
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Group 10–group 14 metal complexes [E–TM]IV: the role of the group 14 site as an L, X and Z-type ligand†
Erik
Wächtler
a,
Robert
Gericke
a,
Erica
Brendler
b,
Birgit
Gerke
c,
Thorsten
Langer
c,
Rainer
Pöttgen
c,
Lyuben
Zhechkov
d,
Thomas
Heine
d and
Jörg
Wagler
*a
aTU Bergakademie Freiberg, Institut für Anorganische Chemie, Leipziger Strasse 29, 09596 Freiberg, Germany. E-mail: joerg.wagler@chemie.tu-freiberg.de; Fax: +49 3731 39 4058; Tel: +49 3731 39 4343
bTU Bergakademie Freiberg, Institut für Analytische Chemie, Leipziger Strasse 29, 09596 Freiberg, Germany
cWestfälische Wilhelms-Universität Münster, Institut für Anorganische und Analytische Chemie, Corrensstrasse 30, 48149 Münster, Germany
dUniversität Leipzig, Wilhelm-Ostwald-Institut für Physikalische und Theoretische Chemie, Linnéstr. 2, 04103 Leipzig, Germany
First published on 8th August 2016
Abstract
A series of new complexes of a general motif [R2E(μ-N,S)2TM–L] (E: metalloid group 14 element; TM: group 10 metal; R: Cl, Ph, pyS, OH, (N,N,O)-chelating ligands; N,S: 1-methylimidazole-2-thiolate (methimazolyl, mt−), pyridine-2-thiolate (pyS−); L: PPh3, PCy3, pyS) was synthesised and characterised by single-crystal X-ray diffraction, multi-nuclear NMR spectroscopy (1H, 13C, 31P, 119Sn), 119Sn Mössbauer spectroscopy and quantum chemical calculations. The E–TM bonding situation in these compounds can be described with various resonance structures which comprise E(II)→TM(II), E(III)–TM(I) and E(IV)←TM(0) features. Thus, in these complexes the atoms of the group 14 based ligand sites reveal L-, X- and Z-type ligand characteristics. A systematic comparison between structural and spectroscopic parameters as well as the results from NLMO analyses of structurally related compounds provided information about the differences in the E–TM bonding situation upon alteration of the metal atoms or ligand patterns. Under investigation are the structurally related compounds [Cl2Sn(μ-pyS)2TM–PPh3] (1: TM = Pd; 2: TM = Ni; 3: TM = Pt), [Cl2Ge(μ-pyS)2Pd–PPh3] (4) and, for in silico analysis, [Cl2Si(μ-pyS)2Pd–PPh3] (5), which indicate a pronounced shift of the E–TM bond electron pair towards TM for TM = Pt. Further complexes serve as representatives of these compounds with different bridging ligands {[Cl2Sn(μ-mt)2Pd–PPh3] (8)}, different trans-E–TM-bound ligands {[Cl2Sn(μ-pyS)2Pd–PCy3] (9), [Cl2Sn(μ-pyS)2Pd]4 (10)} and with different substituents at Sn (including penta- and hexacoordinated tin compounds), i.e., [R2Sn(μ-pyS)2Pd–PPh3] with R = Ph (6) and pyS (7), [(O,N,N)Sn(μ-pyS)2Pd–PPh3] (11) and (12) having two different (O,N,N) tridentate ligands, and [(μ-OH)ClSn(μ-pyS)2Pd–PPh3]2 (13). The latter series indicates a shift of the E–TM (= Sn–Pd) bond electron pair towards Pd upon transition from penta- to hexacoordinated tin compounds.
Introduction
In 1995, Green has established the “covalent bond classification” of transition metal (TM) complexes.1 According to this approach, two electron σ-donor ligands (e.g. Lewis bases like PPh3) are classified as L-type ligands, whereas Lewis acidic two electron σ-acceptor ligands are referred to as Z-type ligands (one electron donors are referred to as X-type). Examples of Z-type ligands are contained in compounds I (Scheme 1), in which platinum acts as a lone pair donor towards electron deficient main group metals.2 The probably largest group of Z-type ligands explored to date are those based on boron as the Lewis acidic site. Unlike the situation in complexes I, in the so-called metallaboratranes3 the presence of a bridging ligand is crucial for the stability of the donor–acceptor bond. Using the same bridging ligand (1-methylimidazole-2-thiolate, methimazolyl, mt−) as Hill in his pioneering work on metallaboratranes (II),4 we have shown that silicon5 and tin6 atoms (III), in spite of their formally saturated valence shell, are capable of acting as Z-type ligand sites in the coordination sphere of TMs. In a related two-fold bridged tin palladium heterobinuclear complex (IV) the electronic situation becomes less clear, i.e. the E–TM bond can be described both as of Sn(IV)←Pd(0) (IVZ) and Sn(II)→Pd(II) (IVL) donor–acceptor nature (in addition to the intermediate Sn(III)–Pd(I) case (IVX) with a perfect covalent bond and various other canonical forms which would require the setting of formal charges).7 Gabbaï observed a similar dichotomy between two borderline cases in a set of 2-phosphinoaryl bridged Sb–Au complexes (V), and referred to this as σ-donor/acceptor confused behaviour of the ligand (ligands of L/Z-type, according to Green).8 In IV, this donor/acceptor confusion is supported by the charge delocalisation within the N,S-bridging ligand (which thus gives rise to various resonance structures without formal charge), whereas in V the combination of the presence of a lone pair at Sb and the well known Lewis acidic properties of Sb(III) compounds9 is responsible for this L/Z-type behaviour.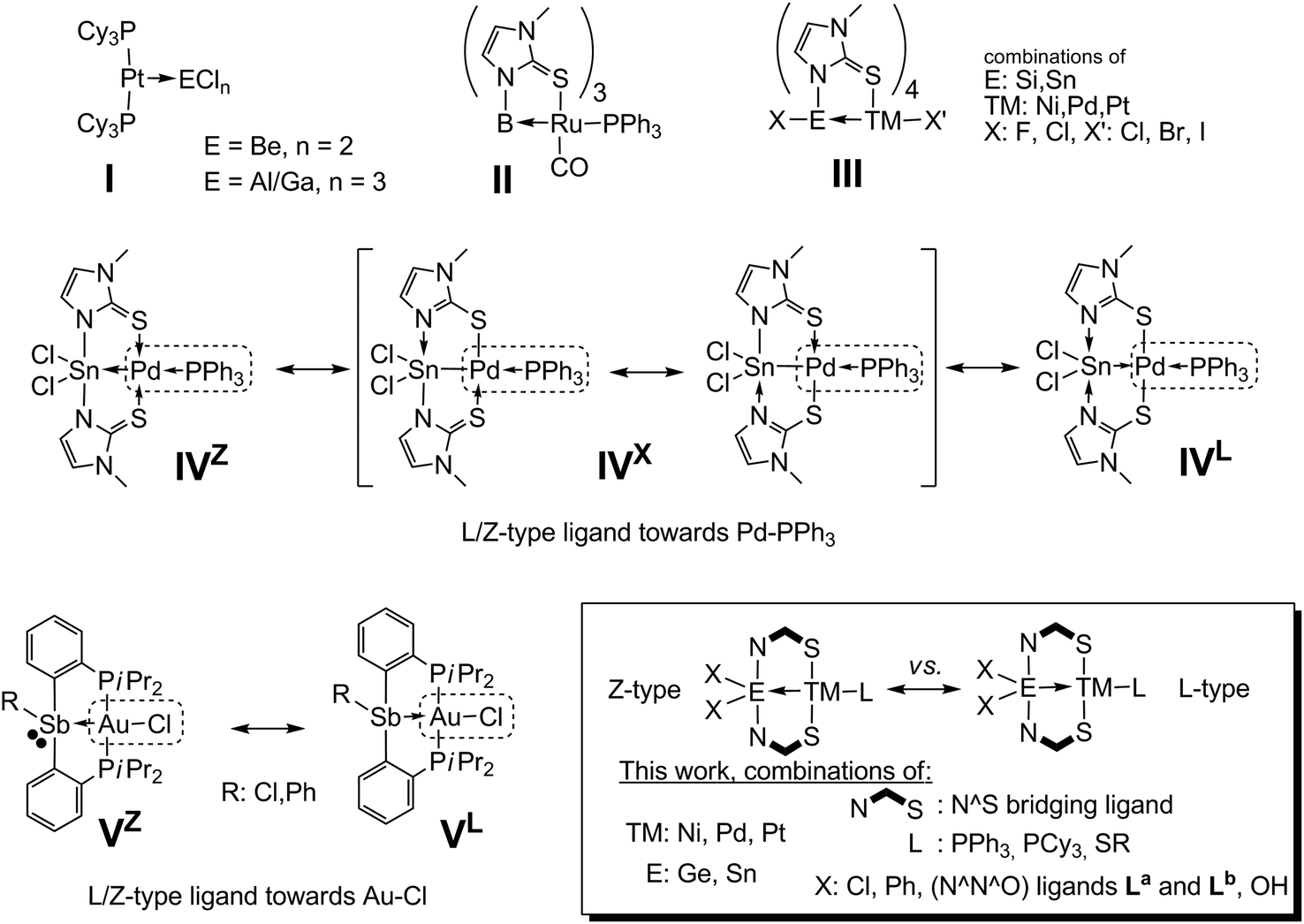 | ||
| Scheme 1 Selected examples of complexes having ligands of the Z-type (I, II, III) and complexes with σ-donor/acceptor confused L/Z-type ligands (IV, V). | ||
The Sb–Au bonding situation in complexes V was found to be highly responsive to the Sb bound substituent R (Ph vs. Cl) with the Z-type character of antimony (contribution of the canonical form VZ) increasing upon formal substitution of Ph for Cl. In a related study of a Sb–Pt system with similar bridging ligands Gabbaï et al. have shown that upon changing the Sb coordination number from 4 via 5 to 6 the Sb–Pt bond electron pair is shifted towards Pt.10 Furthermore, in a greater set of studies Gabbaï et al. have shown the effect of two-electron oxidation/reduction of [E–TM] complexes on the directionality of the intermetallic bond electron pair, i.e., for systems [Te–Au]III/V,11 [Sb–Ni]III/V,12 [Sb–Pt]V/VII,13 [Sb–Au]IV/VI (ref. 14) and [Te–Pt]IV/VI.15 To the best of our knowledge, a comprehensive study of the effects of variations of all possible parameters in a system [E–TM] with constant oxidation number of the bimetallic core on the directionality of the intermetallic bond electron pair has not been reported yet. Recently, we reported on the synthesis and electronic features of Sn–Pd complexes similar to complex IV with the N,S-bridging pyridine-2-thiolato ligand system (pyS−) and we also observed the response of the metal–metal bond towards different SnR2 substitution patterns.16 With the pyridine-2-thiolate (pyS−) ligand we found that those complexes can be generated along different reaction protocols from starting materials with various oxidation states of the metal atoms. Furthermore, the pyS− ligand was also successfully used as a bridging ligand by others for the syntheses of metallaboratranes17 or Sn–Pt complexes.18 With this knowledge, we were able to synthesise a variety of new heterobinuclear complexes with systematic variations of metal atoms (Ni, Pd, Pt), main group metalloid atoms (Ge, Sn) and ligand patterns. Thus, around the parent motif of [Cl2Sn(μ-pyS)2Pd–PPh3] (1) we have characterized a variety of related complexes by varying one parameter (Table 1). Herein we report a systematic comparison of their corresponding crystallographic, spectroscopic and quantum chemical data for a deeper understanding of the influence of a formal substitution of atoms or atomic groups in compounds of the general structure motif shown in Scheme 1 on their heterobimetallic bonding situation.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 , | 9 | 10 | 11 | 12 | 13 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Taken from ref. 16. b Structure optimised by DFT. c Taken from ref. 7. d In addition to mt we aimed at introducing another bridging ligand, i.e., bztzS (the anion of 2-mercaptobenzothiazole), but only succeeded in synthesizing the Ni-complex [Cl2Sn(μ-bztzS)2Ni–PPh3]. Its spectroscopic and electronic features are similar to those of compound 2 and therefore details of its synthesis and characterisation are only listed in the ESI. | |||||||||||||
| TM | Pd | Ni | Pt | Pd | Pd | Pd | Pd | Pd | Pd | Pd | Pd | Pd | Pd |
| E | Sn | Sn | Sn | Ge | Si | Sn | Sn | Sn | Sn | Sn | Sn | Sn | Sn |
| L | PPh3 | PPh3 | PPh3 | PPh3 | PPh3 | PPh3 | PPh3 | PPh3 | PCy 3 | pyS | PPh3 | PPh3 | PPh3 |
| N,S | pyS | pyS | pyS | pyS | pyS | pyS | pyS | mt | pyS | pyS | pyS | pyS | pyS |
| X | Cl | Cl | Cl | Cl | Cl | Ph | pyS | Cl | Cl | Cl | L a | L b | Cl, OH |
Syntheses
For the heterobimetallic complex [Cl2Sn(μ-pyS)2Pd–PPh3] (1) we have established different synthesis routes, comprising the reactions of Cl2Sn(pyS)2 with [Pd(PPh3)4], i.e., Sn(IV)/Pd(0) starting materials, and Sn(pyS)2 with [PdCl2(PPh3)2], i.e., Sn(II)/Pd(II) starting materials.16 According to these reaction protocols, we have now succeeded in the syntheses of the nickel and platinum analogues [Cl2Sn(μ-pyS)2Ni–PPh3] (2) and [Cl2Sn(μ-pyS)2Pt–PPh3] (3) by the reaction of Cl2Sn(pyS)2 with [Ni(PPh3)4] and Sn(pyS)2 with [PtCl2(PPh3)2], respectively (Scheme 2). The reaction of Sn(pyS)2 with [NiCl2(PPh3)2] did not afford complex 2 and treatment of Cl2Sn(pyS)2 with [Pt(PPh3)4] gave 3 only in poor yields. In the same fashion we have synthesised complex [Cl2Ge(μ-pyS)2Pd–PPh3] (4) by reaction of Cl2Ge(pyS)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 with [Pd(PPh3)4] (Scheme 2). Complex [Cl2Si(μ-pyS)2Pd–PPh3] (5) could not be obtained by treatment of Cl2Si(pyS)219 with [Pd(PPh3)4] and also the reaction of [Pd(pyS)2(PPh3)2] with Me3Si–SiCl3 failed. (Under heating and availability of lone pair donors the disilane is expected to liberate “SiCl2” with formation of volatile ClSiMe3, and the silylene SiCl2 should react with [Pd(pyS)2(PPh3)2] to furnish 5.) Therefore, the structure of this compound was optimised by DFT calculations for further computational analyses (see the ESI†). The syntheses of compounds 616 and 716 (with different SnR2 moieties) and 87 (with different bridging ligands) have already been reported in the literature. For modifications at the trans-E–TM coordination site we aimed at substitution reactions of [Cl2Sn(μ-pyS)2Pd–PPh3] (1) with different phosphine ligands and found that the PPh3 ligand in 1 can be replaced by the better σ-donating PCy3 ligand to furnish 9, but not by PCl2Ph or P(OPh)3 (which are weaker σ-donor and stronger π-acceptor ligands). Furthermore, a reaction in the absence of a phosphine donor ligand was performed by treatment of Sn(pyS)2 with an equimolar amount of [PdCl2(MeCN)2]. This reaction was expected to yield either [ClSn(μ-pyS)2Pd–Cl] in which one chloro ligand remains at the palladium atom in order to stabilise the metal centre (compounds of the type [RSn(μ-C^P)Pd–Cl], with C^P = an ortho-metalated tertiary phosphine, are known in the literature20) or a MeCN-analogue of 1, i.e., [Cl2Sn(μ-pyS)2Pd–NCMe]. Instead, this reaction furnished the tetrameric complex [Cl2Sn(μ-pyS)2Pd]4 (10) in which the palladium atom of each [Cl2Sn(μ-pyS)2Pd] moiety forms a Pd–S bond to the next monomeric unit. An amine base supported substitution of the chloro ligands of 1 by the dianionic, tridentate (N,N,O)-ligands La and Lb in ethanol furnished complexes 11 and 12, respectively, with hexacoordinated tin and a formally covalent N–Sn bond trans to palladium (in 11) or a formally dative N–Sn bond trans to palladium (in 12). Related substitution reactions with dianionic tetradentate (O,N,N,O)-ligands (of salen type) failed. Instead, the (μ-OH)ClSn-bearing complex 13 (also with a hexacoordinated tin atom) was obtained in acceptable yield and was thus included in our systematic study. In compound 13 hexacoordination of the Sn atom results from dimerization of [(OH)ClSn(μ-pyS)2PdPPh3] via the Sn(μ-OH)2Sn bridge, a frequently encountered motif in tin chemistry.21
19 with [Pd(PPh3)4] (Scheme 2). Complex [Cl2Si(μ-pyS)2Pd–PPh3] (5) could not be obtained by treatment of Cl2Si(pyS)219 with [Pd(PPh3)4] and also the reaction of [Pd(pyS)2(PPh3)2] with Me3Si–SiCl3 failed. (Under heating and availability of lone pair donors the disilane is expected to liberate “SiCl2” with formation of volatile ClSiMe3, and the silylene SiCl2 should react with [Pd(pyS)2(PPh3)2] to furnish 5.) Therefore, the structure of this compound was optimised by DFT calculations for further computational analyses (see the ESI†). The syntheses of compounds 616 and 716 (with different SnR2 moieties) and 87 (with different bridging ligands) have already been reported in the literature. For modifications at the trans-E–TM coordination site we aimed at substitution reactions of [Cl2Sn(μ-pyS)2Pd–PPh3] (1) with different phosphine ligands and found that the PPh3 ligand in 1 can be replaced by the better σ-donating PCy3 ligand to furnish 9, but not by PCl2Ph or P(OPh)3 (which are weaker σ-donor and stronger π-acceptor ligands). Furthermore, a reaction in the absence of a phosphine donor ligand was performed by treatment of Sn(pyS)2 with an equimolar amount of [PdCl2(MeCN)2]. This reaction was expected to yield either [ClSn(μ-pyS)2Pd–Cl] in which one chloro ligand remains at the palladium atom in order to stabilise the metal centre (compounds of the type [RSn(μ-C^P)Pd–Cl], with C^P = an ortho-metalated tertiary phosphine, are known in the literature20) or a MeCN-analogue of 1, i.e., [Cl2Sn(μ-pyS)2Pd–NCMe]. Instead, this reaction furnished the tetrameric complex [Cl2Sn(μ-pyS)2Pd]4 (10) in which the palladium atom of each [Cl2Sn(μ-pyS)2Pd] moiety forms a Pd–S bond to the next monomeric unit. An amine base supported substitution of the chloro ligands of 1 by the dianionic, tridentate (N,N,O)-ligands La and Lb in ethanol furnished complexes 11 and 12, respectively, with hexacoordinated tin and a formally covalent N–Sn bond trans to palladium (in 11) or a formally dative N–Sn bond trans to palladium (in 12). Related substitution reactions with dianionic tetradentate (O,N,N,O)-ligands (of salen type) failed. Instead, the (μ-OH)ClSn-bearing complex 13 (also with a hexacoordinated tin atom) was obtained in acceptable yield and was thus included in our systematic study. In compound 13 hexacoordination of the Sn atom results from dimerization of [(OH)ClSn(μ-pyS)2PdPPh3] via the Sn(μ-OH)2Sn bridge, a frequently encountered motif in tin chemistry.21
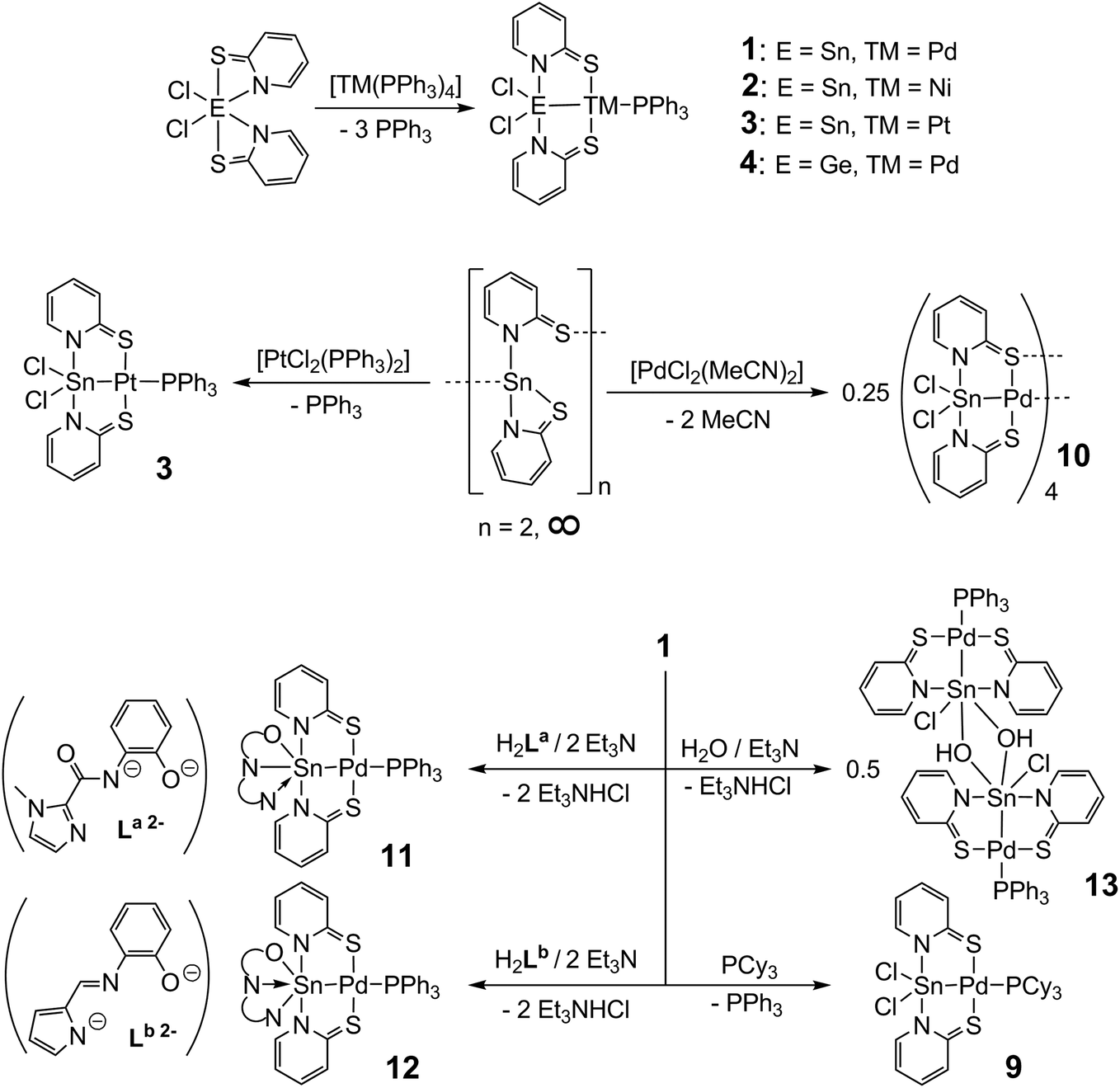 | ||
| Scheme 2 Syntheses of complexes 1, 2, 3, 4, 9, 10, 11, 12 and 13. | ||
Molecular structures, X-ray diffraction analyses
The herein presented compounds 2, 3, 4, 9, 10, 11, 12, and 13 were characterised by single-crystal X-ray diffraction analysis (Fig. 1) and, together with the data of compounds 6, 7 and 8, Table 2 lists selected corresponding bond lengths in the coordination spheres of their metal atoms. In general, these compounds have an almost square-planar coordinated group 10 atom with one of the substituents being the group 14 element in a distorted trigonal-bipyramidal (2, 3, 4, 9, 10) or octahedral (11, 12, 13) coordination sphere. In 11 and 12 we find the expected pattern of shorter (2.181(1) Å) vs. longer (2.198(1) Å) N–Sn bonds trans to the Sn–Pd bond, but their bond length difference is marginal. This is rather unexpected because the Sn(IV) and Sn(II) model compounds Sn(L)2 and Sn2(L)2, respectively, reveal more pronounced differences between the corresponding Sn–N bond lengths: Sn(La)2 2.097(3) and 2.106(4) Å vs. Sn(Lb)2 2.135(1) and 2.136(1) Å; Sn2(La)2 2.206(1) Å vs. Sn2(Lb)2 2.234(1) Å. (Crystallographic data of Sn2(La)2 and Sn(La)2 were taken from ref. 7, data from Sn2(Lb)2 and Sn(Lb)2 are listed in the ESI.†) Whereas for 2 and 4 we observe shortening of the E–TM bond with respect to 1 as expected (as a result of the significantly smaller group 10 or group 14 element, respectively), the Sn–Pd bonds of compounds 1, 6–13 cover the rather narrow range between 2.50 and 2.54 Å with 10 being an outlier with a rather short bond (2.48 Å) that can be interpreted as a result of the weaker trans-disposed donor (a long Pd–S bond), whereas the other complexes have Pd–P bonds of a similar length (between 2.35 and 2.38 Å). Penta- vs. hexacoordination of the tin atom exerts only marginal influence on the Sn–Pd separation. For the Pd–S bonds we observe a similarly narrow range (from 2.29 to 2.33 Å), whereas the Sn–N bond lengths appear to be more variable. The shortest Sn–N bonds to the bridging ligands are found in 8, this can be attributed to the different heterocycles of the bridging ligand (i.e., a five-membered ring with a more acute C–N–C angle, thus exerting less steric demand in the Sn coordination sphere). Correspondingly, the Pd–S bonds in 8 are slightly longer than in the other Sn–Pd compounds. With the resonance structures of the L- and Z-type borderline cases in mind, an increase in the Z-type characteristics is accompanied with an increase in the covalent character (strengthening) of the Sn–N bonds and the dative character (weakening) of the Pd–S bonds. Considering that covalent bonds are longer than formal dative bonds,22 compound 8 (having the shorter Sn–N and longer Pd–S bonds) should exhibit enhanced Z-type characteristics of the L/Z-type Sn-ligand (Scheme 3).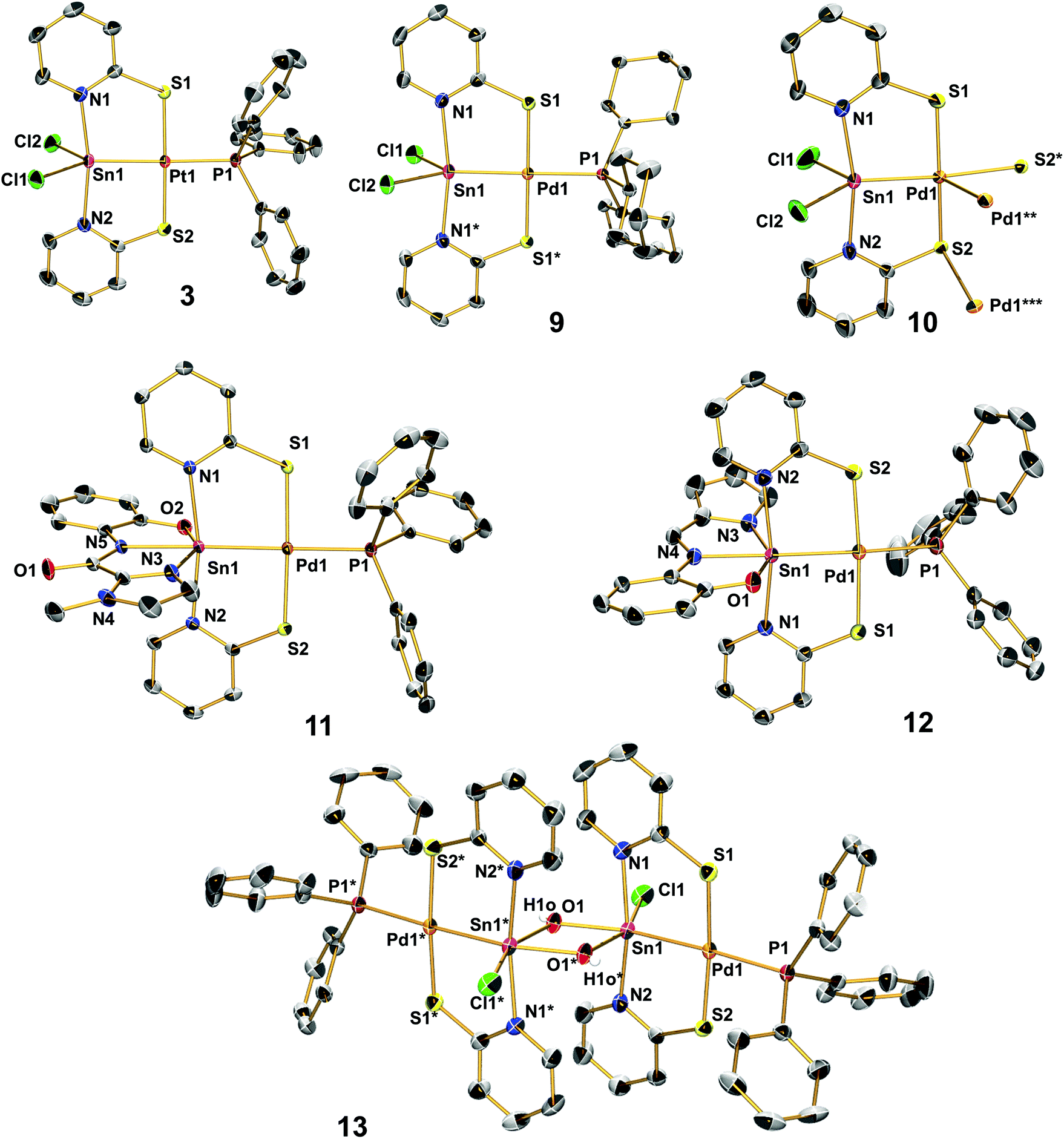 | ||
| Fig. 1 Molecular structures of complexes 3 (the molecular conformation of compounds 2 and 4 is similar to 3), 9, 10 (in 10·2 THF), 11 (in 11·CH2Cl2), 12 and 13 (in 13·2 EtOH·CH2Cl2) in the crystal. Ellipsoids are set at 50% probability, C-bound hydrogen atoms are omitted. Asterisked labels indicate symmetry equivalent positions according to x, 0.5 − y, z (for 9), * = 2 − y, x, 2 − z; ** = 2 − x, 2 − y, z; *** = y, 2 − x, 2 − z (for 10) and 2 − x, 1 − y, −z (for 13). | ||
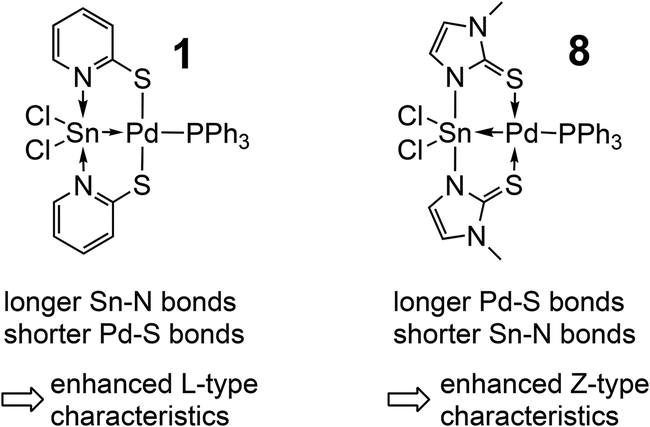 | ||
| Scheme 3 Comparison of 1 and 8 with respect to the Sn–N and Pd–S bond lengths. | ||
| E, TM | E–TM | E–N | TM–S | TM–P | |
|---|---|---|---|---|---|
| a Taken from ref. 16. b Data from two crystallographically independent molecules in the asymmetric unit. c Taken from ref. 7. d Pd–S(trans-Sn) 2.4250(6) Å. | |||||
| 1 | Sn, Pd | 2.5051(2) | 2.287(2), 2.292(2) | 2.2989(5), 2.3019(5) | 2.3751(4) |
| 2(1)b | Sn, Ni | 2.4304(3) | 2.298(2), 2.289(2) | 2.1537(5), 2.1613(6) | 2.2482(5) |
| 2(2)b | Sn, Ni | 2.4143(3) | 2.283(2), 2.287(2) | 2.1573(5), 2.1658(6) | 2.2320(5) |
| 3 | Sn, Pt | 2.5172(2) | 2.288(2), 2.278(3) | 2.2989(7), 2.2963(7) | 2.3255(6) |
| 4 | Ge, Pd | 2.3775(2) | 2.206(1), 2.146(1) | 2.3060(4), 2.2817(4) | 2.3667(3) |
| 6 | Sn, Pd | 2.5370(2) | 2.347(2), 2.387(2) | 2.2965(5), 2.3121(5) | 2.3786(5) |
| 7 | Sn, Pd | 2.5328(2) | 2.335(2), 2.386(2) | 2.3071(5), 2.3105(5) | 2.3531(5) |
| 8 | Sn, Pd | 2.5382(1) | 2.210(1), 2.231(1) | 2.3123(2), 2.3189(2) | 2.3703(2) |
| 9 | Sn, Pd | 2.5228(5) | 2.329(3) | 2.2941(8) | 2.383(1) |
| 10 | Sn, Pd | 2.4798(3) | 2.257(3), 2.356(3) | 2.2999(8), 2.3282(6) | (Pd–S)d |
| 11 | Sn, Pd | 2.5219(2) | 2.327(1), 2.298(1) | 2.2882(4), 2.2863(4) | 2.3736(4) |
| 12 | Sn, Pd | 2.5130(2) | 2.310(2), 2.322(2) | 2.3108(5), 2.2936(5) | 2.3785(5) |
| 13 | Sn, Pd | 2.5227(3) | 2.292(3), 2.311(3) | 2.3089(9), 2.3058(9) | 2.3687(8) |
The Pd–P bonds within the series of Sn–Pd compounds, ranging between 2.35 and 2.38 Å, do not reveal any remarkable response to the different Sn coordination spheres. Interestingly, the Pd–P bond to the stronger σ-donor phosphine (PCy3, in compound 9) is at the longer end of this range and cannot be interpreted as a result of a stronger trans-disposed donor, because the Sn–Pd bond in 9 is also longer than that in the corresponding PPh3 substituted compound 1. Instead, compound 9 reveals slightly shorter Pd–S and longer Sn–N bonds than 1, thus hinting at the compensation of the electron density shifts within the heterometallic core by the bridging pyridine-2-thiolate ligand. This electron density shift could be the shift of the Sn–Pd bond electron pair towards Sn, thus supporting enhanced L- (or lowered X- or Z-) type of Sn in this compound.
Hence, as the bond length characteristics of the herein studied compounds do not reveal clear trends for electron density shifts upon substitution of various parts of the complex (only some hints in the case of few compounds), the following two electron density features of the heterometallic core (1) valence shell population of the group 10 and group 14 element and (2) bond polarization of the intermetallic σ-bond were analysed with the aid of computational methods (NLMO analyses) and, in the case of the tin compounds, with 119Sn Mössbauer spectroscopy. In 2014 Gabbaï et al. and Limberg et al. have reported different descriptions of bond formation for exactly the same compound [ClBi(o-C6H4-PPh2)2AuCl].23 Whereas one paper reports Au→Bi σ-donation and non-bonding features of the Bi(6s) lone-pair, the other paper reports a combination of Au(5dx2−y2)→Bi(6py) σ-donation and Au(6s)←Bi(6s) σ-backdonation (of different magnitude, of course, to eventually yield the same final electron density distribution). Hence, in the following we will exclusively focus on the “as is” situation rather than employing any “electron flow” models.
Valence shell population (119Sn Mössbauer spectroscopy and Natural Electron Configuration analyses)
119Sn Mössbauer spectroscopy, as an experimental probe for the 5s orbital population of tin atoms, proved the (5s) electronic situation of the herein studied tin compounds to be situated almost midway between Sn(II) and Sn(IV) compounds. According to the resonance structures mentioned in Scheme 1 this points at the X-type ligand characteristics of the Sn containing ligand in the transition metal coordination sphere, i.e., covalent Sn(III)–TM(I) bonding situations. Interestingly, the quadrupole splittings (Δ) of the 119Sn Mössbauer signals of the hexacoordinated Sn compounds (1.86–2.06 mm s−1) are found in the same range as those of the pentacoordinated Sn compounds (1.59–2.86 mm s−1). The rather broad range of 119Sn isomer shifts δ (1.27–1.81 mm s−1), however, hints at tendencies towards notable contributions of the Z- or L-type ligand contributions, i.e., shifts towards Sn(IV) or Sn(II) characteristics, respectively. Pronounced shifts to Sn(IV) characteristics were found with compounds 11, 12 and 13, which comprise hexacoordinated Sn atoms. This effect can be caused by an increase of the ionicity of the Sn–Pd bond by an electron density shift towards Pd (and similar effects on the other Sn–X bonds) upon enhancing the Sn coordination number or by general lowering of the 5s orbital contribution in bond formation with a simultaneous increase of the 5p orbital contributions to bond formation. For a closer inspection of these effects the Natural Electron Configurations (NECs) of the metal atoms were computed and analysed (see details in the ESI†). The calculated 5s populations of the tin atoms correlate very well with the isomer shifts observed in the 119Sn Mössbauer spectra (Fig. 2), thus providing a foundation of reliability for the calculated data (Table 3). The NECs are very similar for the transition metal atoms of the herein investigated series of compounds (except of the Pt compound 3, the higher valence shell s orbital population can be rationalised by relativistic considerations), and therefore their Natural Charges (NCs) are very similar. The 5s and 5p populations (and also the s/p population ratio) of the tin atoms vary noticeably. As tin(II) compounds comprise an Sn-located lone pair with high s-orbital contributions and thus exhibit a noticeably higher 5s/5p ratio than Sn(IV) compounds, we had a closer look at this feature. Thus, we have analysed the NEC for some Sn(II) (5s/5p for Sn2(La)2 2.00, SnCl3− 1.50, SnCl42− 1.69) and some Sn(IV) benchmark compounds (5s/5p for Sn(La)2 0.77, SnCl4 0.76, SnCl5− 0.74, SnCl62− 0.72). Most of the pentacoordinated tin compounds exhibit a similar 5s and 5p population (in the ranges 1.20–1.32 and 1.38–1.51 electrons, respectively) and therefore a similar s/p ratio (0.86–0.94). In compounds 5 and 6, however, the lower valence shell s orbital population is compensated by a higher p orbital population, thus reflected by a noticeably lower s/p ratio but similar NCs of the group 14 element. Whereas in the case of compound 5 this lowering of the s orbital population originates from a different group 14 element (Si vs. Sn). In compound 6 the less electronegative Sn-bound substituents (phenyl groups) can be considered relevant as their lower electronegativity is in support of an enhanced p orbital contribution to the bonding situation of the other bonds (according to Bent's rule24).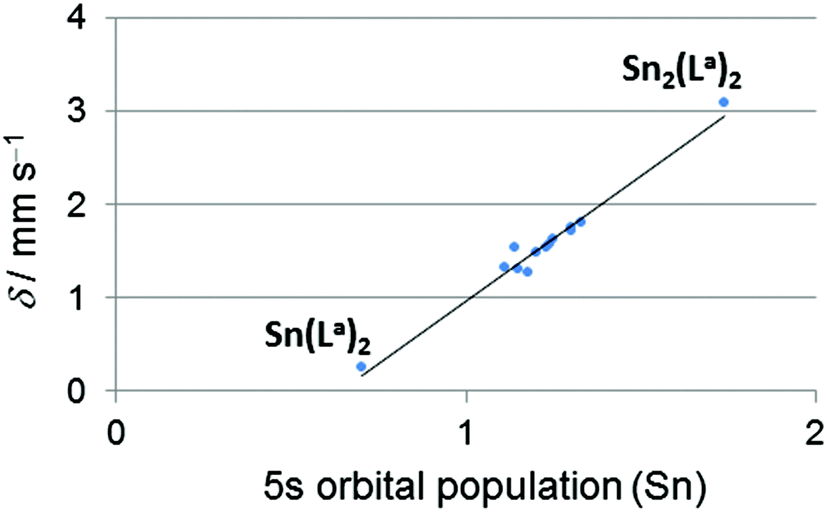 | ||
| Fig. 2
119Sn Mössbauer isomer shifts (δ) vs. calculated 5s orbital population from the tin atom's calculated Natural Electron Configuration of compounds 1–3 and 6–13 as well as the transition metal free Sn(II) and Sn(IV) reference compounds Sn2(La)27 and Sn(La)2,7 respectively, which contain the same (O,N,N) tridentate ligand as compound 11. | ||
| Compound | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Group 10 | Pd | Ni | Pt | Pd | Pd | Pd | Pd | Pd | Pd | Pd | Pd | Pd | Pd | Pd | Pd |
| Group 14 | Sn | Sn | Sn | Ge | Si | Sn | Sn | Sn | Sn | Sn | Sn | Sn | Sn | Sn | Sn |
| δ/mm s−1 | 1.63 | 1.71 | 1.58 | n/a | n/a | 1.54 | 1.81 | 1.48 | 1.75 | 1.53 | 1.32 | 1.27 | 1.30 | ||
| Δ/mm s−1 | 1.67 | 1.68 | 1.83 | n/a | n/a | 2.86 | 2.00 | 1.59 | 1.83 | 1.77 | 2.06 | 1.86 | 1.88 | ||
| NEC Group 14 | |||||||||||||||
| s | 1.25 | 1.30 | 1.24 | 1.32 | 1.13 | 1.14 | 1.33 | 1.20 | 1.30 | 1.23 | 1.11 | 1.18 | 1.15 | 1.38 | 1.35 |
| p | 1.39 | 1.40 | 1.40 | 1.66 | 1.74 | 1.49 | 1.51 | 1.40 | 1.38 | 1.38 | 1.17 | 1.17 | 1.19 | 1.38 | 1.53 |
| s/p | 0.90 | 0.93 | 0.89 | 0.80 | 0.65 | 0.77 | 0.88 | 0.86 | 0.94 | 0.89 | 0.95 | 1.01 | 0.97 | 1.00 | 0.88 |
| NEC Group 10 | |||||||||||||||
| s | 0.53 | 0.53 | 0.74 | 0.51 | 0.52 | 0.52 | 0.49 | 0.53 | 0.52 | 0.48 | 0.50 | 0.51 | 0.51 | 0.45 | 0.50 |
| d | 9.30 | 9.18 | 9.23 | 9.26 | 9.28 | 9.31 | 9.30 | 9.30 | 9.30 | 9.29 | 9.32 | 9.30 | 9.32 | 9.10 | 9.18 |
| NC | |||||||||||||||
| Group 10 | 0.15 | 0.26 | 0.00 | 0.21 | 0.18 | 0.15 | 0.20 | 0.16 | 0.16 | 0.20 | 0.16 | 0.17 | 0.15 | 0.42 | 0.31 |
| Group 14 | 1.30 | 1.24 | 1.31 | 0.96 | 1.04 | 1.34 | 1.11 | 1.35 | 1.27 | 1.34 | 1.67 | 1.61 | 1.60 | 1.18 | 1.06 |
In addition to the lower 5s population, the hexacoordinated tin compounds 11, 12 and 13 exhibit an even more pronounced lowering of the 5p orbital population, reflected by the systematically higher s/p ratio and noticeably more positive NC of the tin atom. In addition to the lower 5s population and the trend of the 119Sn Mössbauer isomer shift arising therefrom, only the general lowering of the valence shell population of the tin atoms in the case of Sn hexacoordination indicates pronounced Z-type ligand characteristics for Sn in compounds 11, 12 and 13. As benchmark compounds with an Sn–Pd bond we have included [Cl3Sn–PdCl3]2− (ref. 25) (14) and [Cl3Sn–Pd(2-methylallyl)(CO)]26 (15) in our analysis (Table 3). In spite of the different Pd substitution patterns the electronic features of these two compounds are surprisingly similar to one another. Between compounds 14 and 15 and the group of the herein investigated Sn–Pd compounds (1, 6–13), however, we find some noteworthy differences. In 14 and 15 the tin atom exhibits a higher 5s orbital population (but a similar s/p ratio though) and less positive NC. Vice versa, the Pd atoms in 14 and 15 are more electron deficient, as reflected by their enhanced positive NC, which mainly arises from a lowered 4d orbital population.
Natural localised molecular orbital (NLMO) analyses
As indicated by the 119Sn NMR shifts of compounds 1–3 and 6–13 (Table 4), the tin atoms are penta- (1–3, 6–10) or hexacoordinated (11–13), and thus the Sn–TM bond can be considered to be a regular two electron bond. The 119Sn NMR shift itself, however, cannot be used as a measure for evaluating the character of the Sn–TM bond, because in addition to the tin coordination number this shift value is strongly influenced by the various dia- and paramagnetic shielding effects as well as relativistic effects of all substituents. Therefore, we need to point out that the presence of the Sn–TM bond is furthermore supported by the 2J(119Sn,31P) coupling observed for the compounds with a Sn–TM–P feature (Table 4) and the 1J(195Pt,119Sn) coupling of 18.6 kHz observed for compound 3. Thus, our further investigation addressed the location and composition of this electron pair (also for the Ge and Si compounds 4 and 5, respectively), which should provide some insights into the influence of substitution patterns on L- vs. X- vs. Z-type ligand characteristics. The bridging ligands used in this study allow for drawing formal E(II)→TM(II), E(III)–TM(I) and E(IV)←TM(0) structures without any charges. Therefore, similar contributions of both bonding partners to the E–TM bond are in support of X-type characteristics (covalent bond); polarisation of this electron pair towards one bonding partner would support the relevance of resonance structures with the electron rich partner as the Lewis base (L-ligand) and the electron deficient partner as the Lewis acid (Z-type). For comparison (as benchmark for stannyl complexes, in which the tin ligand is referred to as an X-type ligand, because this gives rise to the only resonance structure without formal charges, unless the compound itself carries an ionic charge) we have included the NLMO analyses of [Cl3Sn–PdCl3]2− (ref. 25) (14) and of [Cl3Sn–Pd(2-methylallyl)(CO)]26 (15). As shown in Table 4, for each compound under investigation the NLMO of interest is composed of more than 90% orbital contributions of the two core bonding partners (and thus only a minor fraction of delocalised contributions). In the reference compounds 14 and 15 the Sn–Pd bond is significantly polarised towards tin, as reflected by the ratio of the metal contributions to the NLMO (%TM/%E) of 0.35 and 0.53, respectively. The overall appearance of the Sn–Pd NLMO is similar for these two compounds (Fig. 3). The Sn–Pd containing compounds of the series 1–13 reveal significantly higher transition metal contributions, reflected by %TM/%E ranging between 0.63 and 0.83. Furthermore, the overall appearance of the E–TM NLMO in these compounds is noticeably different from those in 14 and 15 but similar within the series of all N,S-bridged complexes 1–13 (for representative examples see Fig. 3, for the other NLMOs see the ESI†). As can be seen for examples 1vs.12, the tin atom's coordination geometry does not reveal any significant impact on the appearance of this NLMO. Thus, considering the Cl3Sn– group (in 14 and 15) as an X-type ligand a trend towards Z-type characteristics of the tin site can be deduced. Even the value for compound 4 (0.56) is still higher, although tin is replaced by the more electronegative germanium. Vice versa, the upper limit is exceeded by compound 3 (0.87) in which palladium is replaced by the more electronegative platinum. With reference to compound 1, most ligand substitutions studied in this series lead to a shift of the atomic contributions to the Sn–Pd NLMO towards palladium, the only exception being compound 9 (%TM/%E = 0.63), in which the reverse shift is expected because of the stronger σ-donor phosphine (PCy3) trans to the Sn–Pd bond.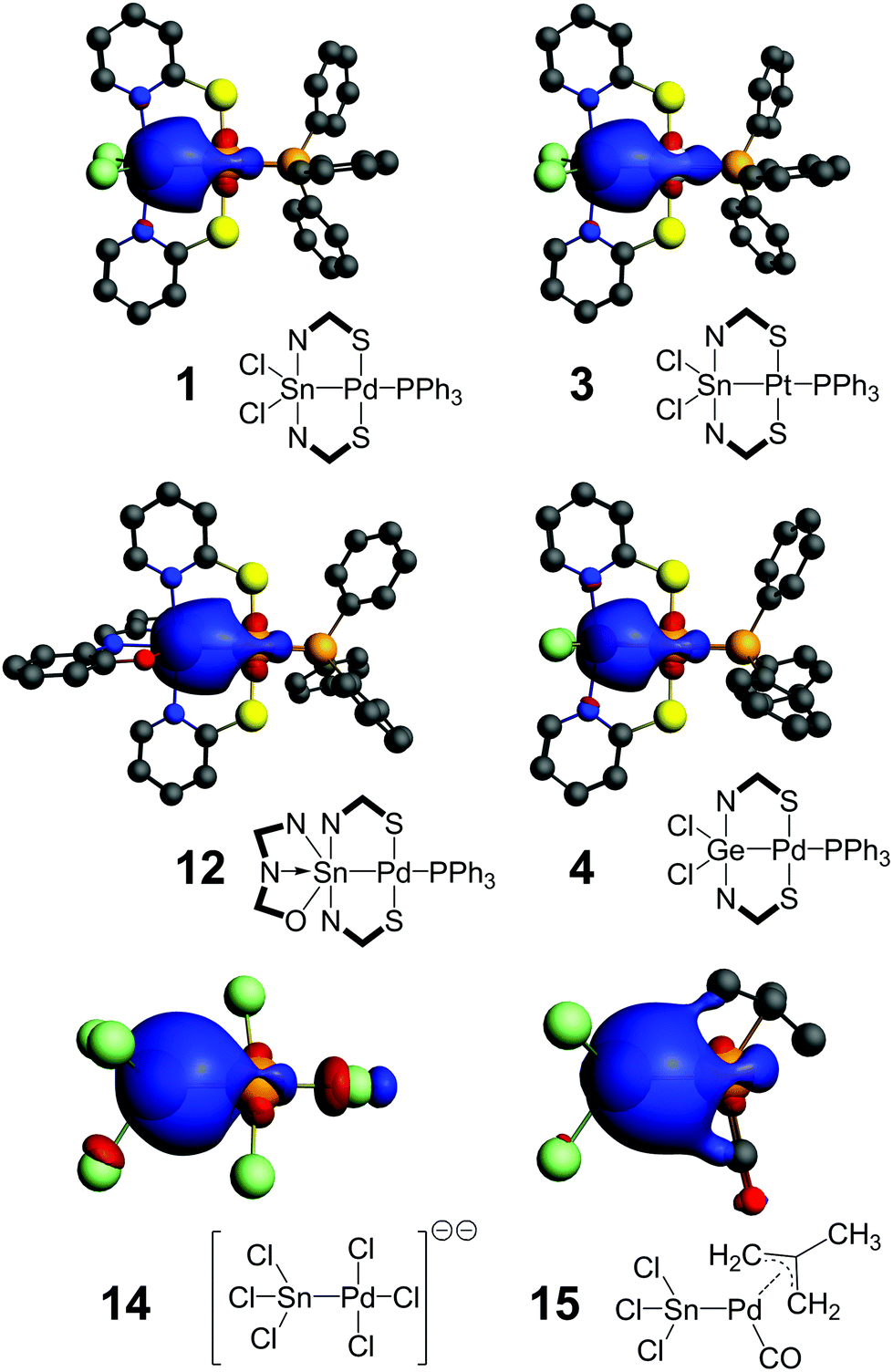 | ||
| Fig. 3 NLMOs of the E–TM bonds in 1, 3, 4, 12, 14 and 15 (plotted at an isosurface value of 0.03 e). | ||
| Compound | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 119Sn NMR (δ/ppm) | −122 | −338 | −30 | n/a | n/a | 1.4 | −212 | −337 | −103 | −160 | −378 | −361 | −448 | ||
| −357 | |||||||||||||||
| 2 J(119Sn,31P)/kHz | 4.31 | 2.81 | 4.14 | n/a | n/a | 2.68 | 4.60 | 4.68 | 4.05 | n/a | 4.55 | 4.40 | 4.73 | ||
| NLMO | |||||||||||||||
| %TM | 39.6 | 37.3 | 45.1 | 34.7 | 43.7 | 44.0 | 39.2 | 44.0 | 37.1 | 41.7 | 42.9 | 39.7 | 43.3 | 24.1 | 32.6 |
| %E | 56.2 | 59.5 | 51.6 | 61.7 | 52.0 | 51.2 | 56.1 | 51.8 | 59.1 | 53.8 | 52.2 | 55.7 | 52.1 | 68.3 | 61.2 |
| SUM | 95.8 | 96.9 | 96.8 | 96.4 | 95.7 | 95.2 | 95.3 | 95.8 | 96.2 | 95.5 | 95.2 | 95.4 | 95.4 | 92.4 | 93.8 |
| %E/SUM | 0.59 | 0.62 | 0.53 | 0.64 | 0.54 | 0.54 | 0.59 | 0.54 | 0.61 | 0.56 | 0.55 | 0.58 | 0.55 | 0.74 | 0.65 |
| %TM/%E | 0.70 | 0.63 | 0.87 | 0.56 | 0.84 | 0.86 | 0.70 | 0.85 | 0.63 | 0.78 | 0.82 | 0.71 | 0.83 | 0.35 | 0.53 |
| % s TM | 26.6 | 30.6 | 36.2 | 26.9 | 25.7 | 24.0 | 26.4 | 26.9 | 27.6 | 25.7 | 22.2 | 24.2 | 24.7 | 27.1 | 42.3 |
| % d TM | 73.2 | 69.1 | 63.7 | 73.0 | 74.2 | 75.9 | 73.4 | 72.9 | 72.2 | 74.2 | 77.6 | 75.6 | 75.2 | 71.9 | 57.2 |
| d/s TM | 2.75 | 2.26 | 1.76 | 2.71 | 2.89 | 3.16 | 2.78 | 2.71 | 2.62 | 2.89 | 3.50 | 3.12 | 3.04 | 2.65 | 1.35 |
| % s E | 69.3 | 71.5 | 67.6 | 67.2 | 56.7 | 55.9 | 69.7 | 66.1 | 71.2 | 69.0 | 69.4 | 72.7 | 73.7 | 74.4 | 66.7 |
| % p E | 30.2 | 28.1 | 32.0 | 32.5 | 42.9 | 43.6 | 30.0 | 33.3 | 28.5 | 30.4 | 30.2 | 26.9 | 25.7 | 25.4 | 32.9 |
| s/p E | 2.29 | 2.54 | 2.11 | 2.07 | 1.32 | 1.28 | 2.32 | 1.98 | 2.50 | 2.27 | 2.30 | 2.70 | 2.88 | 2.93 | 2.03 |
The atomic orbital contributions of the metal atoms towards the E–TM NLMO can be described as hybrids of d and s for TM and s and p for E (with only marginal contributions of p or d, respectively). The s/p ratio of E ranges between 2 and 3 for most of the herein studied compounds, only the silicon compound 5 (s/p 1.32) and the Ph2Sn functionalised compound 6 (s/p 1.28) exhibit pronounced p orbital contributions of their group 14 element, in accord with the lower valence shell s orbital population of these atoms (vide supra). The d/s ratio of the transition metals’ contributions also ranges between 2 and 3 for most of the herein studied compounds, with exceptions found for platinum compound 3 (d/s 1.76) because of the different transition metals and for compounds 6, 11, 12 and 13 (d/s > 3). The latter seems to correlate with their pronounced shift of the 119Sn Mössbauer signal to lower velocities (pronounced Sn(IV) contributions).
Interestingly, the d/s ratio of the Pd atom's contributions to the Sn–Pd NLMO is only 1.35 in the reference compound 15, whereas for compound 14 we find a value similar to those of our N,S-bridged complexes. We interpret this different behaviour of Pd in 15 as a result of the completely different Pd coordination sphere caused by the allyl group.
Summary of computational analyses
As only one of the discrete electronic features such as NLMO composition, Natural Charges or Natural Electron Configurations cannot describe the E–TM bonding situation between Sn(II) (L-type), Sn(III) (X-type) and Sn(IV) (Z-type) in a satisfactory manner, the combination of the features is mapped for the Sn–Pd compounds (and Sn benchmark compounds) in Fig. 4.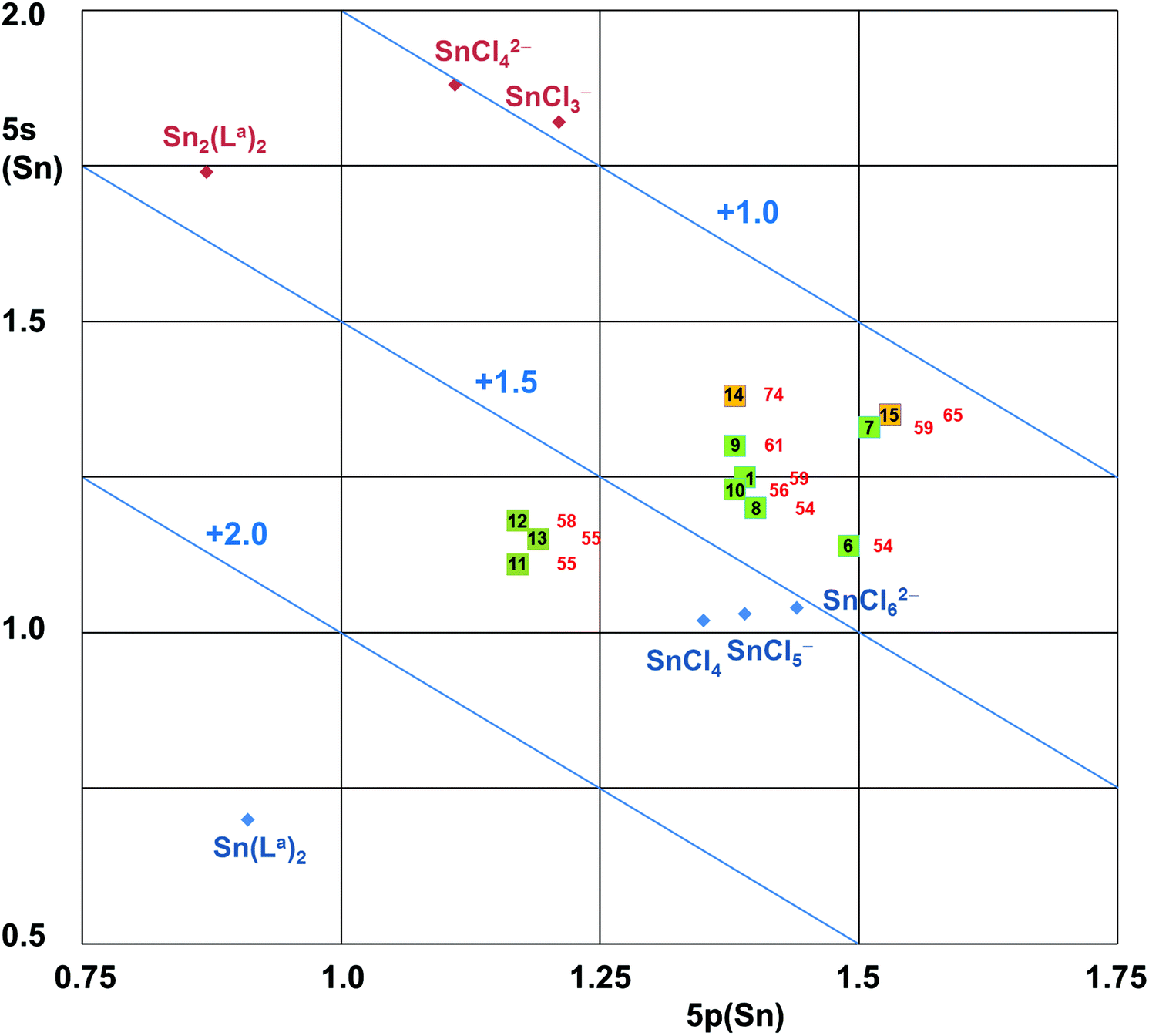 | ||
| Fig. 4 5s vs. 5p orbital populations (from calculated NECs) of Sn-atoms herein reported as compounds with a Sn–Pd bond and selected Sn(II) and Sn(IV) reference compounds. Note: The diagonal lines of charges (+2.0, +1.5, +1.0) correspond to the electron deficiency in 5s and 5p orbitals with respect to four valence shell electrons of Sn. Hence, they are similar (but not equal) to the NCs of the Sn atoms. The bold numbers in the squares correspond to the compounds, the red numbers next to them represent the percentage of Sn contributions (out of Sn and Pd) to the Sn–Pd NLMO (corresponding to the entries in row 7 of Table 4). | ||
The location of the Sn(IV) benchmark compounds reflects the enhanced Sn charge compensation by the electron density of softer ligands (better charge compensation in chlorotin compounds with respect to Sn(La)2 with O,N,N′ donor ligands) while the 5s/5p ratio remains similar. For the Sn(II) benchmark compounds we also found the trend of enhanced charge compensation by the softer ligands, but the already high 5s orbital population required charge compensation by enhanced 5p orbital population (thus lowering the 5s/5p ratio for the chloro compounds).
On this map the Cl3Sn–Pd reference compounds 14 and 15 are located midway between the groups of Sn(II) and Sn(IV) references on the side of pronounced charge compensating ligands. With respect to references 14 and 15 the positions of the other Sn–Pd compounds reported in this paper are shifted towards the group of the Sn(IV) reference compounds. There is a notable separation between the groups of Sn–Pd compounds with a hexacoordinated Sn atom (11, 12, 13) and those with a pentacoordinated Sn atom (1, 6, 7, 8, 9, 10). The relative positioning of these two groups indicates a better charge compensation in the case of the compounds with a pentacoordinated Sn atom (which have SnCl2, SnS2 or SnC2 moieties), which is pronounced for compound 7 (having a SnS2 moiety). On this map one can see that both groups (Sn–Pd compounds with penta- and hexacoordinated Sn atom) are in a similar relative distance to the groups of Sn(II) and Sn(IV) reference compounds, those with an hexacoordinated tin atom only marginally closer to Sn(IV) than those with pentacoordinated Sn.
The relative Sn atom contributions to the Sn–Pd NLMO indicates a further systematic feature about this map, i.e., those compounds with enhanced Sn contributions to the Sn–Pd NLMO are shifted towards the direction of the Sn(II) references. Thus, their enhanced Sn contribution in combination with the increase of the 5s/5p ratio hints at an enhanced lone pair character (i.e., a shift towards L-type characteristics), whereas the opposite direction thus indicates a shift towards X- or Z-type characteristics. The relative positioning of compounds 8 and 9 on this map (i.e., 9 shifted towards Sn(II), 8 shifted towards Sn(IV) relative to compound 1) is in agreement with the prediction made from the analyses of the molecular structures obtained by X-ray crystallography. In addition to compound 8 (having the mt bridges), on this map compound 6 (having SnPh2) is particularly close to the group of Sn(IV) references, while their Sn contributions to the Sn–Pd NLMO (54%) indicates “perfectly covalent” (i.e., X-type) bonding. Thus, these two compounds demonstrate the limits of this analysis: as soon as the Sn lone pair character (L-type feature) vanishes, the tin atom becomes tetravalent (regardless of the tin coordination number). Additional analyses (e.g., contributions to the NLMO of interest) will then be required to differentiate between the X- or Z-type ligand characteristics of Sn in each case. Regarding compounds 14 and 15 as stannyl (X-type Cl3Sn– group) compounds, the other Sn–Pd compounds are then shifted towards Z-type Sn. Regarding the Sn atom's contribution to the NLMO representative of the Sn–Pd bond as the absolute measure, compounds such as 6 and 8 are close to X-type Sn, whereas the other compounds (14 and 15 in particular) exhibit a pronounced L-type Sn character.
Conclusion
In extension of our initial studies of heterobimetallic Sn–Pd complexes which have tin based σ-donor/acceptor confused ligands (L/Z-type ligands), we have now selectively synthesised a library of new compounds with different combinations of group 14 and group 10 metal atoms and ligand patterns. The compounds have been characterised by means of melting point determination, elemental analysis, multinuclear NMR, 119Sn Mössbauer spectroscopy and single crystal X-ray diffraction analysis. Furthermore, NLMO analyses were carried out and a detailed comparison of structural, spectroscopic and quantum chemical data allowed insights into the differences of the ligand behaviours of structurally related compounds, i.e. differences in the ambiguous L-, X-, and Z-type ligand characteristics.Besides the investigation of some recently reported heterobinuclear complexes,27,28 this is the first comprehensive study with special emphasis on the substitution induced variation of the L/X/Z-type behaviour of a ligand system. Whereas clear cut differences between Sn(II) and Sn(IV) (with and without Sn-located lone pair, respectively) in tin oxo compounds can be easily detected by the anisotropic 119Sn NMR properties,29 combinations of 119Sn Mössbauer spectroscopy, crystallography and NLMO analyses allowed for the analyses of the rather continuous changes between electron rich and electron deficient group 14 element ligand sites in ligand bridged heterobimetallic complexes. With respect to the electronic situation in trichlorostannyl palladium compounds, in which the Cl3Sn group is referred to as an X-type ligand, we found: (i) still rather covalent E–TM bonds, suggesting the ligands to be described as X-type, but (ii) noticeable electron density shifts of the Sn–Pd bonds towards Pd in our compounds (which have penta- and hexacoordinated Sn atoms) with respect to the Sn–Pd bonds in two different trichlorostannyl palladium reference complexes and (iii) a pronounced loss of the Sn valence shell electron density in complexes having hexacoordinated E atoms.
Experimental
All reactions were routinely carried out under an atmosphere of dry argon using standard Schlenk and glovebox techniques. Solvents were dried by distillation from Na/benzophenone (THF, diethyl ether and Et3N), distillation over sodium (toluene, pentane), distillation over CaH2 for dichloromethane (DCM) and storage over a molecular sieve 3 Å (chloroform). The compounds [Cl2Sn(μ-pyS)2Pd–PPh3],16 Sn(pyS)2,19 Cl2Sn(pyS)2,19 [PtCl2(PPh3)2],30 H2La,31 H2Lb,32 H2Sal3,33 H2Sal5,33 SnCl2(dioxane),34 and [PdCl2(MeCN)2]35 were prepared according to previously published methods; all other chemicals were commercially available and used as received. (NH4)2SnCl6 was synthesised from the reaction of SnCl2 and NH4Cl with Cl2 in diluted hydrochloric acid. The melting points of the compounds were determined using a microscope “Polytherm A” (Wagner & Munz); they are not corrected. Elemental analyses were measured using a C/H/N/S analyser “vario micro cube” (Elementar). 1H, 13C, 31P and 119Sn NMR spectra were recorded on a DPX 400 spectrometer (Bruker) and were referenced against internal SiMe4 (1H, 13C), external H3PO4, 85% (31P) and external SnMe4 (119Sn). The 31P and 119Sn MAS NMR spectra were recorded on an Avance 400 WB spectrometer (Bruker) using 4 mm or 2.5 mm ZrO2 rotors. The spectra were referenced against external H3PO4, 85% (0 ppm, 31P) and SnO2 (−603 ppm relative to SnMe4, 119Sn). The δiso values in the 119Sn MAS NMR spectra were determined by recording spectra at two different rotation frequencies.A Ca119mSnO3 source was available for the 119Sn Mössbauer spectroscopic investigation. The sample was placed within a PMMA container (2 cm diameter) at a thickness of about 10 mg Sn cm−2 (if necessary, the sample was diluted with quartz powder). A palladium foil of 0.05 mm thickness was used to reduce the tin K X-rays concurrently emitted by this source. The measurement was conducted in the usual transmission geometry at 78 K. Fitting of the spectra was performed with the Normos-90 program system.36 In some of the spectra we observed asymmetric signals caused by the Goldansky–Karyagin effect. As this effect is temperature dependent, we have recorded the spectrum of complex 2 (which revealed the most asymmetric signal of the complexes studied) at 5 K and found a decrease of the asymmetry of the signal (see the ESI†). For the determination of the X-ray crystal structures, a crystal of the appropriate size was selected under inert oil and mounted on a glass capillary by applying a small amount of silicone grease. The dataset was collected on an IPDS 2(T) diffractometer (STOE) using monochromated Mo-Kα radiation (0.71073 Å). The structures were solved by direct methods (ShelXS) and refined in full-matrix least-squares cycles against F2 (ShelXL).37 Hydrogen atoms were refined isotropically in geometrically idealised positions with constrained C–H distances. The hydrogen atoms of the OH group in the structure of 16·2 THF·DCM were detected on the Fourier map and refined without positional constraint. The Uiso values were set to 1.2Ueq (or in the case of Me and OH groups to 1.5Ueq). Parameters of data collection and structure refinement of the crystal structures discussed in this paper are reported in the ESI.† CCDC 999931 (2), 999939 (3), 999940 (4), 999932 {O[ClGe(μ-pyS)2Pd-PPh3]2},38999998 [Cl2Sn(μ-bztzS)2Ni-PPh3],38999936 (9), 1000003 (10·2 THF), 999935 (11·DCM), 999933 (12), 1000002 (13·2 EtOH·DCM) 999937 (Sn2(Lb)2), 999938 (Sn(Lb)2·MeOH) and 999934 ([Ni2(bztzS)4]).
Syntheses
MP: 158–163 °C.
1H NMR (400.13 MHz, CDCl3): δ = 7.00 (m, 2H, H-5 pyS), 7.36 (m, 2H, H-4 pyS), 7.41–7.50 (mm, 11H, H-3 pyS/Phmeta/Phpara), 7.74 (m, 6H, Phortho), 8.59 ppm (d, 3J1H–1H = 5.9 Hz, 2H, H-6 pyS);
13C{1H} NMR (100.63 MHz, CDCl3): δ = 117.9 (C-5 pyS), 126.1 (C-3 pyS), 128.2 (Phmeta), 129.7 (d, 1J13C–31P = 40 Hz, Phipso), 130.6 (Phpara), 134.8 (Phortho), 137.7 (C-4 pyS), 145.2 (C-6 pyS), 159.3 ppm (C-2 pyS);
31P{1H} NMR (161.98 MHz, CDCl3): δ = 29.5 (broad) ppm;
31P MAS NMR (162.02 MHz, vrot: 15 kHz): δ = 30.1 ppm (satellites: 2J31P–(119/117)Sn = 2808 Hz);
119Sn MAS NMR (149.17 MHz, vrot: 14 and 15 kHz): δ = −337.9, −356.9 ppm (2 d);
119Sn Mössbauer (Ca119mSnO3, 78 K): δ = 1.71(1) mm s−1, Δ = 1.66(1) mm s−1.
Anal. calcd for C28H23Cl2N2NiPS2Sn (730.91 g mol−1): C 46.01, H 3.17, N 3.83; found: C 46.06, H 3.19, N 3.84%.
:1) and dried in vacuo. Yield: 67 mg (77 μmol, 62%); method b. [Pt(PPh3)4] (150 mg, 0.35 mmol) was suspended in THF (15 mL) and solid Cl2Sn(pyS)2 was added. The mixture was refluxed for 2 h. Thereafter, the mixture was filtered and diethyl ether was diffused into the solution over the gas phase at rt. Yellow crystals of [Pt(pyS)2(PPh3)2]39 formed within 3 d. The supernatant was decanted off and the volume was reduced in vacuo to about 5 mL. Diethyl ether was slowly diffused into the solution over the gas phase and the orange crystals of the product formed within a week, which were isolated by decantation, washed two times with 1 mL of a mixture of THF/diethyl ether (2:1) and dried in vacuo. Yield: 54 mg (63 μmol, 18%).
MP: not observed (stable up to 250 °C).
1H NMR (400.13 MHz, CDCl3): δ = 7.00 (m, 2H, H-5 pyS), 7.31 (m, 2H, H-4 pyS), 7.44–7.51 (mm, 9H, Phmeta/Phpara), 7.60 (d, 3J1H–1H = 8.0 Hz, 2H, H-3 pyS), 7.66–7.72 (m, 6H, Phortho), 8.63 ppm (d, 3J1H–1H = 5.4 Hz, 2H, H-6 pyS);
13C{1H} NMR (100.63 MHz, CDCl3): δ = 118.2 (C-5 pyS), 125.0 (C-3 pyS), 128.2 (d, 3J13C–31P = 11 Hz, Phmeta), 128.9 (d, 1J13C–31P = 53 Hz, Phipso), 131.1 (Phpara), 134.9 (d, 2J13C–31P = 11 Hz, Phortho), 138.1 (C-4 pyS), 145.7 (C-6 pyS), 156.3 ppm (C-2 pyS);
31P{1H} NMR (161.98 MHz, CDCl3): δ = 24.3 ppm (satellites: 2J31P–(119/117)Sn = 4080 Hz; 2J31P–195Pt = 3330 Hz);
119Sn{1H} NMR (CDCl3, 149.21 MHz): δ = −30.0 ppm (d, 2J119Sn–31P = 4137 Hz, satellites: 1J119Sn–195Pt = 18580 Hz);40
119Sn Mössbauer (Ca119mSnO3, 78 K): δ = 1.58(1) mm s−1, Δ = 1.83(1) mm s−1.
Anal. calcd for C28H23Cl2N2PPtS2Sn (867.29 g mol−1): C 38.78, H 2.67, N 3.23; found: C 38.77, H 2.58, N 3.21%.
:1) and dried in vacuo. The crystals were suitable for X-ray diffraction analysis. Yield: 38 mg (52 μmol, 40%).
MP: 268–271 °C.
1H NMR (400.13 MHz, CDCl3): δ = 6.99 (m, 2H, H-5 pyS), 7.36 (m, 2H, H-4 pyS), 7.43–7.52 (mm, 11H, H-3 pyS/Phmeta/Phpara), 7.64–7.69 (m, 6H, Phortho), 8.66 ppm (d, 3J1H–1H = 5.4 Hz, 2H, H-6 pyS);
13C{1H} NMR (100.63 MHz, CDCl3): δ = 117.6 (C-5 pyS), 124.3 (C-3 pyS), 128.4 (d, 3J13C–31P = 10 Hz, Phmeta), 129.6 (d, 1J13C–31P = 42 Hz, Phipso), 130.9 (Phpara), 134.8 (d, 2J13C–31P = 12 Hz, Phortho), 137.3 (C-4 pyS), 144.9 (C-6 pyS), 157.2 ppm (C-2 pyS);
31P{1H} NMR (161.98 MHz, CDCl3): δ = 25.8 ppm;
Anal. calcd for C28H23Cl2GeN2PPdS2 (732.54 g mol−1): C 45.91, H 3.17, N 3.82; found: C 46.15, H 3.26, N 3.80%.
MP: not observed (stable up to 250 °C).
31P MAS NMR (162.02 MHz, vrot: 10 kHz): δ = 31.0 ppm (satellites: 2J31P–119Sn = 4045 Hz, 2J31P–117Sn = 3911 Hz);
119Sn MAS NMR (149.17 MHz, vrot: 13 and 13.5 kHz): δ = −103 ± 5 ppm;
119Sn Mössbauer (Ca119mSnO3, 78 K): δ = 1.75(1) mm s−1, Δ = 1.83(1) mm s−1.
Anal. calcd for C28H41Cl2N2PPdS2Sn (796.78 g mol−1): C 42.21, H 5.19, N 3.52; found: C 42.53, H 5.11, N 3.45%.
119Sn MAS NMR (149.17 MHz, vrot: 13 and 15 kHz): δ = −160 ± 5 ppm;
119Sn Mössbauer (Ca119mSnO3, 78 K): δ = 1.53(1) mm s−1, Δ = 1.77(1) mm s−1.
Anal. calcd for H47.2C47.6N8O1.9S8Sn4Pd4Cl8 ([Cl2Sn(pyS)2Pd]4·1.9 THF]): C 25.96, H 2.16, N 5.09; found: C 25.84, H 2.13, N 5.09%.
MP: 269–272 °C.
1H NMR (400.13 MHz, CDCl3): δ = 4.11 (s, 3H, CH3), 6.61 (m, 2H, H-5 pyS), 6.73–6.74 (mm, 3H, Aryl), 6.89–6.93 (mm, 2H, Aryl), 7.15 (m, 2H, H-4 pyS), 7.43–7.49 (mm, 11H, Phmeta/Phpara/H-3 pyS), 7.67–7.71 (m, 6H, Phortho), 7.79 (d, 3J1H–1H = 5.1 Hz, 2H, H-6 pyS), 8.85 ppm (d, 3J1H–1H = 7.3 Hz, 1H, O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N–C–CH);
C–N–C–CH);
13C{1H} NMR (100.63 MHz, CDCl3): δ = 35.7 (CH3), 116.2, 116.3 (Aryl), 117.7 (C-5 pyS), 119.2, 123.4, 124.5, 125.6 (Aryl), 126.5 (C-3 pyS), 128.3 (d, 3J13C–31P = 10 Hz, Phmeta), 130.3 (Phpara), 131.0 (d, 1J13C–31P = 35 Hz, Phipso), 132.6 (Aryl), 134.7 (d, 2J13C–31P = 13 Hz, Phortho), 137.0 (C-4 pyS), 140.4 (Aryl), 144.4 (C-6 pyS), 155.4 (CO), 157.2 (C-2 pyS), 161.2 ppm (d, 4J13C–31P = 10 Hz, C–O);
31P{1H} NMR (161.98 MHz, CDCl3): δ = 23.7 ppm (satellites: 2J31P–119Sn = 4539 Hz; 2J31P–117Sn = 4350 Hz);
119Sn{1H} NMR (149.21 MHz, CDCl3): δ = −378.1 ppm (d, 2J119Sn–31P = 4593 Hz);
119Sn Mössbauer (Ca119mSnO3, 78 K): δ = 1.32(1) mm s−1, Δ = 2.06(1) mm s−1.
Anal. calcd for C39H32Cl2N5O2PPdS2Sn (993.84 g mol−1): C 47.13, H 3.25, N 7.05; found: C 47.43, H 3.43, N 7.04%.
MP: not observed (stable up to 250 °C).
1H NMR (400.13 MHz, CDCl3): δ = 6.21 (m, 1H, Aryl), 6.54 (m, 2H, H-5 pyS), 6.71 (m, 1H, Aryl), 6.82–6.83 (mm, 2H, Aryl), 7.03 (m, 1H, Aryl), 7.12 (m, 2H, H-4 pyS), 7.26 (m, 1H, Aryl), 7.44–7.45 (mm, 10H, Phmeta/Phpara/Aryl), 7.49 (d, 3J1H–1H = 8.1 Hz, 2H, H-3 pyS), 7.59 (d, 3J1H–1H = 5.0 Hz, 2H, H-6 pyS), 7.67–7.72 (m, 6H, Phortho), 8.81 ppm (d, 4J1H–1H = 1.1 Hz, 1H, HCN, satellites: 3J1H–(117/119)Sn = 81 Hz);
13C{1H} NMR (100.63 MHz, CDCl3): δ = 112.8, 115.0, 115.9 (Aryl), 117.3 (C-5 pyS), 118.8, 120.3 (Aryl), 127.2 (C-3 pyS), 128.3 (d, 3J13C–31P = 9 Hz, Phmeta), 128.3, 129.6 (Aryl), 130.2 (Phpara), 131.3 (d, 1J13C–31P = 35 Hz, Phipso), 133.8 (Aryl), 134.7 (d, 2J13C–31P = 13 Hz, Phortho), 136.8 (C-4 pyS), 138.0 (Aryl), 141.6 (CN), 143.3 (C-6 pyS), 157.6 (C-2 pyS), 162.2 ppm (d, 4J13C–31P = 10 Hz, C–O);
31P{1H} NMR (161.98 MHz, CDCl3): δ = 22.7 ppm (satellites: 2J31P–119Sn = 4398 Hz; 2J31P–117Sn = 4202 Hz);
119Sn{1H} NMR (149.21 MHz, CDCl3): δ = −361.3 ppm (d, 2J119Sn–31P = 4399 Hz);
119Sn Mössbauer (Ca119mSnO3, 78 K): δ = 1.27(1) mm s−1, Δ = 1.86(1) mm s−1.
Anal. calcd for C39H31N4OPPdS2Sn (891.93 g mol−1): C 52.52, H 3.50, N 6.28; found: C 52.44, H 3.39, N 6.29%.
N–(CH2)5–NCMe–(o-C6H4–OH) (H2Sal5, 22 mg, 63 μmol) were dissolved in DCM (2 mL) and triethylamine (26 μL, 19.3 mg, 191 μmol) was added to the orange solution via a micro syringe. After stirring for 30 min at room temperature, the mixture was evaporated to dryness in vacuo. The residue was dissolved in 4 mL of a DCM/ethanol mixture (3:1) and evaporated slowly in air. After one day orange crystals suitable for single crystal X-ray diffraction had formed (solvate containing two molecules of ethanol and one molecule of DCM per parent molecule). The solution was decanted off and the orange product was washed with ethanol (2 mL) and dried in air. Yield: 42.3 mg (25 μmol, 79%). In a related synthesis [again attempting the synthesis of a compound of the type [(O,N,N,O)Sn(μ-pyS)2Pd-PPh3] the tetradentate ligand H2Sal3 [(o-HO–C6H4)–MeCN–(CH2)3–NCMe–(o-C6H4–OH)] was employed, but again compound 13 was formed as the product.
31P MAS NMR (162.02 MHz, vrot: 20 kHz): δ = 21.4 ppm (satellites: 2J31P–(119/117)Sn = 4730 Hz);
119Sn MAS NMR (149.17 MHz, vrot: 25 and 30 kHz): δ = −448 ppm;
119Sn Mössbauer (Ca119mSnO3, 78 K): δ = 1.30(1) mm s−1, Δ = 1.88(1) mm s−1.
Anal. calcd for C61H62Cl4N4O4P2Pd2S4Sn2 (1697.45 g mol−1): C 43.16, H 3.68, N 3.30; found: C 43.24, H 3.56, N 3.39%.
Notes and references
- M. L. H. Green, J. Organomet. Chem., 1995, 500, 127–148 CrossRef CAS.
- (a) H. Braunschweig, K. Gruss and K. Radacki, Angew. Chem., Int. Ed., 2009, 48, 4239–4241 CrossRef CAS PubMed; (b) H. Braunschweig, K. Gruss and K. Radacki, Angew. Chem., Int. Ed., 2009, 46, 7782–7784 CrossRef PubMed; (c) H. Braunschweig, K. Gruss and K. Radacki, Inorg. Chem., 2008, 47, 8595–8597 CrossRef CAS PubMed.
- For reviews addressing metallaboratranes see: (a) H. Braunschweig and R. D. Dewhurst, Dalton Trans., 2011, 40, 549–558 RSC; (b) A. Amgoune and D. Bourissou, Chem. Commun., 2011, 47, 859–871 RSC; (c) G. R. Owen, Chem. Soc. Rev., 2012, 41, 3535–3546 RSC.
- A. F. Hill, G. R. Owen, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1999, 38, 2759–2761 CrossRef CAS.
- (a) J. Wagler and E. Brendler, Angew. Chem., Int. Ed., 2010, 49, 624–627 CrossRef CAS PubMed; (b) L. A. Truflandier, E. Brendler, J. Wagler and J. Autschbach, Angew. Chem., Int. Ed., 2011, 50, 255–259 CrossRef CAS PubMed; (c) J. Autschbach, K. Sutter, L. A. Truflandier, E. Brendler and J. Wagler, Chem. – Eur. J., 2012, 18, 12803–12813 CrossRef CAS PubMed.
- J. Wagler, A. F. Hill and T. Heine, Eur. J. Inorg. Chem., 2008, 4225–4229 CrossRef CAS.
- E. Brendler, E. Wächtler, T. Heine, L. Zhechkov, T. Langer, R. Pöttgen, A. F. Hill and J. Wagler, Angew. Chem., Int. Ed., 2011, 50, 4696–4700 CrossRef CAS PubMed.
- I.-S. Ke and F. Gabbaï, Inorg. Chem., 2013, 52, 7145–7151 CrossRef CAS PubMed.
- S. Benjamin, W. Levason, G. Reid and R. P. Warr, Organometallics, 2012, 31, 1025–1034 CrossRef CAS.
- J. S. Jones, C. R. Wade and F. P. Gabbaï, Organometallics, 2015, 34, 2647–2654 CrossRef CAS.
- H. Yang, T.-P. Lin and F. P. Gabbaï, Organometallics, 2014, 33, 4368–4373 CrossRef CAS.
- J. S. Jones, C. R. Wade and F. P. Gabbaï, Angew. Chem., Int. Ed., 2014, 53, 8876–8879 CrossRef CAS PubMed.
- I.-S. Ke, J. S. Jones and F. P. Gabbaï, Angew. Chem., Int. Ed., 2014, 53, 2633–2637 CrossRef CAS PubMed.
- C. R. Wade and F. P. Gabbaï, Angew. Chem., Int. Ed., 2011, 50, 7369–7372 CrossRef CAS PubMed.
- T.-P. Lin and F. P. Gabbaï, J. Am. Chem. Soc., 2012, 134, 12230–12338 CrossRef CAS PubMed.
- E. Wächtler, R. Gericke, L. Zhechkov, T. Heine, T. Langer, B. Gerke, R. Pöttgen and J. Wagler, Chem. Commun., 2014, 50, 5382–5384 RSC.
- A. Zech, M. F. Haddow, H. Othman and G. R. Owen, Organometallics, 2012, 31, 6753–6760 CrossRef CAS.
- (a) J. Martincová, L. Dostál, S. Herres-Pawlis, A. Růžička and R. Jambor, Chem. – Eur. J., 2011, 17, 7423–7427 CrossRef PubMed; (b) J. Martincová, L. Dostál, A. Růžička, S. Herres-Pawlis and R. Jambor, Z. Anorg. Allg. Chem., 2012, 638, 1672–1675 CrossRef.
- E. Wächtler, R. Gericke, S. Kutter, E. Brendler and J. Wagler, Main Group Met. Chem., 2013, 36, 181–191 CrossRef.
- J. Takaya, S. Nakamura and N. Iwasawa, Chem. Lett., 2012, 41, 967–969 CrossRef CAS.
- (a) M. Bouška, M. Novák, L. Dostál, A. Růžička, T. Mikysek, R. Metelka and R. Jambor, Eur. J. Inorg. Chem., 2014, 310–318 CrossRef; (b) M. Mehring, I. Vrasidas, D. Horn, M. Schürmann and K. Jurkschat, Organometallics, 2001, 20, 4647–4653 CrossRef CAS; (c) J. Vicente, M.-T. Chicote, M. del C. Ramírez-de-Arellano and P. G. Jones, J. Chem. Soc., Dalton Trans., 1992, 11, 1839–1845 RSC; (d) H. Puff and H. Reuter, J. Organomet. Chem., 1989, 364, 57–65 CrossRef CAS.
- A. Haaland, Angew. Chem., Int. Ed. Engl., 1989, 28, 992–1007 CrossRef.
- (a) T.-P. Lin, I.-S. Ke and F. P. Gabbaï, Angew. Chem., Int. Ed., 2014, 51, 4985–4988 CrossRef PubMed; (b) C. Tschersich, C. Limberg, S. Roggan, C. Herwig, N. Ernsting, S. Kovalenko and S. Mebs, Angew. Chem., Int. Ed., 2014, 51, 4989–4992 CrossRef PubMed.
- H. A. Bent, Chem. Rev., 1961, 61, 275–311 CrossRef CAS.
- E. N. Yurchenko, T. S. Khodashova, M. A. Porai-Koshits and V. P. Nikolaev, Koord. Khim., 1980, 6, 1290–1303 CAS.
- M. Grassi, S. V. Meille, A. Musco, R. Pontellini and A. Sironi, J. Chem. Soc., Dalton Trans., 1989, 615–621 RSC.
- H. Yang, T.-P. Lin and F. P. Gabbaï, Organometallics, 2014, 33, 4368–4373 CrossRef CAS.
- N. Mirzadeh, M. Bennett, J. Wagler, E. Wächtler, B. Gerke, R. Pöttgen and S. Bhargava, Eur. J. Inorg. Chem., 2013, 1997–2001 CrossRef CAS.
- J. S. Knyrim, F. M. Schappacher, R. Pöttgen, J. Schmedt auf der Günne, D. Johrendt and H. Huppertz, Chem. Mater., 2007, 19, 254–262 CrossRef CAS.
- F. J. Ramos-Lima, A. G. Quiroga, J. M. Pérez, M. Font-Bardía, X. Solans and C. Navarro-Ranninger, Eur. J. Inorg. Chem., 2003, 1591–1598 CrossRef CAS.
- J. Wagler and A. F. Hill, Organometallics, 2008, 27, 6579–6586 CrossRef CAS.
- S. Sawusch, N. Jäger, U. Schilde and E. Uhlemann, Struct. Chem., 1999, 10, 105–119 CrossRef CAS.
- U. Dinjus, H. Stahl and E. Uhlig, Z. Anorg. Allg. Chem., 1980, 464, 37–44 CrossRef CAS.
- J. S. Morrison and H. M. Haendler, J. Inorg. Nucl. Chem., 1967, 29, 393–400 CrossRef CAS.
- M. Rimoldi, F. Ragaini, E. Gallo, F. Ferretti, P. Macchi and N. Casati, Dalton Trans., 2012, 41, 3648–3658 RSC.
- R. A. Brand, Normos Mössbauer fitting Program, Universitat Duisburg, 2002 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- These structures were refined with an updated version: ShelXL 2014/7.
- T. S. Lobana, R. Verma and A. Castineiras, Polyhedron, 1998, 17, 3753–3758 CrossRef CAS.
- For related ligand bridged [Sn–Pt]IV complexes with Pt in an equatorial position of the trigonal bipyramidal Sn coordination sphere similar 1J(195Pt,119Sn) coupling (in the range 17 kHz–20 kHz) has been reported: S. Warsink, E. J. Derrah, C. A. Boon, Y. Cabon, J. J. M. de Pater, M. Lutz, R. J. M. Klein Gebbink and B.-J. Deelman, Chem. – Eur. J., 2015, 21, 1765–1779 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 999931–999940, 999998, 1000002 and 1000003. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01621a |
| This journal is © The Royal Society of Chemistry 2016 |