
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarving a 1D CoII-carboranylcarboxylate system by using organic solvents to create stable trinuclear molecular analogues: complete structural and magnetic studies†‡
Mònica
Fontanet
a,
Montserrat
Rodríguez
a,
Xavier
Fontrodona
a,
Isabel
Romero
*a,
Francesc
Teixidor
*b,
Clara
Viñas
b and
Núria
Aliaga-Alcalde
bc
aDepartament de Química and Serveis Tècnics de Recerca, Universitat de Girona, Campus de Montilivi, E-17071 Girona, Spain. E-mail: marisa.romero@udg.edu
bInstitut de Ciencia de Materials de Barcelona, ICMAB-CSIC, Campus UAB, E-08193 Bellaterra, Spain. E-mail: teixidor@icmab.es
cICREA (Institució Catalana de Recerca i Estudis Avançats), Passeig Lluís Companys, 23, 08018, Barcelona, Spain
First published on 2nd June 2016
Abstract
This work presents a straightforward methodology to achieve small linear trinuclear molecules based on the CoII-carboranylcarboxylate system obtained by carving a 1D polynuclear analogous system with the use of diethylether. The reaction of the carboranylcarboxylic ligand, 1-CH3-2-CO2H-1,2-closo-C2B10H10 (LH) with different cobalt salts leads to the polynuclear compound [Co2(μ-H2O)(1-CH3-2-CO2-1,2-closo-C2B10H10)4(THF)4], 1 and the polymeric [Co(μ-H2O)(1-CH3-2-CO2-1,2-closo-C2B10H10)2]n(H2O)n2. This latter 1D chain has been obtained by an unprecedented synthetic strategy for the isolation of cobalt(II) compounds. [Co3(μ-H2O)2(1-CH3-2-CO2-1,2-closo-C2B10H10)6(H2O)2(C4H10O)2], 3 is formed by the dissociation of the polymeric structure that forms 2 when a mild coordinating solvent such as diethylether is added. These compounds have been characterized by analytical and spectroscopic techniques. X-ray analysis of 1 and 3 revealed that 1 presents a dinuclear structure whereas 3 is trinuclear; in both cases a six-coordinated CoII compound with water molecules bridging each of the two CoII centres has been observed. The magnetic properties of 1 and 3 show a weak antiferromagnetic behaviour, respectively, between the CoII centres mediated by two carboxylate ligands and a molecule of water.
Introduction
One major current challenge in materials science and nanotechnology is the association of different properties in the same material which can function either in independent or concerted (synergic) ways.1 In this respect the study of coordination compounds provides effective solutions allowing a clear understanding of their functioning and improving the fundamental and potential applied research; also, these compounds could be used as building blocks in more elaborate structures.2 The presence of a metal–ligand association in metal coordination compounds can lead to the generation of suitable materials with a particular usefulness or applicability; the metal can provide redox and magnetic properties3 among others and the appropriate ligand may determine the functionality of the material. In this sense, coordination polymers are currently of great interest and represent an active area of coordination chemistry because of their special roles in fields such as ion exchange, gas storage, chemical separation, sensor technology, magnets, optoelectronics, energy conversion and storage and catalysis.4 Also, polynuclear metal compounds attract great interest owing to their relevance to many important naturally occurring processes. The cooperative action of closely coupled dinuclear or multinuclear centres is required for several enzymes to carry out their biological functions as for example in the photosystem II.5 The successful behaviour of these structures consists of the use of adequate building blocks that allow the synthesis of 1D linear or twisted chains, 2D squares and polygons and 3D cubes and polyhedra.6The magnetic properties of 1D/polymetallic compounds derive from the cooperative exchange interaction between the paramagnetic metal ions through the bridging ligands. In this aspect the ligand design is crucial to efficiently transmit exhaustive interactions between the metal in a controller manner.7 Concerning organic spacers, carboxylic ligands are frequent choices for metal–organic networks, one reason being their rich modes of coordination.8 With reference to Co compounds containing bridging carboxylates and bridging O atoms (μ-aquo or μ-hydroxo), the structural details of Co core compounds, the nature of the role of coordinating water, the coordinating ligands and the hydrogen bridges are crucial to account for the magnetic properties of polynuclear Co compounds.9 Most of the studies on carboxylate based bi- and trinuclear cobalt metal compounds with bridging water molecules have been performed with trifluoro- or trichloroacetate bridging carboxylates10 and pivalates,11 however the research on new carboxylate Co compounds with different electronic and structural parameters is necessary to develop new systems with potential applications in nanoscience.
Closo boron clusters are tridimensional aromatic compounds electronically related to the conventional Hückel (4n + 2)π (ref. 12) planar aromatic compounds; therefore they are highly stable tridimensional structures very suitable for chemically harsh conditions.13,14 One of the most remarkable examples is [B12H12]2−. This is a dianionic species, highly soluble in water. The substitution of one B− by one carbon lowers the species’ negative charge and hydrophilicity, e.g. [CB11H12]−. If a further B− substitution takes place the charge is further lowered producing a lipophilic species. Therefore the physicochemical properties of structurally very similar borane structures can be tuned according to the physico-chemical needs.15 As is the case with aromatic heterocyclic compounds that are more reactive than their aromatic homocyclic analogs, closo-heteroboranes are more reactive than their closo-homoboranes. Therefore the extensive derivative chemistry of ortho-C2B10H12 is not surprising.16 As a consequence of the electronic polarization originating in the two heteroatoms, in this particular case the adjacent carbon atoms, different substitutions are possible on C and B.17 Therefore the ortho-C2B10H12 offers more possible derivatizations in a small volume than the vast majority of the available chemical scaffoldings; further, it also offers very distinct environments in a very short distance, e.g. one lipophilic environment next to a hydrophilic one. Consequently, using carboranes in supramolecular chemistry is a field to be explored for their particular characteristics13b,18 that shall induce unconventional properties in the supramolecular structures in which they are inserted.19 Boron clusters can form supramolecular polymeric structures not only due to the bridging of polymetallic centres by various non-boron ligands, but also via hydrophobic interactions or halogen or dihydrogen bonding of polyhedral carboranes.20
In earlier studies, we have studied the coordination of the 1-CH3-2-CO2H-1,2-closo-C2B10H10 ligand to CuII and MnII ions together with their physical and chemical properties.21 In the case of MnII, the carboranylcarboxylate ligands lead to highly uncommon water soluble inorganic polymers with water molecules bridging the Mn atoms; in contrast, for CuII the generated compounds do not differ substantially from the related organic based carboxylate ligands. We have also studied the molecular magnetism for these compounds containing carboranylcarboxylate ligands; a strong antiferromagnetic interaction has been observed in CuII compounds while there is a weak antiferromagnetic interaction in the corresponding MnII ones. In this work, we extend these studies to CoII with the aim to learn whether the carboranyl fragment produces more uncommon coordination and nuclearity around the metal centre in the way it did with MnII or, contrastingly the performance is more classical as it is the case for CuII.
With all this in mind, in this work we describe the synthesis of new air-stable di-, tri- and polynuclear CoII compounds using the carboranylcarboxylate ligand 1-CH3-2-CO2H-1,2-closo-C2B10H10 (Chart 1); an exhaustive characterization of the obtained compounds as well as their magnetic properties is also presented in this work.
 | ||
| Chart 1 Drawing of the ligand used in this work. | ||
Results and discussion
Synthesis and structure
The synthetic strategy for the preparation of the CoII compounds, 1–3, containing the carboranylcarboxylate ligand 1-CH3-2-CO2H-1,2-closo-C2B10H10 (LH) and THF, aqua or diethyl ether ligands is outlined in Schemes 1 and 2.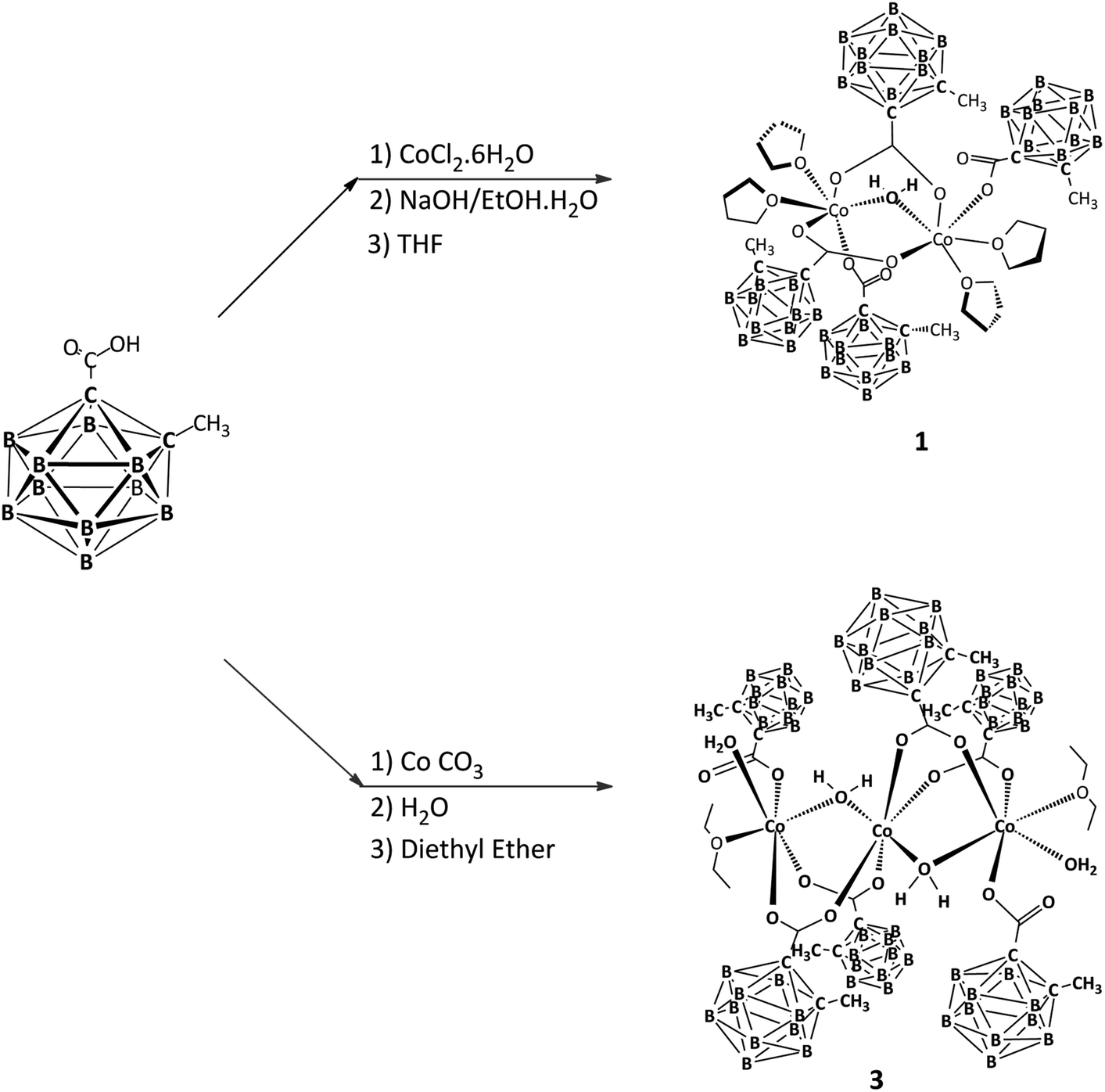 | ||
| Scheme 1 Synthetic strategy for the preparation of Co(II) compounds 1–3. | ||
 | ||
| Scheme 2 Cleavage of 2 by Et2O and formation of the trinuclear complex 3. | ||
The dinuclear cobalt compound [Co2(μ-H2O)(1-CH3-2-CO2-1,2-closo-C2B10H10)4(THF)4], 1, was obtained by neutralization of carboranylcarboxylic acid, LH, with NaOH followed by reaction with CoCl2·6H2O in ethanol/H2O and subsequent extraction in THF. Compound 2 was synthesized by reaction of a suspension of 1-CH3-2-CO2H-1,2-closo-C2B10H10, LH, with CoCO3 in water. The trinuclear compound [Co3(μ-H2O)2(1-CH3-2-CO2-1,2-closo-C2B10H10)6(H2O)2(C4H10O)2], 3, was obtained by recrystallization of 2 in diethyl ether. All attempts to obtain crystals of compound 2 in non-coordinating solvents or water were unsuccessful but its X-ray powder diffraction (XRD) was performed and compared with the calculated XRD for compound 3 (Fig. 1). Fig. 1 shows that both diagrams are different, proving that 2 and 3 display different structures. This led us to tentatively propose that complex 2 presents a polymeric structure (Scheme 2) where each CoII atom is coordinated by four carboxylate oxygen atoms and two aqua oxygen atoms and is bridged to other Co atoms by two carboranylcarboxylate ligands and by an aqua ligand, similar to the polymer manganese compound described by us,21b which displays water molecules bridging every two Mn centres, an unusual feature in 1-D oligomer MnII compounds. In support of this, the elemental analysis of 2 is in agreement with the polymeric structure proposed (see the Experimental section). Compound 2 is broken in coordinating solvents such as diethylether, leading to a linear trinuclear CoII compound 3.
 | ||
| Fig. 1 a) X-ray powder diffraction (XRPD) for 2 and b) calculated XRPD for 3. | ||
The crystal structures of 1 and 3 have been solved by X-ray diffraction analysis. Crystallographic data and selected bond distances and angles for 1 and 3 are presented in Tables 1 and 2, respectively. ORTEP plots with the corresponding atom labels for the X-ray structures of both compounds are presented in Fig. 2.
 | ||
| Fig. 2 X-ray structures (ORTEP plots with ellipsoids at 40% probability level) and the labelling scheme for compounds a) 1 and b) 3. | ||
| 1 | 3 | |
|---|---|---|
| a R 1 = ∑‖Fo − Fc‖/∑Fo. b wR2 = [∑{w(Fo2 − Fc2)2}/∑{w(Fo2)2}]½, where w = 1/[σ2(Fo2) + (0.0042P)2] and P = (Fo2 + 2Fc2)/3. | ||
| Empirical formula | C32H86B40O13Co2 | C48H146B60O22Co3 |
| Formula weight | 1229.27 | 1901.04 |
| Crystal system | Orthorhombic | Triclinic |
| Space group | Pccn |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a [Å] | 12.392(5) | 14.420(9) |
| b [Å] | 22.830(10) | 14.806(10) |
| c [Å] | 23.802(10) | 15.656(10) |
| α [°] | 90 | 63.006(12) |
| β [°] | 90 | 73.533(13) |
| γ [°] | 90 | 67.443(11) |
| V [Å3] | 6734(5) | 2726(3) |
| Formula units/cell | 4 | 1 |
| ρ calc. [g cm−3] | 1.212 | 1.158 |
| μ [mm−1] | 0.542 | 0.506 |
R
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a, [I > 2σ(I)] a, [I > 2σ(I)] |
0.0636 | 0.0979 |
| wR2b [all data] |
0.2183 | 0.2603 |
| 1 | 3 | 3 | |||
|---|---|---|---|---|---|
| Co(1)–O(3) | 2.039(2) | Co(1)–O(1) | 2.099(5) | Co(2)–O(6) | 2.052(4) |
| Co(1)–O(6) | 2.083(2) | Co(1)–O(3) | 2.059(5) | Co(2)–O(4) | 2.072(4) |
| Co(1)–O(10) | 2.0832(17) | Co(1)–O(5) | 2.027(5) | Co(2)–O(8) | 2.134(4) |
| Co(1)–O(5) | 2.092(2) | Co(1)–O(7) | 2.021(5) | Co(2)–O(6)#1 | 2.052(4) |
| Co(1)–O(4) | 2.092(2) | Co(1)–O(8) | 2.217(4) | Co(2)–O(4)#1 | 2.072(4) |
| Co(1)–O(1) | 2.121(2) | Co(1)–O(9) | 2.111(5) | Co(2)–O(8)#1 | 2.134(4) |
| O(3)–Co(1)–O(6) | 90.64(10) | O(7)–Co(1)–O(8) | 88.09(16) | O(4)–Co(2)–O(4)#1 | 180.0(2) |
| O(3)–Co(1)–O(10) | 91.76(8) | O(1)–Co(1)–O(9) | 90.96(19) | O(4)–Co(2)–O(6) | 91.91(19) |
| O(6)–Co(1)–O(10) | 176.61(9) | O(3)–Co(1)–O(9) | 89.45(19) | O(4)#1–Co(2)–O(6) | 88.09(19) |
| O(3)–Co(1)–O(5) | 178.25(10) | O(5)–Co(1)–O(9) | 88.78(18) | O(4)–Co(2)–O(6)#1 | 88.09(19) |
| O(6)–Co(1)–O(5) | 88.23(10) | O(7)–Co(1)–O(9) | 92.40(19) | O(4)#1–Co(2)–O(6)#1 | 91.91(19) |
| O(10)–Co(1)–O(5) | 89.42(8) | O(8)–Co(1)–O(9) | 178.67(19) | O(6)–Co(2)–O(6)#1 | 180.0(1) |
| O(3)–Co(1)–O(4) | 90.10(10) | O(1)–Co(1)–O(3) | 175.49(19) | O(4)–Co(2)–O(8)#1 | 90.43(17) |
| O(6)–Co(1)–O(4) | 89.01(9) | O(1)–Co(1)–O(5) | 92.1(2) | O(4)#1–Co(2)–O(8)#1 | 89.57(17) |
| O(10)–Co(1)–O(4) | 93.39(8) | O(3)–Co(1)–O(5) | 92.42(19) | O(6)–Co(2)–O(8)#1 | 84.84(16) |
| O(5)–Co(1)–O(4) | 88.54(9) | O(1)–Co(1)–O(7) | 87.08(19) | O(6)#1–Co(2)–O(8)#1 | 95.16(16) |
| O(3)–Co(1)–O(1) | 90.37(10) | O(3)–Co(1)–O(7) | 88.4(2) | O(4)–Co(2)–O(8) | 89.57(17) |
| O(6)–Co(1)–O(1) | 88.95(9) | O(5)–Co(1)–O(7) | 178.56(18) | O(4)#1–Co(2)–O(8) | 90.43(17) |
| O(10)–Co(1)–O(1) | 88.63(8) | O(1)–Co(1)–O(8) | 87.83(16) | O(6)–Co(2)–O(8) | 95.16(16) |
| O(5)–Co(1)–O(1) | 90.95(9) | O(3)–Co(1)–O(8) | 91.79(16) | O(6)#1–Co(2)–O(8) | 84.84(16) |
| O(4)–Co(1)–O(1) | 177.91(8) | O(5)–Co(1)–O(8) | 90.71(16) | O(8)#1–Co(2)–O(8) | 180.0(1) |
Dinuclear compound 1 displays a structure with the two cobalt(II) atoms holding together through two syn,syn η1:η1:μ2-carboxylate bridges and one bridged aqua molecule as shown in Fig. 2. The distorted octahedral geometry around each metal ion is completed by one additional monodentate carboranylcarboxylate ligand and two oxygen donor atoms from two THF ligands leading to a local Co(OTHF)2(Ocarboxy)3(OH2O) coordination. Although there are many reported examples with a CoO6 environment10b,22 in which the metal ion is CoII, compound 1 is the first one that contains the carboranyl fragment with structural and electronic parameters different from those previously reported in the literature. The molecule is located in a crystallographically imposed two-fold axis which passes through the oxygen O(10) belonging to the μ2-bridged aqua molecule. The hydrogen atoms of the bridged aqua molecule are involved in intramolecular O(10)–H⋯O–C hydrogen bonds with the uncoordinated O atoms of the monodentate carboranylcarboxylate ligands (O(10)–H⋯O(2)–C(10), 1.607 Å) (see Fig. 3). The bond length Co(1)–O(10)w distance is 2.083(17) Å while the corresponding Co(1)–O(10)–Co(1)#1 angle is 117.82(14)°. This bond distance is shorter than the distance observed for similar carboxylate compounds containing pyridine as the terminal ligand in spite of THF.22c–e The Co–OTHF bond length corresponding to the THF molecule in the trans position to the bridged aqua oxygen atom is significantly shorter (2.083 Å) than the same Co–OTHF distance for the THF molecule trans to the bridged carboranylcarboxylate oxygen atom (2.092 Å). The structural packing of this compound (Fig. 4a) along the c axis shows the formation of channels in which the carboranylcarboxylate ligands are orientated to these channels and opposite to the water bridged molecules, leading to the existence of a hydrophobic environment in these cavities. Interactions B–H⋯H–B of 2.284 Å between neighbouring molecules and B–H⋯H–CTHF (2.3–2.4 Å) between carboxylate ligands and THF molecules along the channels have been observed.
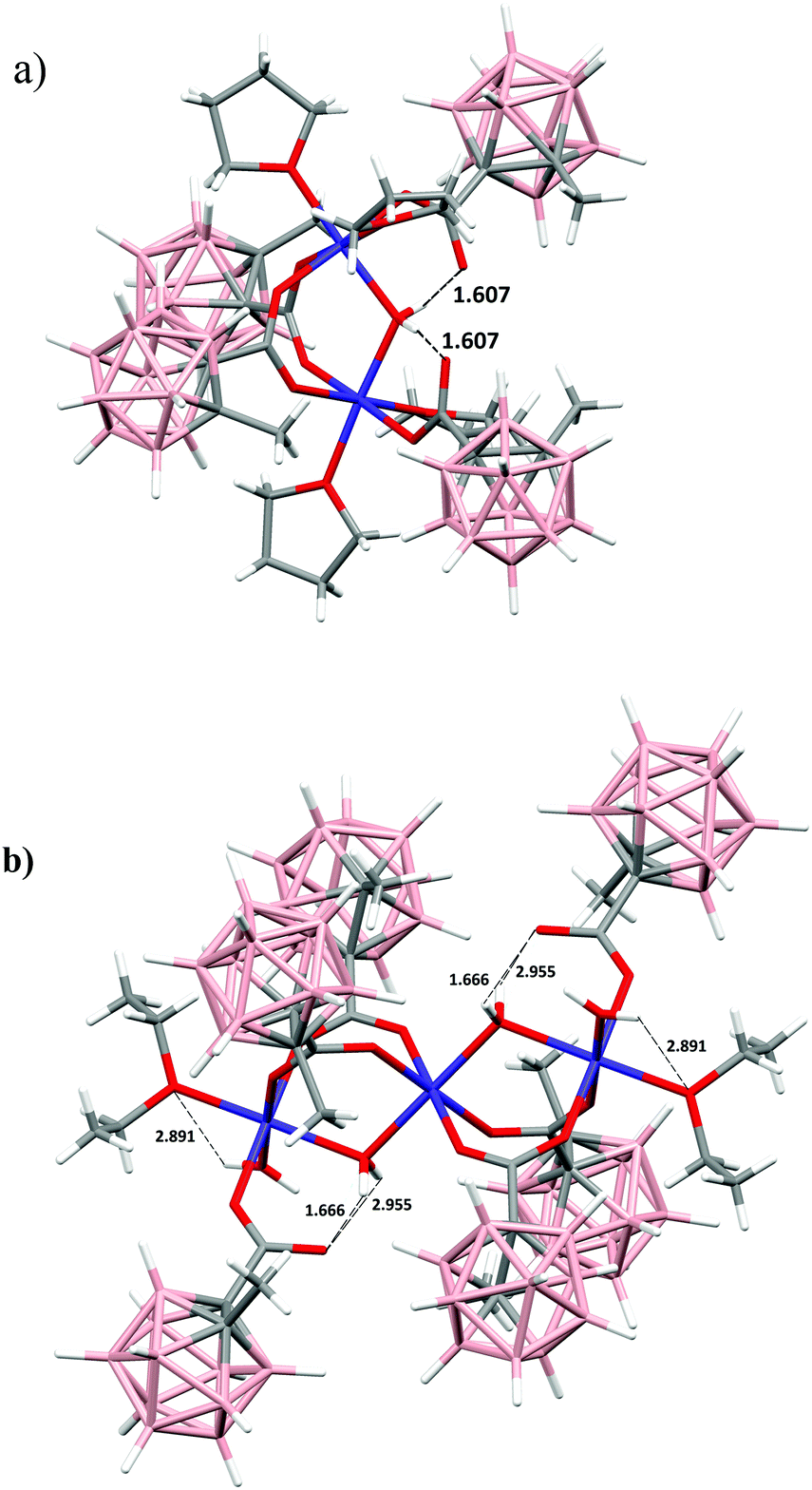 | ||
| Fig. 3 X-ray structures showing the intramolecular hydrogen bonds, a) for 1 and b) for 3. | ||
 | ||
| Fig. 4 Packing diagram of compounds a) 1 along the c axis b) 3 along the c axis. | ||
The X-ray diffraction of 3 discloses a trinuclear cobalt compound with aqua bridged entities that is made up of a linear array of three hexacoordinated CoII ions with a crystallographic inversion centre located on the central Co(2). This central Co(2) is coordinated by four carboxylate and two aqua oxygen atoms and is bonded to both terminal Co(1) ions by two carboranylcarboxylate ligands and by an aqua ligand; the distorted octahedral geometry of the central Co atom is probably similar to the one that is most probably presented by CoII atoms in the polymeric structure of 2. The two outer Co atoms fulfill their hexacoordination with one oxygen atom from one diethyl ether ligand, one terminal H2O ligand, and one monodentate carboranylcarboxylate ligand that probably originally could be shared with another CoII atom in the polymer structure (Scheme 2).
Cobalt centers display a tetragonal distortion that means that the average of the equatorial Co–O distances for terminal Cobalt ions is 2.06 Å and the average of the axial distances is 2.13 Å. The distances and angles observed in this compound are similar and comparable to other similar trinuclear compounds described in the literature most of which form in part either linear chains23a or coordination network systems.23c,d It is worth mentioning that our structure is stabilized by terminal ligands as diethyl ether, which is never observed in cobalt trimer compounds containing carboxylate bridged ligands. It is well known that carboranes induce unconventional properties in the structures in which they are inserted.19 This effect is caused by their volume and their rigid geometry that provide a higher atomic efficiency if compared to aromatic phenyl ligands. These steric properties together with the higher electron-withdrawing character of the carboranylcarboxylate cluster stabilize the unique carboranyl structures presented in this paper. A similar situation could occur in the case of compound 1 where two terminal THF ligands are present around each cobalt atom. The existence of two intramolecular hydrogen bonds between the uncoordinated oxygen atom of the monodentate carboxylate (O(2)) and H8A and H8B of the bridged aqua (H(8A)–O(2), 1.666 Å; H(8B)–O(2), 2.955 Å) is also noticeable. Another intramolecular weak hydrogen bond is observed between H7B of the terminal aqua ligand and O(9) of the terminal diethyl ether molecule, H(9B)–O(7), 2.891 Å (Fig. 3b).
The packing arrangement of compound 3 along the c axis shows parallel lineal chains where the terminal diethyl ether ligands of adjacent molecules from the same chain are facing each other, Fig. 4b.
Spectroscopic properties
The IR spectra of the compounds described display typical ν(B–H) absorption at frequencies above 2590 cm−1 (Fig. S1‡), characteristic of closo carborane derivatives.24 Differences between the frequencies of the symmetric and antisymmetric stretches of the carboxylate ligands lie within the ranges quoted for bidentate bridged ligands.24c The 1H{11B}-, 11B-, 11B{1H }- and 13C{1H}-NMR spectra of these compounds were recorded and are presented in the ESI (Fig. S2‡). It can be observed that the 1H{11B}- spectra exhibit resonances around δ = 2 ppm attributed to the Cc–CH3 protons and the resonances of the protons bonded to the B atoms appear as broad singlets over a wide chemical-shift range in the region from δ = 0 to +5 ppm. The 11B{1H}-NMR resonances for all the compounds featured similar patterns in the range from δ = 2 to −15 ppm that agrees with a closo cluster.25The electronic UV-vis spectra of compounds 1 and 2 are shown in Fig. S3‡ and display two characteristic bands, one in the range 300–400 nm and the other in the range 420–570 nm. The pink binuclear compound 1 revealed two bands at 353 nm and 534 nm, whereas that the spectrum for the solid 2 presents one band at 510 nm, and two shoulders at 356 and 472 nm. The higher energy bands observed in these compounds can be assigned to ligand to metal charge transfer (LMCT) transitions. Absorptions in the visible region at 534 nm for 1 and at 472 and 510 nm for 3 are assigned to the two spin-allowed d–d transitions 4T1g(F) → 4A2g(F) and 4T1g(F) → 4T1g(P).22b,e,26
Magnetic properties
Magnetic structural correlations were performed for compounds 1 and 3, where in both cases the cobalt centres are linked to each other through [1-CH3-2-CO2-1,2-closo-C2B10H10]− ligands and a single molecule of H2O. The two compounds contain hexacoordinated CoII centres with distorted octahedral geometries and in both (1, 3) the bulky nature of the ligand prevents intermolecular interactions among neighbouring molecules (distances CoII⋯CoII above 9 Å). Altogether, 3 can be seen as the extension of 1 by the addition of one extra CoII centre and therefore, it can be predicted that magnetically both systems will behave in a similar manner. Variable-temperature magnetic susceptibility measurements for 1 and 3 were performed in the 2–300 K temperature range applying a 0.5 T dc field. Fig. 5 shows together the two plots of χMT vs. T for 1 and 3. The χMT values at 300 K for compounds 1 and 3 are 7.26 and 13.26 cm3 mol−1 K, respectively. Both values are much higher than the spin-only value expected for non-interacting S = 3/2 centres (3.75 cm3 mol−1 K and 5.62 cm3 mol−1 K, in that order, when g = 2). This fact is a clear indication of the significant orbital contribution of the metallic ions, well-known for high-spin octahedral CoII (HS Oh CoII) centres.27 The continuous decrease observed in χMT for 1 and 3 with decreasing temperature may be due to weak antiferromagnetic interactions and/or to the depopulation of spin–orbit split levels arising from the 4T1g state in the Oh CoII centres (the 4T1g term in HS Oh CoII ions splits into a sextet: a quartet and a Kramers’ doublet by spin–orbit coupling).28 At 2 K, the χMT of 1 arrives at a value of 2.41 cm3 mol−1 K whereas the χMT of 3 reaches a value of 4.10 cm3 mol−1 K at the same temperature. At such temperatures, only the Kramers’ doublets are populated and each CoII centre could be considered to have an effective spin S′ = 1/2. The M/Nμβ values at 2 K and 5 T are of 4.7, and 7.7 for compounds 1 and 3, respectively, in the range of what should be expected for two and three S′ = ½ systems with relevant spin orbital contributions (Fig. S4‡). | ||
| Fig. 5 Experimental data including χMT vs. T of 1 (black squares) and 3 (black triangles) between 2.0 and 300.0 K. The red and blue lines correspond to the theoretical values from the fitting, also in that order. | ||
M vs. H/T data were also recorded at different applied external fields (0.5–5 T) in the 1.8–6.8 K temperature range (Fig. S5‡). The magnetization sharply increases upon cooling the sample to 1.8 K and sweeping field up to 5 T, for the two systems, and clear saturation was observed in the reduced magnetization plots of both (1–3) at the highest achievable field and lowest temperature with values of 4.71 and 7.30Nμβ, respectively. No superposition of the isofield lines was observed, suggesting the existence of relevant anisotropy.29
Our goal in the present work was the estimation of the J parameter using the whole temperature range. This has only been possible in recent years through sophisticated computer programs due to the magnetic complexity of cobalt species.30 Here, the data were fitted by diagonalization of the spin Hamiltonian matrix, using the program PHI,31 which allows the correlation of experimental magnetic data of orbitally degenerate systems using multiple sources; in this case, χMT vs. T data together with M vs. H/T results were used simultaneously. This way, the χMT vs. T and M vs. H/T data fittings in the whole 2–300 K range (Fig. 1) afforded the parameters: J = −0.30 cm−1, g = 2.18, TIP = 490 × 10−6 cm3 mol−1 and ρ = 0.6% for 1 and J1 = J2 = −0.40 cm−1, g = 2.40, TIP = 633 × 10−6 cm3 mol−1 and ρ = 0.5% for 3. As Fig. 5 depicts, a more reliable fitting was obtained in the case of 3, thus the final data achieved for 1 should be taken as illustrative although, due to the structural similarities among the two compounds the resulting magnetic parameters for the latter may be close to reality.
As mentioned above, magnetic measurements for bridged CoII compounds and more precisely, for μ-OH2, μ-O2CR dinuclear/trinuclear CoII systems, are scarce in the literature. However, some worth noting publications have appeared in the last few years.10a,32 Dinuclear CoII systems, described with a molecule of H2O and two carboxylates as bridging ligands between the two metallic centres, are more abundant than trinuclear species. Magnetically, the characterization in former times was mostly based on the oxidation state and description of the nature of the exchange (always antiferro) but, excellent studies have already provided exchange parameter values between −1 and −3 cm−1.10b,22b,33 In the case of Co3 species, only two systems have been well-characterized magnetically: Calvo-Pérez et al.10a performed exhaustive analyses of a system related to 3, finding that the J value was −0.4 cm−1 and similar values where described in additional Co3 systems by some of the same authors some years later.32 The comparison of the data accomplished in this work and those previously published allows some preliminary information. This way, our results confirm what has been described in the past: structures containing strictly two carboxylate ligands and one H2O as bridging units among CoII centres provide small and negative exchange couplings (related to the syn–syn conformation of the carboxylate ligands and the existence of one molecule of H2O as a linker). On the other hand, even though the range of J values is very small (from −0.4 to −3 cm−1) and the number of systems is not that extensive, it is worth mentioning that the lowest values (−0.4 cm−1) have been accomplished with carboxylates of an electron-withdrawing nature (–O2CCF3)10a and ours, the carboranylcarboxylate ligand [1-CH3-2-CO2-1,2-closo-C2B10H10]−. In the past, we observed similar effects regarding this feature in dinuclear paddle-wheel CuII structures.21a Summarizing, in this work, compounds 1 and 3 exhibit weak antiferromagnetic behaviours among the CoII centres as it has been reported for analogous systems of MnII published in the past by the group in ref. 21c.
Conclusions
An unprecedented strategy for the synthesis of a 1D CoII carboranylcarboxylate polymer together with a straightforward methodology to achieve trinuclear molecules based on this 1D system has been presented in this work. The reaction of the carboranylcarboxylate ligand, 1-CH3-2-CO2H-1,2-closo-C2B10H10 (LH) with CoCO3 in water leads easily to the isolation of the polymer. Its crystallization in slightly coordinating solvents, such as diethylether, leads to the trinuclear compound with diethyl ether acting as the ancillary ligand on the two terminal CoII ions. This structure containing diethyl ether as terminal ligands has never been observed in cobalt trimer compounds containing carboxylate bridged ligands; this effect is caused by the unconventional properties of the carboranyl ligand such as its volume, and rigid geometry together with its electron-withdrawing character that stabilizes the unique structure presented in this paper. This crystal shows the inner Co⋯Co pairs with the bridging water unit and the two bridging carboxylate ligands while the terminal Co centres contain a monodentate carboranylcarboxylate ligand, one terminal water molecule and one terminal fragmenting solvent, Et2O, evidencing their origin as a result of polymer fragmentation. A new dinuclear structure has also been presented in this work, where Co atoms held together through two carboxylate bridges and one bridged aqua molecule and one additional monodentate carboranylcarboxylate ligand and two oxygen donor atoms from two THF ligands complete the octahedral geometry. Interestingly, the packing of this compound shows the formation of hydrophobic cavities due to the orientation of the carboranylcarboxylate ligands in the crystal. Magnetic measurements of polynuclear compounds were carried out, and showed, in all cases, weak antiferromagnetic interactions between the cobalt atoms. This methodology to achieve small molecules by carving 1D CoII carboranylcarboxylate systems with organic solvents could be an important challenge in the development of new molecular building blocks with different ancillary solvent ligands. These molecules tend to impart a dynamic behaviour to the potential resultant coordination networks and consequently improve their properties. More studies in this sense are being developed in our laboratory.Experimental section
Materials
All reagents used in the present work were obtained from Aldrich Chemical Co and were used without further purification. Reagent grade organic solvents were obtained from SDS and high purity de-ionized water was obtained by passing distilled water through a nano-pure Mili-Q water purification system. 1-CH3-1,2-closo-C2B10H11 was purchased from Katchem.Instrumentation and measurements
FT-IR spectra were recorded on a Mattson-Galaxy Satellite FT-IR spectrophotometer containing a MKII Golden Gate Single Reflection ATR System. Elemental analyses were performed using a CHNS-O Elemental Analyser EA-1108 from Fisons. UV-Vis spectroscopy was performed on a Cary 50 Scan (Varian) UV-Vis spectrophotometer with 1 cm quartz cells or with an immersion probe of 5 mm path length. NMR spectra have been recorded with a Bruker ARX 300 or a DPX 400 instrument equipped with the appropriate decoupling accessories. 1H and 1H{11B} NMR (300.13/400.13 MHz), 13C{1H} NMR (75.47/100.62 MHz) and 11B and 11B{1H} NMR (96.29/128.37 MHz) spectra were recorded in d6-acetone and D2O. Chemical shift values for 11B NMR spectra were referenced to external BF3 ← OEt2 and those for 1H, 1H{11B} and 13C{1H} NMR spectra were referenced to SiMe4. Chemical shifts are reported in units of parts per million downfield from the reference, and all coupling constants are in Hz. The deconvolution of 11B{1H} spectra has been performed with the software OriginPro 8 SR0, v. 8.0724.X-ray structure determination
Measurement of the crystals was performed on a Bruker Smart Apex CCD diffractometer using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) from an X-ray tube. Data collection, Smart V. 5.631 (BrukerAXS 1997–02); data reduction, Saint+ Version 6.36A (Bruker AXS 2001); absorption correction, SADABS version 2.10 (Bruker AXS 2001) and structure solution and refinement, SHELXL-2013 (Sheldrick, 2013). For structure 3, two disordered ethyl ether solvent molecules per asymmetric unit were removed using the SQUEEZE option in PLATON.34 The crystallographic data as well as details of the structure solution and refinement procedures are reported in Table 1. CCDC 1471844 (1) and 1471845 (3) contain the supplementary crystallographic data for this paper.Magnetic susceptibility studies
Magnetic susceptibility measurements were carried out on polycrystalline samples with a DSM5 Quantum Design susceptometer working in the range 2–300 K under a magnetic field of 0.5 T. TIP and ρ stand for the temperature-independent paramagnetism and impurities (in %), respectively. Diamagnetic corrections were estimated from Pascal Tables.Preparations
1-CH3-2-CO2H-1,2-closo-C2B10H10 was prepared according to the literature procedures.35 The synthetic manipulations of cobalt compounds were routinely performed under ambient conditions.Acknowledgements
This research has been financed by MINECO of Spain and Generalitat de Catalunya through projects, (2014-SGR-149) and Spanish Ministry of Economy and Competitiveness (CTQ2010-21532-CO2-01 and CTQ2013-44670-R). MF thanks Generalitat de Catalunya for a FI predoctoral grant. Serveis Tècnics de Recerca (STR) from UdG is also acknowledged for technical support. Dr N. A.-A. thanks the Spanish MCI (MAT2013-47869-C4-2-P). CV thanks the COST CM 1302 project.References
- M.-C. Dul, E. Pardo, R. Lescouëzec, Y. Journaux, J. Ferrando-Soria, R. Ruiz-García, J. Cano, M. Julve, F. Lloret, D. Cangussu, C. L. M. Pereira, H. O. Stumpf, J. Pasán and C. Ruiz-Pérez, Coord. Chem. Rev., 2010, 254, 2281–2296 CrossRef CAS.
- (a) K. S. Pedersen, J. Bendix and R. Clérac, Chem. Commun., 2014, 50, 4396–4415 RSC; (b) T. E. O. Screen, J. R. G. Thorne, R. G. Denning, D. G. Bucknall and H. L. Anderson, J. Mater. Chem., 2003, 13, 2796–2808 RSC.
- (a) M. Verdaguer and V. Robert, Comprehensive Inorganic Chemistry II: From Elements to Applications, Elsevier, Amsterdam, 2013, vol. 8, pp. 131–189 Search PubMed; (b) D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, Oxford, 2006 Search PubMed; (c) I.-R. Jeon and R. Clérac, Dalton Trans., 2012, 41, 9569–9586 RSC.
- (a) M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 3(4), 319–330 CrossRef; (b) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS PubMed; (c) B. Moulton and M. J. Zaworotko, Curr. Opin. Solid State Mater. Sci., 2002, 6, 117–123 CrossRef CAS; (d) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed; (e) A. K. Cheetham, C. N. R. Rao and R. K. Feller, Chem. Commun., 2006, 4780–4795 RSC; (f) U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastre, J. Mater. Chem., 2006, 16, 626–636 RSC; (g) M. Andruh, Chem. Commun., 2007, 2565–2577 RSC; (h) B. Wang, A. P. Cote, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Nature, 2008, 453, 207–211 CrossRef CAS PubMed.
- (a) S. Mukhopadhyay, S. K. Mandal, S. Bhaduri and W. H. Armstrong, Chem. Rev., 2004, 104, 3981–4026 CrossRef CAS PubMed; (b) D. C. Weatherburn, S. Mandal, S. Mukhopadhyay, S. Bahduri and L. F. Lindoy, Manganese, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier Pergamon, Oxford, 2004, vol. 5, p. 1 Search PubMed; (c) A. J. Wu, J. E. Penner-Hahn and V. L. Pecoraro, Chem. Rev., 2004, 104, 903–938 CrossRef CAS PubMed; (d) C. S. Mullins and V. L. Pecoraro, Coord. Chem. Rev., 2008, 252, 416–443 CrossRef CAS PubMed; (e) M.-N. Collomb and A. Deronzier, Eur. J. Inorg. Chem., 2009, 2025–2046 CrossRef CAS.
- (a) M. Fujita, J. Yazaki and K. Ogura, J. Am. Chem. Soc., 1990, 112, 5645–5647 CrossRef CAS; (b) G. F. Swiegers and T. J. Malefetse, Coord. Chem. Rev., 2002, 225, 91–121 CrossRef CAS; (c) Z. He, C. He, Z.-M. Wang, E. Q. Gao, Y. Liu and C.-H. Yan, Dalton Trans., 2004, 502–504 RSC; (d) C. Janiak, Dalton Trans., 2003, 2781–2804 RSC; (e) R. Chakrabarty, P. S. Mukherjee and P. Stang, Chem. Rev., 2011, 111`, 6810–6918 CrossRef PubMed.
- E. Pardo, R. Ruiz-García, J. Cano, X. Ottenwaelder, R. Lescouëzec, Y. Journaux, F. Lloret and M. Julve, Dalton Trans., 2008, 2780–2805 RSC.
- (a) X. Lin, J. Jia, P. Hubberstey, M. Schroeder and N. R. Champness, CrystEngComm, 2007, 9, 438–448 RSC; (b) C. L. Cahill, D. T. de Lill and M. Frisch, CrystEngComm, 2007, 9, 15–26 RSC.
- U. Turpeinen, R. Hämäläinen and J. Reedijk, Polyhedron, 1987, 6, 1603–1610 CrossRef CAS.
- (a) V. Calvo-Pérez, S. Ostrovsky, A. Vega, J. Pelikan, E. Spodine and W. Haase, Inorg. Chem., 2006, 45, 644–649 CrossRef PubMed; (b) Z. Tomkowicz, S. Ostrovsky, S. Foro, V. Calvo-Perez and W. Haase, Inorg. Chem., 2012, 51, 6046–6055 CrossRef CAS PubMed.
- G. Aromí, A. S. Batsanov, P. Christian, M. Helliwell, A. Parkin, S. Parsons, A. A. Smith, G. A. Timco and R. E. P. Winpenny, Chem. – Eur. J., 2003, 9, 5142–5161 CrossRef PubMed.
- (a) J. Poater, M. Solà, C. Viñas and F. Teixidor, Chem. – Eur. J., 2013, 19, 4169–4175 CrossRef CAS PubMed; (b) J. Poater, M. Solà, C. Viñas and F. Teixidor, Angew. Chem., Int. Ed., 2014, 53, 12191–12195 CrossRef CAS PubMed.
- (a) M. F. Hawthorne, Advances in Boron Chemistry, the Royal Society of Chemistry, Cornwall, U.K., 1997, pp. 261–272 Search PubMed; (b) R. N. Grimes, Carboranes, Academic Press, Burlington, MA, 2nd edn, 2011 Search PubMed.
- (a) H. Kimura, K. Okita, M. Ichitani, T. Sugimoto, S. Kuroki and I. Ando, Chem. Mater., 2003, 15, 355–362 CrossRef CAS; (b) M. K. Kolel-Veetil and T. M. Keller, J. Polym. Sci., 2006, 44, 147–155 CrossRef CAS; (c) A. González-Campo, B. Boury, F. Teixidor and R. Núñez, Chem. Mater., 2006, 18, 4344–4353 CrossRef; (d) E. Hao, B. Fabre, F. R. Fronczek and M. G. H. Vicente, Chem. Mater., 2007, 19, 6195–6205 CrossRef CAS; (e) O. K. Farha, A. M. Spokoyny, K. L. Mulfort, M. F. Hawthorne, C. A. Mirkin and J. T. Hupp, J. Am. Chem. Soc., 2007, 129, 12680–12681 CrossRef CAS PubMed; (f) A. M. Spokoyny, O. K. Farha, K. L. Mulfort, J. T. Hupp and C. A. Mirkin, Inorg. Chem. Acta, 2010, 364, 266–271 CrossRef CAS.
- (a) C. Masalles, S. Borros, C. Viñas and F. Teixidor, Adv. Mater., 2002, 14, 449–452 CrossRef CAS; (b) S.-Y. Lu and I. Hamerton, Prog. Polym. Sci., 2002, 27, 1661–1712 CrossRef CAS; (c) M. Nieuwenhuyzen, K. R. Seddon, F. Teixidor, A. V. Puga and C. Viñas, Inorg. Chem., 2009, 48, 889–901 CrossRef CAS PubMed; (d) J. Mola, E. Mas-Marza, X. Sala, I. Romero, M. Rodríguez, C. Viñas, T. Parella and A. Llobet, Angew. Chem., Int. Ed., 2008, 47, 5830–5832 CrossRef CAS PubMed; (e) K. Kokado and Y. Chujo, J. Org. Chem., 2011, 76, 316–319 CrossRef CAS PubMed; (f) K. Kokado and K. Y. Chujo, Macromolecules, 2009, 42, 1418–1420 CrossRef CAS.
- (a) F. Teixidor and C. Viñas, Sci. Synth., 2005, 6, 1235 Search PubMed; (b) V. I. Bregadze, Chem. Rev., 1992, 92, 209–223 CrossRef CAS.
- (a) F. Teixidor, R. Núñez, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., Int. Ed., 2000, 39, 4290–4292 CrossRef CAS; (b) R. Núñez, P. Farrás, F. Teixidor, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., Int. Ed., 2006, 45, 1270–1272 CrossRef PubMed; (c) F. Teixidor, G. Barbera, A. Vaca, R. Kivekäs and R. Sillanpää, J. Am. Chem. Soc., 2005, 127, 10158–10159 CrossRef CAS PubMed.
- (a) N. S. Hosmane, Boron Science: New Technologies and applications, CRC Press, Taylor & Francis Group, New York, 2012 Search PubMed; (b) A. Olah, G. K. S. Prakash, K. Wade, A. Molnar and R. E. Williams, Hypercarbon Chemistry, Wiley, New York, 2nd edn, 2011 Search PubMed.
- (a) M. F. Hawthorne, Angew. Chem., Int. Ed., 1993, 32, 950–984 CrossRef; (b) A. H. Soloway, W. Tjarks, B. A. Barnum, F.-G. Rong, R. F. Barth, I. M. Codogni and J. G. Wilson, Chem. Rev., 1998, 98, 1515–1562 CrossRef CAS PubMed; (c) J. F. Valliant, K. J. Guenther, A. S. King, P. Morel, P. Schaffer, O. O. Sogbein and K. A. Stephenson, Coord. Chem. Rev., 2002, 232, 173–230 CrossRef CAS; (d) I. B. Sivaev, V. Bregadze and S. Sjöberg, in Research and Development in Neutron Capture Therapy, ed. W. Sauerwein, R. Moss and A. Wittig, Monduzzi Editore S.p.A., Bologna, 2002, pp. 19–23 Search PubMed; (e) M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7065 CrossRef CAS PubMed; (f) R. Julius, O. Farha, J. Chiang, L. Perry and M. F. Hawthorne, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4808–4813 CrossRef CAS PubMed.
- (a) A. V. Puga, F. Teixidor, R. Sillanpää, R. Kivekäs and C. Viñas, Chem. – Eur. J., 2009, 15, 9764–9772 CrossRef CAS PubMed; (b) G. Barberà, C. Viñas, F. Teixidor, G. M. Rosair and A. J. Welch, J. Chem. Soc. Dalton Trans., 2002, 3647–3648 RSC; (c) H. Lee, C. B. Knobler and M. F. Hawthorne, Chem. Commun., 2000, 24, 2485–2486 RSC; (d) J. G. Planas, C. Vinas, F. Teixidor, A. Comas-Vives, G. Ujaque, A. Lledos, M. E. Light and M. B. Hursthouse, J. Am. Chem. Soc., 2005, 127, 15976–15982 CrossRef CAS PubMed; (e) M. J. Hardie, J. Chem. Crystallogr., 2007, 37, 69–80 CrossRef CAS; (f) F. Di Salvo, B. Camarago, Y. Garcia, F. Teixidor, C. Vinas, J. G. Planas, M. E. Light and M. B. Hursthouse, CrystEngComm, 2011, 13, 5788–5806 RSC; (g) O. A. Filippov, N. V. Belkova, L. M. Epstein and E. S. Shubina, J. Organomet. Chem., 2013, 747, 30–42 CrossRef CAS; (h) M. A. Fox and A. K. Hughes, Coord. Chem. Rev., 2004, 248, 457–476 CrossRef CAS.
- (a) M. Fontanet, A. R. Popescu, X. Fontrodona, M. Rodriguez, I. Romero, F. Teixidor, C. Viñas, E. Ruiz and N. Aliaga-Alcalde, Chem. – Eur. J., 2011, 17, 13217–13229 CrossRef CAS PubMed; (b) M. Fontanet, M. Rodriguez, I. Romero, X. Fontrodona, F. Teixidor, C. Viñas, N. Aliaga-Alcalde and P. Matějíček, Dalton Trans., 2013, 42, 7838–7841 RSC; (c) M. Fontanet, M. Rodriguez, X. Fontrodona, I. Romero, F. Teixidor, C. Viñas, N. Aliaga-Alcalde and P. Matějíček, Chem. – Eur. J., 2014, 20, 13993–14003 CrossRef CAS PubMed.
- (a) D. Lee, P.-L. Hung, B. Spingler and S. J. Lippard, Inorg. Chem., 2002, 41, 521–531 CrossRef CAS PubMed; (b) D. A. Brown, W. K. Glass, N. J. Fitzpatrick, T. J. Kemp, W. Errington, G. J. Clarkson, W. Haase, F. Karsten and A. H. Mahdy, Inorg. Chim. Acta, 2004, 357, 1411–1436 CrossRef CAS; (c) T. J. Prior and J. C. Burley, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m1422 Search PubMed; (d) A. Karmakar, R. J. Sarma and J. B. Baruah, Eur. J. Inorg. Chem., 2007, 5, 643–647 CrossRef; (e) A. Karmakar, R. J. Sarma and J. B. Baruah, Polyhedron, 2007, 26, 1347–1355 CrossRef CAS.
- (a) V. Calvo-Pérez, S. Ostrovsky, A. Vega, J. Pelikan, E. Spodine and W. Haase, Inorg. Chem., 2006, 45, 644–649 CrossRef PubMed; (b) P. Nockemann, B. Thijs, K. V. Hecke, L. V. Meervelt and K. Binnemans, Cryst. Growth Des., 2008, 8, 1353–1363 CrossRef CAS; (c) Z. Hulvey, D. S. Wragg, Z. Lin, R. E. Morris and A. K. Cheetham, Dalton Trans., 2009, 1131–1135 RSC; (d) X.-P. Zhou, Z. Xu, M. Zeller, A. D. Hunter, S. S.-Y. Chui and C.-M. Che, Inorg. Chem., 2011, 50, 7142–7149 CrossRef CAS PubMed.
- (a) L. A. Leites, Chem. Rev., 1992, 92, 279–323 CrossRef CAS; (b) A. Laromaine, F. Teixidor, R. Kivekäs, R. Sillanpää, M. Arca, V. Lippolis, E. Crespo and C. Viñas, Dalton Trans., 2006, 5240–5247 RSC; (c) R. C. Mehrotra and R. Bohra, Metal Carboxylates, Academic, New York, 1983, p. 396 Search PubMed.
- L. J. Todd and A. R. Siedle, Prog. Nucl. Magn. Reson. Spectrosc., 1979, 13, 87–176 CrossRef.
- (a) K. B. Gudasi, S. A. Patil, R. S. Vadavi and R. V. Shenoy, Transition Met. Chem., 2006, 31, 586 CrossRef CAS; (b) R. V. S. S. N. Ravikumara, K. Ikedaa, A. V. Chandrasekharb, Y. P. Reddyc, P. S. Raod and J. Yamauchie, J. Phys. Chem. Solids, 2003, 64, 2433–2436 CrossRef.
- A. P. S. Pannu, P. Kapoor, G. Hundal, R. Kapoor, M. Corbella, N. Aliaga-Alcalde and M. S. Hundal, Dalton Trans., 2011, 40, 12560–12569 RSC.
- (a) O. Kahn, Molecular Magnetism, VCH Publishers, New York, 1993 Search PubMed; (b) B. N. Figgis, M. Gerloch, J. Lewis, F. E. Mabbs and G. A. Webb, J. Chem. Soc. A, 1968, 2086–2093 RSC.
- I. C. Lazzarini, A. V. Funes, L. Carrella, L. Sorace, E. Rentschler and P. Alborés, Eur. J. Inorg. Chem., 2014, 2561–2568 CrossRef CAS.
- MAGPACK Program (a) J. J. Borrás-Almenar, J. M. Clemente-Juan, E. Coronado and B. S. Tsukerblat, Inorg. Chem., 1999, 38, 6081–6088 CrossRef; (b) J. J. Borrás-Almenar, J. M. Clemente-Juan, E. Coronado and B. S. Tsukerblat, J. Comput. Chem., 2001, 22, 985–991 CrossRef.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 116–123 CrossRef PubMed.
- Z. Tomkowicz, S. Ostrovsky, H. Müller-Bunz, A. J. Hussein Eltmimi, M. Rams, D. A. Brown and W. Haase, Inorg. Chem., 2008, 47, 6959–6963 CrossRef PubMed.
- (a) F. P. Pruchnik, U. Dawid and A. Kochel, Polyhedron, 2006, 25, 3647–3652 CrossRef CAS.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2005 Search PubMed.
- U. Venkatasubramanian, D. J. Donohoe, D. Ellis, B. T. Giles, S. A. Macgregor, S. Robertson, G. M. Rosair, A. J. Welch, A. S. Batsanov, L. A. Boyd, R. C. B. Copley, M. A. Fox, J. A. K. Howard and K. Wade, Polyhedron, 2004, 23, 629–636 CrossRef CAS.
Footnotes |
| † In memory of our good friend and excellent boron chemist, Igor Chizhevsky, who passed away on May 23 2016. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1471844 and 1471845. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01744d |
| This journal is © The Royal Society of Chemistry 2016 |