
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multinuclear Ni(II), Cu(II) and Zn(II) complexes of chiral macrocyclic nonaazamine†
Marta
Löffler
 *,
Janusz
Gregoliński
,
Maria
Korabik
,
Tadeusz
Lis
and
Jerzy
Lisowski
*,
Janusz
Gregoliński
,
Maria
Korabik
,
Tadeusz
Lis
and
Jerzy
Lisowski
Faculty of Chemistry, University of Wrocław, 14 F. Joliot-Curie, 50-383 Wrocław, Poland. E-mail: marta.loffler@chem.uni.wroc.pl
First published on 2nd September 2016
Abstract
The chiral macrocyclic amines R-L and S-L derived from the 3 + 3 condensation of 2,6-diformylpyridine and (1R,2R)-1,2-diaminocyclohexane or (1S,2S)-1,2-diaminocyclohexane form enantiopure trinuclear Ni(II) and Cu(II) complexes [Ni3(L)(H2O)2Cl5]Cl and [Cu3(L)Cl4]Cl2 and form the dinuclear complex [Zn2(L)Cl2](ZnCl4) with Zn(II). The X-ray crystal structures of these complexes indicate remarkably different conformations of the ligand and different binding modes of the chloride anions. The structure of the copper(II) derivative [Cu3(R-L)Cl4]Cl2·CH3CN·7.5(H2O) indicates unsymmetrical conformation of the macrocycle with three dissimilar pentacoordinate copper(II) ions bridged by chloride; the structure of [Ni3(R-L)(H2O)2Cl5]Cl·0.4CH3CN·4.2H2O is somewhat more symmetrical, with three Ni(II) ions of distorted octahedral geometry, also bridged by a common chloride anion. On the other hand, the macrocycle is highly folded in [Zn2(R-L)Cl2](ZnCl4)·CHCl3·0.8CH3OH·3.7H2O, forming a cleft where the third Zn(II) ion is held via electrostatic interactions as the ZnCl42− anion. The magnetic data for [Cu3(R-L)Cl4]Cl2 indicate the coexistence of antiferromagnetic and ferromagnetic interactions within the quasi isosceles tricopper(II) core (J = −85.6 cm−1, j = 77.1 cm−1). Compound [Ni3(R-L)(H2O)2Cl5]Cl shows the presence of weak antiferromagnetic coupling (J = −2.56 cm−1, j = −1.54 cm−1) between the three Ni(II) ions.
Introduction
Trinuclear and dinuclear complexes, including macrocylic complexes, are attracting attention due to their magnetic and catalytic properties and because they are models for multinuclear metalloenzymes.1–3 For example, many nucleases and esterases utilize two or three metallic centers for cooperative catalysis, which has inspired the synthesis of multinuclear transition metal complexes1 that mimic these enzymes. Research on trinuclear Cu(II) complexes has also aimed at mimicking the trinuclear metalloenzyme sites present in copper oxidases1 such as ascorbate oxidase, ceruplasmin and laccase. It should be mentioned that one form of these copper oxidases corresponds to an antiferromagnetically coupled trinuclear Cu(II) cluster. This form has triggered interest in synthetic trinuclear Cu(II) complexes. In particular, the chiral 3 + 3 macrocycle R-L (Fig. 1) forms an interesting trinuclear Cu(II) complex, [Cu3(R-L)(μ3-OH)2]Cl2(ClO4)2, which exhibits intramolecular ferromagnetic interactions.3 In this complex, three Cu(II) ions of distorted trigonal bipyramidal geometry are bound in the interior of the macrocycle and are additionally bridged by two hydroxo anions; thus, a Cu3(μ3-OH)2 cluster is bound by the macrocycle. For comparison, the same ligand forms mononuclear complexes with lanthanide(III) ions,4 while the Schiff base analogue of the amine macrocycle of L forms dinuclear complexes with Cd(II) ions.5 Trinuclear metal complexes are also formed by a similar chiral macrocyclic 3 + 3 amine, R-L′ (Fig. 1), which has three phenolic fragments instead of three pyridine fragments. The macrocycle L′ (or its Schiff base analogue) is able to form trinuclear complexes with Cu(II) ions,6 Zn(II) ions,7,8 and lanthanide(III) ions.9 The cooperative action of zinc(II) ions in complexes with 3 + 3 macrocycles was utilized in catalytic cleavage of DNA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 and the nitroaldol reaction.10
7 and the nitroaldol reaction.10
 | ||
| Fig. 1 Macrocycles R-L and R-L′. | ||
Inspired by the unusual trinuclear complex [Cu3(R-L)(μ3-OH)2]Cl2(ClO4)2, we have undertaken systematic study of the complexing properties of L towards transition metal ions. Here, we present the synthesis, characterization and crystal structures of trinuclear Ni(II) complex [Ni3(R-L)(H2O)2Cl5]Cl, trinuclear Cu(II) complex [Cu3(R-L)Cl4]Cl2 and dinuclear Zn(II) complex [Zn2(R-L)Cl2](ZnCl4), as well as their enantiomers: [Ni3(S-L)(H2O)2Cl5]Cl, [Cu3(S-L)Cl4]Cl2 and [Zn2(S-L)Cl2](ZnCl4), respectively. We show the structural flexibility of R-L in complex formation; in particular, complex [Cu3(R-L)Cl4]Cl2 substantially differs from complex [Cu3(R-L)(μ3-OH)2]Cl2(ClO4)2 in its coordination mode and ligand conformation. We also discuss the behavior of these complexes in solution and the magnetic properties of [Ni3(R-L)(H2O)2Cl5]Cl and [Cu3(R-L)Cl4]Cl2.
Results and discussion
Synthesis and spectroscopic properties
The [Ni3(L)(H2O)2Cl5]Cl, [Cu3(L)Cl4]Cl2 and [Zn2(L)Cl2](ZnCl4) complexes were obtained by refluxing ligand R-L or S-L with NiCl2·6H2O, CuCl2·2H2O and ZnCl2, respectively, in methanol or acetonitrile.The chiral nature of the synthesized complexes, [Ni3(R-L)(H2O)2Cl5]Cl, [Ni3(S-L)(H2O)2Cl5]Cl, [Cu3(R-L)Cl4]Cl2, [Cu3(S-L)Cl4]Cl2, [Zn2(R-L)Cl2](ZnCl4) and [Zn2(S-L)Cl2](ZnCl4), was confirmed by CD measurements. The CD spectra of the respective enantiomers are mirror images of one another, which reflects the opposite chirality of both compounds (Fig. 1S–3S†).
The 1H NMR spectrum of [Ni3(R-L)(H2O)2Cl5]Cl consists of 10 very broad, paramagnetically shifted lines (Fig. 4S†) and is in accord with an effective D3 symmetry of a high-spin trinuclear Ni(II) complex, which is higher than that observed in the crystal structure (vide infra). This symmetry may arise, for example, from the dynamic exchange of axial ligands in [Ni3(R-L)(H2O)2Cl5]Cl, which effectively averages the coordination spheres of the octahedral Ni(II) ions on the NMR time scale. At elevated temperatures, the lines are narrower, better defined and less shifted, as expected for a paramagnetic complex (Fig. 5S†). Unlike the trinuclear Ni(II) complex of L, the 1H NMR spectrum of trinuclear Cu(II) complex [Cu3(R-L)Cl4]Cl2 indicates lower symmetry, in contrast to that observed for the previously reported trinuclear Cu(II) complex [Cu3(R-L)(μ3-OH)2]Cl2(ClO4)2.3 The spectrum consists of 19 paramagnetically shifted lines, which are relatively narrow for Cu(II) complexes as a result of magnetic interactions between the Cu(II) ions (Fig. 6S†). On the other hand, the effective C3 symmetry observed in solution is higher than the C1 symmetry observed in the crystal structure (vide infra). It follows that axial ligand exchange also operates for complex [Cu3(R-L)Cl4]Cl2 or that the structure of the complex in solution is different from that observed in the solid. The temperature dependence of the chemical shifts of this complex (Fig. 7S†) is typical for paramagnetic species and does not exhibit anti-Curie behavior, which would be observed in the case of very strong antiferromagnetic interactions.
The NMR spectra of the Zn(II) complex [Zn2(R-L)Cl2](ZnCl4) (Fig. 8S†) indicates C1 symmetry of the macrocycle in solution, in accord with the structure observed in the solid state (although the macrocycle is folded, it has no Cs symmetry plane due to the presence of the chiral cyclohexane fragments, vide infra). Thus, the COSY and HMQC spectra indicate that the aromatic region of the 1H NMR spectrum consists of six doublets coupled to three triplets (two of which are ideally overlapped, see ESI Fig. 11S†). This indicates the presence of three different unsymmetrical pyridine rings, in agreement with the crystal structure. Similarly, the C1 symmetry is confirmed by the observation of six different methylene fragments, and six different >CHN positions are seen in the COSY, HMQC and 13C NMR spectra (Fig. 9S–14S†).
The titration of ligand L with ZnCl2, monitored with NMR spectroscopy, indicates initial formation of a presumably mononuclear species with signals broadened by chemical exchange, followed by formation of the dinuclear complex and formation of yet another species (probably trinuclear complexes) in the presence of excess zinc(II) ions (Fig. 15S and 16S†). The signals of the dinuclear complex appear when 1.25 equivalents of ZnCl2 are used and seem to dominate the spectrum even when 3 equivalents are added. The positions of the lines vary slightly as the concentration of added ZnCl2 increases, probably indicating fast chemical exchange corresponding to ion-pair formation between the cationic dinuclear macrocyclic complex and chloride anion or [ZnCl4]2− anion.
X-ray crystal structures
The crystal structures of the [Ni3(R-L)(H2O)2Cl5]Cl·0.4CH3CN·4.2H2O, [Cu3(R-L)Cl4]Cl2·CH3CN·7.5(H2O), and [Zn2(R-L)Cl2](ZnCl4)·CHCl3·0.8CH3OH·3.7H2O complexes indicate the very flexible nature of the ligand L with respect to its conformation and the binding mode of the macrocycle (Fig. 17S†). In the trinuclear complexes [Ni3(R-L)(H2O)2Cl5]Cl·0.4CH3CN·4.2H2O, and [Cu3(R-L)Cl4] Cl2·CH3CN·7.5(H2O), the ligand R-L binds each metal ion via the nitrogen atom of one of the pyridine rings and two nitrogen atoms of the adjacent diaminocyclohexane fragment (Fig. 2). In the Ni(II) complex [Ni3(R-L)(H2O)2Cl5]Cl·0.4CH3CN·4.2H2O, the macrocycle R-L exhibits a relatively flat, helically twisted conformation (Fig. 2 and 3). This conformation is similar to some extent to that reported previously for the trinuclear Cu(II) complex [Cu3(R-L)(μ3-OH)2]Cl2(ClO4)2·5H2O.3 The macrocycle R-L in the latter complex is, however, more compact and resembles the protonated form of the macrocycle.5 The less compact form of [Ni3(R-L)(H2O)2Cl5]Cl·0.4CH3CN·4.2H2O is reflected in the larger radius of the macrocycle and the smaller extent of the helical twist of the pyridine rings. In addition, the macrocycle is partly domed; therefore, its approximate symmetry is C3, in contrast to the approximate D3 symmetry of this macrocycle in complex [Cu3(R-L) (μ3-OH)2]Cl2(ClO4)2·5H2O.3 In contrast, in complex [Cu3(R-L)Cl4]Cl2·CH3CN·7.5(H2O) the ligand R-L adopts a highly distorted, irregular saddle-type conformation (Fig. 3), reflecting both the helical twist and considerable folding of the macrocycle. This distortion towards an irregular conformation of R-L upon complexation is similar to the distortion of a similar macrocycle, R-L′, caused by formation of a mononuclear Eu(III) complex.11 An even more distorted conformation of macrocycle R-L is observed for the dinuclear Zn(II) complex [Zn2(R-L)Cl2](ZnCl4)CHCl3·0.8CH3OH·3.7H2O (Fig. 4). This time, the ligand conformation is dominated by strong bending and is completely different to that observed for the free macrocycle. This complex also differs from complexes [Ni3(R-L)(H2O)2Cl5]Cl·0.4CH3CN·4.2H2O, [Cu3(R-L)Cl4]Cl2·CH3CN·7.5(H2O), and [Cu3(R-L)(μ3-OH)2]Cl2(ClO4)2·5H2O3 in the mode of binding of the metal ion by the macrocycle. In [Zn2(R-L)Cl2](ZnCl4)·CHCl3·0.8CH3OH·3.7H2O, each Zn(II) ion is coordinated by four nitrogen atoms of the macrocycle: a nitrogen atom of the pyridine ring, two nitrogen atoms of the adjacent diaminocyclohexane fragment and one nitrogen atom of the other adjacent diaminocyclohexane fragment; one of the pyridine rings remains uncoordinated.
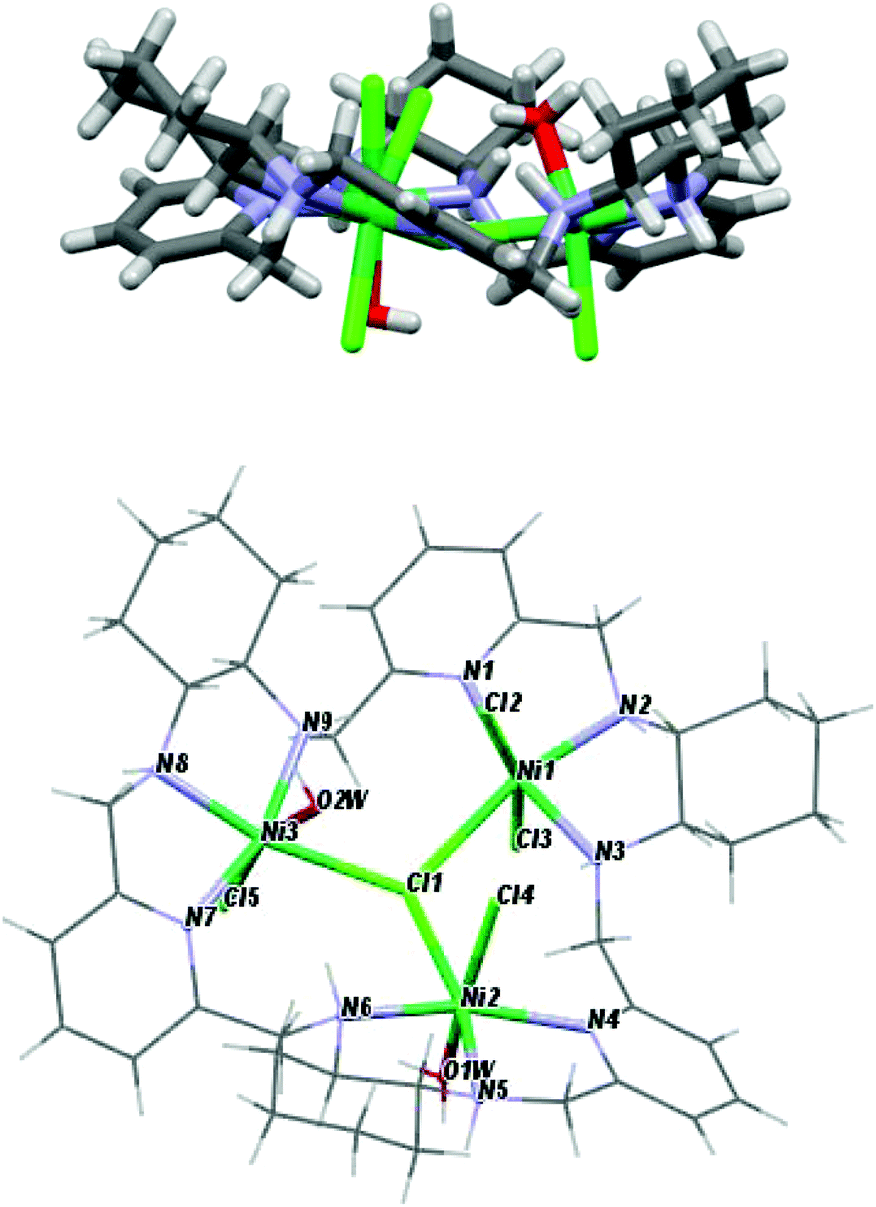 | ||
| Fig. 2 Side and top views of [Ni3(R-L)(H2O)2Cl5]Cl·0.4CH3CN·4.2H2O with the highlighted coordination sphere of the Ni(II) ions. | ||
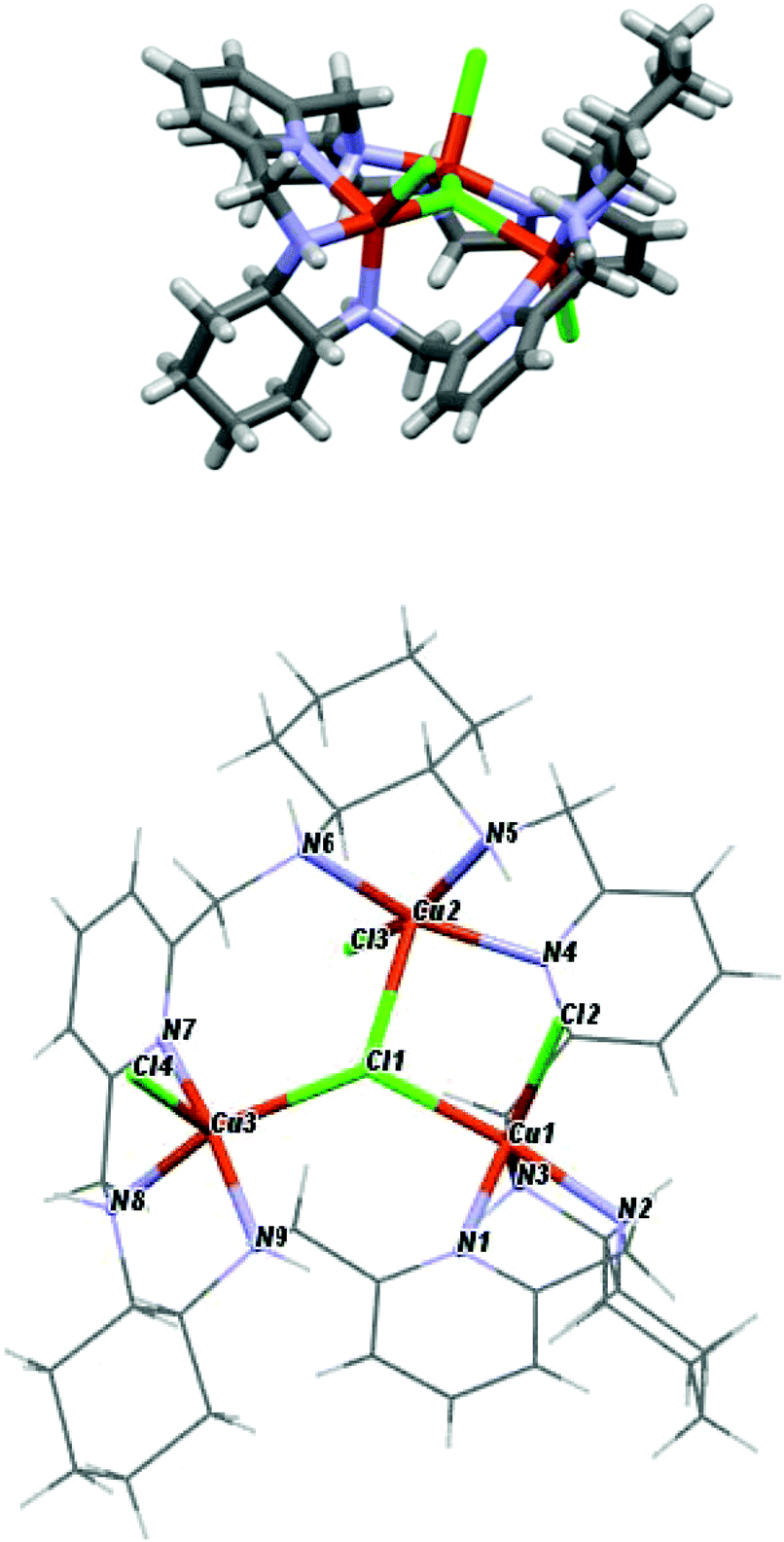 | ||
| Fig. 3 Side and top views of [Cu3(R-L)Cl4]Cl2·CH3CN·7.5(H2O) with the highlighted coordination sphere of the Cu(II) ions. | ||
 | ||
| Fig. 4 Side and top views of [Zn2(R-L)Cl2](ZnCl4)·CHCl3·0.8CH3OH·3.7H2O with the highlighted coordination sphere of the Zn(II) ions. | ||
The crystal structures of the [Ni3(R-L)(H2O)2Cl5]Cl·0.4CH3CN·4.2H2O, [Cu3(R-L)Cl4]Cl2·CH3CN·7.5(H2O) and [Zn2(R-L)Cl2](ZnCl4)·CHCl3·0.8CH3OH·3.7H2O complexes also demonstrate the ability of macrocycle L to adopt metal centers of different geometries with different sets of additional monodentate ligands. The remarkable feature of complexes [Ni3(R-L)(H2O)2Cl5]Cl·0.4CH3CN·4.2H2O and [Cu3(R-L)Cl4]Cl2·CH3CN·7.5(H2O) is the presence of a central μ3-Cl bridge connecting the three metal ions. In complex [Ni3(R-L)(H2O)2Cl5]Cl·0.4CH3CN·4.2H2O, all three Ni(II) ions adopt distorted octahedral geometries. Selected bond distances and angles are listed in ESI Table 1S.† The equatorial plane for all three Ni atoms is created by three nitrogen atoms from the macrocyclic ligand and the bridging chloride atom, with Ni1–(μ3-Cl1), Ni2–(μ3-Cl1) and Ni3–(μ3-Cl1) bond distances equal to 2.745(3), 2.642(3) and 2.554(3) Å, respectively. The apical positions are occupied by two terminal chloride atoms in the case of Ni1 or one chloride atom and one water molecule in the cases of Ni2 and Ni3. The [Ni3] unit is strictly a scalene triangle; however, it can be considered as an approximate isosceles triangle with Ni1⋯Ni2, Ni2⋯Ni3 and Ni3⋯Ni1 distances of 4.579(2), 4.468(3) and 4.496(2) Å, respectively. The chloride atom μ3-Cl1, which is trapped in the middle of the molecule, lies 0.460(3) Å out of the plane defined by the nickel atoms. In complex [Cu3(R-L)Cl4]Cl2·CH3CN·7.5(H2O), each Cu(II) ion is coordinated by three nitrogen atoms of the macrocycle, by the central bridging chloride anion and by additional chloride. Despite this, the complex is much less symmetrical in comparison with [Cu3(R-L)(μ3-OH)2]Cl2(ClO4)2·5H2O,3 and each Cu(II) ion is clearly different; all of them are pentacoordinate and have distorted geometries. Selected bond distances and angles of [Cu3(R-L)Cl4]Cl2·CH3CN·7.5(H2O) are listed in ESI Table 2S.† Two of the Cu(II) ions are closer to square-pyramidal geometry, while the third Cu(II) ion has distorted trigonal bipyramid geometry. The distortions from the ideal geometries are reflected in the values of the angular structural parameter τ (index of trigonality);12τ = (β − α)/60°, where α and β are the two largest angles in the coordination sphere. The values of τ for the Cu(II) ions are equal to 0.57, 0.19 and 0.15 for Cu1, Cu2 and Cu3, respectively. The limit value of τ = 0 corresponds to an ideal square pyramid (α = β ∼ 180°) and τ = 1 corresponds to an ideal trigonal bipyramid (α = 120° and β = 180°). For Cu1, the basal plane of the bipyramid consists of one terminal chloride atom and two nitrogen atoms, one from diaminocyclohexane and one from the pyridine ring. The apical positions are occupied by a second nitrogen atom from diaminocyclohexane and a bridging chloride ligand μ3-Cl1, with a Cu1–(μ3-Cl1) bond distance equal to 2.345(2) Å. The square-planar base of Cu2 is defined by three nitrogen atoms from the macrocyclic ligand and a terminal chloride atom. The apical position is occupied by the bridging chloride ligand μ3-Cl1, with a distance of 2.675(1) Å. In the case of Cu3, the axial position is occupied by the terminal chloride atom, while the equatorial plane consists of three nitrogen atoms from the macrocyclic ligand and the bridging chloride ligand μ3-Cl1, with a Cu3–(μ3-Cl1) bond distance equal to 2.438(1) Å. The [Cu3] unit is strictly a scalene triangle but can be considered as an approximate isosceles triangle with Cu1⋯Cu2, Cu2⋯Cu3 and Cu3⋯Cu1 distances of 3.970(3), 4.424(2) and 4.358(2) Å, respectively. The chloride atom μ3-Cl1, which is trapped in the middle of the molecule, lies 0.301(2) Å out of the plane defined by the copper atoms. The two Zn(II) ions bound by macrocycle R-L in complex [Zn2(R-L)Cl2](ZnCl4)·CHCl3·0.8CH3OH·3.7H2O have coordination spheres with distorted square-pyramidal geometry completed by chloride anions (selected bond distances and angles of [Zn2(R-L)Cl2](ZnCl4)·CHCl3·0.8CH3OH·3.7H2O are listed in ESI Table 3S†). The third Zn(II) present in this crystal is not coordinated by the macrocycle, but forms a tetrahedral [ZnCl4]2− complex counter-anion. The dinuclear Zn(II) macrocyclic cationic complex [Zn2(R-L)Cl2]2+ and the [ZnCl4]2− anion form a tight ion pair; the anion is positioned in a cleft formed by a bend in the macrocycle in [Zn2(R-L)Cl2](ZnCl4)·CHCl3·0.8CH3OH·3.7H2O. Thus, the dinuclear unit of the cationic macrocyclic complex can be regarded as a host for the anionic [ZnCl4]2− guest.
Magnetic properties
The magnetic properties of complexes [Ni3(R-L)(H2O)2Cl5]Cl and [Cu3(R-L)Cl4]Cl2 were examined in the temperature range of 1.8 to 300 K. The temperature dependences of the χmT product (χm being the magnetic susceptibility per trinuclear unit) of the nickel [Ni3(R-L)(H2O)2Cl5]Cl and copper [Cu3(R-L)Cl4]Cl2 compounds are shown in Fig. 5 and 6. Spin–orbit coupling, characteristic for Ni(II) compounds,13 gives rise to the room temperature χmT value for compound [Ni3(R-L)(H2O)2Cl5]Cl of 3.75 cm3 mol−1 K, which is higher than theoretical spin-only value of 3.02 cm3 mol−1 K for the three S = 1 uncoupled centers. Upon cooling, the χmT product decreases continuously, finally reaching a value of 0.88 cm3 mol−1 K at 1.8 K. This behavior is characteristic when three S = 1 paramagnetic metal centers are antiferromagnetically coupled. A similar relationship for triangular Ni3 compounds has been observed by P. Chaudhuri et al.14 and V. V. Pavlishchuk et al.15 This behavior is different from what one would expect for isolated Ni3 molecules, presenting a Curie's law regime and a plateau at the lowest temperature corresponding to the S = 1 ground state (χmT = 1 cm3 K mol−1).16,17 Taking into account differences in the coordination atoms of nickel ions in [Ni3(R-L)(H2O)2Cl5]Cl (a NNNClClμ3-Cl coordination environment for Ni1 and NNN(H2O)Clμ3-Cl coordination environments for Ni2 and Ni3), the quasi-isosceles core of these compounds and the two J and j parameters were assumed by applying the derived Hamiltonian (eqn (1)):| H = −J(S1S2 + S1S3) − j(S2S3). | (1) |
 | ||
| Fig. 5 Temperature dependence of experimental χm (○) and χmT (●) vs. T for complex [Ni3(R-L)(H2O)2Cl5]Cl. Solid lines show the best obtained fit. | ||
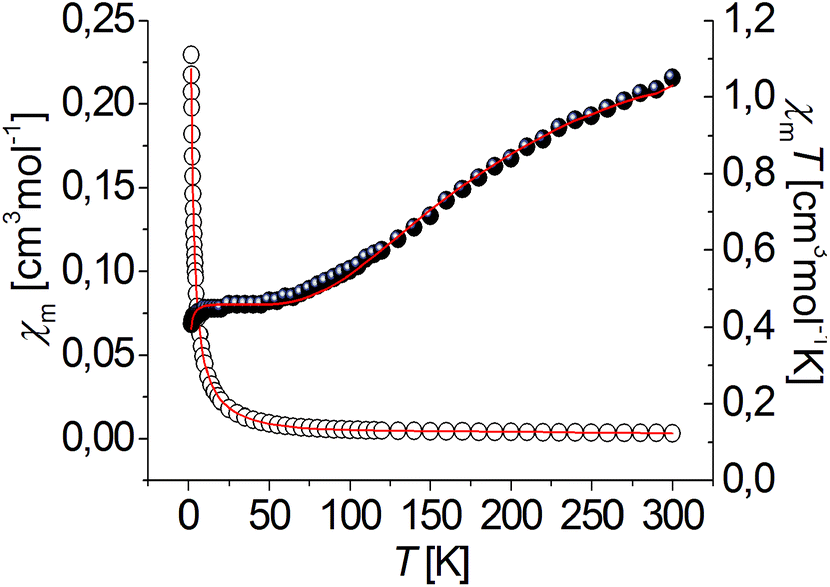 | ||
| Fig. 6 Temperature dependence of experimental χm (○) and χmT (●) vs. T for complex [Cu3(R-L)Cl4]Cl2. Solid lines show the best obtained fit. | ||
However, the distances between the nickel ions are comparable (ESI Table 1S†). The experimental data were fitted using the PHI program,18 including the ZFS parameter D of Ni(II) ion and a term zJ′ for intertrimer exchange. The best-fit parameters are: J = −2.56 cm−1, j = −1.54 cm−1, zJ′ = −0.09 cm−1, g = 2.21 and R = 1.26 × 10−4, R = ∑(χexpT − χcalcT)2/∑(χexpT)2. The S = 0 ground state was found for complex [Ni3(R-L)(H2O)2Cl5]Cl from a j/J ratio13 equal to 0.6. The ground state S = 0 was proposed for ratios between 0.5 and 2.0 and the ground state S = 1 should be observed for ratios less than 0.5 and greater than 2.0.13,17
The χmT value of complex [Cu3(R-L)Cl4]Cl2 is equal to 1.05 cm3 mol−1 K at room temperature, which is only slightly lower than the expected value for three uncoupled S = 1/2 spins (ca. 1.2 cm3 mol−1 K). This value systematically decreases with decreasing temperature to 0.473 cm3 mol−1 K at 60 K. This behavior is characteristic of a dominant antiferromagnetically coupled system. Between 10 and 60 K, a plateau in the χmT vs. T relation is observed, with a value of ∼0.4 cm3 mol−1 K, as expected for an isolated S = 1/2 ground state. The plateau has been observed for many other Cu(II) triangles;19–21,23,26 it indicates that these compounds follow Curie's law, and only the ground spin doublet (or degenerate spin doublets) is thermally populated. The experimental χmT data decreases below 10 K, which suggests that other kinds of antiferromagnetic interactions are operative; intermolecular interactions through hydrogen N–H⋯Cl bonds are observed in the crystal structure (Fig. 19S†). This observation is characteristic of equilateral as well as isosceles and scalene copper(II) triangles19 and is a result of spin frustration. Spin frustration occurs when only two out of three spins achieve full spin compensation simultaneously.13,22 Triangular, trinuclear Cu(II) complexes can be regarded as geometrically spin-frustrated systems13 where an isotropic Heisenberg–Dirac–van Vleck (HDVV) Hamiltonian formalism is not sufficient to investigate the magnetic properties and an antisymmetric term should be added.18–29
The experimental data were fitted using the PHI program.18 Anisotropic, antisymmetric interactions were included in the program. The best fit to the experimental data of [Cu3(R-L)Cl4]Cl2 results in these parameters: J = −85.6 cm−1, j = 77.1 cm−1, zJ′ = −0.14 cm−1.
Magnetostructural correlation
To our knowledge, the trinuclear compounds [Ni3(R-L)(H2O)2Cl5]Cl and [Cu3(R-L)Cl4]Cl2 are the first examples with Cu3Cl and Ni3Cl cores where experimentally observed magnetic interactions are caused by –(μ3-Cl) coupling. Structural data of compounds [Ni3(R-L)(H2O)2Cl5]Cl and [Cu3(R-L)Cl4]Cl2, important in magnetostructural correlation, are presented in Table 1. It is worth noting that the peripheral ligands that hold the M3Cl core do not participate or participate only slightly in magnetic interactions in both the [Ni3(R-L)(H2O)2Cl5]Cl and [Cu3(R-L)Cl4]Cl2 compounds due to the long path of the interactions (the average values of the total sum of the M–N–C–C–N–M distances are 8.56 and 8.51 Å, respectively, for Ni3 and Cu3). Additionally, the conformation of both complexes is helically twisted. The lack of literature data for similar systems, where three copper ions are connected by one chloride bridge, prevents further comparison.Many trinuclear Cu(II) and Ni(II) compounds are described in the literature;14–37 most of these contain M3O(H) cores and peripheral NO–oxime and NN–pyrazole–triazole bridges. Peripheral ligands in equatorial positions also play important roles in the magnetic interactions of these types of complexes, giving rise to strong antiferromagnetic coupling. Magnetostructural correlation was thoroughly performed23–25 for the Cu3O(H) complexes and confirmed by calculations based on density functional theory combined with the broken-symmetry approach (DFT-BS),24 where the spin delocalization mechanism was used. The relationships presented23 between the magnetic coupling and structural features for trinuclear complexes with [Cu3O] cores, and the principal structural factors, are:
(a) The major factor controlling the spin coupling between the metal centers in hydroxido, alkoxido or phenoxido bridged compounds is the bridging Cu–(μ3-O)–Cu angles. The magnetic coupling interaction is switched from ferromagnetic to antiferromagnetic as the Cu–(μ3-X)–Cu angle changes from 76 to 120°.23–25
(b) A linear correlation was found between the coupling constant J and the deviation of the μ3-O atom from the centroid of the Cu3 triangular motif. A smaller deviation determines strong antiferromagnetic coupling.26,27
(c) A more flattened Cu3O(H) bridge favors stronger magnetic interaction.28
The results obtained for [Cu3(R-L)Cl4]Cl2 complex, with a [Cu3Cl] core, confirm the aforementioned conclusions. After analyzing the magnetism of complex [Cu3(R-L)Cl4]Cl2, we can offer an extra point, dependent on the nature and strength of the interactions: the geometry of the copper ion determines the type of the magnetic orbital. Different surroundings of the coupled Cu(II) ions give rise to both antiferromagnetic (J = −85.6 cm−1) and ferromagnetic (j = 77.1 cm−1) interactions. Because the three Cu(II) ions of [Cu3(R-L)Cl4]Cl2 are structurally different, the bonding pathways between the adjacent Cu(II) ions are also different, as is the arrangement of magnetic orbitals with respect to the central chloride bridge. According to the τ parameters, the geometries of Cu2 and Cu3 are square pyramidal (τ = 0.19 and 0.15, respectively), while Cu1 could be regarded as a distorted trigonal bipyramid (τ = 0.59). For that reason, in accordance with the orbital model for magnetic interactions,13 different magnetic orbitals with interacting unpaired electrons could be engaged in magnetic interactions (dx2−y2 in the cases of Cu2 and Cu3, and a part of orbital dz2 in the case of Cu1). The spin delocalization between the p orbitals of the μ3-Cl bridging ligand and the Cu(II) centers contributes to both the ferromagnetic (j) and antiferromagnetic (J) coupling interactions.
Only a few examples in the literature are concerned with similar Cu3Cl cores, which prevents further comparison and discussion of the data obtained in this work. The first triangular structural Cu3(μ3-Cl)2 motif was presented by R. Boča et al.;30 however, no experimental magnetic data were acquired for this compound. A triangular Cu(II) cluster, doubly capped by two μ3-X ligands (X = O(H), Cl, Br), and progression from strongly antiferro- to ferromagnetic exchange has been presented,31 accompanying the change of the Cu–(μ3-X)–Cu angle (X = O, OH, halogen) from 120° to 80°. X-ray crystal geometries of these compounds were used in DFT-BS24 calculations. In all the above cited compounds, equatorial ligands also play important roles in the magnetic interactions.
Analysis of the structural and magnetic data and DFT calculations of the triangle Ni3O(H) compounds indicates that the antiferromagnetic interaction also depends on the Ni–O–Ni angles.32–37
Magnetostructural data of the complexes analyzed by us, with [Ni3Cl] and [Cu3Cl] cores, are presented in Table 1 (explanation of the signs is given in Scheme 1). The magnetic coupling parameters exert effects through the triply bridged μ3-Cl core, which has not been described to date. The parameters obtained for [Ni3(R-L)(H2O)2Cl5]Cl are equal to J = −2.56 cm−1 and j = −1.54 cm−1; the antiferromagnetic nature of these interactions between the metal centers is caused by Ni–Cl–Ni angles greater than 116°. Although the coupling constants found by J. Esteban et al.32 for a [Ni3O] core with similar Ni–0–Ni angles 116°–117° were greater and amounted to −44 to −55 cm−1, this was caused by additional magnetic interactions through oxime and carboxylate bridges. In both the [Ni3(R-L)(H2O)2Cl5]Cl and [Cu3(R-L)Cl4]Cl2 compounds, deviation of μ3-Cl from the metal3 centroid is observed (Table 1); this confirms previous reports that less deviation determines stronger antiferromagnetic coupling. Although the complexes of [Ni3(R-L)(H2O)2Cl5]Cl and [Cu3(R-L)Cl4]Cl2 form apparently similar structures, the differences observed in their magnetic properties are a result of the different geometries and magnetic natures of Cu(II) and Ni(II) ion.
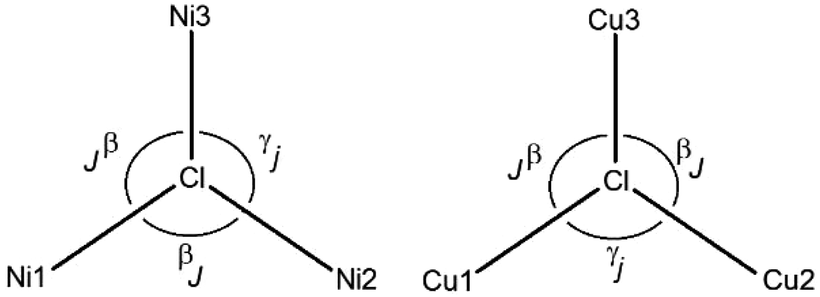 | ||
| Scheme 1 The angle β is defined by the average of the most similar M–Cl–M (M = Ni or Cu) angles with the triangle, whereas the angle γ refers to the most different of these angles. The angle α is defined as the average of the three angles of the complex. | ||
Due to the presence of intermolecular H-bonds in the crystal lattices of both complexes (Fig. 18S and 19S†), very weak intermolecular magnetic interactions through these bonds were found: zJ′ = −0.09 and −0.14 cm−1 for [Ni3(R-L)(H2O)2Cl5]Cl and [Cu3(R-L)Cl4]Cl2, respectively.
Conclusions
Six enantiopure Ni(II), Cu(II) and Zn(II) complexes with chiral nonaazamine have been prepared, and three of the complexes were structurally characterized by X-ray crystallography. As was shown, depending on the metal used, different arrangements of the ligand R-L and different binding modes of the chloride anions were observed. X-ray crystal structures of the trinuclear complexes [Ni3(R-L)(H2O)2Cl5]Cl·0.4CH3CN·4.2H2O and [Cu3(R-L)Cl4]Cl2CH3CN·7.5(H2O) indicate that the six-coordinate Ni(II) or five-coordinate Cu(II) ions are bridged by one Cl− ion lying in the central position of the molecule. In the case of the zinc complex, only two zinc ions are coordinated inside the macrocyclic ligand, which is accompanied by considerable macrocycle ruffling in comparison with the Cu(II) and Ni(II) trinuclear derivatives. The coordination of only two Zn(II) was also confirmed by NMR titration. The variable temperature magnetic measurements of the complexes [Ni3(R-L)(H2O)2Cl5]Cl and [Cu3(R-L)Cl4]Cl2 shows that in complex [Ni3(R-L)(H2O)2Cl5]Cl, three Ni(II) ions are weakly antiferromagnetically coupled through the bridging chloride anion. On the other hand, in complex [Cu3(R-L)Cl4]Cl2, both antiferromagnetic and ferromagnetic interactions are observed within the quasi isosceles tricopper(II) core.Experimental
Methods
The NMR spectra were acquired using a Bruker Advance 500 MHz spectrometer. The electrospray mass spectra (Fig. 20S–22S†) were obtained using Bruker microOTOF-Q and Apex Ultra FT-ICR instruments. The elemental analyses were carried out on a Perkin-Elmer 2400 CHN elemental analyzer. Magnetization measurements in the temperature range of 1.8 to 300 K were carried out for samples of powdered crystals of the complexes [Ni3(R-L)(H2O)2Cl5]Cl·0.4CH3CN·4.2H2O and [Cu3(R-L)Cl4]Cl2·CH3CN·7.5(H2O) (22.13 and 22.18 mg, respectively) at a magnetic field of 0.5 T using a Quantum Design SQUID Magnetometer (type MPMS-XL5). The magnetic data were corrected for the sample holder (Quantum Design Clear Plastic Straws in Paper-AGC2, free of paramagnetic impurities). Corrections for the diamagnetism of the constituting atoms were calculated using Pascal's constants;38 the value of 180 × 10−6 cm3 mol−1 was used to characterize the intermolecular interactions, χtri is the magnetic susceptibility of the trinuclear cluster, and zJ′ is the intermolecular interaction parameter18 used as the temperature-independent paramagnetism of trinuclear Cu(II) complex; the value of 300 × 10−6 cm3 mol−1 was used for the trinuclear Ni(II) complex.13 The effective magnetic moments were calculated from the expression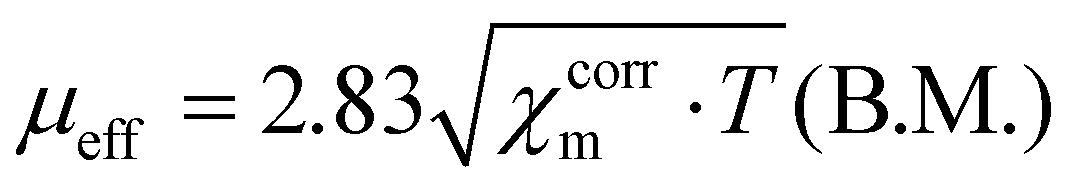
The fitting of the magnetic susceptibility and the simulation of the magnetization were carried out using PHI software.18 The mean-field approximation was calculated as follows:
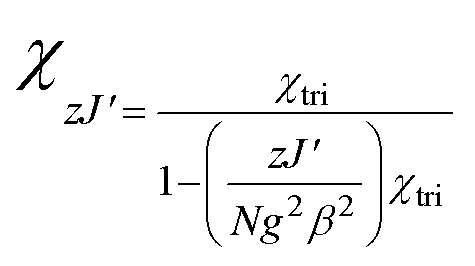
Crystal structure determination
X-ray data for [Ni3(R-L)(H2O)2Cl5]Cl·0.4CH3CN·4.2H2O and [Cu3(R-L)Cl4]Cl2·CH3CN·7.5(H2O) were collected at 100(2) K on an Xcalibur PX instrument (Oxford Diffraction) using Mo-Kα radiation (λ = 0.71073 Å) and CCD; computing cell refinements were performed using CrysAlis RED.39 The X-ray data for [Zn2(R-L)Cl2](ZnCl4)·CHCl3·0.8CH3OH·3.7H2O were collected at 110(2) K using a KM4CCD instrument with Mo-Kα radiation (λ = 0.71073 Å). The images were indexed, integrated and scaled using the Oxford Diffraction data reduction package.40 The structures were solved by direct methods using the SHELXS97 (Sheldrick, 1990) program41 and refined by the full matrix least-squares technique using SHELXL2013 (Sheldrick, 2013).42 All ordered non-hydrogen atoms were refined with anisotropic thermal parameters; the disordered atoms were isotropic. The H atoms attached to C and N atoms were added geometrically and were treated as riding on the concerned parent atoms. H atoms attached to O atoms were located from difference Fourier maps and included in the final refinement cycles on fixed positions. The known absolute configurations of chiral carbon atoms were confirmed on the basis of the value of the Flack parameter. All figures were made using the MERCURY programme.43 Details of the data collection, refinement and crystallographic data are presented in Table 2.| Compound reference | [Ni 3 ( R-L)(H2O)2Cl5]Cl·0.4CH3CN·4.2H2O | [Cu 3 ( R-L)Cl4]Cl2·CH3CN·7.5(H2O) | [Zn 2 ( R-L)Cl2](ZnCl4)·CHCl3·0.8CH3OH·3.7H2O |
|---|---|---|---|
| Chemical formula | C39H61Cl5N9Ni3O2·0.4(C2H3N)·Cl·4.2(H2O) | C39H57Cl4Cu3N9·C2H3N·2Cl·7.5(H2O) | C39H57Cl2N9Zn2·Cl4Zn·(CHCl3)·0.8(CH4O)·3.7(H2O) |
| Formula mass | 1168.88 | 1231.43 | 1259.80 |
| Crystal system | Orthorhombic | Orthorhombic | Monoclinic |
| Space group | P212121 | P212121 | C2 |
| a/Å | 13.592(5) | 14.775 (5) | 26.227(8) |
| b/Å | 15.270(7) | 18.811 (5) | 12.494(3) |
| c/Å | 24.284(12) | 19.639 (6) | 16.625(4) |
| α/° | 90.00 | 90.00 | 90.00 |
| β/° | 90.00 | 90.00 | 92.55(2) |
| γ/° | 90.00 | 90.00 | 90.00 |
| Unit cell volume/Å3 | 5040(4) | 5458 (3) | 5442(2) |
| T/K | 100(2) | 100(2) | 110(2) |
| No. of formula units per unit cell, Z | 4 | 4 | 4 |
| No. of reflections measured | 24969 |
18397 |
27500 |
| No. of independent reflections | 11274 |
13952 |
11499 |
| No. of observed reflections | 5183 | 10484 |
8891 |
| R int | 0.1236 | 0.038 | 0.059 |
| Final R1 value (I > 2σ(I)) | 0.0767 | 0.0595 | 0.0602 |
| Final wR (F2) value (I > 2σ(I)) | 0.0830 | 0.0948 | 0.1492 |
| Final R1 value (all data) | 0.1728 | 0.0910 | 0.0792 |
| Final wR (F2) value (all data) | 0.0943 | 0.1082 | 0.1559 |
| Flack parameter | −0.024(19) | −0.012 (9) | 0.035(11) |
| Largest peak, hole/e Å−3 | 0.95/−0.71 | 0.90/−0.81 | 0.99/−0.82 |
Synthesis
Both enantiomers of macrocyclic ligand L, R-L and S-L, were prepared according to a literature procedure.4The [Ni3(S-L)(H2O)2Cl5]Cl·0.4CH3CN·4.2H2O enantiomer was obtained in a similar fashion. Yield: 78.41 mg (34%) Anal. calc (found) for C39.8H70.6N9.4O6.2Ni3Cl6: C 40.90 (40.78), N 11.26 (11.31), H 6.08 (5.93). CD [MeOH, 298 K, λmax/nm (ε/M−1 cm−1)]: 227 (−6.6), 275 (7), 457 (−0.035), 557 (−0.025), 711 (−0.08).
The [Cu3(S-L)Cl4]Cl2·CH3CN·7.5(H2O) enantiomer was obtained in a similar fashion. Yield: 96.2 mg (41%) Anal. calc (found) for C41H75N10O7.5Cu3Cl6: C 39.99 (40.07), N 11.37 (11.29), H 6.14 (6.23). CD [MeOH, 298 K, λmax/nm (ε/M−1 cm−1)]: 222 (2.7), 238 (0.8), 252 (2.6), 273 (−18.7), 312 (5.6), 361 (−0.8), 420 (−0.14), 500 (0.43), 618 (−0.75), 744 (0.2).
Yield: 99.8 mg (45%). Anal. calc (found) for C39H63N9O3Zn3Cl6: C 42.02 (42.23), N 11.31 (11.15), H 5.70 (5.35). ESI-MS: m/z: 357.7 [LZn]2+; 408.6 [L−HZn2Cl]2+; 426.6 [LZn2Cl2]2+. 1H NMR (500 MHz, CDCl3/CD3OD v/v 2/1) δ 7.88 (t, 2H, α-pyr); 7.82 (t, 1H, α-pyr); 7.42(d, 1H, β-pyr); 7.40(d, 1H, β-pyr); 7.34(d, 1H, β-pyr); 7.31(d, 1H, β-pyr); 7.28(d, 1H, β-pyr); 7.27(d, 1H, β-pyr); 4.78–3.83 (m, 12H, Cγ-pyrCH2NH); 3.55 (m, 1H, NHCHCH2 (Ch)); 3.03 (m, 1H, NHCHCH2 (Ch)); 2.97–2.82 (m, 3H, NHCHCH2 (Ch)); 2.47 (m, 1H, NHCHCH2 (Ch)); 2.44–0.43 (m, 26H, CH2 (Ch)); CD [MeOH, 298 K, λmax/nm (ε/M−1 cm−1)]: 252 (2.4), 273 (−1.6).
The [Zn2(S-L)Cl2](ZnCl4)·3(H2O) complex was obtained in a similar fashion.
Yield: 91.1 mg (41%) Anal. calc (found) for C39H63N9O3Zn3Cl6: C 42.02 (42.01), N 11.31 (11.17), H 5.70 (5.67); CD [MeOH, 298 K, λmax/nm (ε/M−1 cm−1)]: 253 (−1.9), 273 (1.7).
Acknowledgements
This research was supported by MNiSW grant 1506/M/WCH/15.References
- T. Joshi, B. Graham and L. Spiccia, Acc. Chem. Res., 2015, 48, 2366 CrossRef CAS PubMed; J. Serrano-Plata, I. Garcia-Bosch, A. Company and M. Costas, Acc. Chem. Res., 2015, 48, 2397 CrossRef PubMed; E. I. Solomon, A. J. Augustine and J. Yoon, Dalton Trans., 2008, 3921 RSC; E. I. Solomon, J. W. Ginsbach, D. E. Heppner, M. T. Kieber-Emmons, C. H. Kjaergaard, P. J. Smeets, L. Tian and J. S. Woertink, Faraday Discuss., 2011, 148, 11 RSC.
- B. Le Guennic, S. Petit, G. Chastanet, G. Pilet, D. Luneau, N. Ben Amor and V. Robert, Inorg. Chem., 2008, 47, 572 CrossRef CAS PubMed; I. Gautier-Luneau, D. Phanon, C. Duboc, D. Luneau and J.-L. Pierre, Dalton Trans., 2005, 3795 RSC; L. Rigamonti, A. Cinti, A. Forni, A. Passini and O. Piovesana, Eur. J. Inorg. Chem., 2008, 3633 CrossRef; M. U. Anwar, L. K. Thompson and L. N. Dawe, Dalton Trans., 2011, 40, 1437 RSC; R. D. Köhn, L. Tomas Laudo, Z. Pan, F. Speiser and G. Kociok-Köhn, Dalton Trans., 2009, 4556 RSC.
- A. Gonzalez-Alvarez, I. Alfonso, J. Cano, P. Diaz, V. Gotor, V. Gotor-Fernandez, E. Garcia-Espana, S. Garcia-Granda, H. R. Jimenez and G. Lloret, Angew. Chem., Int. Ed., 2009, 48, 6055 CrossRef CAS PubMed.
- J. Gregoliński and J. Lisowski, Angew. Chem., Int. Ed., 2006, 45, 6122 CrossRef CAS PubMed; J. Gregoliński, P. Starynowicz, K. T. Hua, J. L. Lunkley, G. Muller and J. Lisowski, J. Am. Chem. Soc., 2008, 130, 17761 CrossRef PubMed; C. Zhao, J. Ren, J. Gregoliński, J. Lisowski and X. Qu, Nucleic Acids Res., 2012, 40, 8186 CrossRef PubMed.
- A. Gonzalez-Alvarez, I. Alfonso, F. Lopez-Oritz, A. Aguirre, S. Garcia-Granda and V. Gotor, Eur. J. Org. Chem., 2004, 1117 CrossRef CAS.
- M. J. Kobyłka, J. Janczak, T. Lis, T. Kowalik-Jankowska, J. Kłak, M. Pietruszka and J. Lisowski, Dalton Trans., 2012, 41, 1503 RSC.
- S. R. Korupoju, N. Mangayarkarasi, P. S. Zacharias, J. Mizuthani and H. Nishihara, Inorg. Chem., 2002, 41, 4099 CrossRef CAS PubMed.
- A. Sarnicka, P. Starynowicz and J. Lisowski, Chem. Commun., 2012, 48, 2237–2239 RSC; J. Janczak, D. Prochowicz, J. Lewiński, D. Fairen-Jimenez, T. Bereta and J. Lisowski, Chem. – Eur. J., 2016, 22, 598 CrossRef CAS PubMed.
- M. J. Kobyłka, K. Ślepokura, M. Acebrón Rodicio, M. Paluch and J. Lisowski, Inorg. Chem., 2013, 52, 12893 CrossRef CAS PubMed; S.-Y. Lin, Y.-N. Guo, Y. Guo, L. Zhao, P. Zhang, H. Ke and J. Tang, Chem. Commun., 2012, 48, 6924 RSC; M. Paluch, K. Ślepokura, T. Lis and J. Lisowski, Inorg. Chem. Commun., 2011, 14, 92 CrossRef.
- J. Gao, R. A. Zingaro, J. H. Reibenspies and A. Martell, Org. Lett., 2004, 6, 2453 CrossRef CAS PubMed.
- M. Paluch, J. Lisowski and T. Lis, Dalton Trans., 2006, 381 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- O. Kahn, Molecular Magnetism, Willey-VCH, 1993 Search PubMed.
- T. Weyhermüller, R. Wagner, S. Khanra and P. Chaudhuri, Dalton Trans., 2006, 2539 Search PubMed.
- V. V. Pavlishchuk, S. V. Kolotilov, A. W. Addison, M. J. Prushan, R. J. Butcher and L. K. Thompson, Inorg. Chem., 1999, 38, 1759 CrossRef CAS PubMed.
- A. Escuer, R. Vicente, S. B. Kumar, X. Solans, M. Font-Bardía and A. Caneschi, Inorg. Chem., 1996, 35, 3094 CrossRef CAS PubMed.
- A. Escuer, I. Castro, F. Mautner, M. S. El Fallah and R. Vicente, Inorg. Chem., 1997, 36, 4633 CrossRef CAS PubMed; A. Escuer, J. Esteban, J. Mayans and M. Font-Bardía, Eur. J. Inorg. Chem., 2014, 5443 CrossRef.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164 CrossRef CAS PubMed.
- S. Ferrer, F. Lloret, I. Bertomeu, G. Alzuet, J. Borrás, S. García-Granda, M. Liu-González and J. G. Haasnoot, Inorg. Chem., 2002, 41, 5821 CrossRef CAS PubMed; S. Ferrer, F. Lloret, E. Pardo, J. M. Clemente-Juan, M. Liu-González and S. García-Granda, Inorg. Chem., 2012, 51, 985 CrossRef PubMed.
- D. Maity, P. Mukherjee, A. Ghosh, M. G. B. Drew, C. Diaz and G. Mukhopadhyay, Eur. J. Inorg. Chem., 2010, 807 CrossRef CAS.
- A. Escuer, G. Vlahopoulou, F. Lloret and F. A. Mautner, Eur. J. Inorg. Chem., 2014, 83 CrossRef CAS.
- M. U. Anwar, L. K. Thompson and L. N. Dawe, Dalton Trans., 2011, 40, 1437 RSC.
- L. K. Das, M. G. B. Drew, C. Diaz and A. Ghosh, Dalton Trans., 2014, 43, 7589 RSC.
- L.-L. Wang, Y.-M. Sun, Z.-Y. Yu, Z.-N. Qi and Ch.-B. Liu, J. Phys. Chem. A, 2009, 113, 10534 CrossRef CAS PubMed.
- E. Ruiz, P. Alemany, S. Alvarez and J. Cano, J. Am. Chem. Soc., 1997, 119, 1297 CrossRef CAS.
- T. Afrati, C. Dendrinou-Samara, C. Raptopoulou, A. Terzis, V. Tangoulis, A. Tsipis and D. P. Kessissoglou, Inorg. Chem., 2008, 47, 7545 CrossRef CAS PubMed.
- R. Ishikawa, M. Nakano, A. Fuyuhiro, T. Takeuchi, S. Kimura, T. Kashiwagi, M. Hagiwara, K. Kindo, S. Kaizaki and S. Kawata, Chem. – Eur. J., 2010, 16, 11139 CrossRef CAS PubMed.
- R. J. Butchner, C. J. O'Connor and E. Sinn, Inorg. Chem., 1981, 20, 537 CrossRef.
- M. Angoroni, G. A. Ardizzoia, T. Beringhelli, G. La Monica, D. Gatteschi, N. Masciocchi and M. Moret, J. Chem. Soc., Dalton Trans., 1990, 3305 RSC.
- P. A. Angaridis, P. Baran, R. Boča, F. Cervantes-Lee, W. Haase, G. Mezei, R. G. Raptis and R. Werner, Inorg. Chem., 2002, 41, 2219 CrossRef CAS PubMed.
- R. Boča, L. Dlháň, G. Mezei, T. Ortiz-Pérez, R. G. Raptis and J. Telser, Inorg. Chem., 2003, 42, 5801 CrossRef PubMed.
- J. Esteban, M. Font-Bardia and A. Escuer, Eur. J. Inorg. Chem., 2013, 5274 CrossRef CAS.
- J. Esteban, E. Ruiz, M. Font-Bardia, T. Calvet and A. Escuer, Chem. – Eur. J., 2012, 18, 3637 CrossRef CAS PubMed.
- G. A. Craig, O. Roubeau, J. Ribas-Arińo, S. J. Teat and G. Aromí, Polyhedron, 2012, 52, 1369 CrossRef.
- A. D. Katsenis, V. G. Kessler and G. S. Papaefstathiou, Dalton Trans., 2011, 40, 4590 RSC.
- E. Labisbal, L. Rodrıguez, O. Souto, A. Sousa-Pedrares, J. A. Garcıa-Vazquez, J. Romero, A. Sousa, M. Yanez, F. Orallo and J. A. Real, Dalton Trans., 2009, 8644 RSC.
- A. Burkhardt, E. T. Spielberg, S. Simon, H. Görls, A. Buchholz and W. Plass, Chem. – Eur. J., 2009, 15, 1261 CrossRef CAS PubMed.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS.
- CrysAlis_RED, Version 1.1725, Oxford Diffraction Poland Sp., Copyright 1995–2005 Search PubMed.
- Oxford Diffraction Poland Sp., CCD data collection and reduction GUI, Version 1.173.13 beta (release 14.11.2003) Copyright 1995–2004.
- G. M. Sheldrick, SHELXS-97 Program for Solution of Crystal Structure, University of Goettingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- MERCURY, Ver. 2.4, programme for Crystal Structure Visualisation and Exploration, CCDC Cambridge University Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Comparison of macrocycle conformation, intermolecular H-bonds for Ni(II) and Cu(II) complexes, NMR spectra and CD spectra. CCDC 1486259–1486261. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt02504h |
| This journal is © The Royal Society of Chemistry 2016 |