
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and reactivity of Li and TaMe3 complexes supported by N,N′-bis(2,6-diisopropylphenyl)-o-phenylenediamido ligands†
Trevor
Janes
,
Maotong
Xu
and
Datong
Song
*
Davenport Chemical Research Laboratories, Department of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario M5S 3H6, Canada. E-mail: dsong@chem.utoronto.ca
First published on 2nd June 2016
Abstract
The dilithium complex of N,N′-bis(2,6-diisopropylphenyl)-o-phenylenediamide, [Li2L(thf)3], reacts with TaMe3Cl2 in THF/Et2O to yield [Li(Et2O)(thf)LTaMe3Cl] in which the phenylene backbone of L2− is bound η4 to the Ta centre. This dinuclear species reacts with MeLi to yield the tetramethyltantalum complex [Li(Et2O)(thf)LTaMe4]. Double deprotonation of N,N′-bis(2,6-diisopropylphenyl)(4,5-dimethyl)-o-phenylenediamine (H2L′) in Et2O yielded the dilithium complex [Li2L′(OEt2)2]. The two additional methyl groups on L′2− change the observed reactivity towards TaMe3Cl2: rather than bridging between Ta and Li, ligand oxidation occurs to afford mononuclear [LiL′(OEt2)]. This monolithium radical species, which was characterized by EPR spectroscopy, can also be synthesized using the more conventional oxidant AgBF4. Double deprotonation of H2L with KCH2Ph in toluene followed by reaction with TaMe3Cl2 furnished [TaLMe3]. Preliminary reactivity studies show [TaLMe3] reacts with unsaturated substrates N,N′-dicyclohexylcarbodiimide and mesityl azide to undergo migratory insertion into one of the Ta–C bonds: the corresponding amidinate and triazenido ligands are generated. When subjected to UV irradiation, [TaLMe3] undergoes reduction accompanied by loss of a methyl group to yield the dimeric species [TaLMe2]2.
Introduction
Since Juvinall's 1964 preparation of the first tantalum methyl complex, TaMe3Cl2,1 organometallic chemists have been interested in the synthesis and reactivity of Ta–Me species. Members of the research community have continued to design and invent Ta–Me complexes supported by diverse ancillary ligand sets which have facilitated fascinating reactivity. Schrock's syntheses of the homoleptic TaMe5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2 which violently decomposes via alpha abstraction,3 and the first transition metal methylidene complex, [Cp2Ta(CH3)(CH2)]4 are seminal examples. Fryzuk and coworkers discovered that a TaMe3 fragment chelated by their (PhP(CH2SiMe2NPh)2)2− ligand undergoes hydrogenolysis to yield a dinuclear tetrahydride species,5 a lynchpin in the field of N2 activation. More recently, phosphoramidate ligands have been used to sponsor TaMe3Cl precatalysts for room temperature hydroaminoalkylation of olefins.6 Ta methyls have also been grafted onto silica supports for use as well-defined heterogeneous precatalysts for ethylene trimerization7 and alkane metathesis.8
2 which violently decomposes via alpha abstraction,3 and the first transition metal methylidene complex, [Cp2Ta(CH3)(CH2)]4 are seminal examples. Fryzuk and coworkers discovered that a TaMe3 fragment chelated by their (PhP(CH2SiMe2NPh)2)2− ligand undergoes hydrogenolysis to yield a dinuclear tetrahydride species,5 a lynchpin in the field of N2 activation. More recently, phosphoramidate ligands have been used to sponsor TaMe3Cl precatalysts for room temperature hydroaminoalkylation of olefins.6 Ta methyls have also been grafted onto silica supports for use as well-defined heterogeneous precatalysts for ethylene trimerization7 and alkane metathesis.8
Multiple reports have emerged on the synthesis and reactivity of tantalum complexes of diamido ligands with pendant neutral donor functionalities. Fryzuk's work with diamidophosphine5,9–19 and diamidodiphosphine20–22 ligand families is very well-represented. Other prominent examples include Heyduk's redox active NNN-pincer ligand,23–25 diamidoamines26–31 and diamidoaminopyridines.32,33 Tridentate diamido ligands in which the additional donor is a pyridine,27,34,35 carbene,36 arsine,37 and thioether38 are also known. Compared to this abundance of examples of Ta complexes of decorated diamido ligands, simple diamido ligands have not been as well-studied. Tantalum complexes of ligands based on 1,8-diamidonaphthalene,39–41 1,3-diamidopropane,42,43 1,4-diaza-1,3-butadiene,44–49 and o-phenylenediamide (opda) are known. Of the opda complexes, most employ N,N′-disilyl groups,41,50–53 with one report on neopentyl groups.54 To our knowledge, tantalum complexes of N,N′-diaryl substituted opda ligands are heretofore unknown.
We have been investigating the coordination chemistry of N,N′-bis(2,6-diisopropylphenyl)-o-phenylenediamide,55L2−, and sought to prepare Ta complexes of this simple bulky ligand, which may engender new and complementary reactivity to related ligands mentioned above. Herein we report our efforts to coordinate dipp-substituted opda ligands to Ta.
Results and discussion
Synthesis and structures of heterodinuclear compounds 2 and 3(solvent)n
Addition of an Et2O solution of TaMe3Cl2 to a THF solution of Li2L(thf)3 at −70 °C caused the reaction mixture to gradually turn orange as compound 2 formed (Scheme 1). After removal of LiCl, The 7Li NMR spectrum of 2 still featured a singlet at 2.03 ppm, distinct from the 7Li resonance of 1 (2.62 ppm), which indicated incomplete transfer of the diamido ligand from Li. The 1H NMR resonances of the two sets of equivalent o-phenylene protons shifted upfield from 6.57 and 6.33 ppm in starting material 1 to 5.64 and 4.02 ppm in product 2, which is consistent with coordination of the phenylene backbone to Ta. The nine Ta-bound methyl protons resonate as a broad singlet at 0.91 ppm at room temperature. Single crystals were grown by cooling an Et2O solution of 2 to −25 °C. X-ray crystallography revealed the heterodinuclear nature of 2 (Fig. 1). The two opda nitrogen donor atoms of L2− chelate the Li atom; its distorted tetrahedral coordination geometry is completed by the oxygen of one disordered Et2O molecule and the oxygen of one ligand modelled as a 50:50 mixture of THF/Et2O. L2− forms a bridge to Ta via an η4 interaction with its phenylene backbone. The coordination geometry at Ta is akin to a distorted trigonal bipyramid in which one apical ligand has been replaced with a π-bound diene. Cl− occupies the other apical position and three equatorial methyl groups complete the coordination sphere of Ta. The solid state structure suggests two different environments for Ta–Me protons, but cooling a toluene-d8 solution of 2 to −80 °C did not resolve the 1H NMR signal of the methyl groups, which suggests conformational fluxionality with a low barrier.
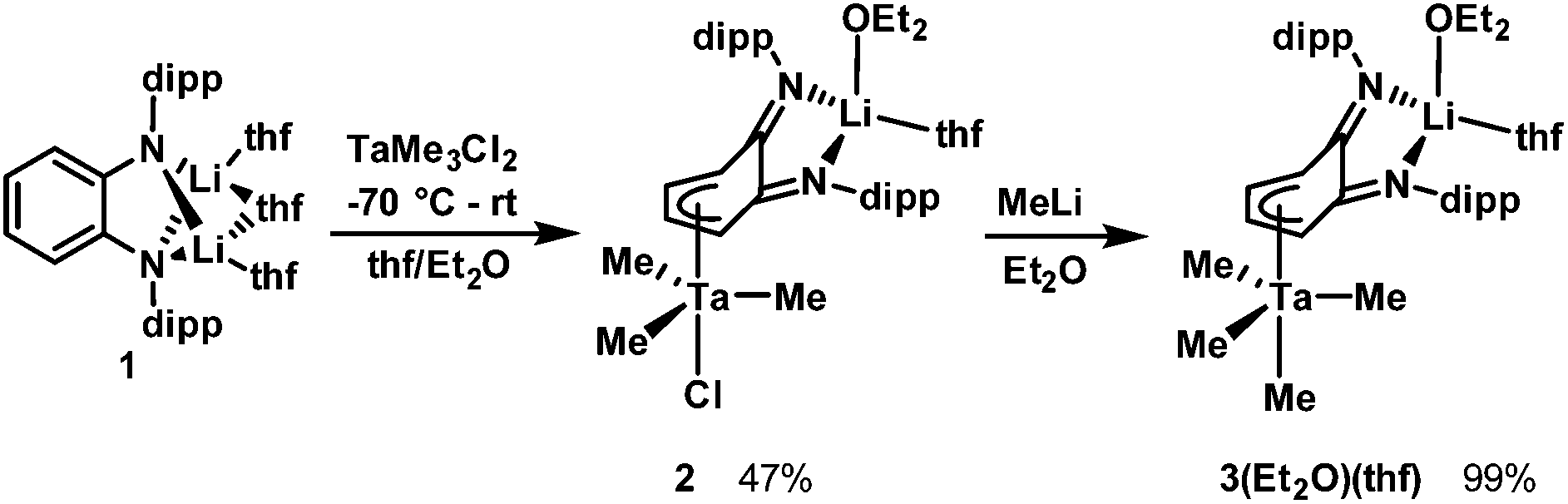 | ||
| Scheme 1 Synthesis of Ta–Li heterodinuclear complexes 2 and 3(Et2O)(thf). | ||
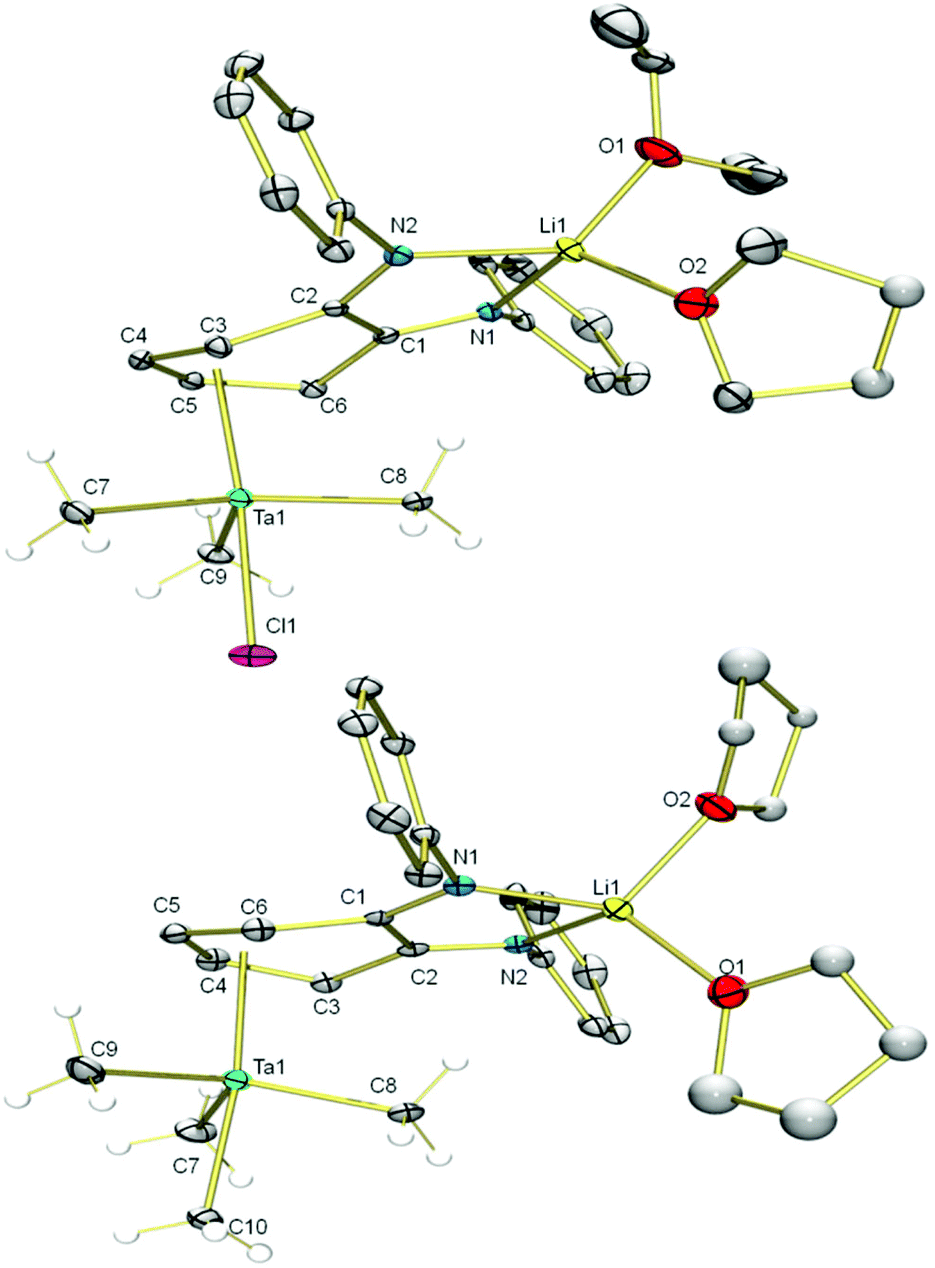 | ||
| Fig. 1 Molecular structures of 2 (top) and 3(thf)2 (bottom). Non-hydrogen atoms are shown as 30% probability ellipsoids except for the disordered portion of Li-coordinated thf. Ta–Me H atoms are shown as spheres of arbitrary radius, the rest of the H-atoms along with the isopropyl groups on L are omitted for clarity. Selected bond angles (°) for 2: N1–Li1–N2 82.1(2), C7–Ta1–C8 118.7(1), C8–Ta1–C9 120.2(1), C7–Ta1–C9 111.8(1), Cl1–Ta1–C7 80.25(9), Cl1–Ta1–C8 80.01(9), Cl1–Ta1–C9 78.96(9). Selected bond angles (°) for 3(thf)2: N1–Li1–N2 82.6(3), C7–Ta1–C8 116.5(2), C8–Ta1–C9 118.3(2), C7–Ta1–C9 114.7(2). C7–Ta1–C10 78.1(2), C8–Ta1–C10 80.6(2), C9–Ta1–C10 78.3(2). | ||
The apical chloride ligand of 2 can be replaced by a methyl group by treatment with MeLi. Addition of MeLi in Et2O to a solution of 2 at −35 °C yielded the tetramethyltantalum complex [Li(Et2O)(thf)LTaMe4], 3(Et2O)(thf). The transformation from 2 to 3(Et2O)(thf) causes a subtle change in the 7Li NMR spectrum: the singlet shifts from 2.03 ppm to 2.26 ppm. Similarly, subtle peak shifts are observed in the 1H NMR spectrum of 3(Et2O)(thf). Distinct from 2, the Ta–Me proton resonance at 0.89 ppm is even more dramatically broadened at room temperature. Single crystals for X-ray analysis were obtained by cooling a pentane solution of 3(Et2O)(thf) to −25 °C (Fig. 1). The solid state data confirmed the replacement of the chloride by a methyl ligand and that the η4 butadiene-type interaction is conserved in the methylation. The solid state structure contains two THF ligands on Li rather than one Et2O and one THF, which is expected based on 1H NMR data. This phenomenon can be explained by ligand exchange on Li and preferential crystallization of 3(thf)2. When 3(Et2O)(thf) is recrystallized from a mixture of pentane and Et2O, resonances from coordinated THF disappear, and only one Et2O ligand is present. This species, 3(Et2O), possesses a simpler alkyl region of the 1H NMR spectrum; upon cooling a toluene-d8 solution to −80 °C, the broad Ta–Me peak splits into three distinct singlets (see ESI†), consistent with the solid state structure. Two of the peaks integrate to three protons each (2.35 and 1.00 ppm) for the two methyl ligands sitting on the mirror plane that bisects the L2− and the third peak integrates to six protons (0.42 ppm) for the two methyl ligands that are related by this mirror plane.
The solid state molecular structures of 2 and 3(thf)2 are very similar. In both, the lithium atom is chelated by the two nitrogen donor atoms of the L2−. The Cphenylene–N bond lengths (1.295(4) Å in 2 and 1.306(5) and 1.307(5) Å in 3(thf)2) are significantly shortened compared to the dilithium complex 1 (Cphenylene–N bond lengths of 1.396(2) and 1.395(2) Å).55 This compression of C–N bonds suggests the diamido ligand in 1 has become a diimine ligand in 2 and 3(thf)2. The tantalum atom in 2 and 3(thf)2 is bound η4- to the o-phenylene ring of the opda ligand in a butadiene-type interaction. In these complexes, the η4-C6 ring is folded along the C3–C6 vector at angles of 31° and 25°, respectively which are very similar to the corresponding angle in the previously reported trimetallic [(Li(thf)2L)2MoCl2] (30°).55 On the continuum between the Chatt-Dewar diene (L2) and metallocyclopentene (LX2) extremes,56 we formulated this MoLi2 species as more of an L2 butadiene-type complex. However, the metric parameters of the η4 ligand used to make this assignment are different from Ta complexes 2 and 3(thf)2. The Δd parameter (where Δd = avg. M–Couter bond length – avg. M–Cinner bond length; Couter refers to C3 and C6 and Cinner refers to C4 and C5) is 0.034(3) Å for 2 and 0.060(4) Å for 3(thf)2, compared to 0.142(6) Å in the MoLi2 species. In the MoLi2 species, the Cinner–Cinner and avg. Cinner–Couter bonds are statistically similar, but in 2 and 3(thf)2, the Cinner–Cinner bonds are 1.377(4) and 1.368(6) Å, respectively. These bonds are significantly shorter than the avg. Cinner–Couter in 2 and 3(thf)2, which are 1.431(4) and 1.427(4) Å. 2 and 3(thf)2 more closely resemble the metallocyclopentene resonance form, which is consistent with the greater electropositivity of Ta relative to Mo. Taken together, the metric parameters of both 2 and 3(thf)2 suggest contribution of L2− resonance form B (Chart 1) is significant (Table 1).
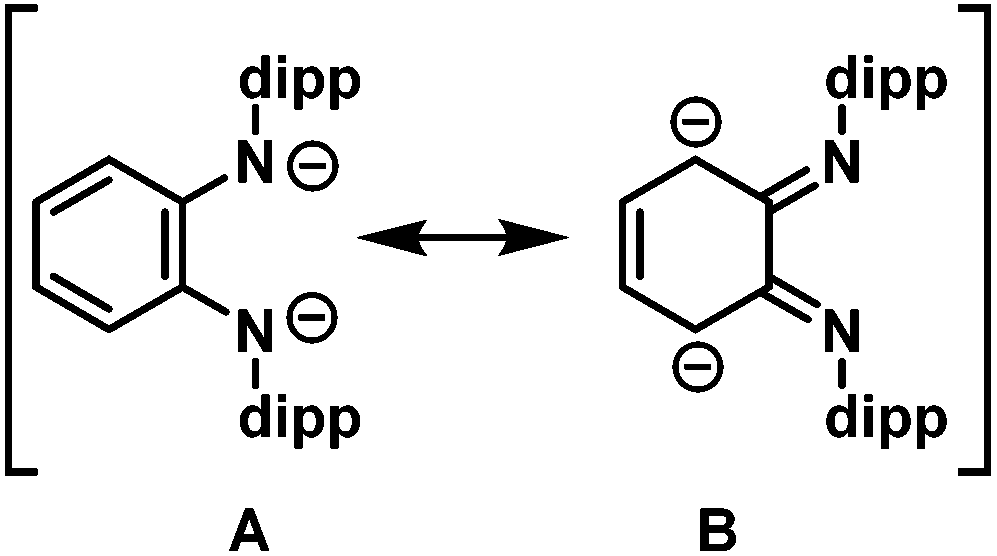 | ||
| Chart 1 Two resonance forms for L2−. | ||
| Bond | Length in 2 (Å) | Length in 3(thf)2 (Å) | Bond | Length in 2 (Å) | Length in 3(thf)2 (Å) |
|---|---|---|---|---|---|
| Ta1–C3 | 2.443(3) | 2.480(4) | C2–C3 | 1.451(5) | 1.428(6) |
| Ta1–C4 | 2.401(4) | 2.422(4) | C3–C4 | 1.426(5) | 1.430(5) |
| Ta1–C5 | 2.400(3) | 2.419(4) | C4–C5 | 1.377(4) | 1.368(6) |
| Ta1–C6 | 2.425(3) | 2.481(4) | C5–C6 | 1.436(5) | 1.424(6) |
| Ta1–C7 | 2.205(3) | 2.199(4) | C1–C6 | 1.443(4) | 1.425(5) |
| Ta1–C8 | 2.209(4) | 2.172(4) | C1–N1 | 1.295(4) | 1.306(5) |
| Ta1–C9 | 2.218(3) | 2.218(6) | C2–N2 | 1.295(4) | 1.307(5) |
| Ta1–Cl1 | 2.428(1) | N1–Li1 | 2.058(5) | 2.043(8) | |
| Ta1–C10 | 2.241(5) | N2–Li1 | 2.076(6) | 2.055(7) | |
| C1–C2 | 1.488(4) | 1.483(6) |
Coordination chemistry of L′
In hopes of disfavouring the phenylene carbons relative to the diamido nitrogens as coordination site for Ta we increased the steric bulk at the phenylene backbone. According to the method of Wenderski et al.,57 we synthesized the doubly methylated diamine proligand H2L′. Double deprotonation was achieved by addition of two equivalents of n-BuLi to a −70 °C diethylether solution of 4 (Scheme 2). After removal of volatiles and precipitation with cold pentane, [Li2L′(Et2O)2], 5, was isolated as a white powder. In the 1H NMR spectrum of 5 in C6D6, the N–H resonance present in the starting material is absent, replaced by a quartet at 2.92 ppm and a triplet at 0.74 ppm corresponding to the ethyl groups on two coordinated Et2O molecules. In the 7Li NMR spectrum, one signal is observed at 0.77 ppm which indicates that the two Li atoms are equivalent in solution. Single crystals for X-ray were obtained by cooling a pentane/Et2O solution to −25 °C (see ESI†).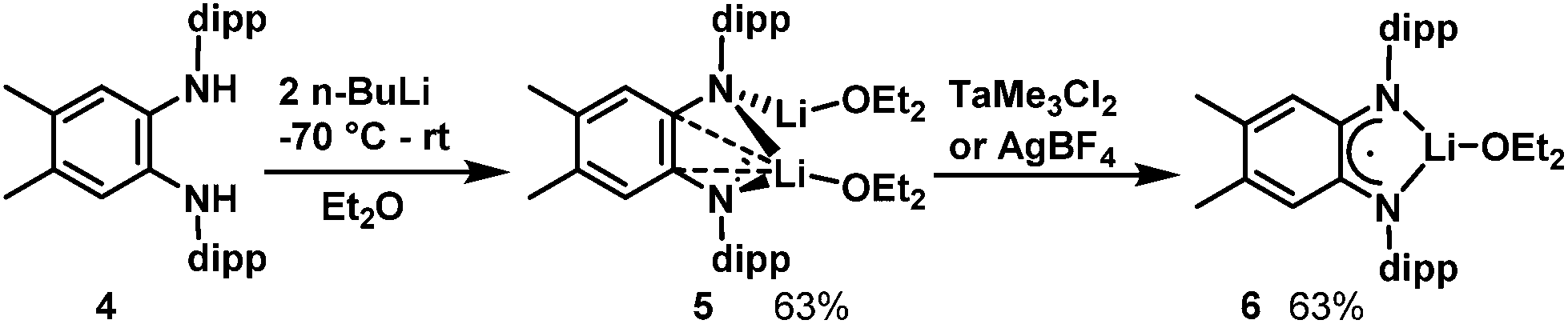 | ||
| Scheme 2 Syntheses of Li complexes 5 and 6. | ||
Reaction of dilithium compound 5 with TaMe3Cl2 causes the solution to darken in colour. After replacing the solvent with pentane and removing insoluble material by filtration, dark green single crystals of 6 grew from the cooled concentrated pentane extract (see ESI† for X-ray structure and EPR spectrum). Unexpectedly, TaMe3Cl2 acted as an oxidant towards the dilithium species 5 and the L′2− complex has lost an electron and a Li ion to become a monolithium diiminosemiquinonate complex. Efforts to structurally characterize the Ta-containing species generated in this reaction have so far not been fruitful. Notably, 6 can also be prepared by oxidation of 5 with a source of Ag+ (see Experimental section).
Synthesis of [TaLMe3], 8
In further efforts to synthesize an NN chelate complex of Ta with dipp-substituted opda ligands we employed dilithium salts of L2− free of coordinated Et2O/THF ligands; we also attempted protonolysis by reacting H2L with TaMe3Cl2 or Ta(NMe2)5, but none of these attempts succeeded. However, the generation of the dipotassium salt of L2− by double deprotonation of H2L with benzylpotassium (for its molecular structure with complexed 1,2-dimethoxyethane ligands see ESI†) followed by reaction with TaMe3Cl2 in toluene yielded the desired [TaLMe3], 8 (Scheme 3). In its 1H NMR spectrum in C6D6 at room temperature, the three methyl ligands resonate as a single peak at 1.03 ppm. Cooling a toluene-d8 solution to −80 °C did not lead to any decoalescence of this signal. X-ray quality single crystals were grown by cooling a saturated pentane solution to −25 °C. The molecular structure of 8 (Fig. 2) reveals L2− chelating a five-coordinate Ta centre, which is bound to three terminal methyl ligands. Ta occupies a distorted square pyramidal geometry, with N1, N2, C7 and C8 forming the square base and C9 in the apical position. The diamido ligand binds Ta with Ta1–N1 and Ta1–N2 bond lengths of 2.055(3) and 2.052(3) Å, respectively. The diamido's N1–Ta1–N2 bite angle is 75.5(1)°, and the five-membered TaN2C2 chelate ring is essentially planar such that the electron pairs on the nitrogen atoms are oriented with correct symmetry to engage in π-bonding with Ta. All o-phenylene C–C bond lengths are statistically similar and both Cphenylene–N bond lengths are 1.413(5) Å, statistically similar to the Cphenylene–N bond lengths in 5. These data suggest the potentially redox-active L2− maintains its dianionic charge, with resonance form A in Chart 1 as the major contributor. | ||
| Scheme 3 Synthesis of [TaLMe3]. | ||
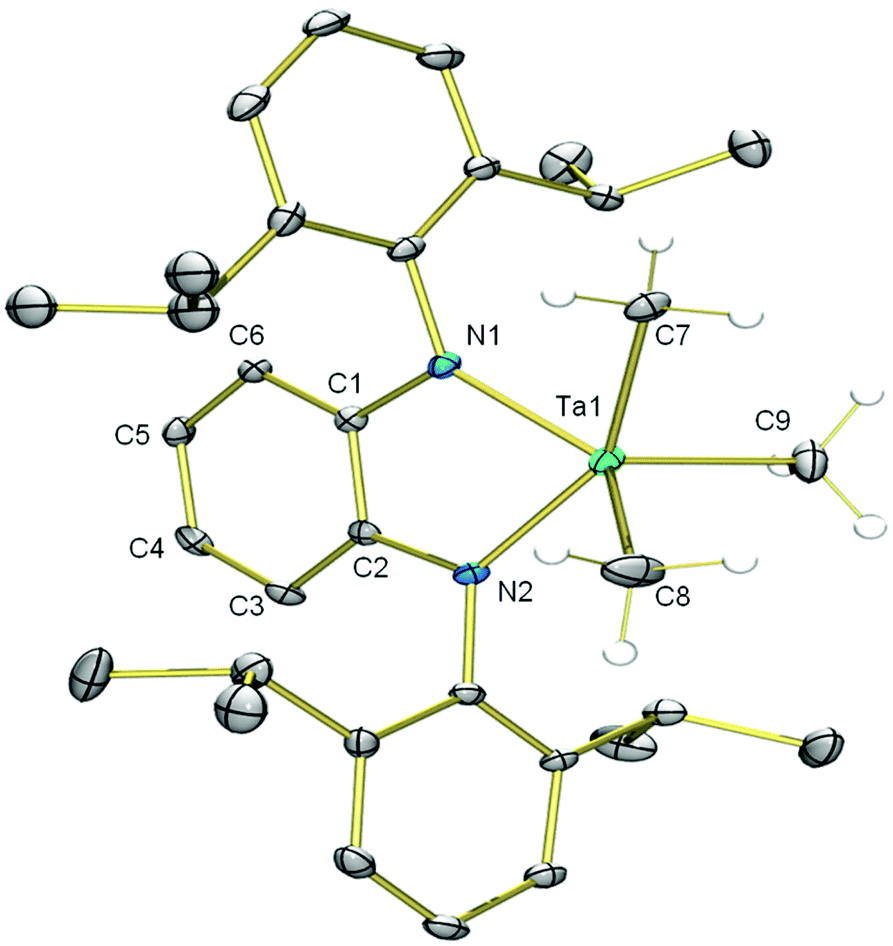 | ||
| Fig. 2 Molecular structure of [TaLMe3], 8. Non-hydrogen atoms are shown as 30% probability ellipsoids. Hydrogen atoms on Ta–Me groups are shown as spheres of arbitrary radius, and the rest of the H-atoms are omitted for clarity. Only one orientation of disordered isopropyl group is shown. Selected bond lengths (Å) and angles (°) for 8: Ta1–C7 2.177(4), Ta1–C8 2.137(6), Ta1–C9 2.137(5), Ta1–N1 2.055(3), Ta1–N2 2.052(3), C1–C2 1.390(6), C2–C3 1.391(6), C3–C4 1.390(6), C4–C5 1.394(6), 1.387(6), C6–C1 1.400(6), C1–N1 1.413(5), C2–N2 1.413(5), N1–Ta1–N2 75.5(1), N1–Ta1–C9 112.3(2), N1–Ta1-C8 138.6(2), N2–Ta1–C7 152.9(1), N2–Ta1–C8 92.3(2), N2–Ta1–C9 105.3(2), C7–Ta1-C8 89.9(2), C7–Ta1–C9 99.5(2), C8–Ta1–C9 109.1(2). | ||
8 could also be generated by thermal loss of LiCl from 2. Heating a solution of 2 in C6D6 at 80 °C for 3 h caused consumption of 2; analysis of the 1H NMR spectrum indicated formation of 8 in a mixture that also contained [TaClLMe2] and 3 (respective ratio of 1: 1.5:2). The formation of [TaClLMe2] can be explained by the exchange of Me and Cl ligands between Ta centres, which is well documented in the literature.58
Reactivity of [TaLMe3]
Insertion of carbodiimides into Ta–CMe bonds has been known since Wilkins’ report in 1974.59 To test the ability of L to sponsor this reaction we subjected 8 to one equivalent of N,N′-dicyclohexylcarbodiimide (DCC) (Scheme 4); the reaction mixture turned cherry red as insertion of DCC into one of the Ta–C bonds occurred. Diagnostic features of the 13C NMR spectrum of 9 are the amidinate NCN and CH3 resonances at 180.18 ppm and 15.12 ppm, respectively. In the 1H NMR spectrum, the amidinate and Ta-bound CH3 protons resonate as singlets integrating to three and six protons at 1.63 ppm and 1.03 ppm, respectively. All other alkyl protons give rise to broadened, overlapped resonances. X-ray analysis of single crystals revealed the molecular structure of 9 (Fig. 3) which includes a newly formed amidinate ligand. The geometry at Ta can be described as distorted trigonal prismatic, with N1–N2–C8 forming one triangular face and N3–N4–C7 forming the other. The amidinate coordination bond lengths are significantly different: N4–Ta1 (2.136(4) Å) is shorter than N3–Ta1 (2.221(4) Å). Its C–N bonds (1.325(6) and 1.344(7) Å) are equal within experimental error. The amidinate ligand chelates the tantalum centre with an N3–Ta1–N4 bite angle of 60.4(1)°, and the four-membered CN2Ta ring is planar. Unlike 8, the five-membered C2N2Ta ring exists in an envelope conformation, with the Ta atom out of plane. The dihedral angle between the o-phenylene plane and the plane defined by N1–Ta1–N2 is ca. 25°. | ||
| Scheme 4 Reactivity of [TaLMe3]: synthesis of 9, 10, and 11. | ||
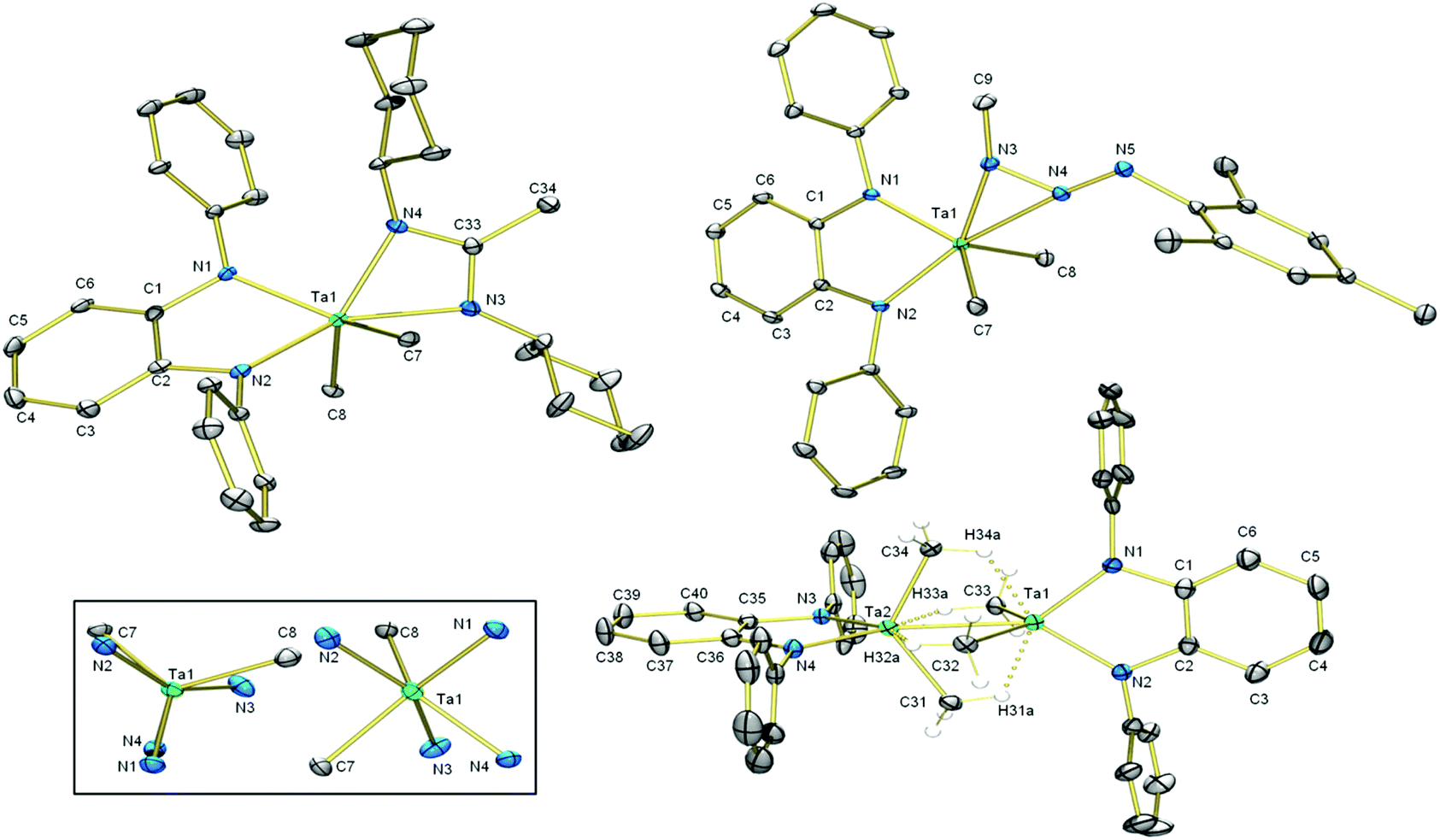 | ||
| Fig. 3 Molecular structures of 9 (top left), 10 (top right), and 11 (bottom right). Inset: Two views of the coordination sphere of 9. Non-hydrogen atoms are shown as 30% probability ellipsoids. Hydrogen atoms on the Ta–Me groups of 11 are shown as spheres of arbitrary radius. The rest of the H-atoms and isopropyl groups on L are omitted for clarity. Selected bond lengths (Å) and angles (°) for 9: Ta1–N3 2.221(4), Ta1–N4 2.136(4), C33–N3 1.325(6), C33–N4 1.344(7). N1–Ta1–N2 77.0(1), N1–Ta1–N3 129.5(2), N1–Ta1–N4 86.3(1), N1–Ta1–C7 136.3(2), N1–Ta1–C8 95.1(2), N2–Ta1–N3 152.4(2), N2–Ta1–N4 139.5(1), N2–Ta1–C7 79.9(2), N2–Ta1–C8 88.2(2), N3–Ta1–N4 60.4(1), N3–Ta1–C7 82.9(2), N3–Ta1–C8 82.2(2), N4–Ta1–C7 87.8(2), N4–Ta1–C8 130.3(2), C7–Ta1–C8 120.9(2). Selected bond lengths (Å) and angles (°) for 10: Ta1–N3 2.049(2), Ta1–N4 2.221(2) N4–N5 1.263(3), Ta1–N3–N4 79.0(1), N1–Ta1–N2 75.57(8), N1–Ta1–N3 83.71(8), N1–Ta1–N4 117.33(8), N1–Ta1–C7 137.13(8), N1–Ta1–C8 106.60(8), N2–Ta1–N3 152.50(8), N2–Ta1–N4 166.45(7), N2–Ta1–C7 85.78(8), N2–Ta1–C8 95.11(8), N3–Ta1–N4 36.16(8), N3–Ta1–C7 97.76(9), N3–Ta1–C8 108.17(9), N4–Ta1–C7 81.63(8), N4–Ta1–C8 85.35(8), C7–Ta1–C8 113.34(9). Selected distances (Å) and angles (°) for 11: Ta1–Ta2 2.7120(5) Ta1–H31a 1.99, Ta1–H34a 1.98, Ta1–C31 2.618(5), Ta1–C34 2.607(4), Ta2–H32a 2.00, Ta2–H33a 1.99, Ta2–C32 2.627(4), Ta2–C33 2.614(4), Ta2–N3 2.033(3), Ta2–N4 2.032(3), C35–N3 1.425(5), C36–N4 1.432(5), C35–36 1.409(6), C36–C37 1.376(6), C37–C38 1.379(7), C38–C39 1.371(6), C39–C40 1.394(6), C35–C40 1.385(6). N2–Ta1–N1 82.1(1), N1–Ta1–H34a 72.2, H34a–Ta1–Ta2 67.5, Ta2–Ta1–H31a 67.7, H31a–Ta1–N2 70.6, C33–Ta1–C32 127.1(2), N3–Ta2–N4 81.9(1), N4–Ta2–H32a 71.8, H32a–Ta2–Ta1 67.7, Ta1–Ta2–H33a, H33a––Ta2–N3 71.3, C31–Ta2–C34 126.7(2). | ||
When mesityl azide was added to a solution of 8, yellow precipitate formed. The 1H NMR resonance associated with the Ta–bound Me groups shifted upfield to 0.67 ppm and its integration was reduced from nine to six protons, which indicated that the azide had reacted with one of the Me ligands on tantalum. Aside from resonances associated with the new mesityl group, an additional methyl singlet appeared at 2.53 ppm. X-ray crystallography on single crystals of 10 revealed that insertion of the azide into the tantalum–carbon bond had taken place, forming a κ2-N2 triazenido ligand. The Ta centre adopts a distorted trigonal bipyramidal geometry, with the equatorial positions occupied by the two Ta–Me groups and N1 of L2−. N2 of L2− and the κ2-N2 interaction occupy the axial positions. The N3–Ta1 bond (2.049(2) Å) is significantly shorter than the N4–Ta1 bond (2.221(2) Å) and the N4–N5 bond (1.263(3) Å) is shorter than the N4–N3 bond (1.335(3) Å), which informs our formulation of 10 shown in Scheme 4. The sum of the angles at N3 (359.8(3)°) indicates its planarity such that its lone pair be oriented perpendicular to this plane with correct symmetry to engage in π-bonding with Ta. The plane of the o-phenylene ring and the plane defined by the three nitrogen atoms of the triazenido ligand meet with a dihedral angle of 32°. For the diamido moiety, intraligand metric parameters are the same as in 8; the diamido behaves as a spectator ligand during the transformation from 8 to 10.
The reactivity most relevant to this transformation is the single and double insertion of aryl azides into the Ta–C bonds of [(ONO)TaMe2] (ONO = bis(phenoxy)amide ligand) to form mono and bis triazenido complexes.60 Distinct from these compounds, the triazenido ligands in 10 are κ2N1,2-bound to Ta. Although this binding mode is well known for f-elements,61–65 to our knowledge this is the first crystallographically characterized example of a triazenido ligand κ2N1,2-bound to Ta.
When a toluene solution of 8 was subjected to UV light, the colour of the solution darkened. In the 1H NMR spectrum in C6D6, the Ta–Me resonance shifted upfield from 1.03 to 0.08 ppm, and there are six Ta–Me protons per diamido ligand, indicating loss of one of the Ta–Me groups. In the 13C NMR spectrum the carbon peak attributable to the Ta–Me groups shifted from 83.50 (1JC–H = 118 Hz) to 49.22 ppm (1JC–H = 116 Hz). Single crystals of 11 were obtained from cold pentane; the molecular structure (Fig. 3, bottom) revealed a dimeric formally Ta(IV) species possessing a Ta–Ta bond length of 2.7120(5) Å. This bond is intermediate between the Ta–Ta bonds found in (μ-H)4 ditantalum complexes of diamidophosphine (2.830(4) Å)9 and diamidodiphosphine (2.6165(5) Å)21 complexes. Each Ta centre is bound to two methyl groups, and each methyl group makes a close contact with its neighbouring Ta centre two bonds away; these distances range from 2.607(4) Å for Ta1–C34 to 2.627(4) Å for Ta1–C32. Although the methyl hydrogens were not located on the Fourier difference map, the proximity of the methyl carbons to both Ta centres and the marked shift of the methyl protons to high field in the 1H NMR spectrum lend support for the presence of agostic interactions between one methyl C–H bond and its opposite Ta centre. Taking these interactions into account, the geometry at each seven-coordinate Ta centre is best understood as distorted pentagonal bipyramidal. On each Ta centre, L2−, the other Ta atom, and two agostic interactions reside in equatorial positions (sum of the bond angles: 360.1 at Ta1 and at Ta2). The two-centre two-electron bound methyl carbons occupy axial positions. This reactivity of [TaLMe3], which contains a simple diamido ligand, is distinct from that of Fryzuk's [(P2N2)TaMe3] (P2N2 = diamidodiphosphine ligand), which undergoes loss of methane to generate a monomeric methyl methylidene complex.20 The authors proposed that the first step of the reaction was the photoinduced Ta–C bond homolysis, generating a methyl radical. Presumably the transformation of 8 to 11 could share the same initial step, where the methyl radical could be scavenged by toluene solvent and the resulting [TaLMe2] species dimerizes to form 11.
Conclusion
Attempts were made to install simple, bulky opda ligands on Ta via salt metathesis with TaMe3Cl2. Use of dilithium complex 1 led to the isolation of heterodinuclear 2 in which the NN chelate site of the diamide ligand was occupied by Li+, and the o-phenylene backbone was engaged in a η4-interaction with Ta. This species underwent clean methylation with MeLi to generate tetramethyl species 3(Et2O)(thf) in which heterodinuclearity is preserved. A new dilithium complex with increased steric bulk at the o-phenylene backbone (5) was synthesized. When reacted with TaMe3Cl2 it underwent oxidation to open-shell monolithium complex 6, which could be synthesized in higher yield and purity using AgBF4. By using a dipotassium derivative of L2−, the desired trimethyltantalum complex (8) could be synthesized. Preliminary reactivity studies show 8 undergoes insertion reactions with DCC and MesN3 to generate compounds 9 and 10. In 10 the newly formed triazenido ligand is bound κ2N1,2, a bonding mode which is unusual for Ta. Irradiation of a toluene solution of 8 leads to isolation of dimeric Ta(IV) species 11, which illustrates the complementary reactivity engendered by the simple bulky opda ligand. Our initial attempts to generate isolable Ta–H species via hydrogenolysis of 8 have not succeeded, but investigation of the reactivity of this and other o-phenylenediamido Ta complexes towards H2 is ongoing in our laboratory.Experimental section
General considerations
Compounds 1,554, 7,57 TaMe3Cl2,4 KCH2Ph,66 and MesN367 were prepared from literature methods. Methyl lithium (1.6 M in ether), n-butyl lithium (1.6 M in hexanes), AgBF4, and N,N′-dicyclohexylcarbodiimide were purchased from commercial sources. All operations were performed using Schlenk techniques under dinitrogen or in a dinitrogen-filled glovebox. All glassware was either flame-dried or dried overnight in a 180 °C oven prior to use except for NMR tubes which were dried overnight in a 60 °C oven. THF, Et2O, toluene, and C6D6 were distilled from Na/benzophenone under N2. Pentane, hexanes, and toluene-d8 were distilled from sodium under dinitrogen. All solvents were then stored over 3 Å molecular sieves prior to use. 1H, 31P, 13C, and 11B NMR spectra were recorded on a Varian 400 MHz, Agilent DD2 500 MHz, or Agilent DD2 600 MHz spectrometer. Electron paramagnetic resonance (EPR) spectra were obtained at 298 K in thf solution using a Bruker ECS-EMX X-band EPR spectrometer equipped with an ER4119HS cavity. Simulation was carried out using PEST WinSIM Software. All chemical shifts are reported in ppm relative the residual protio-solvent peaks 7Li NMR is referenced externally using 9.7 M LiCl in D2O. Elemental analyses were performed by ANALEST at the University of Toronto.[Li(Et2O)(thf)LTaMe3Cl], 2
To [Li2L(thf)3], 1 (115 mg, 0.175 mmol) dissolved in THF (3 mL) and cooled to −70 °C was added a similarly cooled solution of TaMe3Cl2 (48.1 mg, 0.162 mmol) in Et2O (2 mL). The reaction mixture was allowed to warm to room temperature and was stirred for 2 h. Volatiles were removed under reduced pressure. The orange residue was extracted with Et2O (5 mL), filtered, concentrated to ca. 2 mL and cooled to −25 °C overnight. The supernatant was decanted off the crystals that had formed, which were then washed with cold Et2O (3 × 0.5 mL). Residual solvent was removed by briefly applying vacuum to the yellow crystals of [Li(Et2O)(thf)LTaMe3Cl], 2 (63.9 mg, 0.760 mmol, 47%). 1H NMR (600 MHz, benzene-d6) δ 7.21 (dd, J = 7.0, 2.1 Hz, 2H), 7.15–7.10 (m, 4H), 5.66–5.62 (m, 2H), 4.04–4.01 (m, 2H), 3.19 (sept, J = 6.9 Hz, 2H), 3.12–3.08 (m, 4H), 3.06 (sept, J = 6.2 Hz, 2H), 1.34 (d, J = 6.9 Hz, 6H), 1.22 (d, J = 6.9 Hz, 6H), 1.20 (d, J = 6.9 Hz, 6H), 1.10 (d, J = 6.8 Hz, 10H) 1.10 (overlapped, 4H), 0.97–0.94 (m, 6H), 0.91 (br, 9H) 13C NMR (151 MHz, benzene-d6) δ 168.37, 145.38, 142.01, 140.13, 124.84, 123.93, 123.81, 109.94, 86.66, 68.31, 65.99, 28.67, 27.90, 25.30, 24.74, 24.68, 24.54, 24.14, 15.23. We did not observe a resonance attributable to the Ta–Me carbons, presumably due to exchange broadening. 7Li NMR (233 MHz, benzene-d6) δ 2.03. Anal. Calcd for C41H65N2O2LiClTa: C, 58.53; H, 7.79; N, 3.33. Found: C, 58.64; H, 7.60; N, 3.47. Single crystals for XRD were obtained by cooling an Et2O solution to −25 °C.[Li(Et2O)(thf)LTaMe4], 3(Et2O)(thf)
[Li(Et2O)(thf)LTaMe3Cl], 2 (31.5 mg, 0.0374 mmol), was dissolved in Et2O (5 mL) and cooled to −35 °C. Methyl lithium (ca. 0.16 M in diethylether, 0.22 mL) was added dropwise. The reaction mixture was allowed to warm to room temperature protected from light, and was stirred for 60 min, causing the colour of the solution to lighten. Filtration and removal of volatiles yielded yellow microcrystalline [Li(Et2O)(thf)LTaMe4], 3(Et2O)(thf) (30.3 mg, 0.0369 mmol, 99%). Anal. Calcd for C42H68N2O2LiTa: C, 61.45; H, 8.35; N, 3.41. Found: C, 61.72; H, 8.09; N, 3.41. Single crystals of 3(thf)2 for XRD were obtained by cooling a pentane solution to −25 °C. 1H NMR (500 MHz, benzene-d6) δ 7.25 (dd, J = 7.2, 1.8 Hz, 2H), 7.19–7.11 (m, 4H), 5.54–5.45 (m, 2H), 4.26–4.18 (m, 2H), 3.28 (sept, J = 7.0 Hz, 2H), 3.15 (br, 4H), 3.07 (sept, J = 6.9 Hz, 2H), 3.04 (q, J = 7.0 Hz, 4H), 1.34 (d, J = 6.9 Hz, 6H), 1.25 (d, J = 6.9 Hz, 6H), 1.23 (d, J = 6.8 Hz, 6H), 1.16 (d, J = 6.8 Hz, 4H), 1.04 (br, 4H), 0.90 (t, J = 7.0 Hz, 6H), 0.89 (br, 12H) 13C NMR (126 MHz, benzene-d6) δ 164.33, 146.54, 142.45, 140.87, 128.06, 124.31, 123.84, 123.71, 109.26, 89.72, 68.35, 66.01, 28.58, 27.79, 25.19, 24.82, 24.80, 24.60, 24.31, 15.04. We did not observe a resonance attributable to the Ta–Me carbons, presumably due to exchange broadening. 7Li NMR (194 MHz, benzene-d6) δ 2.26. Anal. Calcd for C42H68N2O2LiTa: C, 61.45; H, 8.35; N, 3.41. Found: C, 61.72; H, 8.09; N, 3.41. Single crystals of 3(thf)2 for XRD were obtained by cooling a pentane solution to −25 °C.[Li2L′(Et2O)2], 5
A hexanes solution of nBuLi (1.6 M, 0.29 mL, 0.46 mmol) was added dropwise to a solution of H2L′ (102 mg, 0.223 mmol) in Et2O cooled to −70 °C using a glove box cold well. The reaction was allowed to warm to room temperature and was stirred for 2 h at which point volatiles were removed in vacuo. Pentane (2 mL) was added and the mixture was cooled to −25 °C overnight. The supernatant was decanted off the precipitate which was washed with cold pentane (2 mL) and dried under reduced pressure, leaving fluffy white powder (86 mg, 0.14 mmol, 63%). Due to the highly sensitive nature of this dilithium complex, we could not obtain satisfactory elemental analysis. 1H NMR (300 MHz, benzene-d6) δ 7.37 (d, J = 7.6 Hz, 4H), 7.22 (dd, J = 8.0, 7.1 Hz, 2H), 6.16 (s, 2H), 3.40 (sept, J = 6.8 Hz, 4H), 2.92 (q, J = 7.1 Hz, 4H), 2.03 (s, 6H), 1.36 (d, J = 6.8 Hz, 12H), 1.29 (d, J = 7.0 Hz, 12H), 0.74 (t, J = 7.1 Hz, 12H). 13C NMR (101 MHz, benzene-d6) δ 152.74, 144.83, 143.75, 123.79, 121.29, 120.89, 115.05, 66.22, 28.42, 25.27, 25.22, 19.29, 14.10. 7Li NMR (194 MHz, benzene-d6) δ 0.77. Single crystals for XRD were obtained by cooling a pentane/Et2O solution to −25 °C.[LiL′(Et2O)], 6, Method A
[Li2L′(Et2O)2], 5 (0.213 g, 0.353 mmol) was dissolved in THF (8 mL) and cooled to −70 °C using a glove box cold well. A similarly cooled solution of TaMe3Cl2 (0.104 g, 0.351 mmol) in Et2O (4 mL) was added dropwise, and the mixture was allowed to warm to room temperature and stirred for 16 h. Volatiles were removed in vacuo and pentane (10 mL) was added to the mixture. Insoluble material was filtered off and the filtrate was concentrated and cooled to −25 °C. Crystals formed and were isolated on a fritted funnel; washing with cold pentane (2 × 1 mL) and drying in vacuo yielded dark green crystals (0.104 g, 0.194 mmol, 55%). This material was characterized by EPR and XRD, but even multiple recrystallizations did not lead to satisfactory combustion analysis.[LiL′(Et2O)], 6, Method B
[Li2L’(Et2O)2] (160.8 mg, 0.2607 mmol) was dissolved in toluene (6 mL) and cooled to −25 °C. Under subdued lighting, AgBF4 (50.6 mg, 0.260 mmol) was added as a solid and the mixture was allowed to warm to room temperature with stirring for 2.5 h. At the end of the reaction time, the dark green mixture was filtered through Celite and stripped. Recrystallization of the residue from 5 mL of ca. 5% Et2O in pentane cooled to −25 °C yielded dark green crystals, which were washed with cold pentane and dried in vacuo (87.4 mg, 0.163 mmol, 63%). Anal. Calcd for C36H52N2OLi: C, 80.71; H, 9.78; N, 5.22. Found: C, 80.22; H, 9.77; N, 5.05. Single crystals for XRD were obtained by cooling a pentane/Et2O solution to −25 °C.[TaLMe3], 8
K2L: To a solution of H2L (0.94 g, 2.3 mmol) in toluene (15 mL) cooled to −25 °C was added benzylpotassium (0.61 g, 4.7 mmol)), in one portion. The reaction mixture was allowed to warm to room temperature, and was stirred for 16 h. Pentane (60 mL) was added and the mixture was cooled to −25 °C. The pale green suspension was decanted from any unreacted orange benzylpotassium onto a frit, which left a pale green powder that was then washed with cold toluene (2 × 5 mL) and pentane (2 × 10 mL), and then dried in vacuo leaving a pale green pyrophoric powder 0.97 g, 86%.TaMe3Cl2 (174 mg, 0.586 mmol) was dissolved in toluene (10 mL) and cooled to −70 °C using a glove box cold well. The vial was removed from the cold well and finely ground K2L (330 mg, 0.653 mmol) was added as a solid, in portions over five minutes. The reaction turned gradually turned brownish red as it warmed to room temperature. After 5 h, the mixture was filtered through Celite and the toluene was removed under reduced pressure. Recrystallization of the residue from pentane afforded two crops of crystals (143 mg, 0.219 mmol, 37%). 1H NMR (600 MHz, benzene-d6) δ 7.22–7.21 (m, 6H), 6.52–6.50 (m, 2H), 6.00–5.97 (m, 2H), 3.52 (sept, J = 6.9 Hz, 4H), 1.30 (d, J = 6.9 Hz, 12H), 1.06 (d, J = 6.8 Hz, 12H), 1.03(s, 9H). 13C NMR (151 MHz, C6D6) δ 147.80, 146.09, 144.99, 127.61, 124.69, 120.84, 114.77, 83.50, 28.65, 26.29, 24.26. Anal. Calcd for C33H47N2Ta: C, 60.73; H, 7.26; N, 4.29. Found: C, 60.58; H, 7.19; N, 4.21.
DCC insertion product, 9
[TaLMe3] (66 mg, 0.10 mmol) was dissolved in pentane and cooled to −25 °C. A solution of N,N’-dicyclohexylcarbodiimide (21 mg, 0.10 mmol) in pentane (2 mL) at room temperature was added dropwise. The solution immediately turned cherry red, and was allowed to warm to room temperature with stirring for 16 h. Volatiles were removed under reduced pressure, leaving the insertion product (81 mg, 93%). The analytical sample was washed with pentane. 1H NMR (600 MHz, benzene-d6) δ 7.31 (d, J = 7.5 Hz, 4H), 7.25 (t, 2H), 6.55–6.51 (m, 2H), 6.06–6.02 (m, 2H), 3.71 (br, 2H), 3.63 (br, 2H), 3.40 (br, 2H), 1.63 (s, 3H), 1.55–1.05 (broad, overlapped, 46 H), 1.03 (s, 6H). 13C NMR (151 MHz, C6D6) δ 180.18, 149.05, 146.19, 145.51, 126.75, 125.22 (br), 124.03 (br), 120.40, 115.88, 75.25, 65.92, 59.37, 35.40, 32.81, 28.61, 28.10, 26.51, 25.81, 24.88, 24.30, 15.12. Anal. Calcd for C46H69N4Ta: C, 64.32; H, 8.10; N, 6.52. Found: C, 64.23; H, 8.06; N, 6.69. Single crystals for XRD were obtained by allowing a pentane solution to slowly evaporate at room temperature.MesN3 insertion product, 10
Pentane was added to [TaLMe3] (51.5 mg, 0.0789 mmol) and the brown mixture was cooled to −25 °C. A solution of mesityl azide (13.4 mg, 0.0831 mmol) in pentane (0.5 mL) was added dropwise. The mixture was allowed to warm to room temperature and was stirred for 2 h, causing a precipitate to form. Volatiles were removed under reduced pressure and the residue was washed with cold pentane (3 × 0.5 mL). Drying in vacuo left orange powder (40.7 mg, 0.0500 mmol, 63%). The analytical sample was recrystallized from toluene. 1H NMR (600 MHz, benzene-d6) δ 7.24–7.21 (m, 6H), 6.70 (s, 2H), 6.67–6.61 (m, 2H), 6.29–6.24 (m, 2H), 3.35 (sept, J = 6.9 Hz, 4H), 2.53 (s, 3H), 2.09 (s, 6H), 2.06 (s, 3H), 1.10 (d, J = 6.8 Hz, 12H), 1.09 (d, J = 6.8 Hz, 12H), 0.67 (s, 6H). 13C NMR (151 MHz, benzene-d6) δ 147.94, 145.98, 145.46, 145.25, 135.56, 129.61, 129.40, 127.24, 124.78, 120.53, 114.94, 63.06, 35.02, 28.24, 25.38, 24.83, 20.84, 18.58. Anal. Calcd for C42H58N5Ta: C, 61.98; H, 7.18; N, 8.60. Found: C, 61.76; H, 6.98; N, 8.76. Single crystals for XRD were obtained by cooling an Et2O solution to −25 °C.[TaLMe2]2, 11
A pyrex reaction vessel was charged with a solution of [TaLMe3] (46.7 mg, 0.0716 mmol) in toluene (30 mL) and was sealed with a Teflon screw cap. The bomb was irradiated with a 450 W Ace Glass medium-pressure mercury lamp inside a photochemical reaction cabinet for 3 h. After removal of volatile components, the residue was extracted with pentane and filtered through Celite. Concentration of the filtrate and cooling to −25 °C caused formation of red-orange crystals. The supernatant was decanted and the crystals were washed with cold pentane (3 × 1 mL). Drying in vacuo yielded analytically pure [TaLMe2]2·(pentane)2/3 (9.0 mg, 0.0068 mmol, 19%). 1H NMR (500 MHz, benzene-d6) δ 7.21–7.18 (m, 8H), 7.13 (dd, J = 8.5, 6.7 Hz, 4H), 6.30–6.25 (m, 4H), 5.81–5.75 (m, 4H), 3.66 (sept, J = 6.8 Hz, 8H), 1.19 (d, J = 6.9 Hz, 24H), 1.03 (d, J = 6.7 Hz, 24H), 0.08 (s, 12H). 13C NMR (126 MHz, C6D6) δ 150.06, 145.41, 145.21, 127.51, 124.78, 121.31, 114.73, 49.22, 28.00, 26.24, 24.73. Anal. Calcd for C64H88N4Ta2(C5H12)2/3: C, 61.11; H, 7.31; N, 4.23. Found: C, 61.17; H, 7.29; N, 3.96. Note: the ratio of 11 to pentane was determined by integration of the 1H NMR spectrum. Single crystals for XRD were obtained by cooling a pentane solution to −25 °C.Acknowledgements
We thank NSERC of Canada for funding. T. J. thanks the government of Ontario for an Ontario Graduate Scholarship and the University of Toronto a QEII-GSST scholarship. We thank Digital Specialty Chemicals for providing TaCl5 and PtBu3. Thanks to Dr Timothy Burrow for assistance with EPR. We also acknowledge the Canadian Foundation for Innovation Project #19119, and the Ontario Research Fund for funding the CSICOMP NMR lab at the University of Toronto enabling the purchase of several new spectrometers.References
- G. L. Juvinall, J. Am. Chem. Soc., 1964, 86, 4202–4203 CrossRef CAS.
- R. R. Schrock and P. Meakin, J. Am. Chem. Soc., 1974, 96, 5288–5290 CrossRef CAS.
- R. R. Schrock, J. Organomet. Chem., 1976, 122, 209–225 CrossRef CAS.
- R. R. Schrock and P. R. Sharp, J. Am. Chem. Soc., 1978, 100, 2389–2399 CrossRef CAS.
- M. D. Fryzuk, S. A. Johnson and S. J. Rettig, J. Am. Chem. Soc., 1998, 120, 11024–11025 CrossRef CAS.
- P. Garcia, Y. Y. Lau, M. R. Perry and L. L. Schafer, Angew. Chem., Int. Ed., 2013, 52, 9144–9148 CrossRef CAS PubMed.
- Y. Chen, E. Callens, E. Abou-Hamad, N. Merle, A. J. P. White, M. Taoufik, C. Copéret, E. Le Roux and J. Basset, Angew. Chem., Int. Ed., 2012, 51, 11886–11889 CrossRef CAS PubMed.
- Y. Chen, E. Abou-hamad, A. Hamieh, B. Hamzaoui, L. Emsley and J. Basset, J. Am. Chem. Soc., 2015, 137, 588–591 CrossRef CAS PubMed.
- M. D. Fryzuk, S. A. Johnson, B. O. Patrick, A. Albinati, S. A. Mason and T. F. Koetzle, J. Am. Chem. Soc., 2001, 123, 3960–3973 CrossRef CAS PubMed.
- M. D. Fryzuk, B. A. MacKay, S. A. Johnson and B. O. Patrick, Angew. Chem., Int. Ed., 2002, 41, 3709–3712 CrossRef CAS.
- M. D. Fryzuk, B. A. MacKay and B. O. Patrick, J. Am. Chem. Soc., 2003, 125, 3234–3235 CrossRef CAS PubMed.
- M. P. Shaver, S. A. Johnson and M. D. Fryzuk, Can. J. Chem., 2005, 83, 652–660 CrossRef CAS.
- B. A. MacKay, R. F. Munha and M. D. Fryzuk, J. Am. Chem. Soc., 2006, 128, 9472–9483 CrossRef CAS PubMed.
- B. A. MacKay, B. O. Patrick and M. D. Fryzuk, Organometallics, 2005, 24, 3836–3841 CrossRef CAS.
- J. Ballmann, F. Pick, L. Castro, M. D. Fryzuk and L. Maron, Organometallics, 2012, 31, 8516–8524 CrossRef CAS.
- J. Ballmann, F. Pick, L. Castro, M. D. Fryzuk and L. Maron, Inorg. Chem., 2013, 52, 1685–1687 CrossRef CAS PubMed.
- K. D. J. Parker, D. Nied and M. D. Fryzuk, Organometallics, 2015, 34, 3546–3558 CrossRef CAS.
- J. Ballmann, A. Yeo, B. A. MacKay, S. v. Rijt, B. O. Patrick and M. D. Fryzuk, Chem. Commun., 2010, 46, 8794–8796 RSC.
- J. Ballmann, A. Yeo, B. O. Patrick and M. D. Fryzuk, Angew. Chem., Int. Ed., 2011, 50, 507–510 CrossRef CAS PubMed.
- M. D. Fryzuk, S. A. Johnson and S. J. Rettig, Organometallics, 1999, 18, 4059–4067 CrossRef CAS.
- M. D. Fryzuk, S. A. Johnson and S. J. Rettig, Organometallics, 2000, 19, 3931–3941 CrossRef CAS.
- M. D. Fryzuk, S. A. Johnson and S. J. Rettig, J. Am. Chem. Soc., 2001, 123, 1602–1612 CrossRef CAS PubMed.
- A. I. Nguyen, K. J. Blackmore, S. M. Carter, R. A. Zarkesh and A. F. Heyduk, J. Am. Chem. Soc., 2009, 131, 3307–3316 CrossRef CAS PubMed.
- A. F. Heyduk, R. A. Zarkesh and A. I. Nguyen, Inorg. Chem., 2011, 50, 9849–9863 CrossRef CAS PubMed.
- R. F. Munhá, R. A. Zarkesh and A. F. Heyduk, Inorg. Chem., 2013, 52, 11244–11255 CrossRef PubMed.
- R. R. Schrock, J. Lee, L. Liang and W. M. Davis, Inorg. Chim. Acta, 1998, 270, 353–362 CrossRef CAS.
- J. P. Araujo, D. K. Wicht, P. J. Bonitatebus and R. R. Schrock, Organometallics, 2001, 20, 5682–5689 CrossRef CAS.
- L. P. H. Lopez, R. R. Schrock and P. J. Bonitatebus Jr., Inorg. Chim. Acta, 2006, 359, 4730–4740 CrossRef CAS.
- J. S. Freundlich, R. R. Schrock and W. M. Davis, Organometallics, 1996, 15, 2777–2783 CrossRef CAS.
- J. S. Freundlich, R. R. Schrock and W. M. Davis, J. Am. Chem. Soc., 1996, 118, 3643–3655 CrossRef CAS.
- I. A. Tonks and J. E. Bercaw, Inorg. Chem., 2010, 49, 4648–4656 CrossRef CAS PubMed.
- M. E. G. Skinner, D. A. Cowhig and P. Mountford, Chem. Commun., 2000, 1167–1168 RSC.
- M. E. G. Skinner, T. Toupance, D. A. Cowhig, B. R. Tyrrell and P. Mountford, Organometallics, 2005, 24, 5586–5603 CrossRef CAS.
- F. Guérin, D. H. McConville and J. J. Vittal, Organometallics, 1995, 14, 3154–3156 CrossRef.
- F. Guérin, D. H. McConville, J. J. Vittal and G. A. P. Yap, Organometallics, 1998, 17, 1290–1296 CrossRef.
- L. P. Spencer, C. Beddie, M. B. Hall and M. D. Fryzuk, J. Am. Chem. Soc., 2006, 128, 12531–12543 CrossRef CAS PubMed.
- C. D. Carmichael, M. P. Shaver and M. D. Fryzuk, Can. J. Chem., 2006, 84, 1667–1678 CrossRef CAS.
- R. Fandos, J. Fernández-Gallardo, A. Otero, A. Rodríguez and M. J. Ruiz, Organometallics, 2011, 30, 1551–1557 CrossRef CAS.
- P. Bazinet, G. P. A. Yap and D. S. Richeson, Organometallics, 2001, 20, 4129–4131 CrossRef CAS.
- N. Lavoie, S. I. Gorelsky, Z. Liu, T. J. Burchell, G. P. A. Yap and D. S. Richeson, Inorg. Chem., 2010, 49, 5231–5240 CrossRef CAS PubMed.
- J. M. Decams, S. Daniele, L. Hubert-Pfalzgraf, J. Vaissermann and S. Lecocq, Polyhedron, 2001, 20, 2405–2414 CrossRef CAS.
- S. Feng, G. R. Roof and E. Y. X. Chen, Organometallics, 2002, 21, 832–839 CrossRef CAS.
- T. Chen, C. Xu, T. H. Baum, G. T. Stauf, J. F. Roeder, A. G. DiPasquale and A. L. Rheingold, Chem. Mater., 2010, 22, 27–35 CrossRef CAS.
- K. Mashima, Y. Matsuo and K. Tani, Chem. Lett., 1997, 26, 767–768 CrossRef.
- K. Mashima, Y. Matsuo and K. Tani, Organometallics, 1999, 18, 1471–1481 CrossRef CAS.
- J. Sánchez-Nieves, P. Royo, M. A. Pellinghelli and A. Tiripicchio, Organometallics, 2000, 19, 3161–3169 CrossRef.
- H. Kawaguchi, Y. Yamamoto, K. Asaoka and K. Tatsumi, Organometallics, 1998, 17, 4380–4386 CrossRef CAS.
- H. Tsurugi, T. Ohno, T. Kanayama, R. A. Arteaga-Müller and K. Mashima, Organometallics, 2009, 28, 1950–1960 CrossRef CAS.
- H. Tsurugi, T. Saito, H. Tanahashi, J. Arnold and K. Mashima, J. Am. Chem. Soc., 2011, 133, 18673–18683 CrossRef CAS PubMed.
- K. Aoyagi, P. K. Gantzel and T. D. Tilley, Polyhedron, 1996, 15, 4299–4302 CrossRef CAS.
- G. Jimenez Pindado, M. Thornton-Pett and M. Bochmann, J. Chem. Soc., Dalton Trans., 1998, 393–400 RSC.
- A. Grundmann, M. B. Sárosi, P. Lönnecke, R. Frank and E. Hey-Hawkins, Eur. J. Inorg. Chem., 2013, 3137–3140 CrossRef CAS.
- A. Grundmann, M. B. Sárosi, P. Lönnecke and E. Hey-Hawkins, Eur. J. Inorg. Chem., 2014, 2997–3001 CrossRef CAS.
- V. Tabernero, T. Cuenca, M. E. G. Mosquera and C. R. de Arellano, Polyhedron, 2009, 28, 2545–2554 CrossRef CAS.
- T. Janes, J. M. Rawson and D. Song, Dalton Trans., 2013, 42, 10640–10648 RSC.
- R. H. Crabtree, in The Organometallic Chemistry of the Transition Metals, Wiley, Hoboken, New Jersey, 5th edn, 2009, pp. 122–152 Search PubMed.
- T. Wenderski, K. M. Light, D. Ogrin, S. G. Bott and C. J. Harlan, Tetrahedron Lett., 2004, 45, 6851–6853 CrossRef CAS.
- G. W. A. Fowles, D. A. Rice and J. D. Wilkins, J. Chem. Soc., Dalton Trans., 1973, 961–965 RSC.
- J. D. Wilkins, J. Organomet. Chem., 1974, 80, 349–355 CrossRef CAS.
- R. A. Zarkesh and A. F. Heyduk, Organometallics, 2011, 30, 4890–4898 CrossRef CAS.
- W. J. Evans, J. R. Walensky, J. W. Ziller and A. L. Rheingold, Organometallics, 2009, 28, 3350–3357 CrossRef CAS.
- B. Liu and D. Cui, Dalton Trans., 2009, 550–556 RSC.
- W. J. Evans, T. J. Mueller and J. W. Ziller, J. Am. Chem. Soc., 2009, 131, 2678–2686 CrossRef CAS PubMed.
- E. M. Matson, W. P. Forrest, P. E. Fanwick and S. C. Bart, Organometallics, 2013, 32, 1484–1492 CrossRef CAS.
- S. J. Kraft, P. E. Fanwick and S. C. Bart, Organometallics, 2013, 32, 3279–3285 CrossRef CAS.
- M. Schlosser and J. Hartmann, Angew. Chem., Int. Ed. Engl., 1973, 12, 508–509 CrossRef.
- S. Wiese, M. J. B. Aguila, E. Kogut and T. H. Warren, Organometallics, 2013, 32, 2300–2308 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra and X-ray crystallographic experimental details. CCDC 1479767–1479775. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01908k |
| This journal is © The Royal Society of Chemistry 2016 |