
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intramolecular mobility of η5-ligands in chiral zirconocene complexes and the enantioselectivity of alkene functionalization by organoaluminum compounds†
Lyudmila V.
Parfenova
 *a,
Irina V.
Zakirova
a,
Pavel V.
Kovyazin
a,
Stanislav G.
Karchevsky
b,
Galina P.
Istomina
a,
Leonard M.
Khalilov
a and
Usein M.
Dzhemilev
a
*a,
Irina V.
Zakirova
a,
Pavel V.
Kovyazin
a,
Stanislav G.
Karchevsky
b,
Galina P.
Istomina
a,
Leonard M.
Khalilov
a and
Usein M.
Dzhemilev
a
aInstitute of Petrochemistry and Catalysis, Russian Academy of Sciences, Prospect Oktyabrya, 141, 450075 Ufa, Russian Federation. E-mail: luda_parfenova@mail.ru
bInstitute of Petroleum Refining and Petrochemistry of the Republic of Bashkortostan, Iniciativnaya st., 12, 450065 Ufa, Russian Federation
First published on 14th July 2016
Abstract
The effect of solvent nature (CD2Cl2, d8-toluene, d8-THF) on the conformational behavior of neomenthyl-substituted zirconocenes CpInd*ZrCl2 (Cp = η5-C5H5, Ind* = η5-neomenthylindenyl), CpCp′ZrCl2 (Cp = η5-C5H5, Cp′ = η5-neomenthyl-4,5,6,7-tetrahydroindenyl), and Ind*2ZrCl2 (Ind* = η5-neomenthylindenyl) was shown by means of dynamic NMR spectroscopy, and the constants and thermodynamic parameters of conformer exchange were determined. The experimental conformational composition of the complexes was compared with structures obtained by quantum chemical modeling using the DFT methods PBE/3ζ and M06-2X/cc-pVDZ(H, C, Cl)/cc-pVDZ-PP(Zr), which predicted three rotamers in the case of both CpInd*ZrCl2 and CpCp′ZrCl2, and seven rotational isomers for Ind*2ZrCl2, three of these being C2-symmetric and the others being asymmetric. The enantioselectivity of the conformationally mobile complex Ind*2ZrCl2 in the reactions of terminal alkenes with AlR3 (R = Me, Et) was compared with that of rigid ansa-complexes, rac-p-S, p-S-[Y(η5-C9H10)2]ZrX2 (Y = SiMe2, C2H4; X = S-binaphtholate). Faster exchange between the conformers of Ind*2ZrCl2 in a chlorinated solvent gives the structural isomer of catalytically active sites, which affords higher substrate conversion and reaction enantioselectivity. Binding of the ligands to ansa-zirconocenes prevents the rotational isomerism of the complexes, providing the same configuration of the β-stereogenic center in the methyl- and ethylalumination products (unlike the conformationally mobile complex Ind*2ZrCl2) with an enantiomeric purity of 50–65%.
Introduction
The synthesis of chiral transition metal η5-complexes is a vigorously developing field of chemistry. The interest in these compounds is first of all due to their possible use as stereoselective catalysts for the construction of C–H, C–C, and metal–C bonds. Among these, chiral Ti and Zr complexes played a crucial role in the development of catalytic methods for the synthesis of stereoregular oligo- and polymers1 and for enantioselective functionalization of alkenes with organomagnesium and -aluminum compounds.2Investigations on the Group IV metallocenes as catalysts for alkene polymerization prompted the idea about the possible contribution of the conformational mobility of η5-ligands at a metal atom to the activity and stereoselectivity of the catalytic systems.3 In these studies, it was demonstrated that the stereoselectivity of zirconocenes (R*Ind)2ZrCl2 containing substituted indenyl ligands (R* = neomenthyl, neoisomenthyl, menthyl, isomenthyl) in propene polymerization is correlated with their structural features, namely, the relative configuration of the stereogenic centers in the η5-ligand, which finally determines the conformational composition of complexes.3a,d It was found that the more homogeneous conformational composition provides higher polypropylene isotacticity.
Carbometalation of alkenes, which can be conceived as the first step of Ziegler–Natta polymerization, is a substantive efficient method for alkene functionalization. The applicability of enantiomerically pure zirconium catalysts for asymmetric carbometalation of alkenes with AlR3 was first demonstrated in ref. 4. Thus, the reaction of 1-octene with AlMe3 in the presence of bis(1-neomenthylindenyl)zirconium dichloride (3), bis(1-neoisomenthylindenyl)zirconium dichloride, or bis(1-neoisomenthyl-4,5,6,7-tetrahydroindenyl)zirconium dichloride afforded 2-methyl-1-octanol with an enantioselectivity of 65–85%ee (S), 50%ee (S), and 6%ee (S), respectively (Scheme 1).4a The rigid ansa-zirconocenes, (R,R)-ethylene-bis-(4,5,6,7-tetrahydro-1-indenyl)zirconium dichloride or (R,R)-ethylene-bis-(4,5,6,7-tetrahydro-1-indenyl)zirconium – (R)-l′,l′′-bi-2-naphtholate, showed low enantioselectivity at a level of 8%ee (R) and 6%ee (R), correspondingly.
 | ||
| Scheme 1 | ||
The study of the enantioselectivity of olefin carboalumination with AlMe3 in the presence of p-S, p-S-enantiomers of ansa-zirconocenes containing ethanediyl or dimethylsilylene bridges showed that the use of allylbenzene as the substrate results, after oxidation of the reaction mixture, in 2-methyl-3-phenyl-1-propanol with an enantiomeric excess of 25–33%ee, which scarcely depends on the catalyst structure.5 In this study, the highest enantioselectivity (about 80%ee) was achieved in styrene methylalumination, catalyzed by the [Ph3C][B(C6F5)4]-activated p-S, p-S-ethylene-bis-(1-indenyl)zirconium dichloride.
Investigation of the reaction of terminal alkenes with AlR3 (R = Me, Et) catalyzed by enantiomerically pure Zr complexes, (cyclopentadienyl)(1-neomenthylindenyl)zirconium dichloride (1), (cyclopentadienyl)(1-neomenthyl-4,5,6,7-tetrahydroindenyl)zirconium dichloride (2), and bis-(1-neomenthylindenyl)zirconium dichloride (3), demonstrated6 that the chemo- and enantioselectivity of these reactions are substantially affected by the catalyst and alkene structures, OAC nature, and reaction conditions (temperature, reactant ratio, solvent). Presumably, the key factor determining the dependence of enantioselectivity on the solvent nature and OAC structure is the conformational behavior of the η5-ligands in bimetallic Zr,Al-intermediates, which control the reaction pathways.
Thus, in order to elucidate the influence of the intramolecular dynamics of ligands in zirconocene complexes on the activity and stereoselectivity of the catalyst systems in the reactions of OAC with alkenes, we studied the conformational composition of complexes 1–3 depending on the solvent nature by dynamic NMR spectroscopy (CH2Cl2, toluene, THF). The structures of the possible conformers of the complexes were simulated by DFT calculations. The obtained results were compared with the catalytic activity and enantioselectivity of ansa-zirconocenes 4–7 with a fixed geometry (Scheme 2) in carbo- and cycloalumination of terminal alkenes.
 | ||
| Scheme 2 | ||
Results and discussion
Conformational analysis of complexes 1–3
The conformations of CpInd*ZrCl2 (1) were investigated by low-temperature NMR spectroscopy in CD2Cl2 and d8-toluene. In both solvents, lowering the temperature led to broadening of the Cp and indenyl H2 and H3 signals in the downfield region of the 1H NMR spectrum of complex 1 (Fig. 1). In dichloromethane at 175 K, structures with discrete signals of Cp ligands could be observed. | ||
| Fig. 1 Variable temperature 1H NMR study of complex 1 in CD2Cl2 (a) and d8-toluene (b). | ||
In order to elucidate the structures of the possible conformers of complex 1, we carried out a quantum chemical study using the DFT methods PBE/3ζ7,8 and M06-2X/cc-pVDZ(H, C, Cl)/cc-pVDZ-PP(Zr),9–12 which demonstrated their applicability to the description of the Zr-containing systems. Analysis of the potential energy surface for the molecule revealed three key rotation isomers 1A, 1B, and 1C (Fig. 2, Table 1). According to the calculations using PBE/3ζ, rotamer 1B predominates, whereas the populations of 1C and 1A are considerably lower. The population ratio for rotamers 1B and 1C is 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Calculations in M06-2X showed the prevalence of 1B and 1C with a ratio 6:4.
:1. Calculations in M06-2X showed the prevalence of 1B and 1C with a ratio 6:4.
 | ||
| Fig. 2 Structure and dihedral angle α for conformers 1A–C and 2A–C. | ||
| Complex | Conformer |
|
P 180, % | δ (1H), ppm | ||
|---|---|---|---|---|---|---|
| H2 | H3 | Δδ(H2–H3) | ||||
| PBE/3ζ | ||||||
| 1 | 1A | 1.51 | 0.5 | |||
| 1B | 0.00 | 90.0 | ||||
| 1C | 0.64 | 9.5 | ||||
| 2 | 2A | 0.00 | 98.9 | |||
| 2B | 1.29 | 1.1 | ||||
| 2C | 2.53 | 0.0 | ||||
| 3 | 3A | 2.58 | 0.0 | |||
| 3B | 0.00 | 100.0 | ||||
| 3C | 4.47 | 0.0 | ||||
| 3D | 6.87 | 0.0 | ||||
| 3E | 3.40 | 0.0 | ||||
| 3F | 12.58 | 0.0 | ||||
| 3G | 12.86 | 0.0 | ||||
| M06-2X | ||||||
| 1 | 1A | 1.99 | 0.0 | 7.05 | 6.12 | 0.93 |
| 1B | 0.00 | 57.9 | 7.86 | 6.28 | 1.58 | |
| 1C | 0.09 | 42.1 | 7.84 | 7.10 | 0.74 | |
| 2 | 2A | 0.00 | 99.3 | 6.70 | 5.54 | 1.16 |
| 2B | 1.44 | 0.6 | 7.45 | 5.79 | 1.66 | |
| 2C | 2.01 | 0.1 | 7.50 | 6.54 | 0.96 | |
| 3 | 3A | 1.58 | 0.4 | 7.68 | 6.73 | 0.95 |
| 3B | 0.00 | 99.6 | 7.66 | 5.03 | 2.62 | |
| 3C | 6.77 | 0.0 | 8.05 | 6.56 | 1.49 | |
| 8.44 | 7.18 | 1.26 | ||||
| 3D | 9.23 | 0.0 | 7.14 | 4.84 | 2.30 | |
| 7.97 | 7.48 | 0.49 | ||||
| 3E | 3.26 | 0.0 | 5.56 | 6.88 | -1.32 | |
| 8.19 | 6.64 | 1.55 | ||||
| 3F | 15.35 | 0.0 | 4.38 | 6.00 | -1.63 | |
| 7.72 | 7.54 | 0.18 | ||||
| 3G | 15.19 | 0.0 | 7.41 | 6.73 | 0.69 | |

The observed differences between the Cp chemical shifts for the conformers of complex 1 (Fig. 1a, 175 K) can be attributed to the changes in dihedral angles between the η5-ligand planes (α) in the rotamers (Fig. 2). According to our calculations, this difference can be as large as 16°. The smallest α angles among the conformers were found for the most populated 1B and 1C.
A similar investigation of complex CpCp′ZrCl2 (2) in the same solvents demonstrated the splitting of the 1H NMR signals of characteristic protons at C2 and C3 carbon atoms of the substituted tetrahydroindenyl ligand at temperatures below 220 K (Fig. 3). In toluene, the conformers differed also by the Cp-ring chemical shifts (Fig. 3b, 197–210 K). In dichloromethane at 197 K, the spectrum exhibited two proton signals at δH 6.83 and 6.18 ppm and proton signals at δH 5.59 and 5.67 ppm in 88:12 ratio (Fig. 3). This type of splitting was found in both toluene and THF, the ratio of the conformers remaining at the same level (Table 2). In all cases, the difference between the chemical shifts of the H2 and H3 protons was greater for the major conformer than for the minor one.
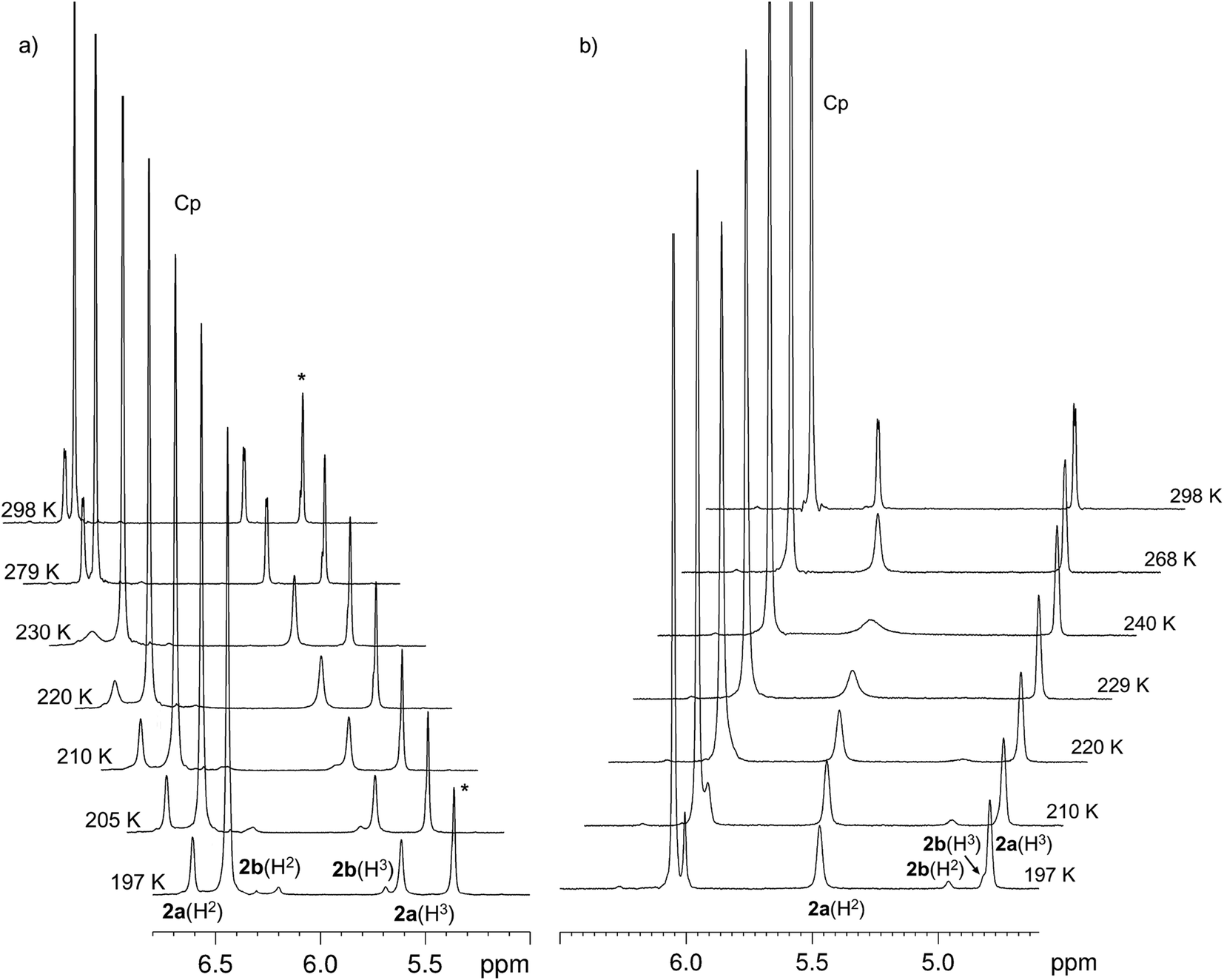 | ||
| Fig. 3 Variable temperature 1H NMR study of complex 2 in CD2Cl2 (a) and d8-toluene (b) (*-solvent). | ||
| Complex | Solvent | Conformer | Conformer ratio | δ (1H), ppm | ||
|---|---|---|---|---|---|---|
| H2 | H3 | Δδ(H2–H3) | ||||
| 2 | CD2Cl2 (190 K) | 2a | 88 | 6.83 | 5.59 | 1.24 |
| 2b | 12 | 6.18 | 5.67 | 0.51 | ||
| d8-Toluene (190 K) | 2a | 90 | 5.47 | 4.79 | 0.68 | |
| 2b | 10 | 4.96 | 4.82 | 0.14 | ||
| d8-THF (190 K) | 2a | 87 | 7.01 | 5.82 | 1.19 | |
| 2b | 13 | 6.51 | 5.89 | 0.62 | ||
| 3 | CD2Cl2 (180 K) | 3a | 70 | 7.15 | 6.32 | 0.83 |
| 3b | 15 | 6.63 | 4.80 | 1.83 | ||
| 3c | 15 | 7.04 | 6.43 | 0.64 | ||
| 6.74 | 6.36 | 0.31 | ||||
| d8-Toluene (180 K) | 3a | 20 | 6.13 | 5.87 | 0.26 | |
| 3b | 80 | 6.07 | 4.40 | 1.67 | ||
| 3c | 0 | — | — | — | ||
| d8-THF (185 K) | 3a | 18 | 7.41 | 6.35 | 1.06 | |
| 3b | 82 | 6.85 | 4.83 | 2.02 | ||
| 3c | 0 | — | — | — | ||
For this complex, the rate constants for the rotamer interconversion were calculated, and activation parameters were derived from the Eyring equation (ln(k/T) vs. 1/T plot, Fig. S8†): in dichloromethane, k190 = 12.5 s−1, ΔG≠190 = 10.24 ± 0.06 kcal mol−1, ΔH≠ = 7.47 ± 0.52 kcal mol−1, and ΔS≠ = −14.89 ± 10.76 cal (mol K)−1; in toluene, k195 = 3.1 s−1, ΔG≠195 = 11.08 ± 0.02 kcal mol−1, ΔH≠ = 7.20 ± 0.24 kcal mol−1, and ΔS≠ = −19.85 ± 4.90 cal (mol K)−1; in THF, k195 = 5.1 s−1, ΔG≠195 = 10.88 ± 0.05 kcal mol−1, ΔH≠ = 7.89 ± 0.67 kcal mol−1, ΔS≠ = −15.57 ± 13.67 cal (mol K)−1.
The exchange rate between the conformers of complex 2 increases in the series toluene < THF < dichloromethane, which is attributable to the decrease in the Gibbs activation energy due to solvation effects.
The quantum chemical study on the structure of the possible conformers of complex 2 revealed the same rotamers as in complex 1 (Fig. 2, Table 1). PES scanning for complex 2 using both calculation methods showed rotamer 2A to predominate (∼99% population). Particularly this form participates in the crystallization of the complex.13a Compared with conformer 2A, populations of conformers 2B and 2C were much lower (Table 1).
Evaluation of the solvation effect on the conformer distribution of complexes 1 and 2 within the conductor-like polarisable continuum model (CPCM)14 exhibited an increase of the relative content of minor conformer 1A up to ∼2% in the case of more polar solvents – THF and dichloromethane (Table S1†), and substantial growth of the conformer 2B fraction up to ∼14% as well. It should be noted that theoretical populations of complex 2 rotamers practically do not depend on the nature of the solvent, which corresponds to the NMR experiment. Moreover, we attempted to assign the structure of conformers 2a,b on the basis of calculated NMR chemical shifts of characteristic protons H2 and H3 in the theoretically possible rotamers 2A–C. Thus, as follows from the calculations shown in Table 1, the proximity of H2 and/or H3 with the Cl atom should provide a downfield shift of the NMR signals of these protons. Only the main conformer 2a satisfies this condition, in which the H2 proton experiences a deshielding effect (Table 2), however the theoretical population of the corresponding rotamer 2B in all three solvents is 7–15%. Thus, a more detailed theoretical studies of the conformer solvation process and its influence on the NMR chemical shifts are required, namely, consideration of specific solvation probably with the gradual inclusion of solvent molecules.
Quantum chemical calculations for the complex Ind*2ZrCl2 (3) showed the existence of seven conformers, three of them having C2 symmetry (3A, 3B, 3G) and the others having the C1 symmetry (Fig. 4). Conformer 3B was found to be energetically most favorable. The relative Gibbs energy difference between the major and minor conformers is more than 1.5 kcal mol−1 (Table 1); therefore, the population of 3B is greater than 99%. According to X-ray diffraction data,3a the complex in the crystal occurs in conformation 3A.
 | ||
| Fig. 4 Structure of conformers of complex Ind*2ZrCl2 (3). | ||
A variable temperature NMR study of solutions of Ind*2ZrCl2 (3) in CD2Cl2, d8-toluene, and d8-THF showed that (i) the dynamic pattern in the NMR spectra is associated with the mobility of indenyl ligands relative to each other and the Zr atom, (ii) mobility of the η5-ligand and, hence, the conformational composition of the complex depends on the solvent nature.
For example, lowering the temperature of a solution of complex 3 in CD2Cl2 down to 180 K induces splitting of the resonance lines of protons at C2 and C3 in the 1H NMR spectra into three groups of signals, indicating the existence of at least three conformers. Two of them (3a,b) refer to C2-symmetry and one (3c) has C1-symmetry, their ratio in CD2Cl2 being 70:15:15 (Fig. 5, Table 2). This result is consistent with published data.3a,d The analysis of the chemical shifts of the observed conformers indicates that the C2-symmetric rotamer 3a can be related to the theoretically predicted structures 3A,G. The upfield shift of the H3 signal of the other conformer of the same symmetry (3b)3a,d is apparently caused by the shielding effect of the benzene moiety of the opposing indenyl ligand, which is consistent with the results on chemical shift calculations showed in Table 1; this provides grounds for assigning the theoretical structure 3B to conformer 3b. The signals of the H2 and H3 protons of the C1-symmetric conformer 3c are located at 6–7 ppm, which is indicative of the absence of the shielding effect from the benzene ring of the opposing ligand. This is the only case of theoretical structure 3C.
 | ||
| Fig. 5 Variable temperature 1H NMR study of complex 3 in CD2Cl2 (a) and d8-toluene (b). | ||
Unlike CD2Cl2, in d8-toluene, complex 3 was found to comprise only two C2-symmetric conformers, 3a and 3b, in a ratio of 20:80 (Table 2). The same pattern was observed in THF. Thus, conformer 3B most probably predominates in these solvents, as has been theoretically predicted.
The rate constants and thermodynamic parameters for the exchange between the rotamers of complex 3 in toluene were determined on the basis of lineshape analysis for the H2 proton signals in the 1H NMR spectrum: k190 = 18.7 s−1, ΔG≠190 = 10.09 ± 0.06 kcal mol−1, ΔH≠ = 8.15 ± 0.38 kcal mol−1, ΔS≠ = −10.30 ± 7.88 cal (mol K)−1 (Fig. S8†). It can be stated that the interconversion rate for complex 3 is higher than that in complex 2. Thus, going from neomenthyl-substituted tetrahydroindenyl to the indenyl ligand accelerates the intramolecular dynamic process, apparently, due to a decrease in the activation barrier.
As follows from Fig. 6, gradual addition of dichloromethane to a toluene solution of complex 3 induces a change in the conformer ratio, so that at a high content of CD2Cl2, conformer 3a actually starts to predominate.
 | ||
| Fig. 6 1H NMR of complex 3 in d8-toluene (0.05 M l−1) and admixtures of CD2Cl2 (T = 188 K): (a) 0 mmol CD2Cl2, (b) 1.6 mmol CD2Cl2, (c) 3.0 mmol CD2Cl2, (d) 4.9 mmol CD2Cl2. | ||
Theoretical modelling of the solvent effect on the conformer 3A–G distribution demonstrated the possibility of the increase in the 3A content to the ratio 3A:3B ≈ 1:1 (Table S1†) both in dichloromethane and THF, however, the model that is used for the system description does not explain the exceptional influence of the chlorine-containing solvent on the complex 3 conformational behavior.
Thus, the conformational composition and the mobility of the η5-ligand largely depend on the nature of the solvent, which may play a considerable role in the control of the catalytic activity and enantioselectivity of chiral zirconocenes in the alkene functionalization by OAC.
Catalytic action of enantiomerically pure zirconocenes in the reactions of alkenes with trialkylalanes
The results of our previous studies on the activity and enantioselectivity of catalysts 1–3 in the alkene carbo- and cyclometalation by organoaluminum or -magnesium compounds,6 which are summarized in Scheme 3 and Table 3, gave an idea on the substantial contribution of the conformational mobility of η5-ligands. Thus, we found that the best results for alkene carbometalation, catalyzed by complexes 1–3 are achieved in dichloromethane.6a,c It was demonstrated that replacement of AlMe3 by AlEt3 in the reaction catalyzed by complex (3) results in the R to S change of the absolute configuration of the β-stereogenic center in the carboalumination products.6a Indeed, if one considers the conformational mobility of η5-ligands in the key intermediates of alkene carbometallation [L2ZrR(μ-Cl)AlR3],15 the dependence of the stereochemical result of the reaction on the type of AlR3 becomes evident.6a Further, the cycloalumination of terminal alkenes gives aluminacyclopentanes in up to 85% yield and 24–57%ee;6b,c the highest enantioselectivity (∼57%ee) in cycloalumination was found in the reaction of vinyl-substituted hydrocarbons with AlEt3 conducted in CH2Cl2.6c Terminal alkenes react with organomagnesium compounds in Et2O or THF in the presence of catalysts 2 or 3 with low conversion and low enantioselectivity (<8%ee).6d The cyclometalation of substituted allylbenzenes with EtAlCl2 (Et2AlCl) and the Mg metal in THF in the presence of catalytic amounts of complexes 1–3 gives trans-2,3-dibenzyl-substituted aluminacyclopentanes with enantiomeric purity of not more than 16%ee.6e | ||
| Scheme 3 | ||
| Catalyst | Reagents | Solvent | Alkene conversion, % | Product yield,a % (ee%, R/S) | Ref. | |
|---|---|---|---|---|---|---|
| Carbometalation | Cyclometalation | |||||
| a The reaction runs in the presence of MAO only (30 mol%) and gives 95% of oligomers after 3 h. | ||||||
| 1, 2 mol% | AlEt3, 1-hexene | CH2Cl2 | 85 | 30 (27, S) | 39 (20, R) | 6a |
| 1, 2 mol% | AlMe3, 1-octene | Toluene | 59 | 58 (3, R) | 6a | |
| 1, 5 mol% | EtMgBr, 1-octene | THF | 60 | 51 (3, S) | 4 (5, S) | 6d |
| 1, 5 mol% | EtMgBr, 1-octene | Toluene | 32 | 18 (8, S) | 6 (2, S) | 6d |
| 1, 5 mol% | EtAlCl2, allylbenzene | THF | 83 | — | 21 (13) | 6e |
| 2, 2 mol% | AlMe3, 1-hexene | CH2Cl2 | 53 | 51 (41, S) | 6a | |
| 2, 2 mol% | AlEt3, 1-hexene | CH2Cl2 | 99 | 36 (13, R) | 38 (11, R) | 6a |
| 3, 8 mol% | AlMe3, 1-hexene | CH2Cl2 | 94 | 81 (69, R) | 6a | |
| 3, 8 mol% | AlEt3, 1-octene | CH2Cl2 | 99 | 92 (59, S) | 7 (3, S) | 6a |
| 3, 8 mol% | AlEt3, 1-octene | Toluene | 99a | 3 (7, S) | 1 | |
| 3, 8 mol% | AlEt3, vinylcyclohexane | CH2Cl2 | 85 | 34 (62, S) | 42 (57, S) | 6c |
| 3, 8 mol% | AlEt3, vinylcyclohexane | C6H6 | 30 | 4 | 23 (24, R) | 6b |
| 3, 5 mol% | MgEt2, 1-octene | THF | 10 | 4 | 4 | 6d |
| 3, 5 mol% | EtMgBr, 1-octene | Toluene | 7 | 3 | 4 | |
| 3, 5 mol% | EtAlCl2, allylbenzene | THF | 65 | — | 34 (16) | 6e |
In order to determine the degree of influence of the ligand intramolecular mobility on the reaction enantioselectivity, we studied the catalytic properties of enantiomerically pure complexes with a fixed geometry. It was found that ansa-complexes 4–7 provide somewhat lower enantioselectivity than the conformationally mobile catalyst 3 (Table 4) both in carbo- and cycloalumination of alkenes. The application of neither binaphtholate (6) nor dichloride (4) complexes with Si-bonded ligands in the reaction of 1-octene with AlMe3 resulted in the formation of products. The MAO activation of the catalyst 4 afforded carbometallation product (8) in ∼66% yield and an enantiomeric excess of 65%, R (Scheme 4). Moreover, functionally substituted oligomers 11 were also formed in up to 34% yield.
 | ||
| Scheme 4 | ||
:alkene:AlEt3 = 1:50:60, CH2Cl2, 72 h, 20 °C)
| Catalyst | Alkene conversion, % | Product yield,a % (ee%, R/S) | ||
|---|---|---|---|---|
| 12 | 13 | 14 | ||
| a Determined by GC of deuterolysis products. | ||||
| p-S, p-S-4 | 99 | 64 (50, R) | 15 (12, S) | 13, n = 2–6, [α]25D(19) = +3.3° |
| p-S, p-S-5 | 95 | 62 (51, R) | 18 (11, S) | 10, n = 2, [α]25D(19) = +0.35° |
| S, p-S, p-S-6 | 99 | 15 (15, R) | 30 (20, S) | 54, n = 2 |
| S, p-S, p-S-7 | 98 | 60 (30, R) | 21 (26, S) | 12, n = 2, [α]25D(19) = −1.5° |
In the reaction of AlEt3 with 1-hexene, the highest enantioselectivity towards carboalumination (12) at a level of 50–51%ee, R, was found for dichloride complexes 4 and 5 (Scheme 5). The catalyst structures containing a binaphtholate moiety (6, 7) provide much lower enantiomeric excess of 12. The enantiomeric purity of cyclic OAC (13) upon the use of catalysts 4–7 was 12–26%ee, S. The reaction of 1-hexene with AlEt3 catalyzed by these complexes also resulted in the regioselective formation of enantiomerically enriched functionally substituted oligomers 14 with up to 6 units (Table 4). Analysis of the isolated functionally substituted dimers 11 and 19 demonstrated that the reaction gave a single diastereomer. The result is consistent with published data indicating a high stereoselectivity of ansa-complexes in the alkene polymerization.16
 | ||
| Scheme 5 | ||
Thus, fixing the geometry of the zirconocene complex results in the formation of methyl- and ethylalumination products with the same configuration of the β-stereogenic center (unlike the conformationally mobile complexes). This result confirms once again the hypothesis stating that in the case of conformationally flexible complexes (e.g., 3), the reaction enantioselectivity is dictated by the intramolecular mobility of η5-ligands in the Zr,Al-bimetallic intermediates (in particular, distribution of conformers of the active sites and the activation energies of alkene insertion).6a
Apparently, the predominance of the R-enantiomers of carbometalation products in the reactions catalyzed by complexes 4–7 occurs upon substrate re-coordination to the Zr–C bond of the active complex having the p-S, p-S-configuration of the conformationally rigid ligand environment (Scheme 6). In the case of complex 3, the formation of R-enantiomers in the reaction of alkenes with AlMe3 can be due to the involvement of the active site originated from conformer 3G, whereas S-isomers are produced via a 3A-like intermediate (Scheme 7).
 | ||
| Scheme 6 | ||
 | ||
| Scheme 7 | ||
The solvent can make an additional contribution to the enantioselectivity control by changing the interconversion rate between the conformers of catalytically active sites and thus forming a particular conformational composition. The higher enantioselectivity of the reaction of alkenes with AlEt3 catalyzed by 3 in a chlorine-containing solvent4b,6a than in the case of hydrocarbon or oxygen-containing solvent may be attributable to the more diverse conformational composition, which, as we have shown in relation to the initial zirconocene dichloride, is possible in this case. The use of toluene or THF as the solvent causes the domination of sterically hindered conformer 3B and, hence, the activity and enantioselectivity of the catalyst system decrease. Thus, it follows that a homogeneous conformational composition and slow interconversion of conformers are not necessary conditions for the reaction to proceed with high yield and enantioselectivity (unlike the stereoselective polymerization catalyzed by conformationally homogeneous complexes3a,d). A significant issue is the ligand mobility: the more variable is a complex (to a certain limit), the more versatile it is. In other words, the greater the number of “keys” (i.e., catalytically active sites with a certain geometry) it can form, the greater the number of “locks” (substrates) that can be subjected to chemo- and enantioselective transformation. As a result, the chemo- and enantioselectivity of the reactions are determined by the kinetic factor, namely, the rate of the key–lock pair reaction. Provided a good match of pair components, high rate and high stereoselectivity along a particular reaction route can be achieved.
Conclusions
The conformational composition and intramolecular exchange between the conformers of neomenthyl-substituted zirconocenes, CpInd*ZrCl2, CpCp′ZrCl2, and Ind*2ZrCl2, depending on the nature of the solvent (CD2Cl2, d8-toluene, d8-THF) were studied by dynamic NMR spectroscopy and DFT calculations.Quantum chemical modeling demonstrated the possibility of the existence of three rotamers of complexes CpInd*ZrCl2 and CpCp′ZrCl2. According to dynamic NMR, even at low temperatures (170–180 K), the exchange between CpInd*ZrCl2 conformers in CD2Cl2 and d8-toluene is not completely retarded. In the case of the complex CpCp′ZrCl2, slow interconversion of two rotamers was found below 220 K, the ratio of the rotamers (∼90:10) being virtually independent of the solvent nature. In the chlorinated solvent, the exchange between the conformers of CpCp′ZrCl2 is accelerated.
For the complex Ind*2ZrCl2, the existence of seven rotamers is theoretically possible, three of them being C2-symmetric and the others being asymmetric. Experimentally we found no more than three rotamers of the complex. The replacement of dichloromethane by toluene or THF results in a considerable change in the conformational composition, namely, in the number and the ratio of rotamers: only two C2-symmetric conformers in an ∼80:20 ratio were found both in toluene and in THF. Switching from the mixed CpCp′ZrCl2 complex containing the substituted tetrahydroindenyl ligand to the bisindenyl Ind*2ZrCl2 complex is accompanied by increase in the intramolecular ligand mobility and decrease in the interconversion barriers for the conformers.
The catalytic properties of conformationally rigid complexes, rac-p-S, p-S-[Y(η5-C9H10)2]ZrX2 (Y = SiMe2, C2H4; X = S-binaphtholate), in reactions of alkenes with AlR3 (R = Me, Et) were studied. The reaction of alkenes with AlMe3 catalyzed by rac-p-S, p-S-[Me2Si(η5-C9H10)2]ZrCl2 in the presence of MAO gave methylalumination products in 66% yield and with enantiomeric purity of 65%ee, R. The alkene carboalumination with AlEt3 catalyzed by rac-p-S, p-S-[Y(η5-C9H10)2]ZrCl2 (Y = SiMe2, C2H4) afforded 2-ethyl-substituted derivatives in 62–64% yield and an enantiomeric excess of 50–51%ee, R. The reaction of AlR3 with alkenes in the presence of enantiomerically pure Zr ansa-complexes provided enantiomerically enriched functionally substituted oligomeric products as well.
A comparison of the catalytic action of conformationally mobile neomenthyl-substituted bisindenyl complex Ind*2ZrCl2 and rigid ansa-zirconocenes in the reactions of alkenes with AlR3 (R = Me, Et) and the relationship between the conformational composition of flexible complexes and their activity and enantioselectivity in the same reactions depending on the solvent nature provide the conclusion about a substantial contribution of the conformational composition and ligand mobility to the activity of catalytic systems and the degree of asymmetric induction. In this connection, further optimization of the ligand environment, namely, the search for appropriate conformers that could be formed via either introduction of suitable substituents into the indenyl ligand or upon binding of ligands could advance these studies towards the design of more efficient catalysts for alkene functionalization by organomagnesium and -aluminum reagents.
Experimental
General procedures
All operations for organometallic compounds were performed under argon according to the Schlenk technique. The zirconocenes 1–3, and racemic 4 and 5 were prepared using the standard procedures from ZrCl4 (99.5%, Aldrich) (1, 2,13,6a3,3a,6brac-4,17rac-518). Kinetic resolution of rac-4 and rac-5 was carried out using lithium S-binaphtholate according to the method described in ref. 19. The solvents (hexane, benzene, toluene) were distilled from i-Bu2AlH immediately prior to use; THF and diethyl ether were dried and distilled from sodium/benzophenone before use. Dichloromethane was dried over P2O5. Commercially available 98% AlEt3, 97% AlMe3 (Aldrich) and 10 wt% MAO in toluene (Aldrich) were involved in the reactions. Terminal alkenes 1-hexene (97%, Acros) and 1-octene (99%, Acros) were used.
The 1H, 13C, and 77Se NMR spectra were recorded on a Bruker AVANCE-400 spectrometer (400.13 MHz (1H), 100.62 MHz (13C), and 76.35 MHz (77Se)). A low temperature NMR study on the conformational behavior of complexes 1–3 was carried out in the interval of 170–300 K. The constants and activation parameters of conformer exchange were found using lineshape analysis of NMR spectra in program Bruker TopSpin 3.0. d8-Toluene, d8-tetrahydrofuran and CD2Cl2 were employed as the solvent and internal standard. The 77Se chemical shifts are referred to Me2Se. 1D and 2D NMR spectra (COSY HH, HSQC, HMBC) were recorded using standard Bruker pulse sequences. The optical rotation [α]20D was measured on a Perkin Elmer-341 polarimeter. Chromatographic analysis was carried out on a CARLO ERBA 1150 instrument in a helium flow, a 50000 × 0.32 mm column, an ULTRA-1 stationary phase, and a flame ionization detector. The deuterated products were analyzed by GC/MS on an automated QP 2010 Ultra GC/MS assembled with a TD-20 Shimadzu thermal desorber. The enantioselectivity of 1-alkene carbo- and cyclometallation was estimated from the enantiomeric purity of the alcohols resulting from oxidation and hydrolysis of the reaction products. The alcohols isolated in a pure state were involved in the reaction with R-(+)-phenylselenopropionic acid (R-PSPA)20 to give diastereomeric esters, which were then analyzed by 1H, 13C, and 77Se NMR.
Computational details
All calculations were carried out at the DFT level. The primary conformer screening by η5-ligand rotation, subsequent geometry optimization of the minima, vibrational frequency analysis, and calculation of thermodynamic corrections to the total energy of the compounds were carried out using the PRIRODA 06 program7 with the Perdew–Burke–Ernzerhof (PBE) functional8 in combination with a 3ζ basis set7b,c (orbital basis sets of contracted Gaussian-type functions of size (5s1p)/[3s1p] for H, (11s6p2d)/[6s3p2d] for C, (15s11p2d)/[10s6p2d] for Cl, and (20s16p11d)/[14s11p7d] for Zr, which were used in combination with the density-fitting basis sets of uncontracted Gaussian-type functions of size (5s2p) for H, (10s3p3d1f) for C, (14s3p3d1f1g) for Cl, and (22s5p5d4f4g) for Zr). Further DFT calculations were performed using the Gaussian 09 software package, Revision D.01 program.21 The density functional M06-2X10 with the cc-pVDZ basis set for the main group elements (H, C, Cl)11 and the cc-pVDZ-PP basis set for the transitional metal (Zr)12 was employed as the method applicable for the description of Zr-containing systems.9 We used the Conductor-like Polarisable Continuum Model (CPCM)14 for the estimation of solvent effects on the conformer distribution. Calculated thermodynamic parameters were determined at 180 K. NMR shifts were calculated using the GIAO method.22 Visualization of quantum chemical data was carried out using the program ChemCraft.23Synthesis of enantiomerically pure ansa-complexes 4 and 6
The complexes were synthesized according to ref. 17 and 18 and derivatized by the procedure described in ref. 19. A 50 mL glass reactor mounted on a magnetic stirrer was charged under argon with S-binaphthol (1.1 mmol, 0.32 g) and toluene (20 mL). The mixture was cooled to −78 °C and butyllithium (0.88 ml, 2.5 M in hexane) was added. The temperature of the reaction mixture was brought to 20 °C and the mixture was stirred until a white lithium S-binaphtholate precipitate formed. Complex rac-4 (2.2 mmol, 1.0 g) was preliminary mixed with toluene (20 ml) and the resulting solution of the complex was added in small portions with cooling (down to −78 °C) to a mixture of lithium S-binaphtholate. The reaction mixture was warmed up to room temperature (22 °C) and stirred for 12 h until a white precipitate was formed. After solvent evaporation, dichloromethane (20 mL) was added to the residue and the solution was filtered through a glass filter. The filtrate was concentrated to give 6 as a microcrystalline precipitate.Binaphtholate complex 6 (0.05 g, 0.075 mmol) was dissolved in dichloromethane (20 mL), and Me3SiCl (0.15 mmol) was added. The reaction mixture was stirred for 1 h. By 1H NMR monitoring, the formation of dichloride complex p-S, p-S-4 (90%, 99%ee) was observed. The complex was isolated by recrystallization from toluene.
The NMR spectra of compounds 4, 5, 7 corresponded to published data (4,175,18b,c719).
Reaction of alkenes with AlR3 (R = Me, Et) in the presence of complexes 4–7
A 25 mL glass reactor mounted on a magnetic stirrer and filled with argon was charged with catalyst (4–7) (0.03 mmol), CH2Cl2 or C6H5CH3 (10 ml), alkene (1.5 mmol), and AlR3 (1.8–7.2 mmol). In the reaction with AlMe3, MAO (1.5 mmol, 10 wt% in toluene) was added. The reaction was carried out at 20 °C with continuous stirring for 72–120 hours. After completion of the reaction, a part of the reaction mixture was quenched with 10% DCl at 0 °C. The products were extracted with benzene and filtered, and the organic layer was dried with Na2SO4. The product yield (8, 9, 12–14) was determined by analyzing the hydrolysis and deuterolysis products by GLC and GC/MS.The remaining reaction mixture was cooled to 0 °C and oxidized by bubbling O2 for 2 h, and then kept in an oxygen atmosphere for 24 hours more. The products were quenched with HCl and extracted into diethyl ether, and the organic layer was dried with Na2SO4, filtered, and concentrated. Functionally substituted dimers (19) were isolated by column chromatography on silica gel using a 6:1 hexane:diethyl ether system. Monohydric alcohols (10, 17) were isolated by column chromatography on silica gel in a 4:1 hexane:diethyl ether system. Dihydric alcohol (18) was isolated as an oily liquid after 17 upon the subsequent washing of the column with acetone. The organic fractions were dried with Na2SO4.
The NMR spectra of compounds 17 and 18 and their PSPA esters corresponded to published data.6a,d
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). 77Se NMR (CDCl3) δ 452.36 (SR), 452.66 (RR).
O). 77Se NMR (CDCl3) δ 452.36 (SR), 452.66 (RR).
Acknowledgements
The authors thank the Russian Foundation of Basic Research (Grants No. 15-03-03227a, 16-33-00162mol_a, 16-33-60203mol_a_dk) for financial support.Notes and references
- (a) Y. Okamoto and T. Nakano, Chem. Rev., 1994, 94, 349–372 CrossRef CAS; (b) C. Janiak, Coord. Chem. Rev., 2006, 250, 66–94 CrossRef CAS; (c) S. Ito and K. Nozaki, in Catalytic Asymmetric Synthesis, ed. I. Ojima, John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 2010, pp. 931–985 Search PubMed.
- (a) A. H. Hoveyda and J. P. Morken, Angew. Chem., Int. Ed., 1996, 35, 1263–1284 CrossRef CAS; (b) U. M. Dzhemilev and A. G. Ibragimov, Russ. Chem. Rev., 2000, 69, 121–135 CrossRef CAS; (c) E.-i. Negishi, in Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, New York, 2000, pp. 165–189 Search PubMed; (d) A. H. Hoveyda, in Titanium and Zirconium in Organic Synthesis, ed. I. Marek, Wiley-VCH Verlag GmbH & Co. KgaA, Weinheim, Germany, 2002, pp. 180–229 Search PubMed; (e) E.-i. Negishi, Bull. Chem. Soc. Jpn., 2007, 80, 233–257 CrossRef CAS; (f) U. M. Dzhemilev and A. G. Ibragimov, J. Organomet. Chem., 2010, 695, 1085–1110 CrossRef CAS; (g) E.-i. Negishi, ARKIVOC, 2011, 8, 34–53 Search PubMed; (h) I. Ojima, J. J. Kaloko, S. J. Chaterpaul, C. F. Lin and Y. H. G. Teng, in Catalytic Asymmetric Synthesis, ed. I. Ojima, John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 2010, pp. 643–681 Search PubMed; (i) S. Xu and E.-i. Negishi, Heterocycles, 2014, 88, 845–877 CrossRef CAS.
- (a) G. Erker, M. Aulbach, M. Knickmeier, D. Wingbermuhle, C. Krueger, M. Nolte and S. Werner, J. Am. Chem. Soc., 1993, 115, 4590–4601 CrossRef CAS; (b) G. W. Coates and R. M. Waymouth, Science, 1995, 267, 217–219 CrossRef CAS PubMed; (c) E. Hauptman and R. M. Waymouth, J. Am. Chem. Soc., 1995, 117, 11586–11587 CrossRef CAS; (d) M. Knickmeier, G. Erker and T. Fox, J. Am. Chem. Soc., 1996, 118, 9623–9630 CrossRef CAS; (e) J. L. Maciejewski Petoff, M. D. Bruce, R. M. Waymouth, A. Masood, T. K. Lal, R. W. Quan and S. J. Behrend, Organometallics, 1997, 16, 5909–5916 CrossRef; (f) C. D. Tagge, R. L. Kravchenko, T. K. Lal and R. M. Waymouth, Organometallics, 1999, 18, 380–388 CrossRef CAS; (g) S. Knuppel, J.-L. Faure, G. Erker, G. Kehr, M. Nissinen and R. Frohlich, Organometallics, 2000, 19, 1262–1268 CrossRef; (h) R. L. Halterman, D. R. Fahey, E. F. Bailly, D. W. Dockter, O. Stenzel, J. L. Shipman, M. A. Khan, S. Dechert and H. Schumann, Organometallics, 2000, 19, 5464–5470 CrossRef CAS; (i) T. Dreier, F. Hannig, G. Erker, K. Bergander and R. Frohlich, J. Phys. Org. Chem., 2002, 15, 582–589 CrossRef CAS; (j) S. Lin and R. M. Waymouth, Acc. Chem. Res., 2002, 35, 765–773 CrossRef CAS PubMed; (k) M. Finze, S. E. Reybuck and R. M. Waymouth, Macromolecules, 2003, 36, 9325–9334 CrossRef CAS; (l) G. M. Wilmes, M. B. France, S. R. Lynch and R. M. Waymouth, Organometallics, 2004, 23, 2405–2411 CrossRef CAS; (m) S. E. Reybuck and R. M. Waymouth, Macromolecules, 2004, 37, 2342–2347 CrossRef CAS; (n) A. L. Lincoln, G. M. Wilmes and R. M. Waymouth, Organometallics, 2005, 24, 5828–5835 CrossRef CAS.
- (a) D. Y. Kondakov and E.-i. Negishi, J. Am. Chem. Soc., 1995, 117, 10771–10772 CrossRef CAS; (b) D. Y. Kondakov and E.-i. Negishi, J. Am. Chem. Soc., 1996, 118, 1577–1578 CrossRef CAS.
- R. A. Petros, J. M. Camara and J. R. Norton, J. Organomet. Chem., 2007, 692, 4768–4773 CrossRef CAS.
- (a) L. V. Parfenova, T. V. Berestova, T. V. Tyumkina, P. V. Kovyazin, L. M. Khalilov and R. J. Whitby, Tetrahedron: Asymmetry, 2010, 21, 299–310 CrossRef CAS; (b) L. V. Parfenova, P. V. Kovyazin, T. V. Tyumkina, T. V. Berestova, L. M. Khalilov and U. M. Dzhemilev, J. Organomet. Chem., 2013, 723, 19–25 CrossRef CAS; (c) L. V. Parfenova, P. V. Kovyazin, T. V. Tyumkina, A. V. Makrushina, L. M. Khalilov and U. M. Dzhemilev, Tetrahedron: Asymmetry, 2015, 26, 124–135 CrossRef CAS; (d) T. V. Berestova, T. A. Raznitsina, L. V. Parfenova and L.M. Khalilov, Bull. Bashkir Univ., 2011, 16, 1147–1151 Search PubMed; (e) L. V. Parfenova, T. V. Berestova, P. V. Kovyazin, A. R. Yakupov, E. S. Mesheryakova, L. M. Khalilov and U. M. Dzhemilev, J. Organomet. Chem., 2014, 772–773, 292–298 CrossRef CAS.
- (a) D. N. Laikov and Y. A. Ustynyuk, Russ. Chem. Bull., 2005, 54, 820 CrossRef CAS; (b) D. N. Laikov, Chem. Phys. Lett., 1997, 281, 151–156 CrossRef CAS; (c) D. N. Laikov, PhD dissertation, Moscow State University, 2000 (in Russian).
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- Y. Sun and H. Chen, J. Chem. Theor. Comput., 2013, 9, 4735–4743 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- (a) T. H. Dunning, Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef; (b) D. E. Woon and T. H. Dunning Jr., J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS; (c) E. R. Davidson, Chem. Phys. Lett., 1996, 260, 514–518 CrossRef CAS.
- K. A. Peterson, D. Figgen, M. Dolg and H. Stoll, J. Chem. Phys., 2007, 126, 124101 CrossRef PubMed.
- (a) L. Bell, R. J. Whitby, R. V. H. Jones and M. C. H. Standen, Tetrahedron Lett., 1996, 37, 7139–7142 CrossRef CAS; (b) G. Dawson, C. A. Durrant, G. G. Kirk, R. J. Whitby, R. V. H. Jones and M. C. H. Standen, Tetrahedron Lett., 1997, 38, 2335–2338 CrossRef CAS; (c) L. Bell, D. C. Brookings, G. J. Dawson, R. J. Whitby, R. V. H. Jones and M. C. H. Standen, Tetrahedron, 1998, 54, 14617–14634 CrossRef CAS.
- (a) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS; (b) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- L. V. Parfenova, V. Z. Gabdrakhmanov, L. M. Khalilov and U. M. Dzhemilev, J. Organomet. Chem., 2009, 694, 3725–3731 CrossRef CAS.
- (a) H.-H. Brintzinger, D. Fischer, R. Mülhaupt, B. Rieger and R. M. Waymouth, Angew. Chem., Int. Ed. Engl., 1995, 34, 1143–1170 CrossRef CAS; (b) H.-H. Brintzinger and D. Fischer, Adv. Polym. Sci., 2013, 258, 29–42 CrossRef CAS; (c) L. Corradini, G. Guerra and L. Cavallo, Top. Stereochem., 2003, 24, 1–46 Search PubMed; (d) Y. Nakayama and T. Shiono, Molecules, 2005, 10, 620–633 CrossRef CAS PubMed.
- (a) W. A. Herrmann, J. Rohrmann, E. Herdtweck, W. Spaleck and A. Winter, Angew. Chem., Int. Ed. Engl., 1989, 28, 1511–1512 CrossRef; (b) H. J. G. Luttikhedde, R. P. Leino, J. H. Nasman, M. Ahlgren and T. Pakkanen, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 1488–1490 CrossRef.
- (a) F. R. W. P. Wild, L. Zsolnai, G. Huttner and H.-H. Brintzinger, J. Organomet. Chem., 1982, 232, 233–247 CAS; (b) F. R. W. P. Wild, M. Wasiucionek, G. Huttner and H.-H. Brintzinger, J. Organomet. Chem., 1985, 288, 63–67 CrossRef CAS; (c) Sc. Collins, Br. A. Kuntz, N. J. Taylor and D. G. Ward, J. Organomet. Chem., 1988, 342, 21–29 CrossRef CAS.
- (a) W. M. Davis and St. L. Buchwald, J. Am. Chem. Soc., 1991, 113, 2321–2322 CrossRef; (b) B. Chin and St. L. Buchwald, J. Org. Chem., 1997, 62, 2267–2268 CrossRef CAS PubMed.
- N. V. Orlov and V. P. Ananikov, Chem. Commun., 2010, 46, 3212–3214 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- S. K. Wolf and T. Ziegler, J. Chem. Phys., 1998, 109, 895–905 CrossRef.
- G. A. Zhurko and D. A. Zhurko, ChemCraft, version 1.6, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6dt01366j |
| This journal is © The Royal Society of Chemistry 2016 |