
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic analogues of Fe(II)–Fe(III) minerals containing a pentagonal ‘Cairo’ magnetic lattice†
J.
Cumby‡
,
R. D.
Bayliss§
,
F. J.
Berry
and
C.
Greaves
*
School of Chemistry, University of Birmingham, Birmingham B15 2TT, UK. E-mail: c.greaves@bham.ac.uk
First published on 24th June 2016
Abstract
Versiliaite and apuanite are two minerals containing Fe2+ and Fe3+ in a low-dimensional structure exhibiting chains of edge-linked FeO6 octahedra. The chemistry of these minerals has not been fully examined because of their rarity. We demonstrate that chemical synthesis of these minerals is possible to allow measurement of their magnetic properties and a more complete description of their structural features using neutron powder diffraction. We also show that chemical manipulation is possible to provide isostructural phases with different chemical compositions.
Introduction
Versiliaite (Fe12Sb12O32S2) and apuanite (Fe20Sb16O48S4) are two mixed-valent iron (Fe2+ and Fe3+) minerals whose structures, determined from single-crystal X-ray diffraction,1,2 are closely related to that of schafarzikite (FeSb2O4).3,4 The atomic arrangement consists of chains of edge-sharing FeO6 octahedra, similar to those found in the rutile structure. However, these chains are isolated from each other by either FeO3S or SbO3E tetrahedra, where E represents the Sb3+ lone electron pair (Fig. 1a and b). Whereas FeSb2O4 has all tetrahedra of the SbO3E type, in the sulfur-containing analogues, the tetrahedral iron (Fe1) sites are bonded by S located within the structural ‘channel’ to form Fe1–S–Fe1 links (Fig. 1c and d).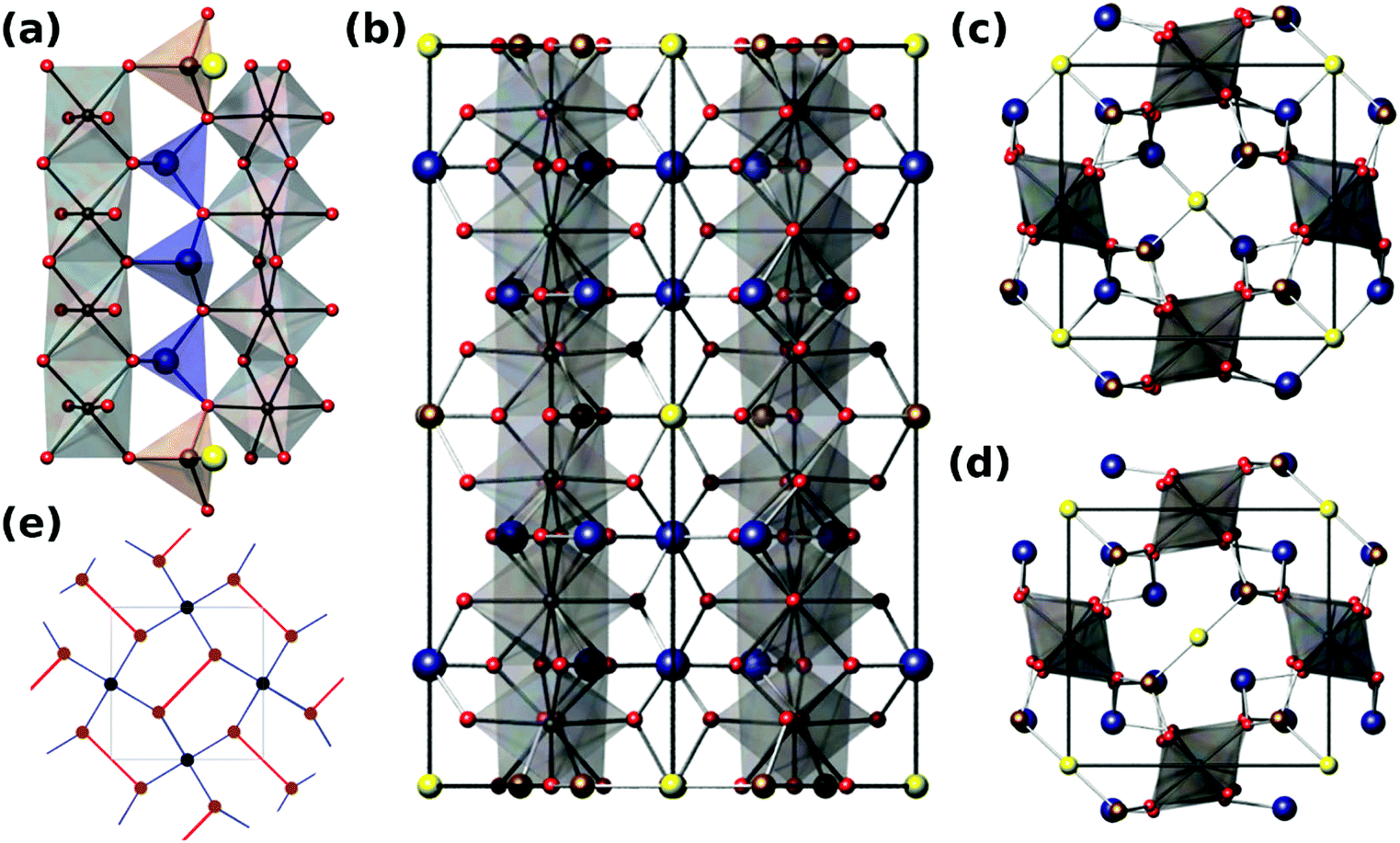 | ||
| Fig. 1 (a) View along [110] direction of versiliaite, showing rutile-like edge sharing octahedra linked by FeO3S and SbO3E tetrahedra; (b) view of apuanite unit cell along [110] showing 3-layer repeat of Fe–S–Fe links; (c) apuanite viewed along [001]; (d) versiliaite viewed along [001]; (e) schematic showing relationship to the Cairo lattice, red lines denote the Fe–S–Fe chain linkages. Grey spheres/polyhedra – octahedral Fe; brown spheres/polyhedra – tetrahedral Fe; blue spheres – Sb; red spheres – O; yellow spheres – S. | ||
The main structural difference between versiliaite and apuanite is in the number of FeO3S tetrahedra and their spacing along the crystallographic c-axis. In versiliaite, every fourth possible tetrahedron is replaced, whereas one in three is substituted in apuanite (Fig. 1a and b). Since each sulfide ion introduces two Fe3+ ions for charge balance, versiliaite and apuanite contain mixed-valent Fe2+/Fe3+ within the octahedral chains in contrast to FeSb2O4 which contains only Fe2+. Within the chains, the cations order: …Fe2+Fe2+Fe3+Fe3+Fe2+Fe2+… in versiliaite and …Fe2+Fe3+Fe3+Fe2+Fe3+Fe3+… in apuanite;1,2 in both cases the Fe3+ cations are adjacent to the layers of interstitial sulfide ions. The tetrahedral iron sites contain only Fe3+.1,2 The structural formulae can be better represented as [Fe8]oct[Fe4Sb12]tetO32S2 and [Fe12]oct[Fe8Sb16]tetO48S4, respectively (cf. [Fe]oct[Sb2]tetO4 for schafarzikite). Since adjacent tetrahedral layers correspond to a 90 degree rotation of the Fe1–S–Fe1 links, versiliaite (with an even repeat) has orthorhombic (Pbam) symmetry, while odd-repeating apuanite has tetragonal (P42/mbc) symmetry.
The parent mineral schafarzikite, FeSb2O4, shows A-type antiferromagnetic (AFM) order where the Fe magnetic moments align …+−+−+−… within a given chain and are directed perpendicular to [001] of the tetragonal structure. We have recently shown that partial oxidation of Fe2+ to Fe3+ by the substitution of Pb2+ for Sb3+, results in a change in magnetic order to C-type where the magnetic moments within a chain are ferromagnetically (FM) aligned parallel to [001], but neighbouring chains are anti-parallel.5 In fact, mixed-valence can provide a range of interesting properties in materials such as metal-insulator transitions, structural phase transitions in addition to unusual magnetic properties. Despite the interesting low-dimensional structures of versiliaite and apuanite, and the presence of Fe2+/Fe3+, their rarity has restricted our knowledge of their chemistry except for their structures. This is unfortunate since a key feature of the structures is the presence of magnetic Fe3+ ions substituting for diamagnetic Sb3+ ions where interstitial S2− ions occupy channel sites: the resulting Fe–S–Fe would be expected to enhance the interchain magnetic exchange interactions which could result in a more complex range of interactions and possibly frustration. Magnetic frustration is found in many materials, and is of great fundamental interest. It generally originates from triangular (or tetrahedral) arrangements of magnetic ions, such as in the Kagomé lattice, which occurs in a number of structures such as pyrochlores.6 An alternative route to magnetic frustration is by tessellation of irregular pentagons such as occurs in the so-called ‘Cairo’ lattice. While widely used in art, this packing is relatively rare in crystallography, the most well-known structure exhibiting this motif being that of mullite (Al6Si2O13). The magnetic analogue Bi2Fe4O9 (and related compounds) are currently the nearest examples of a 2D Cairo lattice exhibiting magnetic frustration.7,8 A number of theoretical magnetic ground states have been predicted for this 2D lattice, but few have been experimentally realised.
Here we report the first chemical synthesis of apuanite and versiliaite which will now allow the exploration of their properties and their ability to undergo chemical manipulation to target specific features. For the first time, we report the magnetic structures of the minerals and compare with our synthetic analogues. As a result of the Fe–S–Fe links, the structures show a rare, pentagonal lattice of magnetic ions which provides a relatively high temperature antiferromagnetism, and a magnetic structure exhibiting geometric frustration. We also demonstrate that atomic substitution is possible within these materials, by synthesising a Mg-containing versiliaite-like phase, Mg4Fe8Sb12O32S2.
Experimental
Samples were synthesised from stoichiometric amounts of Fe2O3, Fe metal, MgO, Sb2O3 and Sb2S3 (all >99%, Aldrich), ground and heated within sealed quartz ampoules between 520 °C and 600 °C for between 48 hours and 1 month, with intermediate grinding. Slow cooling was necessary in order to minimise structural disorder. Phase purity was checked with laboratory X-ray powder diffraction (XRPD) equipment (Bruker D2 [Co Kα] and Bruker D8 [Cu Kα1], both with PSD LynxEye dectector). Neutron powder diffraction (NPD) data were collected at Institut Laue-Langevin (D2B diffractometer, λ = 1.594 Å), Paul-Scherrer Institute (HRPT instrument, λ = 1.494 Å and 1.886 Å) and the ISIS facility, UK (GEM diffractometer, TOF) at a range of temperatures in He cryostats. Refinements were performed using GSAS with the EXPGUI interface.9,10 Mineralogical samples were obtained from Systematic Mineralogy and removed from parent dolomite samples using a mineral drill. XRPD showed a small amount of residual dolomite within the samples.Magnetic susceptibility measurements were obtained using a Quantum Design MPMS under field-cooled (FC) and zero-field-cooled (ZFC) conditions, with an applied magnetic field of 500 Oe. Powdered sample was loaded into a gelatin capsule, and sample movement was suppressed using a small amount of PTFE tape. No diamagnetic correction was applied to the data.
Results and discussion
The synthesised products were analysed initially using XRPD data, which revealed close agreement with data from mineral samples, although the latter were found to be contaminated with small amounts of dolomite. Fig. 2 compares synthetic and naturally-occurring apuanite and highlights the main peak from dolomite. Rietveld refinement profiles against XRPD data are given in Fig. S1 and S2, ESI.† Refinement against NPD data (Fig. 3 and S3 in ESI†) revealed only small impurities: both versiliaite and apuanite samples contained a small amount of Fe3O4 impurity (1.27(4) wt% and 1.55(4) wt%, respectively) whilst apuanite also showed a small α-Fe2O3 impurity (2.23(6) wt%). Structure refinement was based on the reported mineral structures.1,2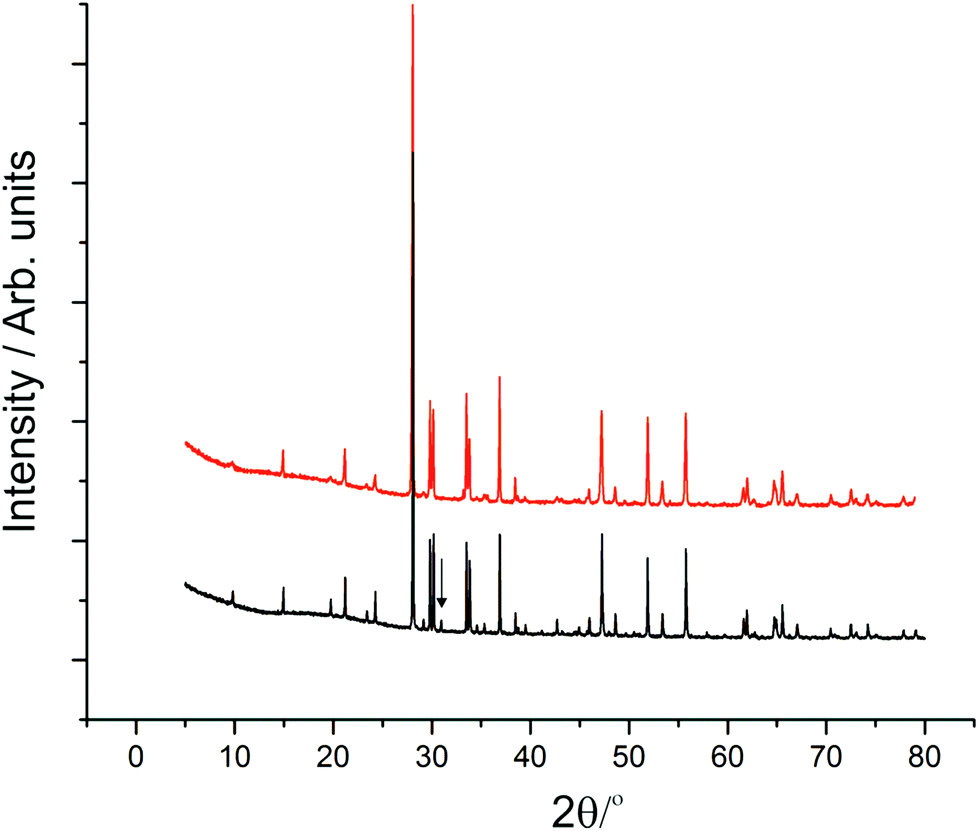 | ||
| Fig. 2 XRPD data from synthetic apuanite (red) and a mineral sample (black). An offset has been applied to the synthetic sample to allow comparison. The mineral contains a small amount of dolomite, the main peak being marked with an arrow. | ||
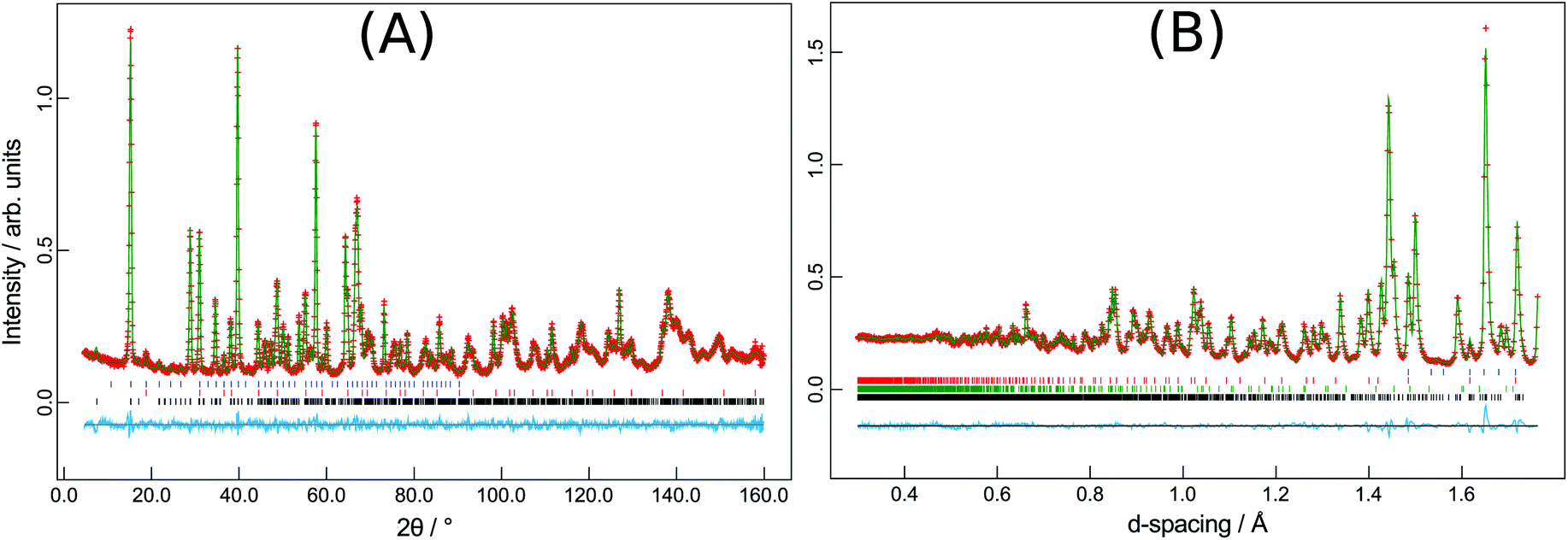 | ||
| Fig. 3 (A) Versiliaite refinement from HRPT NPD data (298 K) and (B) apuanite refinement from GEM (backscattering bank) data (300 K). Black ticks-main phase; red (blue) ticks-Fe3O4 nuclear (magnetic) phases; green ticks-Fe2O3 nuclear and magnetic phase. | ||
Incomplete order of the sulfide ions was suggested by a degree of broadening of some diffraction peaks, anisotropic peak broadening and some weakened reflections. This effect has previously been observed in mineralogical samples.3 The bond valence sums for the Fe sites are also consistent with such partial disorder (vide infra); the order is enhanced by long reaction times. The structural data presented here were from a sample that was heated for ca. 30 days at 530 °C. The disorder was modelled in the refinements using anisotropic peak broadening for high-resolution diffraction data. Correction for anisotropic peak broadening was found to be necessary for HRPT (PSI) and D2B (ILL) data, but not for GEM (ISIS) data.
Refinements show similar results to previous single crystal diffraction studies from mineral samples; slight differences are expected due to the dispersion of impurities in the natural samples. Table 1 shows the unit cell sizes for 298 K data [Tables of coordinates are available in ESI†].
Bond valence sums (BVS)11 (Table 2) clearly show that the tetrahedral iron site (Fe1) is trivalent, as expected. The valences of the other two sites are less clear, but are consistent with local charge-balance considerations: a fully ordered structure has divalent Fe2 and trivalent Fe3 since the latter site is closer to the FeO3S-containing layers. Deviations from ideal BVS values for the Fe2 and Fe3 sites reflect a small degree of disorder with respect to the sulfide ions.
| Versiliaite | Site | 298 K | 5 K | Mineral (298 K) |
|---|---|---|---|---|
| Fe1 | 3.109 | 3.131 | 2.705 | |
| Fe2 | 2.450 | 2.494 | 2.235 | |
| Fe3 | 2.811 | 2.813 | 3.107 |
| Apuanite | 1.5 K | |||
|---|---|---|---|---|
| Fe1 | 2.910 | 3.030 | 2.772 | |
| Fe2 | 2.375 | 2.332 | 2.196 | |
| Fe3 | 2.937 | 2.906 | 3.114 |
Magnetic susceptibility for synthetic compounds is dominated by the Verwey transition in Fe3O4 due to the substantial moment change at this 120 K transition compared to the overall magnetic moment of the main phase (Fig. 4). This is observed despite the rather small level of contamination and this large signal is often seen for materials containing traces of Fe3O4. Nevertheless, it should be noted that for the parent schafarzikite phases, MSb2O4, M can represent a range of magnetic ions for which no Fe3O4 impurities are observed. We would therefore expect it to be possible to synthesise magnetic phases related to apuanite and versiliaite with no magnetic contamination. There is limited evidence of a transition at around 50 K in both samples under zero-field cooled (ZFC) conditions. Susceptibility of mineralogical samples (Fig. 4c and d) show a weak transition at a slightly higher temperature than this, as well as a significant deviation from the Curie–Weiss fit occurring at ca. 170 K and 190 K for versiliaite and apuanite, respectively; these temperatures correspond to the onset of long-range magnetic order from NPD (vide infra). All transitions occur over a broad temperature range, suggesting low-dimensional behaviour. The low-temperature upturn in both mineral samples can be attributed to paramagnetic impurities within the geological material. Curie–Weiss fits to the paramagnetic region give high-temperature effective moments of 5.69μB and 5.13μB per Fe for versiliaite and apuanite, in reasonable agreement with the expected spin-only values (5.58μB and 5.71μB, respectively). Weiss constants of −160 K and −217 K demonstrate dominant antiferromagnetic (AFM) exchange at high temperature.
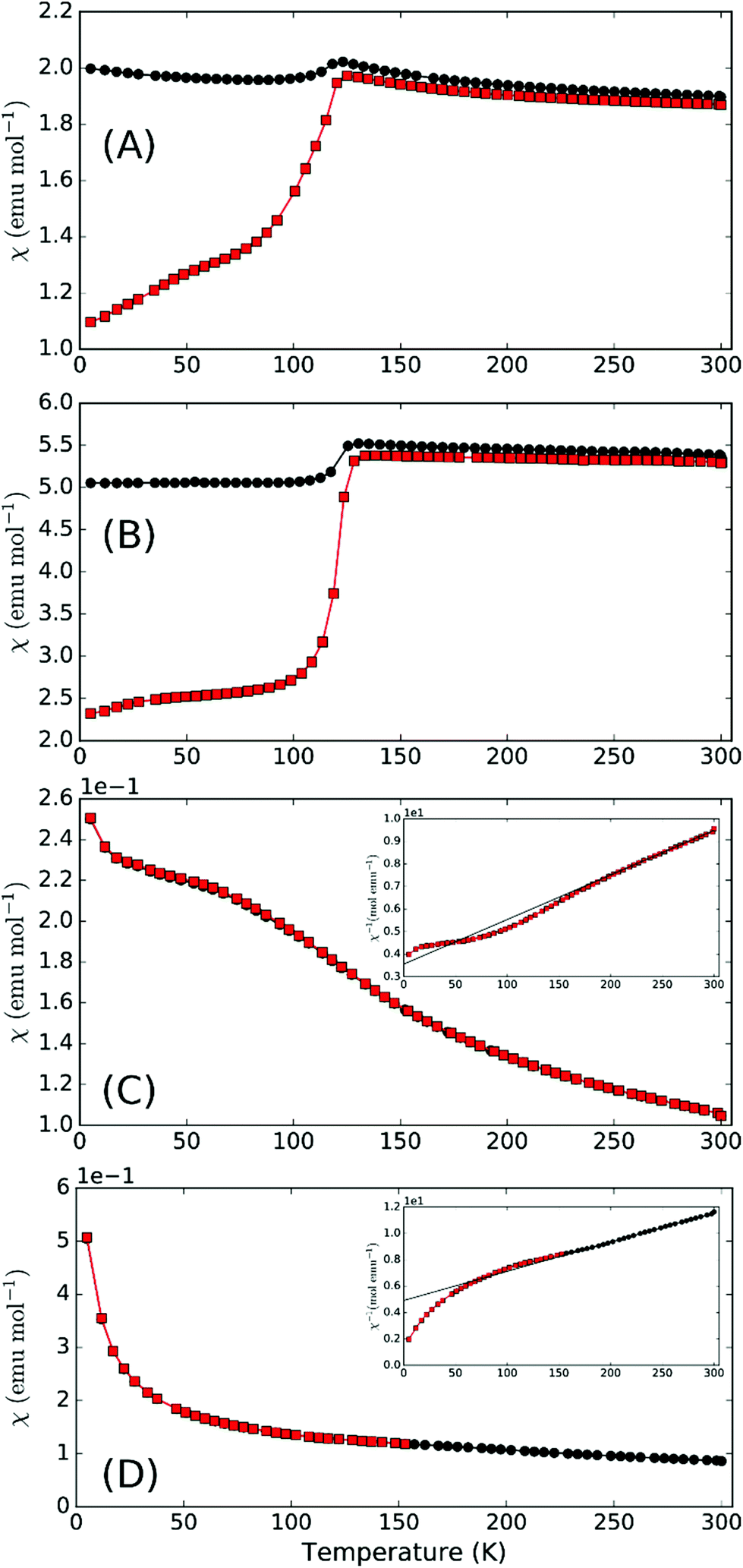 | ||
| Fig. 4 Magnetic susceptibility (inverse inset) for (a) synthetic versiliaite, (b) synthetic apuanite, (c) mineral versiliaite and (d) mineral apuanite, showing ZFC (red squares) and FC (black circles) data. | ||
Neutron diffraction from synthetic samples shows development of long range magnetic order below approximately 160 K for versiliaite, while for apuanite, broad magnetic peaks (corresponding to short-range ordering) are observed up to 210 K (Fig. 5). Both of these temperatures are similar in magnitude to the Weiss constants obtained from magnetic susceptibility data in the minerals. This is somewhat surprising given the presence of Cairo linkages highlighted in Fig. 1e. However, the direct exchange between the Fe centres within the chains appears very strong which may reduce the impact of the inherent frustration caused by the Fe–S–Fe linkages. The magnetic structures of both synthetic compounds can be indexed with a propagation vector k = [0.5, 0.5, 0], with all magnetic moments oriented in the ab-plane. The complex magnetic order is shown in Fig. 6. The Fe1–S–Fe1 linkages show AFM alignment in accordance with simple superexchange arguments; however, the best fit was obtained with adjacent linkages in a given ab-plane arranged at 90° to each other (Fig. S4 and S5, ESI†). In versiliaite, the Fe1 moments point along the nuclear a- or b-axes; in apuanite, the exact orientation is unknown due to the metrically tetragonal symmetry observed from neutron diffraction. During refinement, the Fe1 moments were therefore constrained to align in the same directions as versiliaite. In both structures, adjacent sulfur-containing layers along the c-axis exhibit parallel spins, such that any two Fe1 sites linking the same two octahedral chains are parallel. In both structures, moments within each octahedral chain display ferromagnetic (FM) alignment, directed approximately along an Fe1–S–Fe1 linkage direction. In the refined model, nearest-neighbour octahedral chains show FM order along the nuclear [110]nuc direction, but AFM order along [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10]nuc. The results (Table 3) were obtained using a single constrained moment for both Fe2 and Fe3 sites; without this constraint, the refined moments remained identical within error, but produced a less stable refinement.
10]nuc. The results (Table 3) were obtained using a single constrained moment for both Fe2 and Fe3 sites; without this constraint, the refined moments remained identical within error, but produced a less stable refinement.
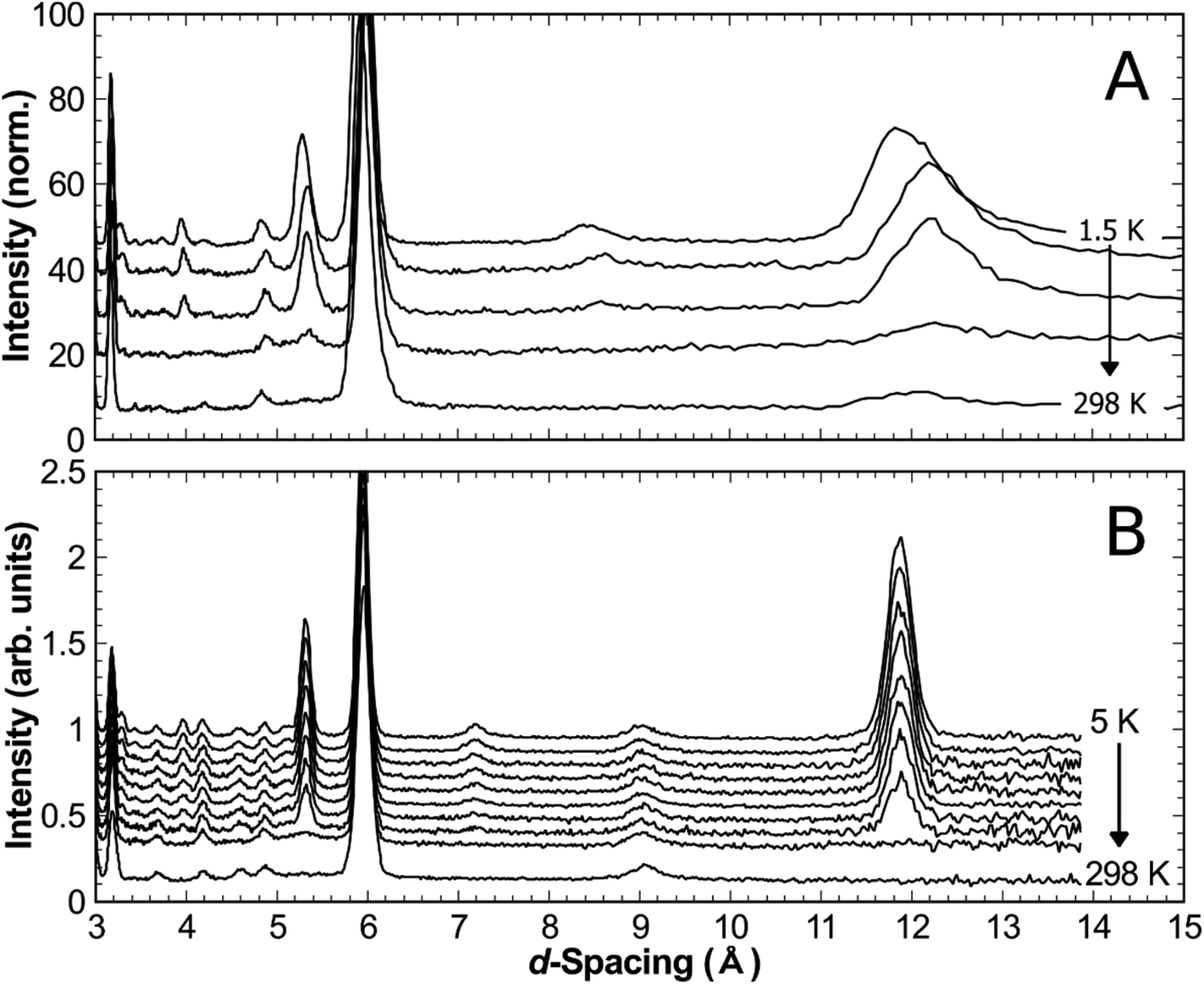 | ||
| Fig. 5 Development of magnetic reflection intensity (measured on warming) in (a) synthetic versiliaite measured at 298 K, 150 K, 50 K, 20 K and 1.5 K, and (b) synthetic apuanite measured at 298 K, 210 K, 170 K, 150 K, 130 K, 120 K, 100 K, 80 K, 40 K and 5 K. | ||
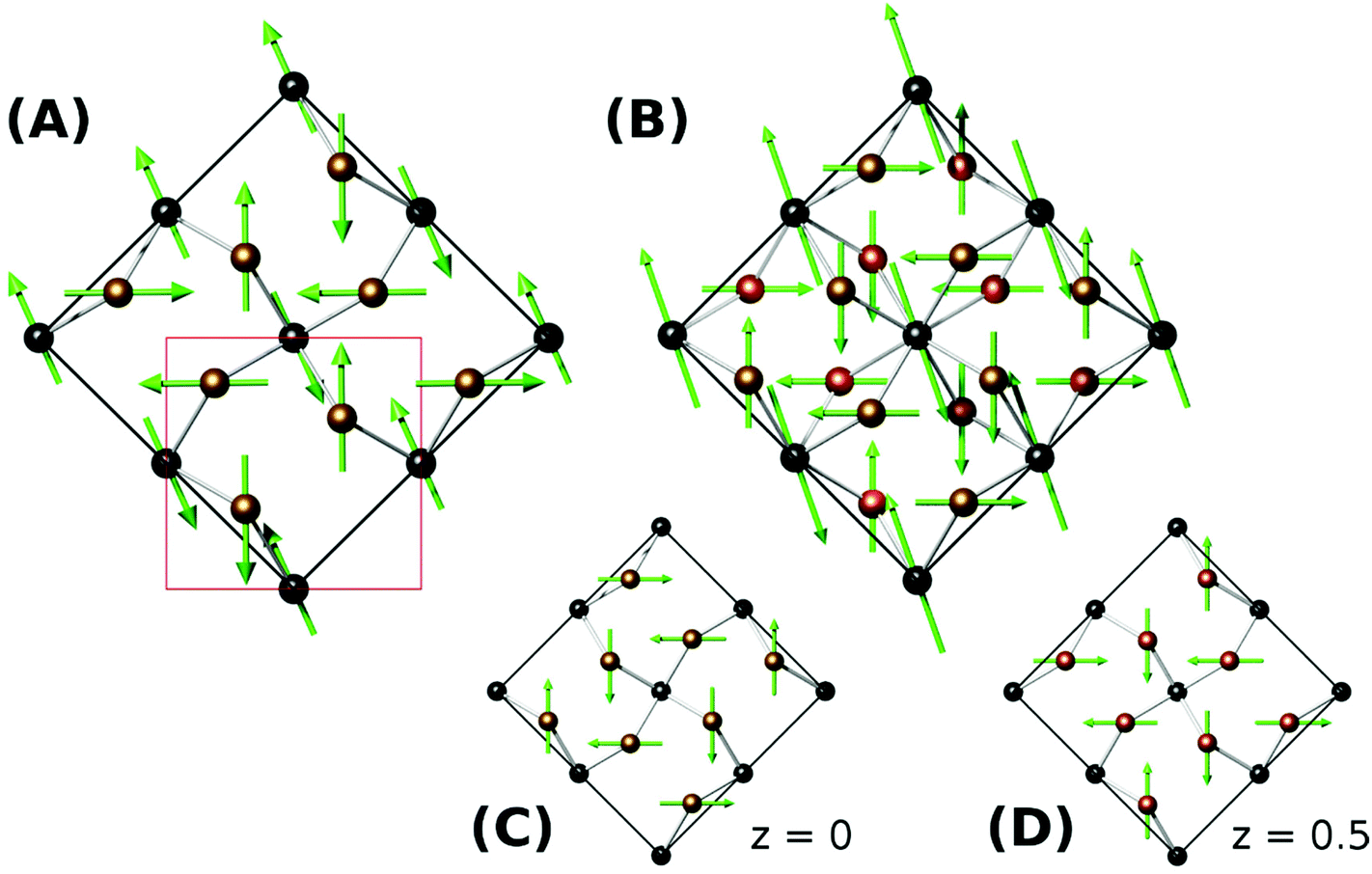 | ||
| Fig. 6 Refined magnetic structures for (a) versiliaite, (b,c,d) apuanite. (c) and (d) show magnetic moment alignment in sulfur-containing layers at z = 0 and z = 0.5, respectively. The nuclear unit cell is marked in red in (a). | ||
| Versiliaite (5 K) | Apuanite (1.5 K) | Mg4Fe8Sb12O32S2 (5 K) | ||
|---|---|---|---|---|
| Fe1 | M x , My (μB) | 2.8(1), 2.68(9) | 1.70(3), 1.70(3) | 2.4(12), 2.4(6) |
| M tot (μB) | 3.89(7) | 2.41(5) | 3.3(4) | |
| Fe2/3 | M x , My (μB) | ±1.25(3), ±3.35(4) | ±3.94(2), ±1.88(3) | ±2.0(1), ±3.0(1) |
| M tot (μB) | 3.57(4) | 4.36(3) | 3.7(1) |
The refined magnetic model is very similar to that found in Bi2Fe4O9 (ref. 7) and recently in Bi2Fe3−xCrxO9.8 However, the moments in the latter are canted away from the ab plane and the chain moments in the former are modulated along both [110]nuc and [10]nuc directions. A similar model applied to the materials studied here does not fit as well, although the primary difference from powder diffraction would be in the (110)m magnetic peak, which unfortunately coincides with the Fe3O4 (001) peak. However, the deduced magnetic model corresponds closely with the magnetic order theoretically predicted if the coupling between the Fe1 (tetrahedral) sites is stronger than between Fe1 and Fe3.12
In addition to synthetic analogues of versiliaite and apuanite discussed above, we have also demonstrated that chemical modification, e.g. cation substitutions, are possible. For example, the versiliaite analogue Mg4Fe8Sb12O32S2, where Mg2+ ions replace all Fe2+ ions, has been successfully synthesised. Characterisation from NPD (Fig. S6 and S7, ESI†) shows a predominantly single phase material, although with a small (3 wt%) Fe3O4 impurity. Lattice parameters (at 298 K) are larger than the other synthetic samples (a = 8.4933(12), b = 8.3897(12) and c = 11.9525(13)) particularly for a and b. This larger ab plane dimension may reflect significant occupation of the tetrahedral site by the slightly larger Mg2+ ion (0.57 Å radius, cf. 0.49 Å for Fe3+).13 Refined site occupancies show that the Fe1 (tetrahedral) site is almost equally occupied by Mg and Fe (51(3)% Fe), while the two octahedral sites show iron occupancies of 78(5)% (Fe2) and 71(5)% (Fe3). Magnetically, this compound shows similar susceptibility behaviour to versiliaite and apuanite, although with a strong Fe3O4 signal (Fig. 7). Neutron diffraction data at 5 K can be fitted to the same model as for versiliaite, and with similar Fe moments (Table 3).
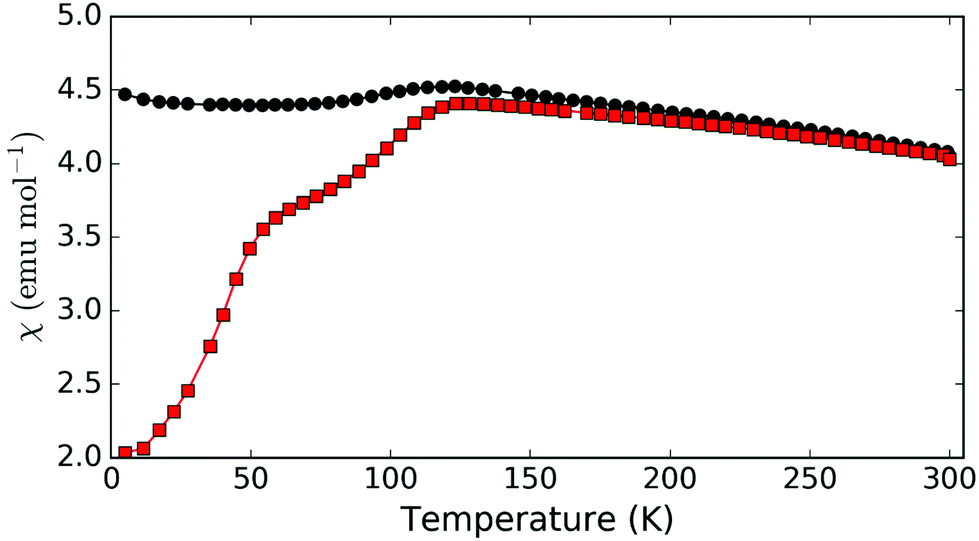 | ||
| Fig. 7 Magnetic susceptibility for Mg-substituted versiliaite, Mg4Fe8Sb12O32S2, showing ZFC (red squares) and FC (black circles) data. | ||
Conclusions
We have shown that the rare minerals versiliaite and apuanite can be synthesised using a simple sealed-tube approach. The crystal structures are in close agreement with mineralogical samples. Moreover the successful synthesis of a Mg-substituted versiliaite confirms that the synthetic method will facilitate chemical manipulations in order to explore how the interesting low-dimensional structure may be utilised to provide specific functional properties. Magnetically, the materials studied here exhibit AFM order at relatively high temperatures, although they show magnetic development over a broad temperature range. The arrangement of magnetic ions represents a new class of compounds containing a Cairo-lattice motif, and orders as expected from theoretical models. The significant difference between these compounds and existing Cairo lattices (such as for Bi2Fe4O9) is that the Cairo layers are much more isolated in versiliaite and apuanite, with interlayer distances of ∼12 Å and ∼9 Å, respectively (compared to ∼3 Å in Bi2Fe4O9). Chemical substitutions may result in greater magnetic isolation between Cairo layers, and a useful route to study frustrated magnetism on a non-triangular lattice.Acknowledgements
We thank EPSRC for financial support of this research (EP/L014114/1 and EP/P505410/1) and EPSRC, EU and PSI for the provision of NPD facilities. We acknowledge the assistance of Dr Emma Suard, Dr Vladimir Pomjakushin and Dr Winfried Kockelmann in collecting the NPD data. The X-ray diffractometers used in this research were obtained through Birmingham Science City: Creating and Characterising Next Generation Advanced Materials (West Midlands Centre for Advanced Materials Project 1), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). Data providing the results in this paper are accessible from the University of Birmingham archive: http://epapers.bham.ac.uk/2175/.References
- M. Mellini, S. Merlino and P. Orlandi, Am. Mineral., 1979, 64, 1230 CAS.
- M. Mellini and S. Merlino, Am. Mineral., 1979, 64, 1235 CAS.
- M. Mellini, Am. Mineral., 1981, 66, 1073 CAS.
- J. R. Gavarri, J. P. Vigouroux, G. Calvarin and A. W. Hewat, J. Solid State Chem., 1981, 36, 81 CrossRef CAS.
- M. J. Whitaker, R. D. Bayliss, F. J. Berry and C. Greaves, J. Mater. Chem., 2011, 21, 14523 RSC.
- R. Moessner and A. P. Ramirez, Phys. Today, 2006, 59, 24 CrossRef CAS.
- E. Ressouche, V. Simonet, B. Canals, M. Gospodinov and V. Skumryev, Phys. Rev. Lett., 2009, 103, 267204 CrossRef CAS PubMed.
- M. G. Rozova, V. V. Grigoriev, I. A. Bobrikov, D. S. Filimonov, K. V. Zakharov, O. S. Volkova, A. N. Vasiliev, A. A. Tsirlin and A. M. Abakumov, Dalton Trans., 2016, 45, 1192 RSC.
- C. Larson and R. B. Von Dreele, General Structure Analysis System (GSAS), Los Alamos National Laboratory Report LAUR 86-748, 2000 Search PubMed.
- H. Toby, J. Appl. Crystallogr., 2001, 34, 210 CrossRef.
- D. Brown and D. Altermatt, Acta Crystallogr, Sect. B: Struct. Sci., 1985, 41, 244 CrossRef.
- I. Rousochatzakis, A. M. Lauchli and R. Moessner, Phys. Rev. B: Condens. Matter, 2012, 85, 104415 CrossRef.
- R. D. Shannon, Acta Crystallogr., 1976, A32, 751 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6dt01672c |
| ‡ Current address: Centre for Science at Extreme Conditions and School of Chemistry, University of Edinburgh, Edinburgh EH9 3FD, UK. |
| § Current address: Department of Chemistry, University of Illinois at Chicago, Chicago 60607, IL, USA. |
| This journal is © The Royal Society of Chemistry 2016 |