
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Kinetic studies of phosphoester hydrolysis promoted by a dimeric tetrazirconium(IV) Wells–Dawson polyoxometalate†
Thi Kim Nga
Luong
a,
Pavletta
Shestakova
b and
Tatjana N.
Parac-Vogt
*a
aDepartment of Chemistry, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: Tatjana.Vogt@chem.kuleuven.be; Web: http://www.chem.kuleuven.be/lbc/
bNMR Laboratory, Institute of Organic Chemistry with Centre of Phytochemistry, Bulgarian Academy of Sciences, Acad. G. Bontchev Str., Bl.9, 1113 Sofia, Bulgaria
First published on 4th July 2016
Abstract
The catalytic hydrolysis of a phosphoester bond in the DNA-model substrate 4-nitrophenyl phosphate (NPP) promoted by Zr(IV)-substituted Wells–Dawson Na14[Zr4(P2W16O59)2(μ3-O)2(OH)2(H2O)4]·57H2O polyoxometalate (ZrWD 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) was followed by means of 1H and 31P NMR spectroscopy. The hydrolytic reaction proceeded with a rate constant of 8.44 (±0.36) × 10−5 s−1 at pD 6.4 and 50 °C, representing a 300-fold rate enhancement in comparison with the spontaneous hydrolysis of NPP (kobs = 2.81 (±0.25) × 10−7 s−1) under the same reaction conditions. The ZrWD 4:2 was also active towards hydrolysis of bis(4-nitrophenyl) phosphate (BNPP) and the RNA model substrate 2-hydroxypropyl-4-nitrophenyl phosphate (HPNP). The pD dependence of kobs shows that the rate constants for NPP hydrolysis decrease significantly when the pD values of the reaction mixtures increase. The formation constant (Kf = 190 M−1) and catalytic rate constant (kc = 6.40 × 10−4 s−1) for the NPP-ZrWD 4:2 complex, activation energy (Ea) of 110.15 ± 7.06 kJ mol−1, enthalpy of activation (ΔH‡) of 109.03 ± 6.86 kJ mol−1, entropy of activation (ΔS‡) of 15.20 ± 2.49 J mol−1 K−1, and Gibbs activation energy (ΔG‡) of 104.32 ± 6.09 kJ mol−1 at 37 °C were calculated from kinetic studies. The recyclability of ZrWD 4:2 was examined by adding an extra amount (5.0 mM) of NPP twice to a fully hydrolyzed mixture of 5.0 mM NPP and 1.0 mM ZrWD 4:2. The interaction between ZrWD 4:2 and the P–O bond of NPP was evidenced by a change in the 31P chemical shift of the 31P atom in NPP upon addition of ZrWD 4:2. Based on 31P NMR experiments and the kinetic studies, a mechanism for NPP hydrolysis promoted by ZrWD 4:2 has been proposed.
:2) was followed by means of 1H and 31P NMR spectroscopy. The hydrolytic reaction proceeded with a rate constant of 8.44 (±0.36) × 10−5 s−1 at pD 6.4 and 50 °C, representing a 300-fold rate enhancement in comparison with the spontaneous hydrolysis of NPP (kobs = 2.81 (±0.25) × 10−7 s−1) under the same reaction conditions. The ZrWD 4:2 was also active towards hydrolysis of bis(4-nitrophenyl) phosphate (BNPP) and the RNA model substrate 2-hydroxypropyl-4-nitrophenyl phosphate (HPNP). The pD dependence of kobs shows that the rate constants for NPP hydrolysis decrease significantly when the pD values of the reaction mixtures increase. The formation constant (Kf = 190 M−1) and catalytic rate constant (kc = 6.40 × 10−4 s−1) for the NPP-ZrWD 4:2 complex, activation energy (Ea) of 110.15 ± 7.06 kJ mol−1, enthalpy of activation (ΔH‡) of 109.03 ± 6.86 kJ mol−1, entropy of activation (ΔS‡) of 15.20 ± 2.49 J mol−1 K−1, and Gibbs activation energy (ΔG‡) of 104.32 ± 6.09 kJ mol−1 at 37 °C were calculated from kinetic studies. The recyclability of ZrWD 4:2 was examined by adding an extra amount (5.0 mM) of NPP twice to a fully hydrolyzed mixture of 5.0 mM NPP and 1.0 mM ZrWD 4:2. The interaction between ZrWD 4:2 and the P–O bond of NPP was evidenced by a change in the 31P chemical shift of the 31P atom in NPP upon addition of ZrWD 4:2. Based on 31P NMR experiments and the kinetic studies, a mechanism for NPP hydrolysis promoted by ZrWD 4:2 has been proposed.
Introduction
Phosphate esters play a number of important functions in biological systems such as information storage (DNA/RNA), cellular signaling (cAMP) and energy transduction (ATP).1 The phosphodiester bonds in these molecules are extremely resistant towards hydrolysis because of the repulsion between the negatively charged backbone and potential nucleophiles.2–5 In the absence of a catalyst and at room temperature the half-life for phosphodiester bond hydrolysis has been estimated to be 130000 years for DNA at neutral pH, and 4 years for RNA at pH 6.0.6,7 This stability makes these compounds excellent systems for information storage.8 However, despite the extreme stability of the phosphodiester bonds, their efficient cleavage is often a required procedure in biochemistry. Although natural nuclease enzymes play a critical role in various biotechnology applications, efficient artificial nucleases are still necessary for several purposes.2 For example, an artificial restriction enzyme can be made sequence specific by encoding sequence specificity into the cleavage agent or by linking it to an appropriate DNA binding agent.2 Such enzymes are not only very useful for the molecular biological field such as DNA cutting at a site not recognized by current restriction enzymes, but also for genomic analysis. Besides, artificial nucleases can be synthesized in large amounts9 and are often used to elucidate the precise role of active metal ions2 or to understand the role of the second coordination sphere in natural nucleases. A large number of metal complexes that act as artificial nucleases have been synthesized up to now, and they have been summarized in some of the recent reviews.10,11 It has been postulated that the metal ions can accelerate the hydrolysis process by different mechanisms that include activation of the substrate through binding, delivery of the nucleophile, or by stabilization of the associative transition state.10–14 In addition, rapid exchange of the bound water ligand can also facilitate the hydrolysis. The structural fragments of the natural nucleases, e.g. amino acids that are not directly involved in interaction with the metal center can also play an important role in the hydrolysis process via second coordination sphere interactions such as hydrogen bonding, electrostatic interactions, hydrophobic and van der Waals interactions. These non-covalent interactions alter the chemical and physical properties of the active sites, resulting in the stabilization of the catalytic transition states and promoting the expulsion of the leaving groups.15–17
Recently, our group discovered that a number of Zr(IV)-substituted polyoxometalate (POM) complexes could act as artificial phosphoesterases. POMs are generally described as a large class of inorganic oxoclusters that contain early transition metals (V, Nb, Ta, Mo and W) in their highest oxidation state. POMs can be used in a broad range of research domains including materials science,18,19 medicine20 and catalysis.21,22 Zr(IV) is the best candidate that acts as an active center in artificial hydrolytic metalloenzymes due to its high Lewis-acidity and oxophilic properties, allowing both the coordination and activation of the substrate and nucleophile. A mononuclear Zr(IV)-substituted Wells–Dawson type POM K15H[Zr(α2-P2W17O61)2]·25H2O (ZrWD 1:2) was shown to be able to catalytically accelerate the hydrolysis of the DNA model substrate 4-nitrophenyl phosphate (NPP) by nearly two orders of magnitude.23 In a previous study we found that the dinuclear Zr(IV)-substituted Keggin type POM (Et2NH2)8[{α-PW11O39Zr(μ-OH)(H2O)}2]·7H2O (ZrK 2:2) efficiently promoted hydrolysis of the extremely stable phosphodiester bis(4-nitrophenyl) phosphate (BNPP), with a 320-fold rate enhancement in comparison with pure BNPP hydrolysis.24 In addition, it was shown that the monomeric [α-PW11O39Zr(μ-OH)(H2O)]4− (ZrK 1:1) species is present in aqueous ZrK 2:2 solution at near-neutral pH and that this species is responsible for the hydrolysis of the phosphodiester bond in BNPP.14 ZrK 1:1 is considered to be more catalytically active when compared to ZrK 2:2 because its Zr(IV) ion has more coordinated water molecules that can be replaced by the substrate or that can act as a nucleophile. Theoretical calculations have shown that when ZrK 1:1 interacts with NPP or BNPP, it prefers to form monodentate complexes which are more stable than the corresponding bidentate complexes.14 More interestingly, we recently also found that ZrK 2:2 showed a high hydrolytic reactivity towards the phosphoester bond in the RNA model substrate 2-hydroxypropyl-4-nitrophenyl phosphate (HPNP).25 Model DNA compounds such as NPP, BNPP and RNA model substrate HPNP are often used when testing the reactivity of newly developed artificial phosphatases and nucleases14,23,24,26–29 because of the presence of good leaving groups in their structures, increasing their reactivity in comparison with extremely stable DNA and RNA biomolecules.
The previous studies with POMs containing one or two Zr(IV) ions revealed that their reactivity is influenced by the number of Zr(IV) centers present in the POM structures, as well as by the number of coordinated water molecules at Zr(IV) ions. In this study we explore the phosphoesterase activity of the Zr(IV)-substituted Wells–Dawson that contains four Zr(IV) centers, Na14[Zr4(P2W16O59)2 (μ3-O)2(OH)2(H2O)4]·57H2O POM (ZrWD 4:2)30 (Fig. 1) towards the DNA model substrate NPP, a stable phosphoester characterized by a half-life of 135 days at pH 5.0 and 50 °C.27
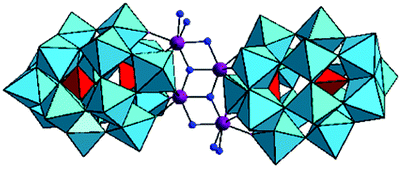 | ||
| Fig. 1 Structure of ZrWD 4:2. The WO6 groups are represented by sky blue octahedra, while the internal PO4 groups are represented by red tetrahedra. Zr(IV) is represented by violet while oxygen atoms of H2O molecules and OH groups are represented by blue balls. | ||
The structure of ZrWD 4:2 consists of a centrosymmetric assembly of two [P2W16O59]12− anions linked by a {Zr4O4}8+ group. This structure contains two different Zr(IV) environments and each Zr(IV) can be considered as 7-coordinate. The two unshared oxygen atoms attached to Zr correspond to water molecules.30 A previous study found that ZrWD 4:2 was still stable at a wide range of pD values (4.0–10.4) after 15-day incubation at 60 °C.31 In this work the kinetics of NPP hydrolysis in the presence of ZrWD 4:2 were performed under different conditions including pD, temperature, POM concentration and ionic strength. A detailed kinetic study combined with multinuclear NMR spectroscopy experiments allowed to propose the detailed mechanism of NPP hydrolysis catalyzed by this POM and to obtain further insight into the role of Zr(IV) centers in the hydrolytic reaction.
Results and discussion
Hydrolysis of NPP in the presence of ZrWD 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :2
:2
The hydrolytic reaction of 5.0 mM of NPP in the presence of 1.0 mM of ZrWD 4:2 (Scheme 1) was conveniently observed by means of 1H and 31P NMR spectroscopy. A five-fold excess of NPP substrate was used, since the reaction with an equimolar ratio of the substrate and ZrWD 4:2 was very fast as compared to the timescale of the NMR experiment and it was not possible to observe simultaneously the signals of the reactant and the product during the course of the reaction. We followed the hydrolysis of the P–O bond based on the disappearance of the aromatic resonances of NPP (8.23 ppm and 7.33 ppm) and the appearance of the aromatic p-nitrophenyl (NP) resonances at 8.17 ppm and 6.78 ppm. An example of the 1H NMR spectra for NPP hydrolysis in the presence of ZrWD 4:2 at pH 6.4 and at different time intervals is shown in Fig. 2A.
 | ||
| Scheme 1 Hydrolysis of NPP promoted by ZrWD 4:2. | ||
 | ||
| Fig. 2
1H spectra (A) and 31P spectra (B) of the hydrolysis of 5.0 mM of NPP in the presence of 1.0 mM of ZrWD 4:2 at different time intervals at pD 6.4 and 50 °C. | ||
The percentage of NPP at different time increments was calculated based on the 1H NMR integration values from Fig. 2A in order to obtain the observed rate constant, and half-life. The percentage of NPP and NP as a function of reaction time is shown in Fig. 3 and the natural logarithm of the concentration of NPP as a function of time (Fig. S1†) was fitted to a first-order linear decay function. At pD 6.4 and 50 °C, an observed rate constant of 8.44 (±0.36) × 10−5 s−1 and half-life of 2.3 h were calculated. The presence of ZrWD 4:2 resulted in a 300-fold rate acceleration in comparison with the hydrolysis of pure NPP (2.81 (±0.25) × 10−7 s−1) under the same reaction conditions. This rate acceleration, achieved in a 1:5 catalyst/substrate ratio is also significantly higher than that of NPP hydrolysis by ZrWD 1:2 which was performed in equimolar amounts of catalysts/substrates, under very similar conditions (kobs = 1.29 × 10−5 s−1, 50 °C, pD 7.2).23
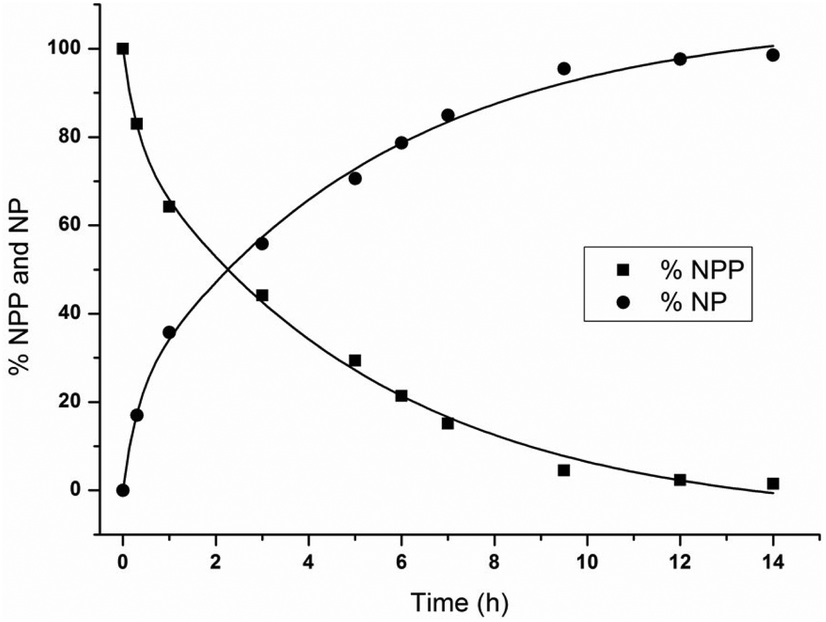 | ||
| Fig. 3 Percentage of NPP and NP as a function of reaction time for the hydrolysis of 5.0 mM of NPP in the presence of 1.0 mM of ZrWD 4:2 at pD 6.4 and 50 °C. | ||
31P NMR spectra (Fig. 2B) recorded during the course of the reaction show that phosphate (P) with a 31P resonance at 0.81 ppm is formed. At the end of hydrolysis, no NPP resonances were detected in both 1H and 31P NMR spectra, indicating full conversion of NPP into phosphate and NP and P.
Aiming to compare the rate constants of the hydrolysis of different substrates catalyzed by ZrWD 4:2 as well as the rate constants of NPP hydrolysis in the presence of different catalysts, several control experiments were performed. First of all, the hydrolysis of the very stable DNA-model substrate BNPP (5.0 mM) in the presence of 1.0 mM of ZrWD 4:2 was also observed at pD 6.4 and 50 °C. The rate constant of 3.49 (±0.22) × 10−7 s−1 was calculated, representing 242-fold decrease in comparison with NPP hydrolysis in the presence of this POM. Similarly, the hydrolysis of RNA-model system HPNP (5.0 mM) in the presence of 1.0 mM ZrWD 4:2 was also examined under identical conditions and the rate constant of 5.06 (±0.18) × 10−6 s−1 was calculated, resulting in a 17-fold decrease in comparison with NPP hydrolysis in the presence of ZrWD 4:2. The ratio between the rate constants of each substrate in the presence (kobs) and in the absence (kuncat) of ZrWD 4:2 was also calculated. The kobs/kuncat for BNPP (386) was nearly 5-fold higher than that for HPNP (83). At pD 6.4 and 50 °C, the reaction between NPP and the monolacunary Wells–Dawson POM indicates that the monolacunary POM does not promote NPP hydrolysis and that the embedded ZrIV ions are responsible for the observed reactivity. Under identical conditions NPP hydrolysis in the presence of ZrCl2O·8H2O was also determined. However, the formation of insoluble ZrIV hydroxyl polymeric gels32,33 was observed, resulting in slower hydrolysis with an observed rate constant of 1.86 (±0.61) × 10−5 s−1. In the current study, the monolacunary Wells–Dawson POM was used to stabilize Zr(IV) ions since aqueous solution of Zr(IV) ions is subject to a solubility problem at pH > 5.0, resulting in polymeric Zr(IV)-hydroxo precipitates.32
Influence of pD on the NPP hydrolysis
As mentioned above, our previous study showed that ZrWD 4:2 is stable in the wide range of pD from 4.0 to 10.4.31 However, as pD plays an important role in the formation of different protonation states of NPP, in this study the effect of pD on the hydrolysis rate of NPP in the absence and presence of ZrWD 4:2 was examined in the pD range from 4.4 to 10.4 and the results are summarized in Table S1.† The pD-rate constant profile for the NPP hydrolysis in the presence of ZrWD 4:2 is shown in Fig. 4. As can be seen from the data from Table S1† and Fig. 4, in the presence of ZrWD 4:2, the rate constants are significantly higher than that in the absence of ZrWD 4:2. In the presence of ZrWD 4:2 the increase of pD values of the reaction mixture leads to the decrease of the observed rate constant. The highest rate constant of 26.6(±0.45) × 10−5 s−1 was observed at pD 4.4 and the value of the rate constant dramatically drops to 2.54 (±0.12) × 10−5 s−1 at pD 10.4. As ZrWD 4:2 preserved its structure in this pH range, this observation is most likely due to the changes in the protonation state of NPP.34 Having a pKa of 5.2,35 NPP exists mostly as a monoanion (C6H5NO6P−) at pH values below 5.2, whereas above 5.2 the dianion (C6H4NO6P2−) species dominates. Therefore, when the pD values of the reaction solution increase, the repulsion between the negative charges of NPP and the negative charges of the POM also increases, resulting in the drop of the rate constant.
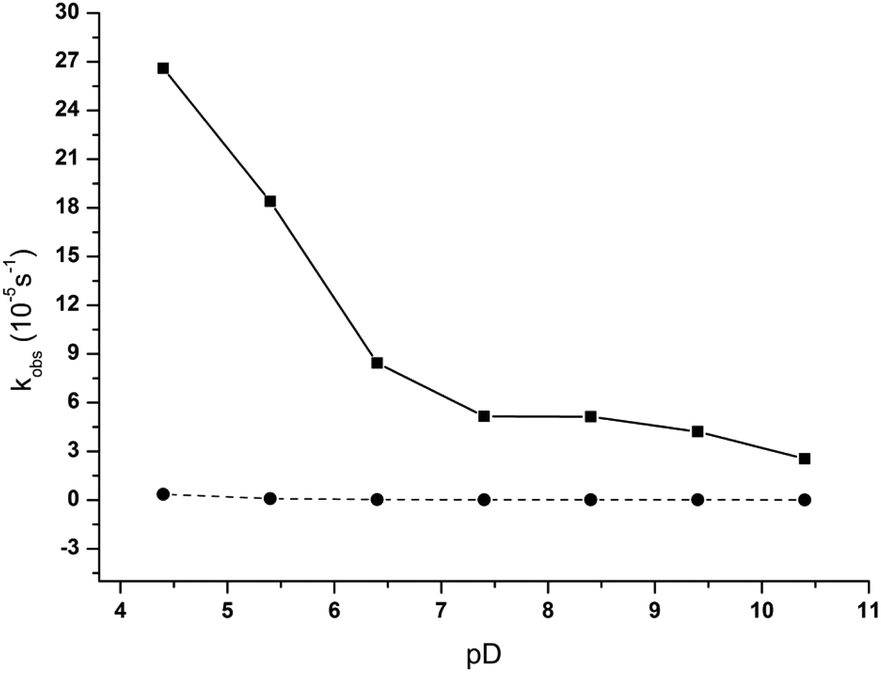 | ||
| Fig. 4 pD dependence profile of kobs for the hydrolysis of 5.0 mM of NPP in the presence (solid line) and in the absence (dashed line) of 1.0 mM of ZrWD 4:2 at 50 °C. | ||
In order to confirm the stability of the POM during the course of the hydrolysis reaction, which was followed at elevated temperatures, 31P NMR spectra were recorded at different time intervals at pD 4.4 (Fig. S2†) and at pD 10.4 (Fig. S3†). As we can see from Fig. S2 and S3† after the reaction was complete (4 hours at pD 4.4, 50 °C and after 96 h at pD 10.4, 50 °C), the signals of two peaks of ZrWD 4:2 at −7.17 ppm and −14.42 ppm were clearly seen with high intensity, indicating that ZrWD 4:2 is indeed stable during the course of the hydrolytic reaction in this range of pD.
Influence of temperature on NPP hydrolysis
With the aim to calculate the kinetic data for the hydrolysis of NPP promoted by ZrWD 4:2, the effect of temperature on the rate constant was examined on a solution containing 5.0 mM of NPP and 1.0 mM ZrWD 4:2 at pD 6.4 in the temperature range from 37 °C to 80 °C and the results are summarized in Table S2.† From these data the activation energy (110.15 ± 7.06 kJ mol−1) was calculated based on the Arrhenius equation (Fig. S4A†). This value is significantly lower than that in the absence of ZrWD 4:2 under identical reaction conditions (147.17 ± 9.86 kJ mol−1). The enthalpy of activation (ΔH‡ = 109.03 ± 6.86 kJ mol−1) and entropy of activation (ΔS‡ = 15.20 ± 2.49 J mol−1 K−1) were calculated based on the Eyring equation (Fig. S4B†). The positive entropy of activation may result from the weak binding of NPP to the ZrK 4:2 POM catalyst. Out of these data, the Gibbs activation energy (ΔG‡) was calculated to be 104.32 ± 6.09 kJ mol−1 at 37 °C. This ΔG‡ value is similar to that of BNPP hydrolysis (111.12 kJ mol−1 at 37 °C) by a binuclear Zr(IV)-substituted Keggin type POM (Et2NH2)8[{α-PW11O39Zr(μ-OH)(H2O)}2]·7H2O24 and of NPP hydrolysis (96.94 kJ mol−1 at 37 °C) by ZrWD 1:2.23
31P NMR spectra in Fig. S5† show that after the reaction at pD 6.4 and 80 °C was complete, the signals of ZrWD 4:2 were unchanged, indicating that ZrWD 4:2 is still stable under these conditions.
Influence of ZrWD 4:2 concentration on NPP hydrolysis
In order to observe the influence of POM concentration on the rate constants, reaction mixtures containing 5.0 mM NPP and increasing amounts of ZrWD 4:2, ranging from 0.5 mM to 3.0 mM, were studied at pD 6.4 and 50 °C and the results are shown in Table S3.† The general catalytic scheme for the hydrolysis of NPP in the presence of ZrWD 4:2 is shown in Scheme 2.
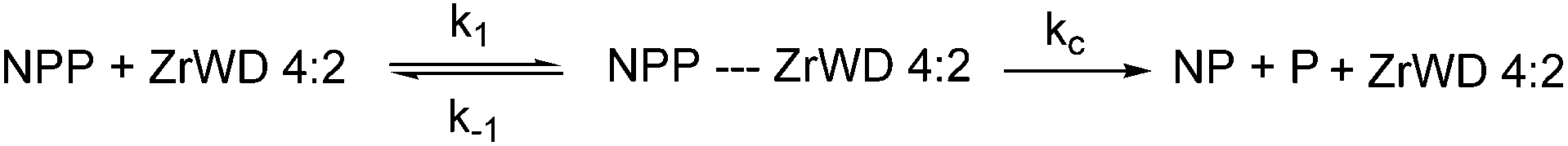 | ||
| Scheme 2 General catalytic scheme for NPP hydrolysis by ZrWD 4:2. | ||
We assume that the formation of the products (kc) is much slower than reaching the equilibrium between NPP and ZrWD 4:2 (e.g. we have k1 + k−1 ≫ kc). Because the formation of nitrophenol (NP) is a first-order reaction, kobs can be written as in eqn (1).23,28,29,36 By fitting the data from Table S3† to eqn (1), the binding constant (Kf = k1/k−1 = 190 M−1) and the catalytic rate constant (kc = 6.40 × 10−4 s−1) were obtained from Fig. 5.
 | (1) |
 | ||
| Fig. 5 Dependence of the observed rate constants on the concentration of ZrWD 4:2 (0.5–3.0) mM for the hydrolysis of 5.0 mM of NPP at pD 6.4 and 50 °C. | ||
The catalytic activity of ZrWD 4:2 is higher than that previously found for ZrWD 1:2 POM (kc = 0.28 × 10−4 s−1).23 This can be explained by the fact that as ZrWD 4:2 POM contains two Zr(IV) ions, which also have more free coordination sites than the Zr(IV) ion in ZrWD 1:2 POM. More interestingly, the catalytic activity of ZrWD 4:2 towards NPP hydrolysis is similar to that of binuclear FeIIIZnII complexes that are biomimetic for purple acid phosphatases and which gave kc values from 4.63 × 10−4 s−1 to 9.20 × 10−4 s−1.37 The catalytic activity of the FeIIIZnII complex was studied towards bis-(2,4-dinitrophenyl) phosphate, which is a phosphodiester but has a better leaving group (2,4-dinitrophenyl) compared to NPP. The detailed studies with the FeIIIZnII complex have revealed that [(OH)FeIII(μ-OH)ZnII(OH2)] is the catalytically active species which has a labile site that first facilitates the monodentate binding of the substrate to the ZnII center, followed by an intramolecular nucleophilic attack of FeIII-bound hydroxide leading to the formation of 2,4-dinitrophenolate. Considering the resemblances in kc of ZrWD 4:2 and FeIIIZnII complexes, it is plausible that they operate by a similar mechanism in promoting the hydrolysis of the phosphoester bond.
As can be seen from Fig. 5 an increase in ZrWD 4:2 concentration leads to an increase in the reaction rate and ZrWD 4:2 can hydrolyse a large excess amount of NPP. Complete hydrolysis of NPP was still observed when 5.0 mM of NPP and 0.5 mM of ZrWD 4:2 were used. This suggests that one equivalent of ZrWD 4:2 hydrolyses at least 10 equivalents of NPP, demonstrating its catalytic activity.
31P NMR spectra of the reaction between 5.0 mM NPP and 3.0 mM ZrWD 4:2 were recorded during the course of the reaction and are shown in Fig. S6 in the ESI.† As can be seen from Fig. S6† after 4 h 45 min when the reaction was complete, no change in the signals of the POM was observed, demonstrating that 3.0 mM ZrWD 4:2 concentration does not affect its stability.
The recyclability of ZrWD 4:2
To evaluate the recyclability of ZrWD 4:2, an extra amount (5.0 mM) of NPP was added to a fully hydrolyzed mixture of 5.0 mM NPP and 1.0 mM ZrWD 4:2 and the pD value of the new reaction mixture was adjusted to 6.4 and the reaction was followed at 50 °C. After the third cycle, the observed rate constants of the three reaction cycles increased nearly 3-fold in comparison with the first reaction (Table S4†). This increase in the rate constant appears unusual at the first glance, and it might originate from the increased ionic strength of solution which is expected after each reaction cycle. Higher ionic strength is a result of increased Na+ concentration from the addition of sodium salt of NPP, and the increase of phosphate ions, one of the products of reaction. In order to explain it we tested the effect of ionic strength on the reaction rate by adding 10 mM NaClO4 to the reaction mixture. Indeed an increase of the reaction rate from 8.44 (±0.36) × 10−5 s−1 to 15.03 (±0.25) × 10−5 s−1 was observed. As both NPP and ZrWD 4:2 are negatively charged it is likely that the presence of additional ions in solution modulates repulsion between these two anions.
The 31P NMR spectrum (Fig. S7†) was recorded after each completed reaction. Fig. S7† shows that there were no new signals as well as no change in the chemical shift of ZrWD 4:2, indicating the stability of ZrWD 4:2 in solution after the third cycle while the intensity of the phosphate signal increased significantly.
The interaction between NPP and ZrWD 4:2
The interaction between NPP and ZrWD 4:2 was studied by the means of 31P NMR spectroscopy. Fig. 6 shows that at pD 6.4 free ZrWD 4:2 is characterized by two peaks at −7.17 and −14.42 ppm, while the free NPP signal has a chemical shift at −1.25 ppm. In the mixture of 5.0 mM NPP and 1.0 mM ZrWD 4:2 the peak of NPP is shifted by 0.18 ppm downfield and no line-broadening was observed while the peaks of ZrWD 4:2 are not changed. The change in the chemical shift of NPP indicates that an interaction between NPP and ZrWD 4:2 occurs.
 | ||
| Fig. 6
31P NMR spectra of (a) 1.0 mM of ZrWD 4:2, (b) 5.0 mM NPP and (c) the mixture of 5.0 mM of NPP and 1.0 mM of ZrWD 4:2 after adjusting the pD to 6.4 (400 MHz, D2O, 293 K, NS = 256, 25% H3PO4). | ||
Proposed mechanism
From the kinetic studies and 31P NMR measurement, the mechanism of NPP hydrolysis in the presence of ZrWD 4:2 is proposed in Scheme 3.14,24,25 In a previous study we found that when NPP interacts with the monomeric ZrIV-substituted Keggin type POM (ZrK 1:1), the coordination of ZrIV of the POM to the oxygen atom of the phosphate group of NPP formed the monodentate complex.14,25 This binding results in a more positive charge on the phosphorus atom of NPP, which facilitates the nucleophilic attack of the OH group of ZrK 1:1 or from a water molecule of the solvent, leading to the cleavage of the P–O bond and the formation of nitrophenol and phosphate. In this study we suggest that the binding between ZrWD 4:2 and NPP would occur in an identical mode. This binding was experimentally supported by the observed shift of the NPP signal in the 31P spectrum (Fig. 6). The ZrIV atom in the POM is essential for hydrolysis as it was evidenced by the control experiments. ZrWD 4:2 contains two different Zr(IV) environments: two ZrIV atoms in which each atom carries two bound water molecules and the other two Zr(IV) ions not having any bound water molecules.30 The former Zr(IV) ions are expected to be more catalytically active since their bound water molecules can be replaced by the substrate or can act as a nucleophile. The binding between ZrIV atom(s) and the oxygen atom of the phosphate group of NPP would result in a more positive charge on the phosphorus atom of NPP, which facilitates the nucleophilic attack of the water molecule of ZrWD 4:2 or from the solvent, resulting in the hydrolysis of the P–O bond and the formation of nitrophenol and phosphate. As there are two Zr(IV) ions in ZrWD 4:2 POM with free coordination sites, it is in principle likely that two NPP molecules bind to this POM.
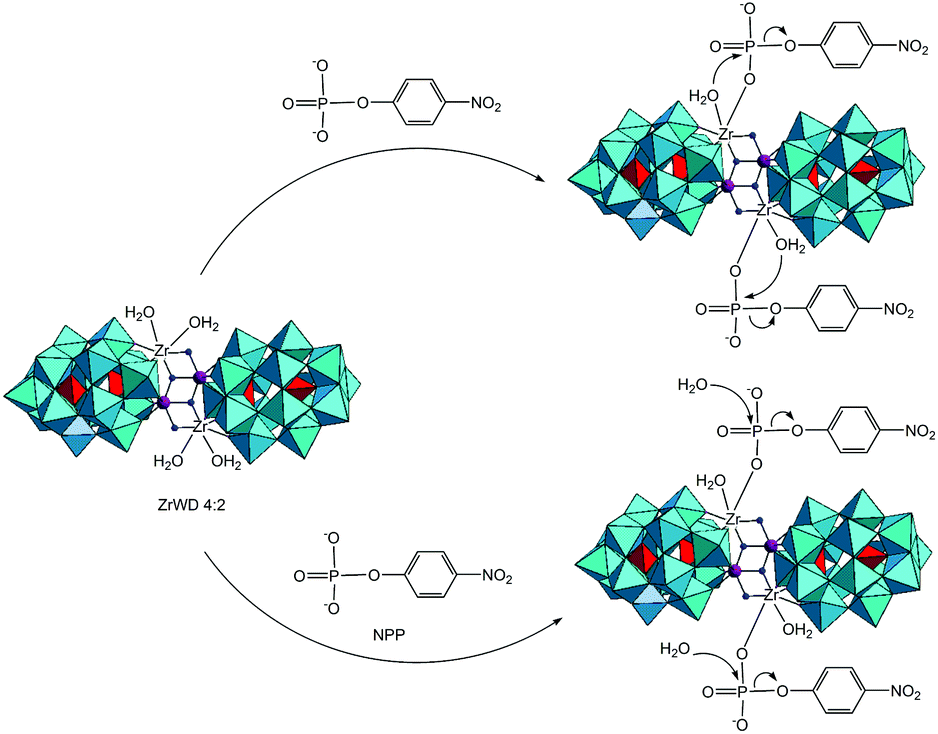 | ||
| Scheme 3 Proposed hydrolytic reaction mechanism of NPP catalyzed by ZrWD 4:2. | ||
The rate acceleration of the NPP hydrolysis promoted by ZrWD 4:2 with a 5:1 ratio in this study is significantly higher than that of NPP hydrolysis promoted by ZrWD 1:2 with a 1:1 ratio in our previous study,17 supporting the hypothesis that both Zr(IV) ions having attached bound water molecules took part in the binding with two NPP molecules.
Conclusions
We report herein the detailed kinetic studies of phosphoester bond hydrolysis promoted by a Zr(IV)-substituted Wells–Dawson type POM (ZrWD 4:2). The study revealed that the presence of multiple Zr(IV) centers in the POM structure is very favorable for the catalytic activity of the POM, most likely due to the possibility of binding and activating more than one molecule of the substrate simultaneously.
The two Zr(IV) ions with free coordination sites are located at the opposite sites of the POM structure, in such a way that the binding of two substrate molecules is sterically possible. The present study demonstrates the potential of ZrWD 4:2 as an artificial phosphatase and contributes to the further development of POMs as Lewis acid catalysts for the hydrolysis of biomolecules.38–42 The results from this study encourage us to further exploit the hydrolytic activity of this POM towards other biologically relevant targets such as phosphoanhydride bonds in ATP, the phosphodiester bonds in complex substrates such as DNA/RNA fragments and the carbon ester bond in p-nitrophenyl acetate.
Experimental
Materials
The Zr(IV)-substituted Wells–Dawson POM Na14[Zr4(P2W16O59)2(μ3-O)2(OH)2(H2O)4]·57H2O30 (ZrWD 4:2), K10[α2-P2W17O61]·20H2O43 and 2-hydroxypropyl-4-nitrophenyl phosphate44 (HPNP) were synthesized and characterized according to the literature. Disodium 4-nitrophenyl phosphate (NPP, C6H4NO6PNa2·6H2O), sodium bis(4-nitrophenyl) phosphate (BNPP, C12H8N2NaO8P), DCl, and NaOD were purchased from Acros and used without any further purification.
Kinetic measurements
Solutions containing 5.0 mM of NPP and 0.5–3.0 mM of ZrWD 4:2 were prepared in D2O. The final pD was adjusted to expected values (4.4–10.4) with minor amounts of 10% DCl and 15% NaOD solutions in D2O. The pH-meter value was corrected by using the equation: pD = pH meter reading + 0.41.45 The pD of the samples was measured at the beginning and at the end of hydrolysis, and the difference was typically less than 0.5 units. The reaction mixture was kept at constant temperature (37–80 °C) and 1H NMR spectra were recorded at certain time intervals during the hydrolytic reaction to calculate the observed rate constant (kobs) by the integral method.
NMR spectroscopy
All of the 1H NMR spectra were recorded on a Bruker Avance 400 and 0.5 mM of 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid (TMSP) was used as an internal standard. All of the 31P NMR spectra were also recorded on a Bruker Avance 400 and 25% phosphoric acid was used as a 31P external reference.Acknowledgements
T. N. P.-V. and P. S. (BOF + fellowship) thank KU Leuven for the financial support and F. W. O. Flanders for a bilateral project. T. K. N. L. thanks the Vietnamese Government and KU Leuven for a doctoral fellowship. The authors acknowledge the CMST COST Action CM1203 (Polyoxometalate Chemistry for Molecular Nanoscience) for the financial support in terms of STSM applications.Notes and references
- P. M. Cullis and E. Snip, J. Am. Chem. Soc., 1999, 121, 6125–6130 CrossRef CAS.
- E. L. Hegg and J. N. Burstyn, Coord. Chem. Rev., 1998, 173, 133–165 CrossRef CAS.
- M. Komiyama, N. Takeda and H. Shigekawa, Chem. Commun., 1999, 1443–1451 RSC.
- R. Kramer, Coord. Chem. Rev., 1999, 182, 243–261 CrossRef.
- A. Blasko and T. C. Bruice, Acc. Chem. Res., 1999, 32, 475–484 CrossRef CAS.
- A. Radzicka and R. Wolfenden, Science, 1995, 267, 90–93 CAS.
- J. E. Thompson, T. G. Kutateladze, M. C. Schuster, F. D. Venegas, J. M. Messmore and R. T. Raines, Bioorg. Chem., 1995, 23, 471–481 CrossRef CAS PubMed.
- J. Rawlings, W. W. Cleland and A. C. Hengge, J. Am. Chem. Soc., 2006, 128, 17120–17125 CrossRef CAS PubMed.
- D. T. Thomas, in Metal-DNA Chemistry, American Chemical Society, Washington, DC, 1989, vol. 402, ch. 1, pp. 1–23 Search PubMed.
- G. Schenk, N. Mitić, G. R. Hanson and P. Comba, Coord. Chem. Rev., 2013, 257, 473–482 CrossRef CAS.
- N. Mitić, S. J. Smith, A. Neves, L. W. Guddat, L. R. Gahan and G. Schenk, Chem. Rev., 2006, 106, 3338–3363 CrossRef PubMed.
- G. Schenk, T. W. Elliott, E. Leung, L. E. Carrington, N. Mitić, L. R. Gahan and L. W. Guddat, BMC Struct. Biol., 2008, 8, 1–13 CrossRef PubMed.
- N. Mitić, K. S. Hadler, L. R. Gahan, A. C. Hengge and G. Schenk, J. Am. Chem. Soc., 2010, 132, 7049–7054 CrossRef PubMed.
- T. K. N. Luong, P. Shestakova, T. T. Mihaylov, G. Absillis, K. Pierloot and T. N. Parac-Vogt, Chem. – Eur. J., 2015, 21, 4428–4439 CrossRef CAS PubMed.
- M. Zhao, H.-B. Wang, L.-N. Ji and Z.-W. Mao, Chem. Soc. Rev., 2013, 42, 8360–8375 RSC.
- M. Zhao, L. Zhang, H.-Y. Chen, H.-L. Wang, L.-N. Ji and Z.-W. Mao, Chem. Commun., 2010, 46, 6497–6499 RSC.
- P. Hu, G.-F. Liu, L.-N. Ji and Z.-W. Mao, Chem. Commun., 2012, 48, 5515–5517 RSC.
- M. Carraro and S. Gross, Materials, 2014, 7, 3956–3989 CrossRef CAS.
- A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605–7622 RSC.
- H. Stephan, M. Kubeil, F. Emmerling and C. E. Müller, Eur. J. Inorg. Chem., 2013, 2013, 1585–1594 CrossRef CAS.
- N. V. Izarova, M. T. Pope and U. Kortz, Angew. Chem., Int. Ed., 2012, 51, 9492–9510 CrossRef CAS PubMed.
- A. Sartorel, M. Bonchio, S. Campagna and F. Scandola, Chem. Soc. Rev., 2013, 42, 2262–2280 RSC.
- S. Vanhaecht, G. Absillis and T. N. Parac-Vogt, Dalton Trans., 2012, 41, 10028–10034 RSC.
- T. K. N. Luong, G. Absillis, P. Shestakova and T. N. Parac-Vogt, Eur. J. Inorg. Chem., 2014, 2014, 5276–5284 CrossRef CAS.
- T. K. N. Luong, G. Absillis, P. Shestakova and T. N. Parac-Vogt, Dalton Trans., 2015, 44, 15690–15696 RSC.
- P. Nunes, A. C. Gomes, M. Pillinger, I. S. Gonçalves and M. Abrantes, Microporous Mesoporous Mater., 2015, 208, 21–29 CrossRef CAS.
- N. Steens, A. M. Ramadan, G. Absillis and T. N. Parac-Vogt, Dalton Trans., 2010, 39, 585–592 RSC.
- G. Absillis, R. Van Deun and T. N. Parac-Vogt, Inorg. Chem., 2011, 50, 11552–11560 CrossRef CAS PubMed.
- E. Cartuyvels, G. Absillis and T. N. Parac-Vogt, Chem. Commun., 2008, 85–87 RSC.
- A. J. Gaunt, I. May, D. Collison, K. Travis Holman and M. T. Pope, J. Mol. Struct., 2003, 656, 101–106 CrossRef CAS.
- H. G. T. Ly, G. Absillis and T. N. Parac-Vogt, Eur. J. Inorg. Chem., 2015, 2015, 2206–2215 CrossRef CAS.
- A. Singhal, L. M. Toth, J. S. Lin and K. Affholter, J. Am. Chem. Soc., 1996, 118, 11529–11534 CrossRef CAS.
- R. A. Moss, J. Zhang and K. G. Ragunathan, Tetrahedron Lett., 1998, 39, 1529–1532 CrossRef CAS.
- K. A. Holbrook and L. Ouellet, Can. J. Chem., 1958, 36, 686–690 CrossRef CAS.
- Z. Y. Zhang, W. P. Malachowski, R. L. Vanetten and J. E. Dixon, J. Biol. Chem., 1994, 269, 8140–8145 CAS.
- J. H. Espenson, in Chemical kinetics and reaction mechanisms, McGraw-Hill, 1995, ch. 4, pp. 86–90 Search PubMed.
- R. A. Peralta, A. J. Bortoluzzi, B. de Souza, R. Jovito, F. R. Xavier, R. A. A. Couto, A. Casellato, F. Nome, A. Dick, L. R. Gahan, G. Schenk, G. R. Hanson, F. C. S. de Paula, E. C. Pereira-Maia, S. de P. Machado, P. C. Severino, C. Pich, T. Bortolotto, H. Terenzi, E. E. Castellano, A. Neves and M. J. Riley, Inorg. Chem., 2010, 49, 11421–11438 CrossRef CAS PubMed.
- K. Stroobants, G. Absillis, E. Moelants, P. Proost and T. N. Parac-Vogt, Chem. – Eur. J., 2014, 20, 3894–3897 CrossRef CAS PubMed.
- K. Stroobants, V. Goovaerts, G. Absillis, G. Bruylants, E. Moelants, P. Proost and T. N. Parac-Vogt, Chem. – Eur. J., 2014, 20, 9567–9577 CrossRef CAS PubMed.
- H. G. T. Ly, G. Absillis, R. Janssens, P. Proost and T. N. Parac-Vogt, Angew. Chem., Int. Ed., 2015, 54, 7391–7394 CrossRef CAS PubMed.
- A. Sap, E. De Zitter, L. Van Meervelt and T. N. Parac-Vogt, Chem. – Eur. J., 2015, 21, 11692–11695 CrossRef CAS PubMed.
- T. Quanten, P. Shestakova, D. Van Den Bulck, C. Kirschhock and T. N. Parac-Vogt, Chem. – Eur. J., 2016, 22, 3775–3784 CrossRef CAS PubMed.
- A. P. Ginsberg, in Inorganic syntheses, John Wiley & Sons, 1990, vol. 27, ch. 3, p. 107 Search PubMed.
- J. S. W. Tsang, A. A. Neverov and R. S. Brown, J. Am. Chem. Soc., 2003, 125, 1559–1566 CrossRef CAS PubMed.
- P. K. Glasoe and F. A. Long, J. Phys. Chem., 1960, 64, 188–190 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 31P NMR spectra, ln[NPP] as a function of time, the data of the kinetic studies. See DOI: 10.1039/c6dt02211a |
| This journal is © The Royal Society of Chemistry 2016 |