
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two new Cu(II) and La(III) 2D coordination polymers, synthesis and in situ structural analysis by X-ray diffraction†
F.
Lundvall
*a,
D. S.
Wragg
ab,
P. D. C.
Dietzel
c and
H.
Fjellvåg
ab
aSMN – Centre for Materials Science and Nanotechnology, Department of Chemistry, University of Oslo, P.O. Box 1126, N-0318 Oslo, Norway. E-mail: fredrik.lundvall@smn.uio.no
binGAP – Innovative Natural Gas Processes and Products, Department of Chemistry, University of Oslo, P.O. Box 1033, N-0315 Oslo, Norway
cDepartment of Chemistry, University of Bergen, P.O. Box 7803, N-5020 Bergen, Norway
First published on 25th July 2016
Abstract
Two new coordination polymers were synthesized solvothermally using 4,4′-dimethoxy-3,3′-biphenyldicarboxylic acid (H2dmbpdc), and di- and trivalent metal salts (Cu(NO3)2·2.5H2O and La(NO3)3·6H2O). Their structures were determined by single-crystal X-ray diffraction analysis, and their thermal stability was evaluated by thermogravimetric analysis. The copper compound Cu(dmbpdc)(DMF; N,N-dimethylformamide), CPO-71-Cu, is based on the well known copper acetate paddlewheel secondary building unit. The asymmetric unit comprises one copper cation with one DMF molecule and one linker molecule coordinated. The lanthanum compound La2(dmbpdc)3(DMF)(H2O)3, CPO-72-La, is formed from a dimer of nine-coordinate, edge sharing lanthanum cations. To this dimer, three water molecules and one DMF molecule are coordinated in an ordered fashion. In addition, the asymmetric unit contains three crystallographically unique linker molecules. Both CPO-71-Cu and CPO-72-La form two-dimensional layered structures, and topological analyses reveal sql topologies with point symbol 44·62 and vertex symbol 4·4·4·4·6(2)·6(2). The thermal behavior of CPO-71-Cu was investigated in an in situ structural analysis by variable temperature powder- and single-crystal X-ray diffraction.
Introduction
Coordination polymers (CPs) and metal–organic frameworks (MOFs) have attracted significant attention in the literature in the last decades. The combination of inorganic metal secondary building units (SBUs) and organic linkers has opened up a world of crystalline one-, two and three-dimensional (1D, 2D and 3D) compounds. The compounds can be both porous and non-porous with interesting properties within highly diverse fields such as gas separation and sorption, catalysis, magnetism and optical applications.1–7Investigating the properties of known MOFs and coordination polymers is a highly active research field. Improving existing knowledge and discovering new details is of great importance to drive the field forward. Equally important are the numerous reports on completely new structures that can inspire and set off new investigations. Notably, the search for new structures has frequently been limited to linkers available from commercial suppliers. As a consequence, many well known MOFs such as MOF-5, HKUST-1, CPO-27/MOF-74 and MIL-53 have in common that they are based on relatively simple commercially available linkers.8–12 Recently, the trend in coordination polymer chemistry is moving towards more specialized linkers, often designed and produced in-house with a specific property in mind.13 A linker can for instance be intended for post-synthetic modification, have a built in photoactive center, or be designed with catalytic properties in mind.
Recently, we reported two series of 3D coordination polymers based on 4,4′-dimethoxy-3,3′-biphenyldicarboxylic acid (H2dmbpdc), a relatively simple yet unexplored linker.14 Here we present the synthesis and characterization of two new coordination polymers based on the same linker. The new coordination polymers are layered 2D structures based on dinuclear metal SBUs, not previously reported in combination with H2dmbpdc. Furthermore this is the first report of a coordination polymer combining H2dmbpdc and a trivalent rare-earth cation.
Experimental
Materials and methods
The linker, 4,4′-dimethoxy-3,3′-biphenyldicarboxylic acid, was synthesized according to our previously reported procedure.14 All other starting materials and solvents were used as received from commercial suppliers (Sigma-Aldrich and VWR).Thermogravimetric analysis and Differential Scanning Calorimetry (TGA/DSC) was performed on a Netzsch STA 449F1 under a flow of N2-gas. The samples were heated from 30 to 800 °C in alumina or platinum crucibles, with a ramp rate of 2 or 5 °C min−1. The TGA/DSC results are displayed in Fig. S2 and S3 of the ESI.†
Single-crystal X-ray diffraction (S-XRD) data were recorded on a Bruker D8 Venture instrument fitted with a PHOTON CCD area-detector, and using monochromatic MoKα1 radiation (λ = 0.7093 Å) from a fine-focus sealed tube source. Data reduction was done using SAINT,15 and absorption correction by SADABS.15 The structures were solved by direct methods using SIR92,16 and refined using SHELX-2014/6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 as implemented in the WinGX18 and OLEX219 program suites. Where necessary for the refinement of the structure, the SQUEEZE protocol of PLATON20 was used to remove residual electron density from the solvent accessible voids (SAV).
17 as implemented in the WinGX18 and OLEX219 program suites. Where necessary for the refinement of the structure, the SQUEEZE protocol of PLATON20 was used to remove residual electron density from the solvent accessible voids (SAV).
Variable temperature (VT) S-XRD was recorded on the Swiss-Norwegian Beamline (BM01A, SNBL) at the European Synchrotron Radiation Facility (ESRF) using monochromatic synchrotron radiation (λ = 0.69396 Å) and a Dectris Pilatus 2 M photon counting pixel area detector.21 Data reduction, absorption correction and structure solution was done using CrysAlis,22 and the structures were refined using SHELX-2014/617 as implemented in the WinGX18 and OLEX219 program suites. Hydrogen atoms were positioned geometrically at distances of 0.95 (CH) and 0.98 Å (CH3), and refined using a riding model with Uiso(H) = 1.2Ueq(CH) and Uiso(H) = 1.5Ueq(CH3).
Powder X-ray diffraction (P-XRD) data were recorded at ambient atmosphere and temperature on a Bruker D8-A25 instrument using monochromatic CuKα1 radiation (λ = 1.5406 Å) from a fine-focus sealed tube source and a LynxEye XE position sensitive detector operated in transmission geometry. Crystals of CPO-71-Cu and CPO-72-La were ground to a powder in a mortar and sealed in glass capillaries for measurement. A Pawley fit was performed on the recorded patterns using the TOPAS23 software with the unit cell parameters determined by S-XRD to confirm the absence of other crystalline phases in the samples (see Fig. S6 and S7 in the ESI†).
VT P-XRD was recorded on the SNBL at the ESRF using monochromatic synchrotron radiation (λ = 0.69396 Å). The experimental setup uses a Huber goniometer and Dectris Pilatus 2 M photon counting pixel area detector. The patterns were analyzed by parametric refinement24 using TOPAS to extract the unit cell parameters as a function of temperature. The patterns were visualized as 2D films using Powder3D25 (see Fig. S1 in the ESI†).
Syntheses
Results and discussion
The solvothermal synthesis of 4,4′-dimethoxy-3,3′-biphenyldicarboxylic acid and either Cu(NO3)2·2.5H2O or La(NO3)3·6H2O resulted in two new CPs; CPO-71-Cu and CPO-72-La (Table 1). Both compounds are layered structures with solvent accessible interlayers. Topological analyses with the TOPOS40 software26 reveal that the underlying topologies of the structures are the same. Both CPO-71-Cu and CPO-72-La are based on cation dimers that form four-connected nodes coupled by linker molecules. This results in a sql topology following the three-letter codes recommended by RCSR27 (Fig. 1d and 2d), with point symbol 44·62 and vertex symbol 4·4·4·4·6(2)·6(2). In both compounds, the organic linker is fully deprotonated. Hence, there is a 1:1 and 2:3 metal to linker ratio in CPO-71-Cu and CPO-72-La to maintain charge balance.
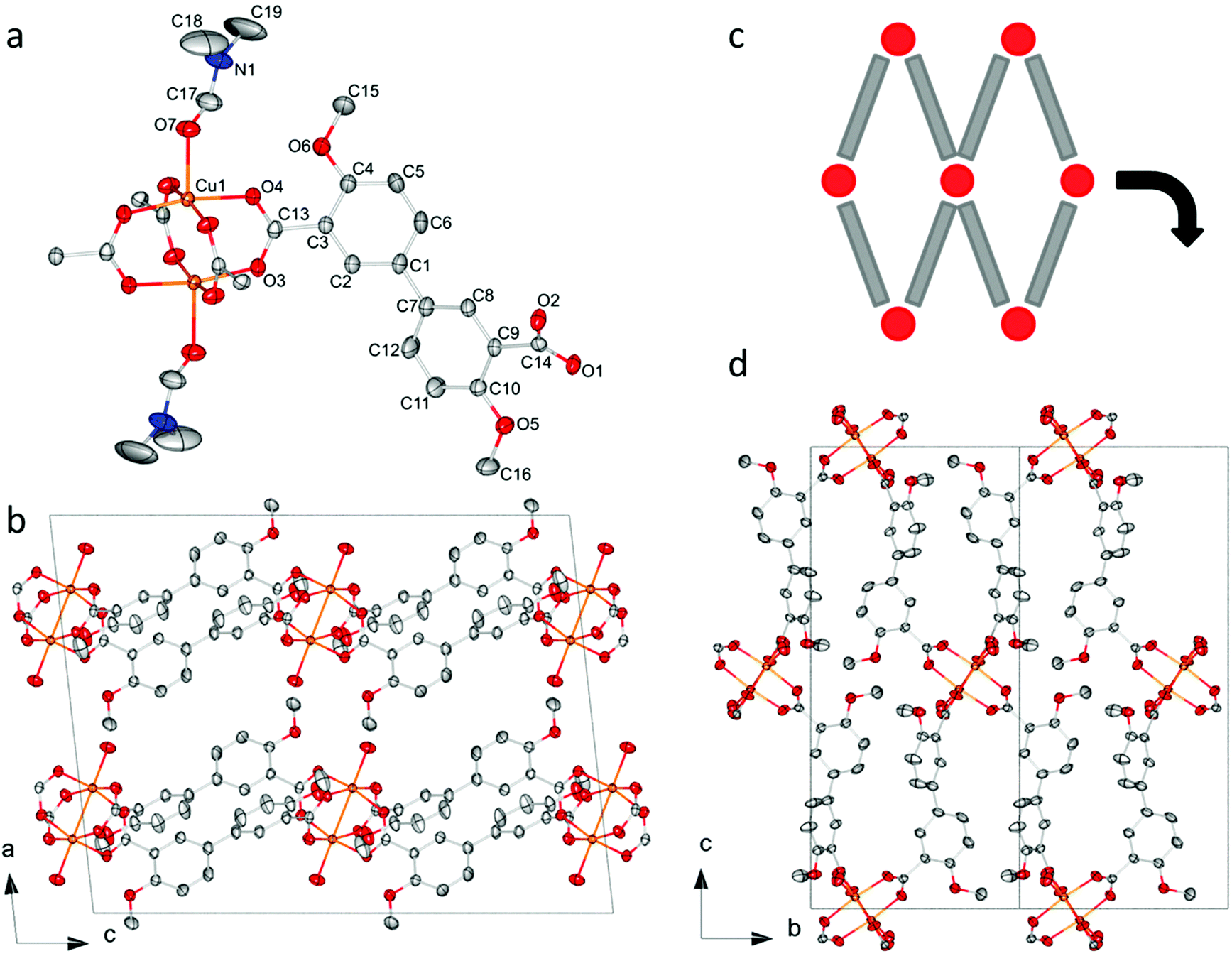 | ||
| Fig. 1 The crystal structure of CPO-71-Cu. (a) The coordination environment around the copper paddlewheel SBU and one linker molecule. (b) Packing diagram showing the layers of the structure, viewed along the b-axis. (c) A cartoon illustrating the underlying sql topology of CPO-71-Cu. (d) Packing diagram viewed along the a-axis, emphasizing the sql topology of the structure. Non-oxygen atoms of the coordinated DMF molecules are omitted for clarity in (b) and (d). All hydrogen atoms are omitted for clarity and thermal ellipsoids are drawn at 50% probability level (Cu: orange, C: grey, O: red, N: blue). | ||
 | ||
| Fig. 2 The six carboxylate to lanthanum coordination modes in CPO-72-La. The two bottom cartoons represent the two different edges that connect the nodes in the sql network. | ||
| Name | CPO-71-Cu | CPO-72-La |
|---|---|---|
| Formula | C19H19NO7Cu | C54H64N2O27La2 |
| Formula weight | 436.89 | 1450.89 |
| T (K) | 105 (2) | 105 (2) |
| Crystal system | Monoclinic | Triclinic |
| Space group (#) | C2/c (15) |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (2) (2) |
| Z | 8 | 2 |
| a (Å) | 18.0477 (7) | 13.4844 (5) |
| b (Å) | 10.3801 (4) | 14.1740 (6) |
| c (Å) | 23.4227 (9) | 16.6448 (6) |
| α (°) | 90 | 90.4630 (10) |
| β (°) | 96.7430 (10) | 105.8700 (10) |
| γ (°) | 90 | 99.8160 (10) |
| V (Å3) | 4357.6 (3) | 2829.36 (19) |
| D c (g cm−3) | 1.332 | 1.703 |
| μ (mm−1) | 1.039 | 1.582 |
| Reflections collected | 34274 |
90198 |
| Reflections unique | 5023 | 14081 |
| Parameters | 257 | 837 |
| Restraints | 0 | 55 |
| R int | 0.0531 | 0.0348 |
| Crystal size (mm3) | 0.26 × 0.14 × 0.14 | 0.75 × 0.30 × 0.22 |
| Crystal color | Green | Pale brown |
| Crystal shape | Needle | Needle |
| F(000) | 1800 | 1464 |
| Residual density max/min (e Å−3) | 0.594/−0.335 | 2.989/−1.316 |
| GOF | 1.059 | 1.103 |
| Final R indices [I > 2σ(I)] | R1 = 0.0405 | R1 = 0.0316 |
| wR2 = 0.0792 | wR2 = 0.0683 | |
| R indices (all data) | R1 = 0.0589 | R1 = 0.0425 |
| wR2 = 0.0841 | wR2 = 0.0720 |
We recently reported two new series of CPs, CPO-68-M and CPO-69-M, that are based on the same linker as CPO-71-Cu and CPO-72-La.14 Although they are based on the same linker, CPO-68-M and CPO-69-M feature a completely different topology (sra). CPO-68-M and CPO-69-M are formed from 1D metal–carboxylate chain SBUs that are separated by linker molecules. This results in 3D structures differing significantly from the layered structures of CPO-71-Cu and CPO-72-La. The synthesis procedures of all four CPs have similarities with regards to synthesis solvent and temperature range. Thus, it is evident that other factors than these determine which SBU is more favourable in the different CPs.
Cu(dmbpdc)(DMF), CPO-71-Cu
The crystal structure of CPO-71-Cu is based on the copper acetate paddlewheel SBU.28–30 The two copper atoms of the SBU are linked together by the carboxylate group of four organic linkers, creating a four-connected node. DMF solvent molecules are coordinated in the axial positions of the SBU (Fig. 1a), and the layers of the structure are oriented parallel to the bc-plane (Fig. 1b). However, due to the non-linear nature of the linker, the axis of the copper SBU is not oriented normal to the layers as might be expected. Moreover, by measuring the angle between bc-plane and the axis of the SBU, one finds that the axis is offset by more than 30° from the normal to the bc-plane. The combination of a rigid copper SBU and a non-linear linker has resulted in a degree of distortion of the linker itself to form the structure. Carboxylate substituted biphenyl compounds often tend towards a near planar configuration to preserve the stabilization energy of an extended π-orbital system. Recently we published the crystal structure of the linker, which exhibits a planar configuration in its uncoordinated state.31 In CPO-71-Cu however, both the carboxylate groups as well as the rings of the biphenyl moiety are rotated significantly out of plane (Table 2). Thus, some stabilization energy in the linker is probably lost during synthesis to preserve the rigid copper SBU.| CPO-71-Cu | |
|---|---|
| (i) −x + 0.5, y − 0.5, −z + 0.5; (ii) x, −y + 1, z + 0.5; (iii) −x + 0.5, −y + 0.5, −z + 1; (iv) phenyl–phenyl torsion; (v) phenyl–carboxylate torsion. | |
| Cu1–O1i | 1.9655 (15) |
| Cu1–O2ii | 1.9620 (15) |
| Cu1–O4 | 1.9477 (15) |
| Cu1–O5iii | 1.9639 (16) |
| Cu1–O1C | 2.1430 (16) |
| (C1–C6)–(C7–C12)iv | 35.85 (12) |
| (C1–C6)–(O1–C13–O2)v | 40.09 (15) |
| (C7–C12)–(O4–C15–O5)v | 31.92 (18) |
Indeed, the stability of the copper paddlewheel SBU provides a possible explanation as to why CPO-71-Cu is not iso-structural to either CPO-68-M or CPO-69-M. One can draw some parallels to the case of Cu2(dhtp),32 a late addition to the CPO-27/MOF-74 family of metal–organic frameworks.10,11 Like CPO-68-M and CPO-69-M, the structure of CPO-27/MOF-74 is based on 1D metal–carboxylate chains. Similarly, the structure also supports a range of divalent metal cations (e.g. Zn2+, Co2+, Ni2+, Mg2+, Fe2+ and Cu2+). However, divalent copper ions have a strong tendency to form the paddlewheel motif in the presence of carboxylates. Hence, the synthesis of the copper analogue of CPO-27/MOF-74 was only recently achieved through careful tuning of the synthesis conditions. Currently there is no reported copper analogue of CPO-68-M or CPO-69-M, but the possibility of synthesizing such a material should not be ruled out.
The structure of CPO-71-Cu includes solvent accessible interlayers. Although the crystallographic data set is of high quality and the coordinated DMF molecules can be refined with high precision, this is not the case for the solvent located in the interlayers. The interlayers contain a disordered mixture of the synthesis solvents DMF and H2O in the as synthesized compound. The electron density originating from the disordered solvent was removed from the structure using SQUEEZE before the final refinement. The calculation from PLATON gives a solvent accessible volume of 856 Å3 in the unit cell (19.6% of total volume) for the dataset collected at 105 K.
La2(dmbpdc)3(DMF)(H2O)3, CPO-72-La
The crystal structure of CPO-72-La is based on a dimer of lanthanum cations forming a four-connected node. Each lanthanum cation is coordinated to nine oxygen atoms. These stem from both the carboxylate groups of the linkers as well as from the synthesis solvents DMF and H2O (Fig. 3a and b). The six unique carboxylate groups of the asymmetric unit have three different coordination modes (Fig. 2). Two carboxylate groups coordinate their oxygen atoms to only one lanthanum cation each, two groups are coordinated in a bridging mode, and two groups are coordinated with three bonds to both lanthanum cations (blue, red and yellow circles respectively in Fig. 2). Thus, two of the oxygen atoms (O1 and O7) in the edge sharing coordination polyhedra are shared between the two lanthanum cations. The remaining two vertices in the nine-coordinated polyhedra are occupied by solvent molecules. A distinct feature of the structure is the non-identical coordination environment around the two cations with regards to solvent molecules. Whereas La1 is coordinated to two H2O molecules, La2 is coordinated to one DMF and one H2O in an ordered fashion. The structure forms layers parallel to (01−1) with solvent accessible interlayers (Fig. 3c). The sql topology of the layers might at first be counter intuitive considering the 2:3 metal to ligand ratio of the structure. However, when the structure is viewed along [01−1], the sql topology becomes obvious as each lanthanum-node is clearly connected to four other nodes (Fig. 3d).
 | ||
| Fig. 3 The crystal structure of CPO-72-La. (a) The asymmetric unit of CPO-72-La, with both the coordinated DMF molecule as well as the DMF trapped in the solvent accessible interlayers. (b) The coordination environment around the lanthanum dimer SBU. (c) Packing diagram showing the layers of the structure, viewed along the a-axis. (d) Packing diagram viewed close to [01−1], emphasizing the sql topology of the structure. Non-oxygen atoms of the coordinated DMF molecule are omitted for clarity in b, c and d. All hydrogen atoms are omitted for clarity and thermal ellipsoids except La in (b) are drawn at 50% probability level (La: pink, C: grey, O: red, N: blue). | ||
Contrary to CPO-71-Cu, the solvent in the interlayers of CPO-72-La is ordered, and one DMF molecule as well as four H2O molecules can be refined in the structure. The single-crystals of CPO-72-La also differ from CPO-71-Cu by being highly sensitive to solvent loss. Treatment in vacuum, thermal treatment or even just exposure to ambient conditions rapidly results in cracking of the single-crystals into smaller crystallites. Polycrystalline P-XRD samples are slightly more resilient, and P-XRD patterns of decent quality can be recorded up to 100 °C. Above this temperature however, the patterns rapidly deteriorate to the point that unit cell parameters can no longer be refined with satisfactory accuracy above 140 °C. This indicates that the solvent molecules in the interlayer stabilize the structure, and that the ordering of the solvent is likely due to interactions between the solvent and the framework. The O–O distances between oxygen atoms in the framework and the interlayer solvent range from 2.848 (3) Å to 2.887 (3) Å. This is consistent with hydrogen bonding interaction (see Table S3 in the ESI†). There also appears to be a hydrogen bonding network between the individual solvent molecules, with O–O distances ranging from 2.737 (4) Å to 2.886 (4) Å. This could be a contributing factor to the ordering. The TGA data indicates some degree of thermal stability until about 300 °C (see Fig. S2 in the ESI†). Unfortunately, the material lacks the long range order necessary for XRD analysis at this temperature, possibly due to turbostratic disorder.33
In the structure, there are a total of 18 La–O bonds, ranging from 2.419 (2) Å to 2.868 (2) Å. These bond lengths are in accordance with the expected values for La3+/O2− in a nine-coordinate environment when applying the bond valence method.34 Selected bond lengths are presented in Table 3, and the full list is available in the ESI (Table S2†). Note that the La–O(DMF) bond is slightly shorter than the La–O(H2O) bonds. Similar to CPO-71-Cu, there is some distortion of the linkers in CPO-72-La. This distortion is however less pronounced, and mainly limited to four of the six carboxylate groups as noted in Table 3. The two remaining carboxylate groups deviate less than 10 degrees from the benzene ring plane.
| CPO-72-La | |
|---|---|
| (i) H2O; (ii) DMF; (iii) phenyl–carboxylate torsion. | |
| La1–O3Ci | 2.504 (2) |
| La1–O4Ci | 2.677 (2) |
| La2–O1Cii | 2.481 (2) |
| La2–O2Ci | 2.641 (3) |
| (C1–C6)–(O1–C7–O2)iii | 59.77 (16) |
| (C9–C14)–(O4–C15–O5)iii | 23.27 (26) |
| (C17–C22)–(O7–C23–O8)iii | 62.23 (17) |
| (C25–C30)–(O10–C31–O11)iii | 26.47 (27) |
In the synthesis procedure, the solvent mixture of DMF and H2O was pre-treated at 80 °C for 72 hours. This step is crucial in the synthesis procedure, although the exact reason and mechanism is currently unknown. What is known is that DMF hydrolyses in the presence of water at elevated temperatures, forming formic acid and dimethyl amine. There are reports in the literature where the in situ formed dimethylammonium and/or formate is incorporated in the resulting structure.35–37 Although no such incorporation is observed for CPO-72-La, it is possible that one or both of these hydrolysis products may have a form of templating effect on the structure during synthesis.38 Another possibility one could imagine is that the formic acid takes on the role as a modulator, regulating the crystal growth.39–41 However, the concentration of formic acid present during synthesis is much lower than common modulator concentrations (e.g. 5–30 mole equivalents).41,42 Further insight into the role of such in situ formed species in low concentration is needed.
In situ structural analysis of the thermal behavior of CPO-71-Cu
CPO-71-Cu is less sensitive to solvent removal when compared to CPO-72-La. The thermal behavior was investigated in situ by variable temperature (VT) P-XRD at the ESRF. TGA data recorded prior to this experiment indicated a significant mass loss starting around 110 °C, likely due to solvent evaporation (see Fig. S2 in the ESI†). Thus, we expected a change in the crystallographic parameters around this temperature. At large scale, the diffraction patterns remain fairly unchanged up to about 230 to 250 °C, where the diffraction patterns start to deteriorate significantly. The TGA data indicate thermal decomposition around the same temperature. We performed a parametric refinement of the P-XRD data that revealed several interesting subtle features with respect to the unit cell parameters (Fig. 4, and Fig. S5 in the ESI†). Note that the precision of the refinements start to decrease around 230 °C, which coincides well with the thermal breakdown indicated by the TGA data (Fig. S4 in the ESI†). | ||
| Fig. 4 Change in unit cell axis and β-angle parameters of CPO-71-Cu plotted against temperature. Unit cell axis parameters have been scaled to show change in percentage relative to the initial room temperature values. | ||
Firstly, the changes in the b- and c-axis parameters are tightly linked, indicating a concerted movement within the layers of the structure upon heating. Secondly, the a-axis initially lengthens, before it starts to contract around 130 °C. Lastly, there are subtle changes to the β-angle throughout the experiment, but most pronounced from 130 °C. The changes in the β-angle seem linked to the changes in the a-axis. The sum of the cell parameter changes is plotted as cell volume as function of temperature (Fig. 5). These observations are interpreted as a thermal expansion followed by contraction of the framework due to solvent evaporation.
 | ||
| Fig. 5 Unit cell volume of CPO-71-Cu plotted against temperature. | ||
A change in the a-axis was expected since the layers of the structure are oriented parallel to the bc-plane. However, the changes in the b- and c-axis were more surprising, and are attributed to a slight reorientation of the linker molecules as the solvent evaporates. The sum of these changes is evident as shifts in certain reflections, and is particularly noticeable in reflections with a high h- and l-Miller index component (Fig. 6).
 | ||
| Fig. 6 Excerpt of the VT measurement of CPO-71-Cu displayed as a 2D film, y-axis = scan number (increasing temperature), x-axis = reflection angle in degrees 2Θ (λ = 0.69396 Å). Selected temperatures and reflections are indicated in red. The full 2D film is available in the ESI.† | ||
To further elucidate on the structural changes, a VT S-XRD experiment was set up to collect full datasets of the crystal structure every 10 °C from 70 to 280 °C. The S-XRD results are consistent with the findings from the P-XRD analysis (see also Table S1 and Fig. S5 in the ESI†). On large scale there are only subtle changes in the main structure upon heating. Furthermore, the electron density in the interlayers that originates from the disordered solvent molecules remains fairly constant up to around 130 °C (Fig. 7). From 130 °C onwards, the electron density decreases steadily until the structure is fully evacuated at around 230 °C.
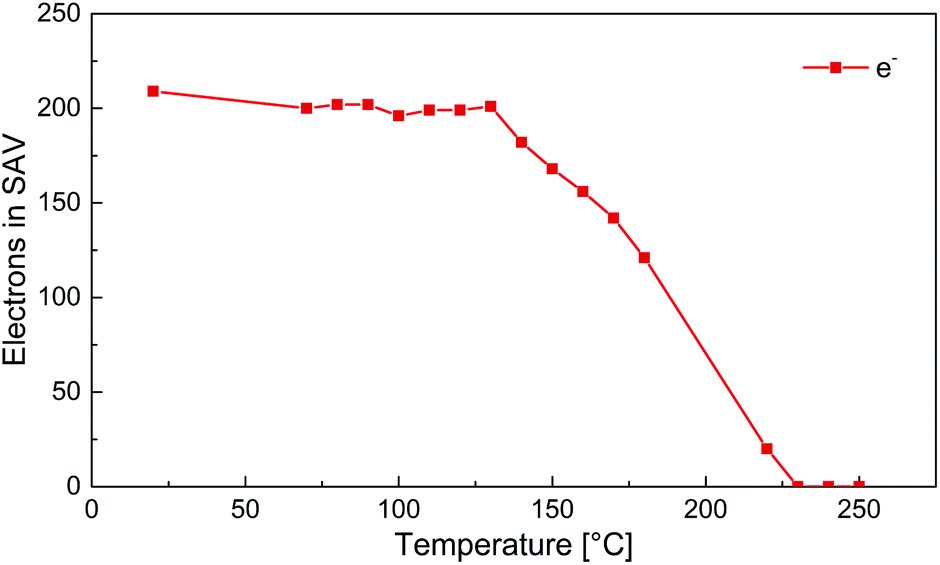 | ||
| Fig. 7 Electrons removed from the solvent accessible voids (SAV) in the unit cell by the SQUEEZE protocol of PLATON, plotted against temperature. | ||
Interestingly, the DMF molecule coordinated to the copper paddlewheel SBU remains in place even at 250 °C, demonstrating that this coordination is very stable. As indicated in Table 2, the Cu–O(DMF) bond length in the structure is 2.1430 (16) Å. This is comparable to Cu–O(H2O/DMF/DEF/MeOH) bonds lengths that are reported in the literature for coordination polymers based on the same copper paddlewheel SBU.9,43,44 In other words, the stability of the coordination can not simply be explained by an unusually short or strong bond. By comparison, the coordinated H2O solvent molecules found in the SBU of the Cu(BTC) MOF (HKUST-1) are very labile. The Cu–O(H2O) bond length is here 2.165 (8) Å, thus only marginally longer than in CPO-71-Cu. Yet the coordinated solvent can easily be removed by heating the MOF to 100 °C in air.9 It is thus likely that other structural factors than the Cu–O(DMF) bond length dictates the stability of this bond.
Once the structure is fully evacuated at 230 to 250 °C, the S-XRD patterns deteriorate rapidly, indicating that the structure only supports partial solvent removal. Additional experiments on ex situ activated samples support this hypothesis. Samples of CPO-71-Cu were activated at 130 and 160 °C under dynamic vacuum, as well as one sample activated at 180 °C under atmospheric conditions. By applying the SQUEEZE protocol to the S-XRD data collected on these samples, we could determine residual electron density comparable to the in situ S-XRD data (169, 80 and 30 electrons per unit cell for the three respective datasets). Note that the crystal quality was slightly diminished in the sample activated at 180 °C when compared to the samples activated at 130 and 160 °C. Solvent exchange does not seem to aid the activation process. A sample exchanged with methanol for 48 hours became visually opaque and non-diffracting in X-rays (see also Fig. S8 in the ESI†). From these experiments it seems that partial activation of CPO-71-Cu is possible. However some residual solvent, probably DMF, is necessary to provide a form of cohesion between the layers in the structure.
Conclusions
In this article we have described the solvothermal synthesis of two new 2D coordination polymers. Both compounds exhibit the sql topology in spite of the difference in cation charge and metal to ligand ratio. CPO-71-Cu was investigated in situ by variable temperature P-XRD and S-XRD. These studies revealed that CPO-71-Cu is thermally robust, and has an interesting structural response to desolvation of the framework. CPO-72-La is sensitive to solvent loss, and was deemed unsuitable for in situ variable temperature measurements. However, the crystal structure itself is interesting with ordered solvent molecules in the interlayers as well as a lanthanide dimer SBU that is unusual for compounds with sql topology. Furthermore, CPO-72-La demonstrates that the H2dmbpdc linker is suitable also for trivalent cations with large radii.Acknowledgements
We acknowledge the support from the Research Council of Norway (project #190980), inGAP, and the Departments of Chemistry at UiO and UiB. The assistance from the staff at the Swiss – Norwegian Beamlines, ESRF, Grenoble, is highly acknowledged. We acknowledge use of the Norwegian national infrastructure for X-ray diffraction and scattering (RECX).Notes and references
- S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- U. Müller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastre, J. Mater. Chem., 2006, 16, 626–636 RSC.
- A. U. Czaja, N. Trukhan and U. Müller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC.
- C. Wang, D. Liu and W. Lin, J. Am. Chem. Soc., 2013, 135, 13222–13234 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2011, 112, 1232–1268 CrossRef PubMed.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- P. D. C. Dietzel, Y. Morita, R. Blom and H. Fjellvåg, Angew. Chem., Int. Ed., 2005, 44, 6354–6358 CrossRef CAS PubMed.
- N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504–1518 CrossRef CAS PubMed.
- F. Millange, C. Serre and G. Férey, Chem. Commun., 2002, 822–823 RSC.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- F. Lundvall, P. Vajeeston, D. S. Wragg, P. D. C. Dietzel and H. Fjellvåg, Cryst. Growth Des., 2016, 16, 339–346 CAS.
- SAINT, SADABS and TWINABS, Bruker AXS, 2011 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- ESRF, Swiss-Norwegian Beamline (BM01A), http://www.esrf.eu/UsersAndScience/Experiments/CRG/BM01/bm01-a/image.htm, (accessed 04/08-2015).
- CrysAlis, Agilent Technologies, 2012 Search PubMed.
- TOPAS 4.2, Bruker AXS, 2009 Search PubMed.
- G. W. Stinton and J. S. O. Evans, J. Appl. Crystallogr., 2007, 40, 87–95 CAS.
- B. Hinrichsen, R. E. Dinnebier and M. Jansen, Z. Kristallogr. Supp., 2006, 231–236 CrossRef.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576–3586 CAS.
- M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782–1789 CrossRef PubMed.
- G. M. Brown and R. Chidambaram, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1973, 29, 2393–2403 CrossRef CAS.
- J. N. van Niekerk and F. R. L. Schoening, Acta Crystallogr., 1953, 6, 227–232 CrossRef CAS.
- M. Eddaoudi, J. Kim, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2002, 124, 376–377 CrossRef CAS PubMed.
- F. Lundvall, D. S. Wragg, P. D. C. Dietzel and H. Fjellvåg, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2014, 70, o615 CAS.
- R. Sanz, F. Martinez, G. Orcajo, L. Wojtas and D. Briones, Dalton Trans., 2013, 42, 2392–2398 RSC.
- A. J. Fletcher, K. M. Thomas and M. J. Rosseinsky, J. Solid State Chem., 2005, 178, 2491–2510 CrossRef CAS.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244–247 CrossRef.
- E. Sletten and L. H. Jensen, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1973, 29, 1752–1756 CrossRef CAS.
- H. F. Clausen, R. D. Poulsen, A. D. Bond, M.-A. S. Chevallier and B. B. Iversen, J. Solid State Chem., 2005, 178, 3342–3351 CrossRef CAS.
- S. M. Hawxwell and L. Brammer, CrystEngComm, 2006, 8, 473–476 RSC.
- A. D. Burrows, K. Cassar, R. M. W. Friend, M. F. Mahon, S. P. Rigby and J. E. Warren, CrystEngComm, 2005, 7, 548–550 RSC.
- T. Tsuruoka, S. Furukawa, Y. Takashima, K. Yoshida, S. Isoda and S. Kitagawa, Angew. Chem., Int. Ed., 2009, 48, 4739–4743 CrossRef CAS PubMed.
- S. Diring, S. Furukawa, Y. Takashima, T. Tsuruoka and S. Kitagawa, Chem. Mater., 2010, 22, 4531–4538 CrossRef CAS.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem. – Eur. J., 2011, 17, 6643–6651 CrossRef CAS PubMed.
- F. Vermoortele, B. Bueken, G. Le Bars, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. Van Speybroeck, C. Kirschhock and D. E. De Vos, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, D. Vodak, A. Sudik, J. Wachter, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4900–4904 CrossRef CAS PubMed.
- H. Furukawa, J. Kim, N. W. Ockwig, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2008, 130, 11650–11661 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 2D representation of the VT experiment of CPO-71-Ca, TGA/DSC, unit cell parameters from the VT P-XRD and S-XRD experiments performed on CPO-71-Cu, P-XRD of the structures and the full table of La–O bond lengths for CPO-72-La. CCDC 1483001 and 1483002. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt02195f |
| This journal is © The Royal Society of Chemistry 2016 |