Walensky, Inorg. Chem., 2015, 54, 8509–8517 CrossRef CAS PubMed.
- C. Ives, E. L. Fillis and J. R. Hagadorn, Dalton Trans., 2003, 527–531 RSC.
- M. Niemeyer, Organometallics, 1998, 17, 4649–4656 CrossRef CAS.
- V. K. Shen, D. W. Siderius, W. P. Krekelberg and Eds., NIST Standard Reference Database Number 101 September 2015 edn., 2015.
- A. C. Lane, M. V. Vollmer, C. H. Laber, D. Y. Melgarejo, G. M. Chiarella, J. P. Fackler, X. Yang, G. A. Baker and J. R. Walensky, Inorg. Chem., 2014, 53, 11357–11366 CrossRef CAS PubMed.
- S. Groysman and R. H. Holm, Inorg. Chem., 2009, 48, 621–627 CrossRef CAS PubMed.
- U. Jayarathne, S. R. Parmelee and N. P. Mankad, Inorg. Chem., 2014, 53, 7730–7737 CrossRef CAS PubMed.
- M. Niemeyer, Z. Anorg. Allg. Chem., 2003, 629, 1535–1540 CrossRef CAS.
- O. Kluge, K. Grummt, R. Biedermann and H. Krautscheid, Inorg. Chem., 2011, 50, 4742–4752 CrossRef CAS PubMed.
- I. G. Dance, L. J. Fitzpatrick and M. L. Scudder, J. Chem. Soc., Chem. Commun., 1983, 546–548 RSC.
- D. Ohlmann, H. Pritzkow, H. Grutzmacher, M. Anthamatten and R. Glaser, J. Chem. Soc., Chem. Commun., 1995, 1011–1012 RSC.
- A. F. Stange and W. Kaim, Chem. Commun., 1998, 469–470 RSC.
- A. Sundararaman, R. A. Lalancette, L. N. Zakharov, A. L. Rheingold and F. Jäkle, Organometallics, 2003, 22, 3526–3532 CrossRef CAS.
- M. Niemeyer and P. P. Power, Inorg. Chem., 1996, 35, 7264–7272 CrossRef CAS PubMed.
- J. J. Ellison, K. Ruhlandt-Senge, H. H. Hope and P. P. Power, Inorg. Chem., 1995, 34, 49–54 CrossRef CAS.
- F. G. Mann, D. Purdie and A. F. Wells, J. Chem. Soc., 1936, 1503–1513 RSC.
- M. Niemeyer and P. P. Power, Inorg. Chim. Acta, 1997, 263, 201–207 CrossRef CAS.
- A. Saednya and H. Hart, Synthesis, 1996, 1455–1458 CrossRef CAS.
-
APEX2 Suite, Bruker AXS Inc., Madison, Wisconsin, USA, 2007 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Cryst., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1491737–1491749. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt02709a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence