
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anisotropic thermal motion in transition-metal carbonyls from experiments and ab initio theory†
Volker L.
Deringer‡
a,
Ai
Wang
a,
Janine
George
a,
Richard
Dronskowski
*ab and
Ulli
Englert
*a
aInstitute of Inorganic Chemistry, RWTH Aachen University, Landoltweg 1, 52056 Aachen, Germany. E-mail: drons@HAL9000.ac.rwth-aachen.de; ullrich.englert@ac.rwth-aachen.de; Fax: +49 241 80 92642, +49 241 80 92288
bJülich-Aachen Research Alliance (JARA-HPC), RWTH Aachen University, 52056 Aachen, Germany
First published on 1st August 2016
Abstract
The thermal motion of atoms in crystals is quantified by anisotropic displacement parameters (ADPs). Here we show that dispersion-corrected periodic density-functional theory can be used to compute accurate ADPs for transition metal carbonyls, which serve as model systems for crystalline organometallic and coordination compounds.
Continuing work to synthesize new chemical compounds is giving rise to an ever-growing number of known structures. For organic and organometallic crystals, this knowledge is collected in the Cambridge Structural Database,1 which is growing not only in size, but also regarding the quality and depth of information. Careful experiments yield reliable intensity data and trustworthy anisotropic displacement parameters (ADPs),2 which allow refinements to be improved and subtle internal inconsistencies identified. ADPs can help to develop or even disprove a candidate structural model and are routinely requested for newly synthesized organometallic compounds. Physically meaningful ADPs convey information about structural dynamics and translate this into instructive ellipsoid drawings; the Oak Ridge Thermal Ellipsoid Plot (ORTEP) program has just reached its 50th anniversary.3
Unfortunately, the most common method of quantifying ADPs – namely, X-ray diffraction (XRD) – has inherent limitations. The visibility of a scattering centre in XRD scales with its electron count. Improved XRD hardware and methodology now available allow charge density studies to be carried out,4 even on systems of lower suitability,5 and accurate ADPs can be determined from XRD measurements for light atoms next to heavy atoms, although this requires a higher resolution.6 For Mo Kα radiation, a recent study identified a minimum of 2θmax > 65° for determining reliable ADPs;7 this target resolution is currently out of experimental reach when using Cu Kα radiation for single-crystal XRD. We will address high-resolution XRD and neutron diffraction as experimental solutions in the quest for reliable ADPs, but at the heart of this work is an emerging, alternative route of predicting ADPs from ab initio theory. Over recent years, quantum-chemical computations in general have proved to be valuable tools for molecular crystallographers in validating structures,8 assisting with H atom localization9 and in ranking or even predicting complex molecular crystal structures.10 And likewise, it has very recently become possible to compute ADPs from first principles.11–13
We used dispersion-corrected density functional theory (DFT-D)18 to compute ADPs for representative carbonyl compounds of transition metals (Table 1) and validated the results against experimental benchmarks. We discuss the compounds in sequence and use each example to address pertinent questions regarding theoretical but also experimental aspects and limitations. This proof-of-concept study may open the way towards a more routine application of ab initio ADPs in organometallic chemistry.
| Compound | Cambridge structural database ref. code | Experimental parameters | RMS (Å) | |
|---|---|---|---|---|
| a There are several previous reports of 1 (see text); for conciseness, we only list the results from an exemplary room-temperature experiment and our own low-temperature data. | ||||
| 1 |
|
FOHCOU | X-rays, room temp. (ref. 15) | 0.04 |
| X-rays, 100 K (this work) | 0.02 | |||
| 2 |
|
BZCRCO | X-rays, 78 K (ref. 16) | 0.11 |
| Neutrons, 78 K (ref. 16) | 0.04 | |||
| 3 |
|
BOTTAF | Neutrons, 12 K (ref. 17) | 0.05 |
We begin with chromium hexacarbonyl Cr(CO)6 (1), a textbook example of a transition-metal carbonyl that has been widely studied from very early, almost historical, experiments15,19 to more recent ones with higher accuracy.20 We performed a separate high-resolution diffraction experiment on 1. The displacement ellipsoids from our measurements and from DFT-D (Fig. 1) are in excellent agreement and practically no difference can be seen with the naked eye.
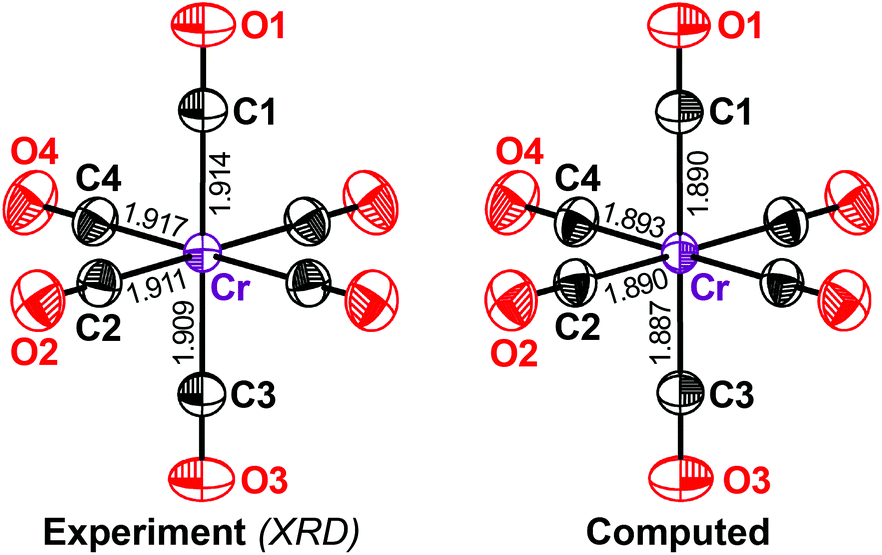 | ||
| Fig. 1 Displacement ellipsoids for crystalline 1 at the 90% probability level, comparing XRD results (left-hand panel) with DFT-D based phonon computations (right-hand panel); both refer to a temperature of 100 K. Symmetry-inequivalent atom labels are given, together with the respective Cr–C distances (in Å). | ||
How close is this agreement numerically? To answer this question, we first inspected the equivalent isotropic displacement parameter, Ueq, for each symmetry-independent atom in 1 (Table 2). We compared the computed values with the experimental values, but before had to answer an even more fundamental question: how accurate is our experimental benchmark?
| Single-crystal XRD | Computed results | ||
|---|---|---|---|
| (2θ ≤ 90°) | (2θ ≤ 110°) | (DFT-D) | |
| Cr | 85.2(3) | 92.8(3) | 97.4 |
| C(1) | 129(1) | 136(1) | 135 |
| C(2) | 125(1) | 132(1) | 131 |
| C(3) | 127(1) | 135(1) | 133 |
| C(4) | 129(1) | 136(1) | 136 |
| O(1) | 196(1) | 203(1) | 201 |
| O(2) | 192(1) | 199(1) | 195 |
| O(3) | 199(1) | 206(1) | 206 |
| O(4) | 203(1) | 209(1) | 208 |
For comparison, refinements of the structural model for 1 were carried out based on data with a resolution better than 1.15 Å−1 (corresponding to an elevated maximum scattering angle of 2θ = 110° with Mo Kα radiation) and also, in parallel, for a purposefully truncated dataset (up to 0.99 Å−1 or 90°). In other words, we report here a “tailored” experiment on 1 in which we probed the effect of resolution. Note that even our truncated dataset exceeded that of most everyday laboratory experiments (usual resolution 0.6–0.7 Å−1). Still, the value of Ueq and thus the ADPs at limited resolution are consistently smaller than those at high resolution (Table 2) and this difference is an order of magnitude larger than the standard uncertainties. This is important for any future benchmarking of computed ADPs.
Fundamentally, this effect is not surprising: X-rays interact with the electron cloud of an atom and hence XRD probes the electronic density. (The obvious alternative is neutron diffraction, to which we will come back when discussing 2.) In XRD, low-order reflections also carry information about the valence electrons between the nuclei, whereas higher-resolution data emphasize the core electrons; the latter will thus better reproduce the properties dominated by nucleic positions, such as lattice vibrations. Experimental ADPs will therefore depend on the resolution and be better described by high-resolution data. This is precisely what is seen in Table 2.
Using higher-order diffraction data only will provide ADPs similar to those from the complete, high-resolution dataset; tentative high-order refinements confirmed this expectation for 1. This is, however, not the norm in many routine laboratory experiments, where such high-order data will be simply unavailable and ADPs from standard refinements with spherical scattering factors will suffer from the limitations seen in Table 2. In principle, using aspherical scattering factors can improve the situation (even if the available resolution does not allow the deconvolution of deformation due to asphericity and thermal motion). Such aspherical scattering factors may be obtained via several approaches based on transferability22 or from Hirshfeld atom refinement.23 Notably, the computationally more demanding iterative Hirshfeld atom refinement has even allowed to refine ADPs for H atoms from XRD data at standard resolution.24
Returning to the computed values of Ueq in 1, our results for the carbonyl ligands are almost without exception within the error range of the high-resolution experiment. Even more importantly for the present study, the value of Ueq for the transition-metal centre is also very well reproduced (Table 2). The DFT-D result for the Cr atom differs from the experimental value by c. 5%, which is less than the effect of resolution alone (c. 8%).
From this encouraging, but admittedly simple, example, we moved to a slightly more complex one: the “piano stool” molecule (η6-benzene)-tricarbonyl-chromium (2), a representative of a half-sandwich compound with a hexahapto benzene ring bonded to the metal atom. It has been studied both by XRD and neutron diffraction experiments,16 which allows for side-by-side comparison. Both previous results are compared with our DFT-D data in Fig. 2a.
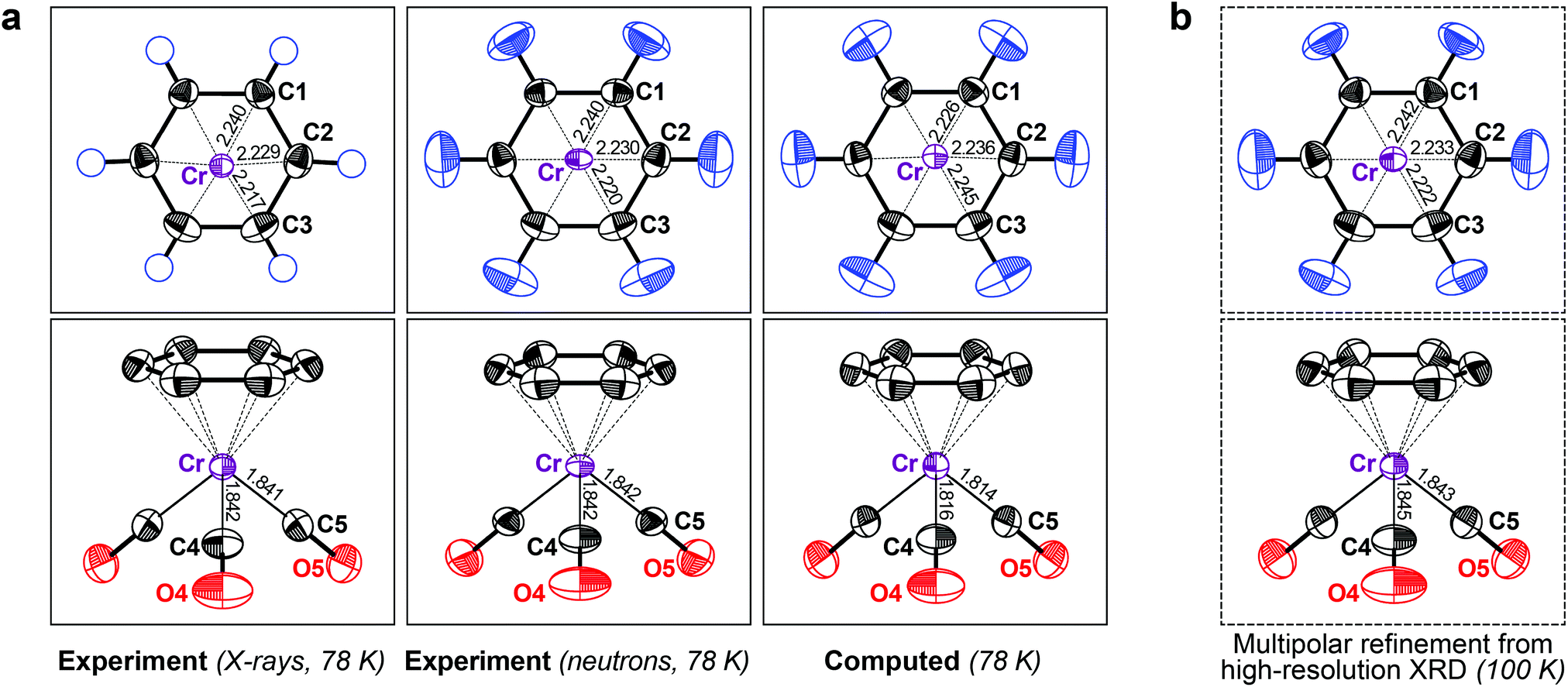 | ||
| Fig. 2 (a) Displacement ellipsoids (90% probability at 78 K) for crystalline 2, comparing X-ray (left) and neutron diffraction results (middle; both datasets from ref. 16) with DFT-D based phonon computations (right). (b) Results of a multipolar refinement of high-resolution XRD data (at 100 K; data from ref. 25; see also ESI†). | ||
We began by assessing the experimental benchmarks. The first difference between the two methods is obvious: no ADPs for H atoms are available from the 1973 XRD experiment. Although neutron diffraction did give hydrogen ADPs (Fig. 2a), its performance is otherwise not a priori better than that of XRD; the standard uncertainties of the neutron experiment by Rees and Coppens16 are rather high and the authors reported a potential problem with the data. The neutron-derived ADPs for Cr are smaller in two axes and larger in the third and thus they are more anisotropic. This is, however, not inherent to the measurement technique: similar shapes for neutron and XRD derived ADPs have been confirmed in two independent experiments, but these studies also found significant differences in magnitude for both parameter sets. The reported discrepancies were 11% (ref. 26) and 5% (ref. 27), again orders of magnitude larger than the respective standard uncertainties.
In this light, the performance of the theoretical method for 2 is even more encouraging. DFT-D readily predicts the elusive hydrogen ADPs (Fig. 2a) in good agreement with the neutron benchmark, both with respect to size and anisotropy (the “rugby-ball-ness”) of the ORTEPs. For further validation and to illustrate the capabilities of state-of-the-art high-resolution XRD, we also show the ADPs obtained from a charge density study at 100 K (Fig. 2b),25 although we note that, in the latter work, the hydrogen ADPs had been re-scaled from the initial neutron study. A more detailed comparison (including theoretical results at the same temperature) is reported in the ESI.† The motion of the Cr atom in 2, as described by DFT-D, almost perfectly matches both XRD results and it is unclear whether the (slight) deviation between theory and neutron diffraction is a result of the former. No significant problem with the theoretical results is seen in either case.
Computed ADPs, if using the present theoretical machinery, inevitably begin to deviate from the experimental values at higher temperatures as anharmonic vibrational contributions come into play.12,13 We previously carried out a temperature-dependent study and found that, for practical purposes, anharmonic contributions were not influential up to 100 K;13 however, to (almost) fully rule them out, it would be interesting to look at a compound that has been studied at very low temperatures. Therefore we next turned to 3, a dinuclear ethylene-bridged complex initially synthesized and routinely characterized by XRD.28 The authors were aware of the contrast problem in their initial study and their experimental answer was neutron diffraction at 12 K.17 Intuitively, this should provide us with an almost perfect benchmark because thermal effects are limited at such low temperatures and neutron diffraction can accurately localize the H atoms.
Fig. 3 shows that the motion of all the H atoms in 3 is well reproduced by computation. The same holds true for the carbonyl ligands, although, in this case, the ORTEPs are slightly underestimated by the theoretical method throughout. By contrast, strong deviations are seen for the metal atoms: both are predicted by theory to move significantly too little. The computed (measured) Ueq for Os(1) and Os(2) are 29 (47) × 10−4 and 28 (50) × 10−4 Å2, respectively and so the theoretical method underestimates them by almost 50%. This is readily seen in Fig. 3.
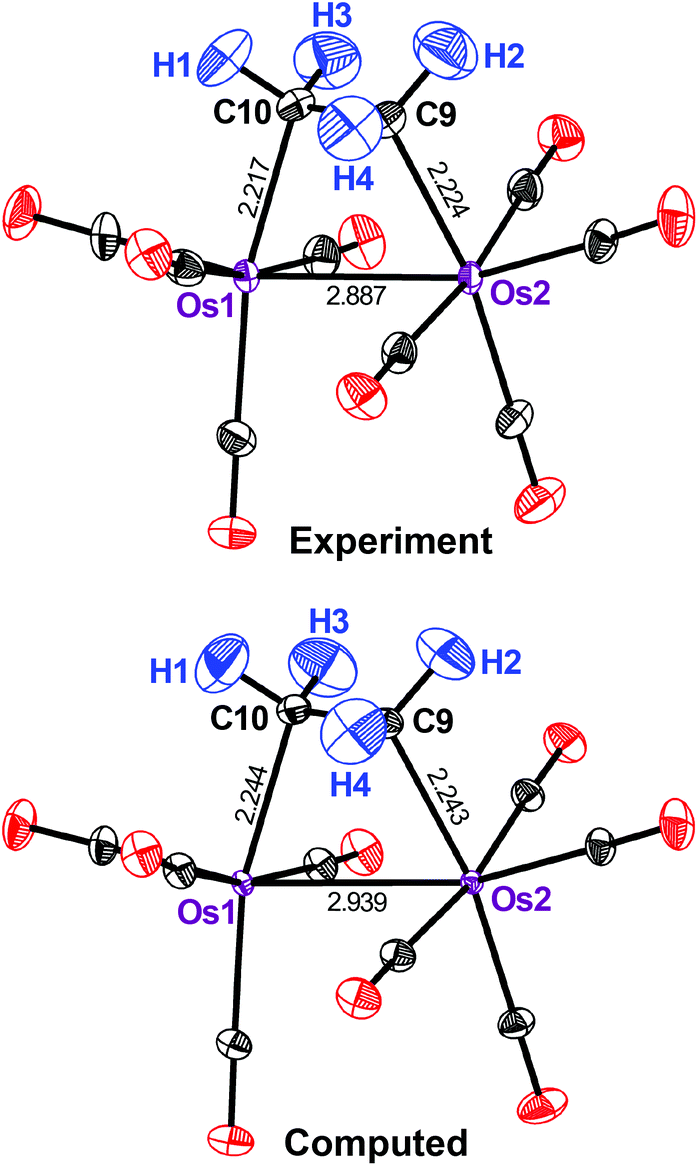 | ||
| Fig. 3 Displacement ellipsoids (90% probability) as in Fig. 2, but for the ethylene-bridged osmium complex 3 at 12 K (experimental data from ref. 17). | ||
How does this difference arise? A possible reason may be found in the higher mass of Os. The vibrations of the heavy nuclei in 3 are strongly localized at low frequencies (ESI†), much more so than for the Cr compounds discussed earlier. As the expression for the ab initio ADPs contains a 1/ω term, uncertainties at low frequencies are the most crucial. A possible solution may be a better description of the electronic structure, especially of the Os dimer species, within the DFT framework used.29 However, we performed test computations for 3, including the M06L functional,30 but found no significant improvement (ESI†). Much validation work, including additional compounds and tailored experiments with improved accuracy, will be required for a definitive answer.
More generally, the theoretical method again emerges as complementary to experiments. In XRD, many-electron scattering centres are reliably localized, with small standard uncertainties for the coordinates and displacement parameters, whereas the light periphery atoms face problems. Theory, by contrast, describes the ligands (including H) with high confidence and could be useful even in those cases where the simulation underestimates the motion of the heavy metal atom(s), as seen in Fig. 3.
Fig. 4 summarizes the results of this study by comparing all the principal elements of the experimental and computed displacement tensors. Ideally, these would all reside on the identity (dashed lines), and the degree of deviation allows us to judge the quality of the DFT-D predictions. Regarding the metal atoms (Fig. 4a), the scatterplot readily exposes the underestimated ADPs in 3, but this trend is not general, as the metal atoms in 1 and 2 are well described by theory. The motion of the peripheral ligand atoms (Fig. 4b) is likewise reliably reproduced; this holds for the carbon, oxygen and hydrogen atoms alike and no systematic deviation is seen. We reiterate that some degree of scattering in the experimental benchmarks must be taken into account, no matter how well these experiments have been carried out. In this light, in particular, the results shown in Fig. 4 are highly encouraging.
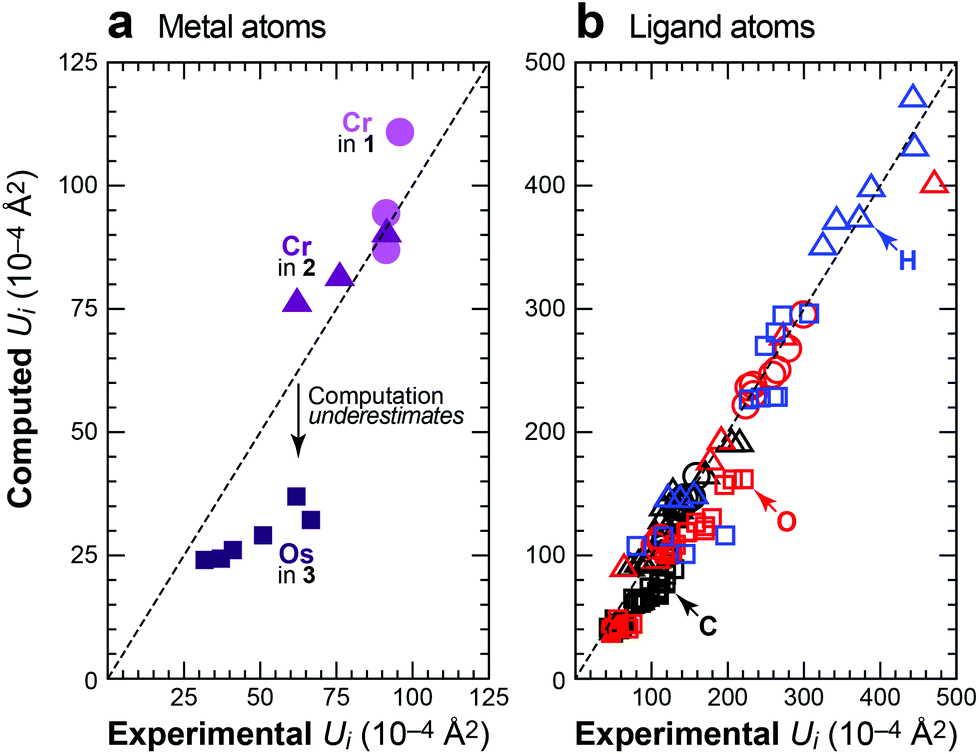 | ||
| Fig. 4 Experimental vs. computed ADPs for crystalline 1 (100 K; circles), 2 (78 K; triangles; experimental data from ref. 16) and 3 (12 K; squares; experimental data from ref. 17), given as main-axis displacement parameters U1, U2, U3 and separately for (a) the metal atoms and (b) the light ligand atoms. For 2, where both XRD and neutron data are available (see text), we benchmarked the computation against the XRD results for the metal atom and against neutron diffraction for the ligand atoms. | ||
Conclusions
DFT-D computations can be used to predict ADPs for organometallic and coordination compounds, which seems to be particularly useful for cases where routine experiments face (inevitable) difficulties. The method is both practical and economic, and predictive within its limits, which have been explored here for selected organometallic model compounds. Notably, theory might even be closer to reality than an experiment of limited resolution (cf.Table 2), as long as the electronic ground state can be correctly described by DFT. A further, much broader study is currently in progress in our laboratories. We note that ab initio ADP computations have now been interfaced with the popular SHADE server,31 which underlines their significant potential for widespread future use.Methods
High-quality crystals were available for 1. The key quality indicators for data at full resolution were: sin(θmax)/λ = 1.152 Å−1; 47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 979 reflections; 5016 independent observations; 67 variables; R1 (all data) = 0.0448; wR2 = 0.1054; and GOF = 1.055. Key quality indicators for data truncated at sin(θmax)/λ = 0.995 Å−1: 35569 reflections; 3287 independent observations; 67 variables; R1 (all data) = 0.0325; wR2 = 0.0758; and GOF = 1.066. Details about data collection and the completeness and quality of diffraction data are given in the ESI.†
979 reflections; 5016 independent observations; 67 variables; R1 (all data) = 0.0448; wR2 = 0.1054; and GOF = 1.055. Key quality indicators for data truncated at sin(θmax)/λ = 0.995 Å−1: 35569 reflections; 3287 independent observations; 67 variables; R1 (all data) = 0.0325; wR2 = 0.0758; and GOF = 1.066. Details about data collection and the completeness and quality of diffraction data are given in the ESI.†
DFT-D computations were carried out using the projector augmented-wave method32 implemented in VASP,33 with a plane-wave cut-off of 500 eV. Reciprocal space was sampled on dense Γ-centred Monkhorst–Pack meshes.34 Strict convergence criteria of ΔE < 10−8 (10−6) eV per cell were applied for electronic (structural) optimization, respectively. Exchange and correlation were treated using the Perdew–Burke–Ernzerhof functional,35 with dispersion corrections applied using the D3 scheme of Grimme et al. and Becke–Johnson damping.18 This method has been thoroughly validated for molecular crystals36 and for ADP computations.13
Crystal structures were taken from experiments and fully optimized, re-starting from scratch several times to ensure a well-converged minimum. The computed structures were assessed using the root mean square (RMS) displacement of atomic coordinates, which has been introduced to validate experiments,8 but can also be used to check the quality of computations.14
ADPs were obtained from phonon computations12,37 based on DFT-D and PHONOPY,38 as detailed in the ESI.† The PHONOPY output was converted into crystallographic Uij values using a custom-written MATLAB code.13,39
Acknowledgements
Crystals of Cr(CO)6 were kindly provided by Professor Guido Pampaloni, University of Pisa, Italy. We gratefully acknowledge financial support from the China Scholarship Council (scholarship for A. W.), Fonds der Chemischen Industrie (scholarship for J. G.) and RWTH Aachen University. Computer time was provided by the Jülich-Aachen Research Alliance (project jara0069).Notes and references
- C. R. Groom and F. H. Allen, Angew. Chem., Int. Ed., 2014, 53, 662–671 CrossRef CAS PubMed.
- (a) D. W. J. Cruickshank, Acta Crystallogr., 1956, 9, 747–753 CrossRef CAS; (b) D. W. J. Cruickshank, Acta Crystallogr., 1956, 9, 754–756 CrossRef CAS.
- C. K. Johnson, OR TEP: A FORTRAN Thermal-Ellipsoid Plot Program for Crystal Structure Illustrations, ORNL-3794 (Rev.), Union Carbide Corp., Oak Ridge Natl. Lab, 1965 Search PubMed.
- We can only provide examples here: (a) A. I. Stash, K. Tanaka, K. Shiozawa, H. Makino and V. G. Tsirelson, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 418–428 Search PubMed; (b) D. Hashizume, N. Suzuki and T. Chihara, Chem. Commun., 2006, 1233–1235 RSC; (c) A. Reisinger, N. Trapp, I. Krossing, S. Altmannshofer, V. Herz, M. Presnitz and W. Scherer, Angew. Chem., Int. Ed., 2007, 46, 8295–8298 CrossRef CAS PubMed; (d) R. Wang, C. W. Lehmann and U. Englert, Acta Crystallogr., Sect. B: Struct. Sci., 2009, 65, 600–611 CAS; (e) R. Wang, T. Dols, C. W. Lehmann and U. Englert, Chem. Commun., 2012, 48, 6830–6832 RSC; (f) C. Merkens, F. Pan and U. Englert, CrystEngComm, 2013, 15, 8153–8158 RSC.
- P. Coppens, Isr. J. Chem., 1977, 16, 144–148 CrossRef CAS.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CAS.
- W. F. Sanjuan-Szklarz, A. A. Hoser, M. Gutmann, A. Ø. Madsen and K. Woźniak, IUCrJ, 2016, 3, 61–70 CAS.
- J. Van de Streek and M. A. Neumann, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 544–558 CAS.
- V. L. Deringer, V. Hoepfner and R. Dronskowski, Cryst. Growth Des., 2012, 12, 1014–1021 CAS.
- (a) M. A. Neumann, F. J. J. Leusen and J. Kendrick, Angew. Chem., Int. Ed., 2007, 47, 2427–2430 CrossRef PubMed; (b) J. van de Streek and M. A. Neumann, CrystEngComm, 2011, 13, 7135–7142 RSC; (c) D. A. Bardwell, et al. , Acta Crystallogr., Sect. B: Struct. Sci., 2011, 67, 535–551 CAS.
- (a) A. M. Reilly, D. A. Wann, C. A. Morrison and D. W. H. Rankin, Chem. Phys. Lett., 2007, 448, 61–64 CrossRef CAS; (b) B. Dittrich, S. Pfitzenreuter and C. B. Hübschle, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2012, 68, 110–116 CAS; (c) A. Ø. Madsen, B. Civalleri, M. Ferrabone, F. Pascale and A. Erba, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2013, 69, 309–321 CrossRef CAS; (d) A. M. Reilly, D. A. Wann, M. J. Gutmann, M. Jura, C. A. Morrison and D. W. H. Rankin, J. Appl. Crystallogr., 2013, 46, 656–662 CAS; (e) C. G. Pozzi, A. C. Fantoni, A. E. Goeta, E. de Matos Gomes, G. J. McIntyre and G. Punte, Chem. Phys., 2013, 423, 85–91 CrossRef CAS.
- V. L. Deringer, R. P. Stoffel, A. Togo, B. Eck, M. Meven and R. Dronskowski, CrystEngComm, 2014, 16, 10907–10915 RSC.
- J. George, A. Wang, V. L. Deringer, R. Wang, R. Dronskowski and U. Englert, CrystEngComm, 2015, 17, 7414–7422 RSC.
- J. George, V. L. Deringer and R. Dronskowski, Inorg. Chem., 2015, 54, 956–962 CrossRef CAS PubMed.
- A. Whitaker and J. W. Jeffery, Acta Crystallogr., 1967, 23, 977–984 CrossRef CAS.
- B. Rees and P. Coppens, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1973, 29, 2515–2528 CrossRef.
- O. P. Anderson, B. R. Bender, J. R. Norton, A. C. Larson and P. J. Vergamini, Organometallics, 1991, 10, 3145–3150 CrossRef CAS.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (b) E. R. Johnson and A. D. Becke, J. Chem. Phys., 2006, 124, 174104 CrossRef PubMed; (c) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- (a) W. Rüdorff and U. Hofmann, Z. Phys. Chem. B, 1935, 28, 351 Search PubMed; (b) L. O. Brockway, R. V. G. Ewens and M. W. Lister, Trans. Faraday Soc., 1938, 34, 1350 RSC.
- (a) A. Jost, B. Rees and W. B. Yelon, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 2649–2658 CrossRef; (b) B. Rees and A. Mitschler, J. Am. Chem. Soc., 1976, 98, 7918–7924 CrossRef CAS; (c) L. J. Farrugia and C. Evans, J. Phys. Chem. A, 2005, 109, 8834–8848 CrossRef CAS PubMed.
- K. N. Trueblood, H.-B. Bürgi, H. Burzlaff, J. D. Dunitz, C. M. Gramaccioli, H. H. Schulz, U. Shmueli and S. C. Abrahams, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1996, 52, 770–781 CrossRef.
- (a) C. Jelsch, V. Pichon-Pesme, C. Lecomte and A. Aubry, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1998, 54, 1306–1318 CrossRef CAS; (b) B. Dittrich, T. Koritsánszky and P. Luger, Angew. Chem., Int. Ed., 2004, 43, 2718–2721 CrossRef CAS; (c) A. Volkov, X. Li, T. Koritsánszky and P. Coppens, J. Phys. Chem. A, 2004, 108, 4283–4300 CrossRef CAS.
- (a) F. L. Hirshfeld, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 239–244 CrossRef; (b) F. L. Hirshfeld, Isr. J. Chem., 1977, 16, 198–201 CrossRef CAS; (c) D. Jayatilaka and B. Dittrich, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 383–393 CrossRef CAS PubMed.
- S. C. Capelli, H.-B. Bürgi, B. Dittrich, S. Grabowsky and D. Jayatilaka, IUCrJ, 2014, 1, 361–379 CAS.
- L. J. Farrugia, C. Evans, D. Lentz and M. Roemer, J. Am. Chem. Soc., 2009, 131, 1251–1268 CrossRef CAS PubMed.
- C. Flensburg, S. Larsen and R. F. Stewart, J. Phys. Chem., 1995, 99, 10130–10141 CrossRef CAS.
- M.-D. Şerb, R. Wang, M. Meven and U. Englert, Acta Crystallogr., Sect. B: Struct. Sci., 2011, 67, 552–559 Search PubMed.
- K. M. Motyl, J. R. Norton, C. K. Schauer and O. P. Anderson, J. Am. Chem. Soc., 1982, 104, 7325–7327 CrossRef CAS.
- Transition-metal dimers, most prominently Cr2, pose well-known challenges to even the most sophisticated quantum-chemical methods. For but two recent examples, see: (a) W. Purwanto, S. Zhang and H. Krakauer, J. Chem. Phys., 2015, 142, 064302 CrossRef PubMed; (b) S. Vancoillie, P. Å. Malmqvist and V. Veryazov, J. Chem. Theory Comput., 2016, 12, 1647–1655 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- A. Ø. Madsen and A. A. Hoser, J. Appl. Crystallogr., 2014, 47, 2100–2104 CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter, 1994, 50, 17953–17979 CrossRef.
- (a) G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1993, 47, 558–561 CrossRef CAS; (b) G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169–11186 CrossRef CAS; (c) G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter, 1999, 59, 1758–1775 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. Moellmann and S. Grimme, J. Phys. Chem. C, 2014, 118, 7615–7621 CAS.
- N. J. Lane, S. C. Vogel, G. Hug, A. Togo, L. Chaput, L. Hultman and M. W. Barsoum, Phys. Rev. B: Condens. Matter, 2012, 86, 214301 CrossRef.
- (a) A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter, 2008, 78, 134106 CrossRef; (b) A. Togo and I. Tanaka, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- J. George, ADP-Toolbox. This code is freely available via the Internet at http://www.ellipsoids.de, together with additional information regarding ADP computation.
Footnotes |
| † Electronic supplementary information (ESI) available: Details concerning the diffraction experiment for 1 (data collection and refinement; diagrams concerning data completeness and quality); extended theoretical methods and data for 3 (integrated partial phonon DOS; results at the LDA and M06L levels of DFT). CCDC 1486566 (1 at full resolution) and 1486567 (1 at truncated resolution). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt02487d |
| ‡ Current address: Engineering Laboratory, University of Cambridge, Trumpington Street, Cambridge CB2 1PZ, UK. |
| This journal is © The Royal Society of Chemistry 2016 |