
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pyrithione-based ruthenium complexes as inhibitors of aldo–keto reductase 1C enzymes and anticancer agents†
Jakob
Kljun
a,
Maja
Anko
b,
Katja
Traven
a,
Maša
Sinreih
b,
Renata
Pavlič
b,
Špela
Peršič
a,
Žiga
Ude
a,
Elisa Esteve
Codina
ac,
Jure
Stojan
b,
Tea
Lanišnik Rižner
*b and
Iztok
Turel
*a
aDepartment of Chemistry and Biochemistry, Faculty of Chemistry and Chemical Technology, University of Ljubljana, Večna pot 113, SI-1000 Ljubljana, Slovenia. E-mail: iztok.turel@fkkt.uni-lj.si; Fax: +386-1-24-19-220; Tel: +386-1-24-19-124
bInstitute of Biochemistry, Faculty of Medicine, University of Ljubljana, Vrazov trg 2, SI-1000 Ljubljana, Slovenia. E-mail: tea.lanisnik-rizner@mf.uni-lj.si; Fax: +386-1-54-37-641; Tel: +386-1-54-37-657
cFaculty of Chemistry, University of Valencia, Carrer Doctor Moliner, 50. 46100, Burjassot, Spain
First published on 20th June 2016
Abstract
Four ruthenium complexes of clinically used zinc ionophore pyrithione and its oxygen analog 2-hydroxypyridine N-oxide were prepared and evaluated as inhibitors of enzymes of the aldo–keto reductase subfamily 1C (AKR1C). A kinetic study assisted with docking simulations showed a mixed type of inhibition consisting of a fast reversible and a slow irreversible step in the case of both organometallic compounds 1A and 1B. Both compounds also showed a remarkable selectivity towards AKR1C1 and AKR1C3 which are targets for breast cancer drug design. The organoruthenium complex of ligand pyrithione as well as pyrithione itself also displayed toxicity on the hormone-dependent MCF-7 breast cancer cell line with EC50 values in the low micromolar range.
Introduction
The discovery of cisplatin paved the way for the introduction of several platinum-based anticancer agents into clinical practice in the 1970s and 80s.1 Despite great efforts both cisplatin and the subsequently developed platinum drugs exhibit great efficiency with several downfalls, mainly resistance development and severe side effects due to general toxicity and non-specific mode of action.2 The development of novel alternatives or complementary anticancer agents is thus urgently needed to reduce the social and economic impact of the growing occurrence of cancer. The hormone-dependent (breast, prostate and uterine) cancers alone comprise more than 20% of all cancers, where breast cancer represents the most common cancer in women worldwide. In 2012, approx. 1.7 million new cases of breast cancer were associated with more than half million deaths.3Isozymes from the aldo–keto reductase 1C subfamily (AKR1C) are implicated in the development of hormone-dependent forms of cancers, other hormone-dependent diseases,4 and also in development of lung, oral, larynx, and bladder cancers.5,6 There is a plethora of data supporting the important roles of AKR1C isozymes in pathophysiology, reinforcing these enzymes, and especially AKR1C1 and AKR1C3, as emerging targets for drug development.4 Furthermore, AKR1C isozymes are related to the resistance to a variety of anticancer drugs including the platinum-based drugs cisplatin, carboplatin and oxaliplatin.7–9 Increased expression of AKR1Cs in these resistant cancers thus calls for drugs that can overcome this phenomenon by either not affecting the AKR1C levels or by inhibiting the AKR1C enzymes.
Transition metal ionophores are considered to be an emerging class of ligands in the design of anticancer drugs and bioactive metal-based compounds.10 Zinc pyrithione (bis(2-thiolatopyridine N-oxide)zinc) has found its medical use as a bacteriostatic and fungistatic agent in dandruff shampoos and baby zinc powder. It acts by disrupting the membrane transport by blocking the proton pumps.11 Structurally related sulfur containing pyridine N-oxides were found to possess anti-inflammatory properties,12 inhibit metalloenzyme carbonic anhydrase13 and pyrithione itself induces increased Zn2+ cell influx resulting in herpes simplex virus replication inhibition.14 Moreover, pyrithione zinc is remarkably effective towards acute myeloid leukemia by triggering apoptosis through NF-κB inhibition,15 and towards cervical tumor cells by activating p53 and its dependent genes resulting in loss of membrane potential and activation of apoptosis.16
Organometallic complexes of other precious metals (Ru, Ir, Os) have recently been shown to be promising alternatives to platinum-based drugs17–19 and two ruthenium compounds have recently completed phase II clinical trials.20,21 Several other compounds are in the preclinical phases of investigations.22–25 Besides conventional chemotherapy, ruthenium-based coordination compounds are being studied in conjunction with electrotherapy (i.e. electrochemotherapy),26,27 thermotherapy28 and photodynamic therapy.29 Structure–activity studies involving ruthenium complexes with diamine,30 polypyridyl,30 phosphine31 and diketone ligands32 were performed during the last 15 years with clear patterns emerging, however proving different modes of action depending on the ligand types. Moreover, a targeted approach can be applied to the development of novel drugs using ruthenium scaffolds as successfully shown by the Meggers group.33
Our previous research focused on the design of novel drug candidates in which clinically used drugs (quinolone antibacterials and azole antifungals) were complexed to different ruthenium species with proven biological activity.34–39 While these complexes displayed various degrees of toxicity in different cancer cell lines, the study of their biological properties such as binding to serum proteins35,40 was coupled with investigations of possible targets including DNA,35,39 cysteine proteases cathepsin B and S,35,36 and, recently, enzymes of the aldo–keto reductase subfamily 1C (AKR1C1–3).41 In our previous study we identified one ruthenium complex and two precursor compounds which inhibited AKR1C1–AKR1C3 enzymes, and one ruthenium complex as a specific inhibitor of AKR1C3 with nM Ki value.41
Continuing our study on ruthenium complexes with clinically used transition metal ionophores, herein we report the study of the biological activity of two types of ruthenium complexes of zinc ionophores pyrithione and its oxygen-containing analog 2-hydroxypyridine N-oxide (Scheme 1). We examined the inhibition of AKR1C enzymes known to be implicated in the pathophysiology of a variety of cancers, including breast cancer, and their potential cytotoxic effects on the model cell line of hormone-dependent breast cancer, MCF-7.
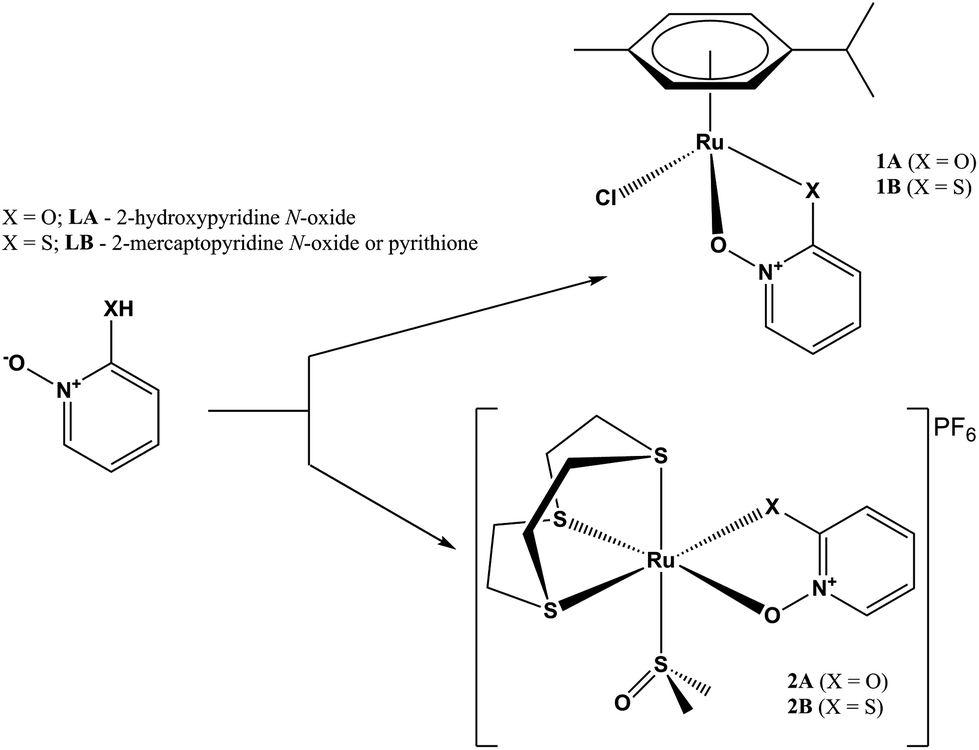 | ||
| Scheme 1 Reaction path for complexes of series 1 and 2. | ||
Experimental part
Synthesis and characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeOH/CHCl3. The reaction mixture was refluxed for 2.5 h during which the color changed from yellow to light orange and was finally allowed to cool while stirring for additional 0.5 h. The solvents were removed and the oily residue was redissolved in 5 mL of CH2Cl2. The insoluble byproduct NaCl was removed by filtration over celite. 10 mL of hexane were added to the CH2Cl2 solution which became turbid. Evaporation of the CH2Cl2 solution resulted in the formation of a light brown precipitate which was filtered and dried at 45 °C overnight. Yield = 22.5 mg (45%). Crystals of 1A were obtained by dissolving 5 mg of 1A in CH2Cl2 and adding 10 mL of n-hexane. The solution was left in an open flask overnight yielding orange needle-like crystals.
:1 MeOH/CHCl3. The reaction mixture was refluxed for 2.5 h during which the color changed from yellow to light orange and was finally allowed to cool while stirring for additional 0.5 h. The solvents were removed and the oily residue was redissolved in 5 mL of CH2Cl2. The insoluble byproduct NaCl was removed by filtration over celite. 10 mL of hexane were added to the CH2Cl2 solution which became turbid. Evaporation of the CH2Cl2 solution resulted in the formation of a light brown precipitate which was filtered and dried at 45 °C overnight. Yield = 22.5 mg (45%). Crystals of 1A were obtained by dissolving 5 mg of 1A in CH2Cl2 and adding 10 mL of n-hexane. The solution was left in an open flask overnight yielding orange needle-like crystals.
1H NMR (CDCl3, 500.10 MHz): δ 7.81 (dd, J(H,H) = 1.5 Hz, 8.5 Hz, 1H, Ar-H LA), 7.16–7.12 (m, 1H, Ar-H LA), 6.75 (dd, J(H,H) = 1.5 Hz, 10 Hz, 1H, Ar-H LA), 6.36 (td, J(H,H) = 2 Hz, 7 Hz, 1H, Ar-H LA), 5.53–5.50 (m, 2H Ar-H cym), 5.31–5.28 (m, 2H Ar-H cym), 2.96–2.91 (m, 1H, Ar-CH(CH3)2 cym), 2.33 (s, 3H, Ar-CH3 cym), 1.34 (dd, J(H,H) = 3 Hz, 7 Hz, 6H, Ar-CH-(CH3)2 cym). IR (cm−1, ATR): 3065, 3030, 2960, 2901, 1615, 1550, 1474, 1450, 1353, 1237, 867, 768. CHN (%) for C15H18NClO2Ru: calc. C 47.31, H 4.77, N 3.68; exp. C 47.24, H 4.42, N 3.49. ESI-HRMS (CH3CN) m/z for (M − Cl)+ (found (calc.)): 346.0616 (346.0381).
:1 MeOH/CHCl3. The reaction mixture was refluxed for 2.5 h during which the color changed from deep yellow to light red and was finally allowed to cool while stirring for additional 0.5 h. The solvents were removed and the oily residue was redissolved in 5 mL of CH2Cl2. The insoluble byproduct NaCl was removed by filtration over celite. 10 mL of hexane were added to the CH2Cl2 solution which became turbid. Evaporation of the CH2Cl2 solution resulted in the formation of a dark orange/brown precipitate which was filtered and dried at 45 °C overnight. Yield = 25.0 mg (48%). Crystals of 1B were obtained by slow evaporation of a 1:1 acetone/hexane solution.
1H NMR (CDCl3, 500.10 MHz): δ 8.05 (d, J(H,H) = 6.5 Hz, 1H, Ar-H LB), 7.45 (d, 1H, Ar-H LB), 7.05–7.02 (m, 1H, Ar-H LB), 6.72 (td, J(H,H) = 2 Hz, 7 Hz, 1H, Ar-H LB), 5.48 (d, 2H, Ar-H cym), 5.28 (d, J(H,H) = 6 Hz, 2H, Ar-H cym), 2.86–2.80 (m, 1H, Ar-CH(CH3)2 cym), 2.35 (s, 3H, Ar-CH3 cym), 1.28 (d, J(H,H) = 7 Hz, 6H, Ar-CH-(CH3)2 cym). IR (cm−1, ATR): 2986, 2901, 1593, 1546, 1454, 1260, 1236, 757. CHN (%) for C15H18NClORuS: calc. C 45.39, H 4.57, N 3.53; exp. C 44.96, H 4.44, N 3.42. ESI-HRMS (CH3CN) m/z for (M − Cl)+ (found (calc.)): 362.0272 (362.0152).
1H NMR (acetone-d6, 500.10 MHz): δ 8.1 (dd, J(H,H) = 1.5 Hz, 6.5 Hz, 1H, Ar-H LA), 7.49–7.45 (m, 1H, Ar-H LA), 6.91 (dd, J(H,H) = 1.5 Hz, 9 Hz, 1H, Ar-H LA), 6.70 (td, J(H,H) = 1.5 Hz, 7 Hz, 1H, Ar-H LA), 3.68–3.44 (m, 12H, 9aneS3), 3.03 (s, 3H, CH3, dmso), 2.99 (s, 3H, CH3, dmso). IR (cm−1, ATR): 2988, 2901, 1620, 1553, 1499, 1458, 1360, 1234, 1074, 1066, 826. CHN (%) for C13H22NF6O3PRuS4: calc. C 25.40, H 3.61, N 2.28; exp. C 25.82, H 3.51, N 2.59. ESI-HRMS (CH3CN) m/z for (M − PF6 + H)+ (found (calc.)): 469.9720 (469.9526).
1H NMR (acetone-d6, 500.10 MHz): δ 8.38 (d, J(H,H) = 6.5 Hz, 1H, Ar-H LB), 7.71 (dd, J(H,H) = 1.5 Hz, 8.5 Hz, 1H, Ar-H LB), 7.42 (td, J(H,H) = 5 Hz, 7 Hz, 1H, Ar-H LB), 7.13 (td, J(H,H) = 1.5 Hz, 6.5 Hz, 1H, Ar-H LB), 3.73–3.68 (m, 12H, 9aneS3), 2.82 (s, 4H, CH3, dmso), 2.58 (s, 2H, CH3, dmso). IR (cm−1, ATR): 2987, 2901, 1598, 1547, 1456, 1237, 1088, 828. CHN (%) for C13H22NF6O2PRuS5: calc. C 24.76, H 3.52, N 2.22; exp. C 25.04, H 3.25, N 2.24. ESI-HRMS (CH3CN) m/z for (M-PF6)+ (found (calc.)): 485.9755 (485.9297).
Stability and reactivity in aqueous media
The stability of complexes in aqueous solution was monitored by 1H NMR by dissolving 2–3 mg of the product in 0.7 mL of D2O (approx. 2–10 mM concentrations) as well as 10% dmso-d6/90% D2O. The spectra show no changes for 72 h. Compound 1B as the most active compound in both enzymatic and toxicity assays was investigated in more detail. Reactivity towards metal-coordinating amino acids histidine (His) and cysteine (Cys) as well as antioxidant and ROS scavenger glutathione (GSH) in view of the proposed binding mode to the AKR1C enzymes was studied. A 500 μL of a 4 mg mL−1 solution of 1B in 20% dmso-d6/80% D2O was mixed with 500 μL of His/Cys/GSH D2O solutions of the same molar concentrations (1:1 ratio between 1B and reactant). The reactions were monitored by 1H NMR.
Materials and methods
Reagents
Precursor P1, [(η6-p-cymene)Ru(μ-Cl)Cl]2, ligands LA and LB are commercially available substances and were purchased from Sigma-Aldrich. Precursor P2, [(9aneS3)Ru(dmso)3](PF6)2, was prepared according to the procedure published in ref 42. All chemicals and solvents were used without further purification or drying.Spectroscopy and elemental analysis
The starting materials were purchased from commercial suppliers and used as received. The progress of all reactions was monitored on Fluka precoated silica gel plates (with fluorescence indicator UV254) using 3–10% MeOH/DCM as the solvent system. 1H NMR spectra were recorded on a Bruker Avance III 500 spectrometer (at room temperature and 500.10 MHz) by using TMS as an internal standard in (CD3)2CO or CDCl3. The splitting of proton resonances is defined as s = singlet, d = doublet, t = triplet, q = quartet, sept = septet, m = multiplet. All NMR data processing was carried out using MestReNova version 8.1.2. Infrared spectra were recorded with a Perkin-Elmer Spectrum 100 FTIR spectrometer, equipped with a Specac Golden Gate Diamond ATR as a solid sample support. Elemental analyses were recorded using a Perkin-Elmer 2400 II instrument (CHN), and HRMS were measured on an Agilent 6224 Accurate Mass TOF LC/MS instrument.X-ray diffraction data for all compounds were collected on an Oxford Diffraction SuperNova diffractometer with a Mo/Cu microfocus X-ray source with mirror optics and an Atlas detector. Data on software used complete with references are given in the ESI.† The crystal structures of compounds 1A, 1B, 2A, and 2B have been submitted to the CCDC and have been allocated the deposition numbers CCDC 1444456–1444459.
Inhibition assays for the AKR1C enzymes
The recombinant AKR1C1–AKR1C3 enzymes were purified as described previously.43 AKR1C enzymes can catalyze the oxidation of an artificial substrate, chiral 1-acenaphthenol, in the presence of a coenzyme NAD+. We therefore, spectrophotometrically followed the increase in NADH absorbance at 340 nm (ελ340 = 6220 M−1 cm−1) in the absence and presence of tested compounds (1A, 1B, 2A, 2B, LA, LB). The assays were carried out in a 0.3 mL volume that included 100 mM potassium phosphate buffer (pH 9.0), 0.005% (v/v) Triton X-114 and 5% (v/v) DMSO as co-solvents. For the assays with AKR1C1, AKR1C2 and AKR1C3, substrate concentrations of 90 μM, 180 μM and 250 μM, respectively, and enzyme concentrations of 50 nM, 120 nM and 1.5 μM, respectively, were used, in the presence of 2.3 mM coenzyme NAD+. High pH and a large surplus of the coenzyme NAD+ allowed us to follow the oxidation of both substrate enantiomers until completion. The analysis of such progress curves provides the information on the inhibitory action of putative inhibitors at continuously decreasing substrate concentrations. The concentrations of the tested Ru compounds and ligands were between 10 and 100 μM, depending on the extent of inhibition. All measurements were performed in duplicate or triplicate on a Biotek (Winooski, VT, USA) PowerWave XS microplate reader.To compare the inhibitory potential of the tested compounds, we used a previously determined reaction mechanism of 1-acenaphthenol oxidation by AKR1C enzymes (Scheme 2, ref. 41). Briefly, it is a sequential bi-bi reaction mechanism comprising the reaction with both enantiomers of 1-acenaphthenol. Due to the large surplus of coenzyme NAD+ during the measurements, the tested compounds are only allowed to react with holoenzyme (enzyme–coenzyme complex), where they compete with the racemic substrate. This competition, however, consists of two phases: one is a fast reversible inhibition and the other, a slow irreversible inactivation. Importantly, the two phases are explained by the subsequent action of two Ru complex molecules.
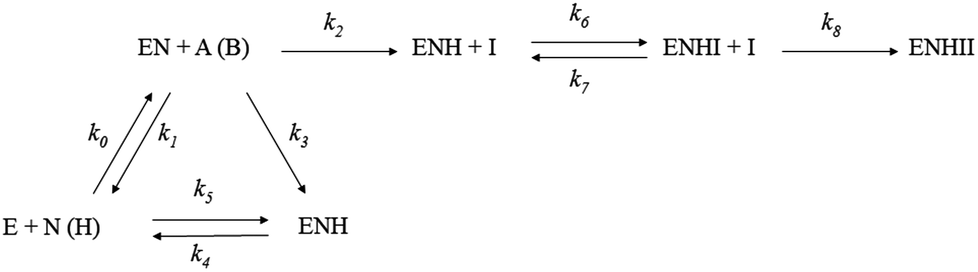 | ||
| Scheme 2 Universal scheme for inhibition/inactivation of AKR1C enzymes by Ru complexes and ligands. E is free enzyme, N is coenzyme NAD+, EN and ENH are enzyme–coenzyme complexes with oxidized and reduced form of coenzyme, respectively, A and B are the enantiomers of 1-acenaphthenol, and I is inhibitor; k0 − k8 are first and second order rate constants. | ||
All measured progress curves were analyzed by ENZO a web tool for easy construction, quick testing and evaluation of kinetic models (cf.: http://enzo.cmm.ki.si/kinetic.php?uwd=140808146&load=true; click Set Parameters, Start). The data acquired for each enzyme type were analyzed for each active compound.
Visualization of enzyme inhibitor complexes
To visualize the interactions for the binding of two 1B Ru complex molecules to AKR1C enzymes, as suggested by kinetic analyses, we first submitted pyrithione and η6-p-cymene molecular models to ParaChem, a web server for the automatic generation of additive force fields in CHARMM (CGenFF).44,45 In the Charmm input script we then combined the obtained stream files with the latest version of the all-atom CGenFF additive parameter set and manually adopted VDW and the electrostatic parameters for the Ru2+ cation, using the crystal structure as a topology standard. Subsequently, the first 1B molecule was docked at the entrance to the AKR1C1 (PDB code 3NTY, devoid of all hetero atoms) active site and the second one, without chloride, was placed on the Nε2 of deprotonated His53. A small optimization run was conducted with constrained heavy atoms of the protein and distances between the Ru2+ cation with all attaching atoms set to the values as determined by crystallography of the complex alone. No constraint was applied to the bond between Ru2+ and the nitrogen from His53.Cell proliferation assay
The influence of ruthenium compounds on cell proliferation was analyzed using the breast cancer cell line MCF-7 and the cell proliferation reagent WST-1 (Roche Diagnostics, Germany) following the manufacturer's instructions. All the experiments were performed in triplicate. Absorbance of the well containing only the non-treated cells was used as a normalization control. The half-maximal effective concentration (EC50) was determined by constructing a dose–response curve (Graph Pad Prism, Version 5.0). More details are given in the ESI.†Results and discussion
We have successfully synthesized and fully characterized (including their X-ray crystal structures; Fig. 1 and 2) four ruthenium complexes of the zinc ionophore pyrithione (2-mercaptopyridine-N-oxide) and its oxygen-containing analog (LB and LA respectively) and investigated their potential as inhibitors of human oxidoreductases from the AKR1C subfamily.7,8,46 The enzymes AKR1C1–AKR1C3 represent emerging targets for development of anti-cancer agents and are also associated with resistance to platinum based chemotherapeutics. Complexes 1A and 1B are organometallic complexes with a pseudo-octahedral structure in which the p-cymene ligand is π-bonded to the metal ion. The remaining three coordination sites are occupied by the N-oxide oxygen and the oxygen/sulfur of the chelating ligands LA/LB as well as the monodentate chlorido ligand. In the case of complexes 2A and 2B the arene ligand is substituted by the sulfur macrocycle trithiacyclononane (9aneS3) and the monodentate ligand is an S-bonded dmso molecule. The crystal structures of the four complexes confirm the proposed structure of the compounds and the bond lengths and angle values are within the range of other reported structures of organoruthenium/Ru-trithiacyclononane complexes bearing a chelating O,O- or O,S- ligand which forms a five-membered ring with the central metal ion. Interestingly the crystal structures of 1A and 2A reveal a partial dearomatisation of the 2-hydroxypyridine N-oxide ligand where the observed C3–C4 and C5–C6 distances are significantly shorter than C4–C5 (see cif files in the ESI†).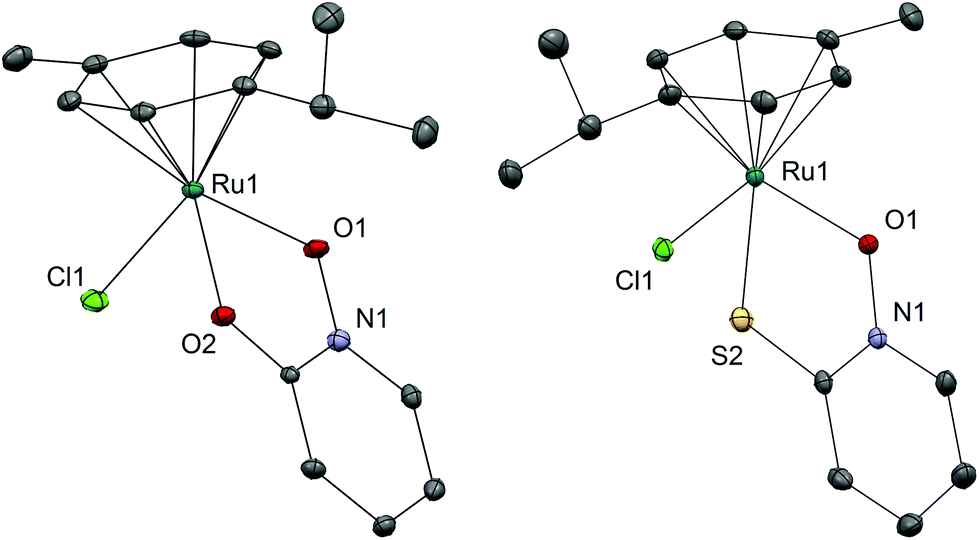 | ||
| Fig. 1 Crystal structures of complexes 1A and 1B with heteroatom labelling. Hydrogen atoms are omitted for clarity of presentation. The ellipsoids are shown at the 35% probability level. Selected bond lengths and angles – 1A: Ru1–O1 2.0768(17) Å; Ru1–O2 2.0762(14) Å; Ru1–Cl1 2.4170(6) Å; Ru1–CYMcentroid 1.6372(11) Å; O1–Ru1–O2 78.05(6)°; O1–Ru1–Cl1 85.87(5)°; O2–Ru1–Cl1 85.46(5)°; 1B: Ru1–O1 2.0808(18) Å; Ru1–S2 2.3555(7) Å; Ru1–Cl1 2.4487(6) Å; Ru1–CYMcentroid 1.6613(12) Å; O1–Ru1–S2 82.73(5)°; O1–Ru1–Cl1 84.53(5)°; S2–Ru1–Cl1 87.34(2)°. | ||
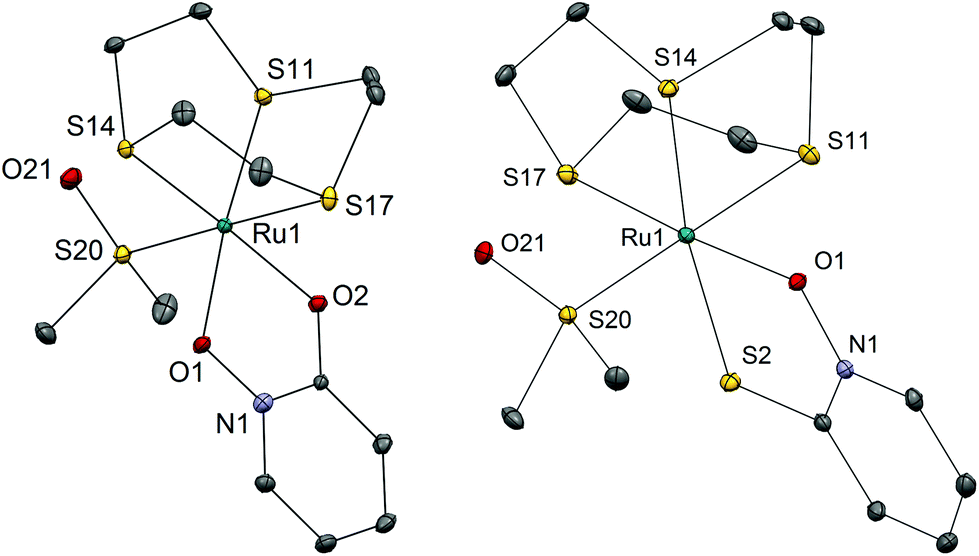 | ||
| Fig. 2 Crystal structures of complexes 2A and 2B with heteroatom labelling. Hydrogen atoms, hexafluorophosphate anions and co-crystallized solvent molecules are omitted for clarity of presentation. The ellipsoids are shown at the 35% probability level. Selected bond lengths and angles – 2A: Ru1–O1 2.0786(1) Å; Ru1–O2 2.0974(1) Å; Ru1–S11 2.2913(5) Å; Ru1–S14 2.2888(1) Å; Ru1–S17 2.3421(5) Å; Ru1–S20 2.2596(5) Å; O1–Ru1–O2 79.71(6)°; 2B: Ru1–O1 2.0857(15) Å; Ru1–S2 2.3703(6) Å; Ru1–S11 2.3465(7) Å; Ru1–S14 2.3278(6) Å; Ru1–S17 2.2982(6) Å; Ru1–S20 2.2633(7) Å; O1–Ru1–S2 83.60(4)°. | ||
All compounds are stable in D2O and 10% dmso-d6/D2O solutions. The most active compound – 1B – was investigated in detail to further support the proposed binding mode to the AKR1C enzymes. Compound 1B readily reacts with histidine, cysteine and glutathione which is also evidenced by color changes of the reaction mixtures (see photograph in the ESI†) distinct for chloride ligand substitution with N-ligand (orange→yellow) in the case of His and with S-ligand (orange→brown) in the case of both Cys and GSH. In the case of His the reaction is quick and complete in few minutes as the starting complex 1B is not observed in the first recorded spectrum (after approx. 4 minutes). In the case of Cys and GSH, NMR shows the formation of multiple species however even after 24 hours the predominant species is the starting complex 1B. We must keep in mind that in biological media the complexes are in the presence of a number of competitive ligands (small molecules and macromolecules) and most importantly high extracellular chloride ion concentrations which generally slow down or, as in the case of cisplatin, even prevent the first aquation step which in turn allows DNA- and protein-binding. Moreover, it is now widely accepted that the interaction with DNA is crucial for the mode of action of cisplatin but it is interesting to note that it was determined that only a small amount (0.33%) of cisplatin that enters the cell is bound to genomic DNA while the majority of the intracellular platinum is bound to proteins or small molecules.2 These model reactions thus serve to substantiate and support the proposed binding mode to the AKR1C enzymes which is described below. As expected, our compounds react at various extent and rates with the ligands present in biological systems as was also observed in many other metal based drugs.
The ruthenium complexes 1A and 1B inhibit AKR1C1–AKR1C3 enzymes
Four ruthenium complexes (1A, 1B, 2A, 2B) and two ligands (LA, LB) were evaluated as inhibitors of the recombinant enzymes AKR1C1, AKR1C2 and AKR1C3. The kinetic measurements (Table 1 and ESI Fig. 1–3†) revealed that the 2-hydroxypyridine-N-oxide ligand LA, is inactive, however, when complexed to an organoruthenium species, the synthesized Ru complex 1A, is a very potent inhibitor of all three AKR1C enzymes, with the highest potency against AKR1C1. On the other hand, its sulfur analogue LB alone inactivates all enzymes, again most efficiently the AKR1C1 enzyme. Similarly, its organoruthenium complex (1B) inhibits all three AKR1C enzymes, with the highest activity seen for AKR1C1. Interestingly, the trithiacyclononane-ruthenium complexes 2A and 2B show only a moderate inhibitory potency which is however selective for the AKR1C1 enzyme as AKR1C2 and AKR1C3 are not affected even at 0.1 mM concentration.| AKR1C1 | AKR1C2 | AKR1C3 | |
|---|---|---|---|
| The constants with added standard errors were fitted from progress curves (ESI Tables 1–3) according to the reaction in Scheme 2.a These ratios were calculated; n.i. – no inhibition. | |||
| LA | |||
| n.i. | n.i. | n.i. | |
| LB | |||
| k 6 | 288.0 ± 1.6 M−1 s−1 | 7.8 ± 0.1 M−1 s−1 | 50.0 ± 0.2 M−1 s−1 |
| 1A | |||
| k 6 | 890 ± 4 M−1 s−1 | 20.3 ± 0.2 M−1 s−1 | — |
| k 7 | 3.3 ± 0.7 × 10−4 s−1 | 3.0 ± 0.3 × 10−4 s−1 | — |
| k 7/k6 | 0.37 μMa | 15 μMa | 2.9 ± 0.1 μM |
| k 8 | 96 ± 25 M−1 s−1 | 4.5 ± 0.3 M−1 s−1 | 8.0 ± 0.1 M−1 s−1 |
| 1B | |||
| k 6 | 21500 ± 470 M−1 s−1 |
129 ± 1 M−1 s−1 | — |
| k 7 | 17.8 ± 0.5 × 10−3 s−1 | 1.7 ± 0.1 × 10−3 s−1 | — |
| k 7/k6 | 0.83 μMa | 13 μMa | 1.80 ± 0.03 μM |
| k 8 | 579 ± 10 M−1 s−1 | 208 ± 14 M−1 s−1 | 22.9 ± 0.3 M−1 s−1 |
| 2A | |||
| k 7/k6 | 3.82 ± 0.06 μM | n.i. | n.i. |
| k 8 | 8.1 ± 0.1 M−1 s−1 | n.i. | n.i. |
| 2B | |||
| k 6 | 55.8 ± 0.4 M−1 s−1 | n.i. | n.i. |
We can compare the potencies of these compounds for the first, fast reversible step, i.e. binding to the holoenzyme, which is characterized by the rate constants k6 and k7, where Ki = k7/k6, and the second slow irreversible step of inhibition, which is characterized by the rate constant k8 (Scheme 2, Table 1). The lowest values of dissociation constants (Ki) were determined for the reversible inhibition of AKR1C1 by complexes 1A (0.37 μM) and 1B (0.83 μM) with about 2–8-fold higher Ki for AKR1C3 and 16–40-fold higher Ki values for AKR1C2. The complex 2A is about 10-fold less potent against AKR1C1 (Ki = 3.8 μM). When we look at the slow irreversible step of inhibition; the complex 1B has the highest k8 for the inactivation of AKR1C1, followed by 3-fold and 25-fold lower k8 values for AKR1C2 and AKR1C3. Complex 1A is a less potent irreversible inhibitor with a 6-fold lower k8 value for AKR1C1 as compared to 1B and 21-fold and 12-fold lower potencies for AKR1C2 and AKR1C3, respectively. Interestingly, compounds LB and 2B show only slow irreversible inactivation characterized by rate constant k6 (Table 1) with the highest values determined for the inhibition of AKR1C1.
Our kinetic measurements thus demonstrated that among the four ruthenium complexes tested, complexes 1A and 1B inhibit all three recombinant human AKR1C enzymes in vitro, with higher potencies for AKR1C1 and AKR1C3, in a mixed manner, by a rapid reversible inhibition followed by a slow irreversible inactivation (Table 1). Visualization of 1B complex in the crystal structure of AKR1C1 revealed that the binding to the active site and the peripheral site is sterically possible (Fig. 3), thus supporting their reversible and irreversible modes of inhibition. On the other hand, 9aneS3 analogues 2A and 2B only affect AKR1C1 with much lower potencies and act in mixed reversible/irreversible and irreversible manners, respectively. For the two ligands it turned out that LB, pyrithione, inactivated all three isoenzymes with irreversible mode of inhibition, while the oxygen analog LA was inactive.
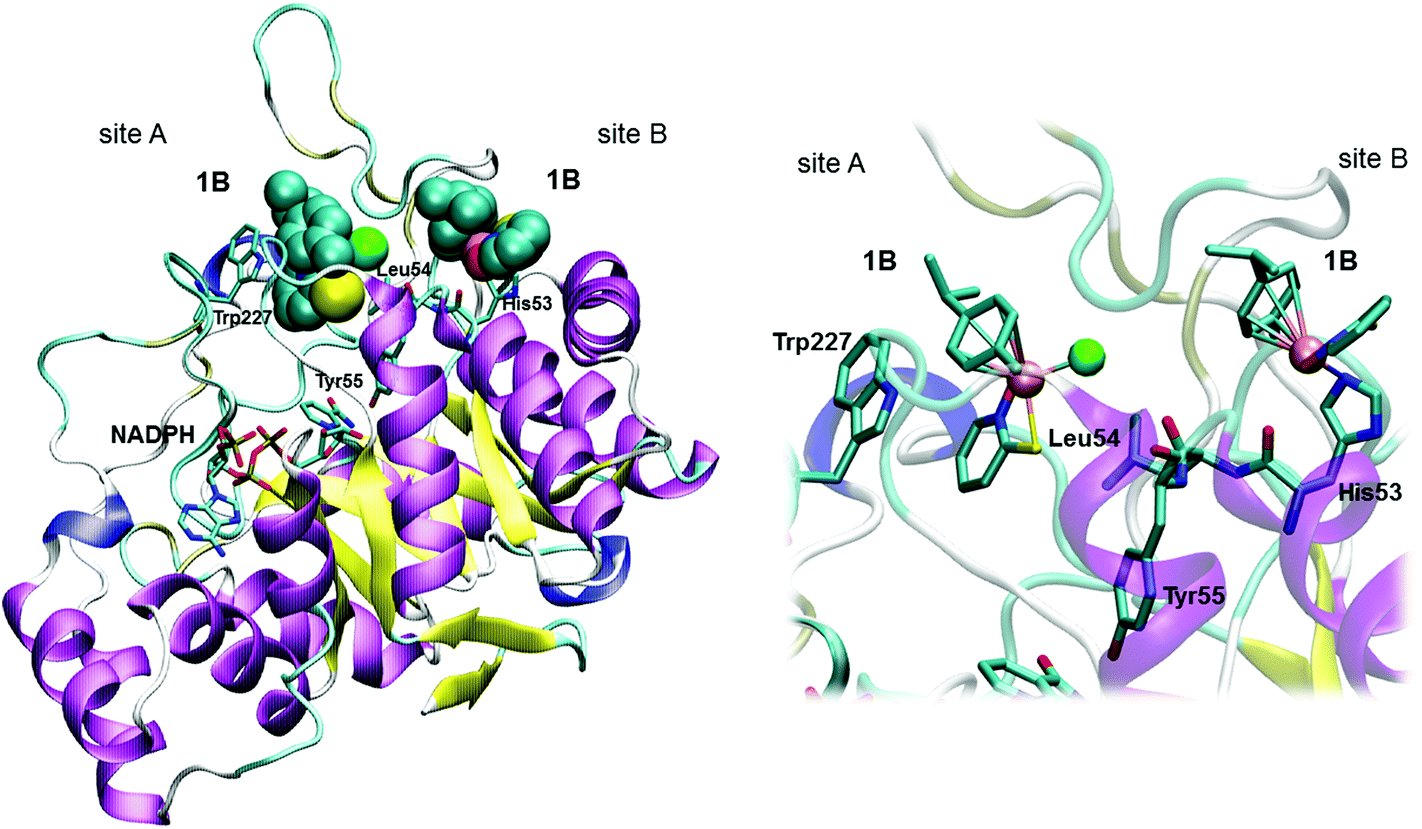 | ||
| Fig. 3 Visualization of 1B binding to AKR1C1 enzyme. Left – complete protein structure with a spacefill model of 1B; right – close-up view of the 1B binding site. Binding of Ru complex 1B can be explained by the rapid competitive inhibition of the first Ru containing molecule to the holoenzyme (Site A), followed by slow substitution of Cl− from the second Ru complex (via aqua species) by a protein nucleophilic side chain in the vicinity of the active site, putatively His53 (Site B). Coenzyme NADPH, the catalytic Tyr55, and His53, Leu54 and Trp227 together with two molecules of 1B are depicted. | ||
Numerous studies show37,38,47–50 that the chlorido ligands undergo instantaneous hydrolysis and formation of the respective aqua species allowing direct interaction between the metal centre and target molecules. On the other hand the hydrolysis of the S-bound dmso ligand is generally very slow and only in marginal fractions (in our case not observed in D2O solution). These facts support our experimental data revealing the higher inhibitory potential of the series 1 complexes for slow quasi-irreversible inhibition according to the proposed mechanism (Scheme 2). With respect to the chelating ligands, O,O-ligands are known to form less stable complexes which slowly (partially) decompose in aqueous solution forming the biologically inactive and very stable hydroxy-bridged dimer [(p-cymene)Ru(μ-OH)3Ru(p-cymene)], while their O,S-analogs remain stable under physiological conditions.35,36,38,49–51 This is also in agreement with the observed higher inhibitory potencies of B series compounds.
The shape of the progress curves for the oxidation of the artificial racemic substrate 1-acenaphthenol catalyzed by the AKR1C enzymes in the absence and presence of active Ru complexes (ESI Fig. 1–3†) revealed a double inhibition character, summarized in Scheme 2: the initial parts of the curves are characteristic for rapid reversible inhibition (k7/k6) and the lower plateaus in the presence of Ru compounds indicate slow irreversible enzyme inactivation (represented by k8). These two steps can only be explained as a subsequent binding of two inhibitor molecules to the two different sites on the enzyme. One plausible explanation for such a behavior under our experimental conditions would be a rapid competitive inhibition of the first Ru containing molecule to the holoenzyme, followed by slow substitution of S-dmso/Cl−/H2O from the second inhibitor molecule by a protein nucleophilic side chain in the vicinity of the active site. A suitable candidate in AKR1C enzymes would be His53 (PDB code 3NTY) which is on the surface, but only two residues away from the main catalytic Tyr55. A quasi-irreversible substitution of Cl− in the Ru complex (via aqua species) by Nε of this histidine would restrict loop movements needed for coenzyme exchange and/or displace the catalytic Tyr55 (Fig. 3). It should be stressed at this point that a similar double binding of a Ru compound and its adduct has already been shown by X-ray crystallography of lysozyme (PDB code 4W94).52 In contrast, to reproduce the curves in the presence of pyrithione (LB) and 2B only one slow irreversible step is needed (k6), since the initial rate is inhibitor independent.
The complexes 1A and 1B show similar potencies for the reversible inhibition of AKR1C enzymes, while the complex 1B was a more efficient inhibitor as compared to complex 1A in an irreversible manner. This observation can be explained by a better positioning of 1B within the peripheral site. It seems that the bulky S atom enables appropriate accommodation of the B series compounds and thus their stabilization by coordination with His53 and effective inhibition of AKR1C enzymes. Importantly, compounds 1A and 1B show 41-fold and 16-fold higher potencies for reversible inhibition of AKR1C1 compared to AKR1C2, which differs from AKR1C1 in only 7 amino acids, of which only one is in the active site (Leu54/Val54). The complexes 1A and 1B are also 5.2-fold and 7.2-fold more potent inhibitors of AKR1C3, which also includes Leu54, compared to AKR1C2. It seems that the specificity of compounds 1A and 1B for binding into the active site of AKR1C1 and AKR1C3 enzymes may be explained by favorable hydrophobic interactions between Leu54 and the pyridine ligand (Fig. 3). These interactions are weaker in AKR1C2 possessing Val54 with a shorter aliphatic side chain.
Ruthenium complexes are known to bind to plasma proteins, especially to the most abundant albumin,53 which led us to evaluate the inhibition of AKR1C enzymes by Ru complexes in the presence of different concentrations of albumin. The most potent reversible/irreversible inhibitor of AKR1C1, compound 1B, was selected for these studies. The original reaction scheme (Scheme 2) for the oxidation and inhibition of acenaphthenol in the presence of a ruthenium compound was expanded by adding subsequent steps for the binding of two molecules of ruthenium compound 1B to albumin (Scheme S1†). Further analysis of progress curves (Scheme S4†) using the ENZO web application revealed that 1B binds to albumin in a reversible manner with dissociation constants of 5.53 μM and 1.76 μM. Although compound 1B binds to AKR1C1 with higher affinity compared to albumin (0.83 μM Ki value versus 5.53 μM and 1.76 μM Kd values), the major difference lies in the quasi-irreversible binding of the second 1B molecule in the case of the target enzyme AKR1C1. The physiological relevance of this irreversible inhibition depends on the availability of the free (non-protein-bound) Ru complexes, similarly as for cisplatin, which binds to albumin, but still affects the target molecules.2
Interestingly, the suggested binding of two 1B molecules to albumin corroborates the binding of cisplatin molecules to His105 and Met329 side chains on the protein surface, as recently determined crystallographically (PDB code 4S1Y54).
As AKR1C enzymes are associated with resistance to a variety of anti-cancer agents, and also have crucial roles in the pathophysiology of a variety of cancers, the inhibitors of these enzymes are highly sought after.4 The inhibitors of AKR1C enzymes reported so far act in reversible manners with the lowest Ki values reported in the low nM range.55 Although the Ru complexes reported in this study are about 100-fold less efficient as reversible inhibitors, they show high potency as irreversible inhibitors and show a relatively high level of specificity for AKR1C1 and AKR1C3 compared to AKR1C2.
The ruthenium complex 1B has cytotoxic effects on breast cancer cell line MCF-7
We also investigated the effect of these six compounds (1A, 1B, 2A, 2B, LA, and LB) on the proliferation and viability of a model cell line of hormone-dependent breast cancer, MCF-7. All the tested compounds had dose-dependent effects on the MCF-7 cell proliferation (Fig. 4). Complexes 1B and 2B and the corresponding ligand LB show cytotoxic effects on MCF-7, where compounds 1B and LB have similar EC50 values, 3.8 and 3.2 μM, respectively, whereas compound 2B has EC50 above 200 μM. Interestingly, the complexes 1A and 2A show a trend of increased proliferation of MCF-7 cells, whereas the corresponding ligand LA slightly inhibits proliferation above 300 μM concentration.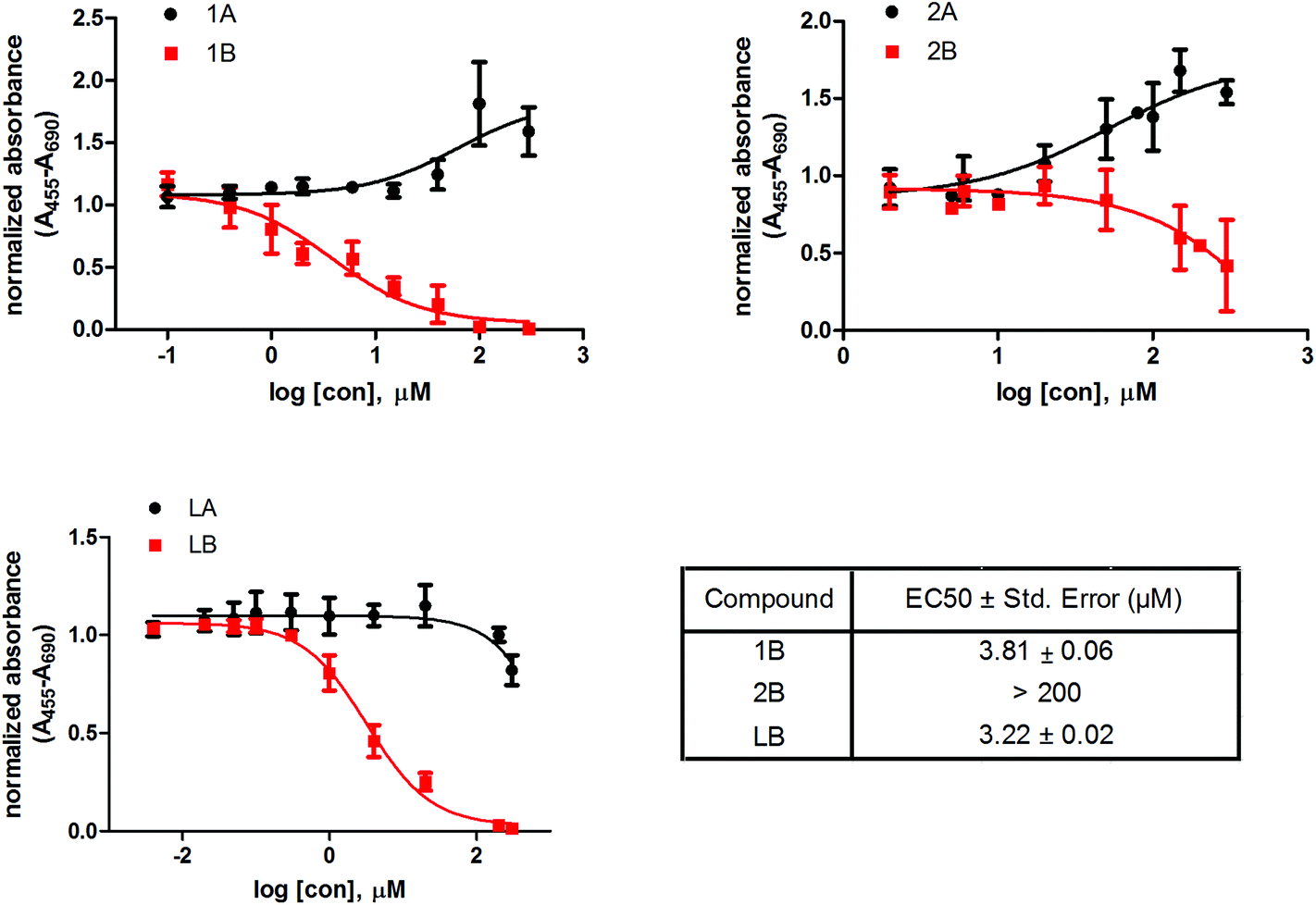 | ||
| Fig. 4 Effect of ruthenium compounds and free ligands on the proliferation and viability of the MCF-7 cell line. Ru complexes (1A, 1B, 2A, 2B) and two ligands (LA, LB) were tested at different concentrations. After the 48 h incubation period, the proliferation was measured using WST-1 assay. The results are expressed as mean ± SD (n ≥ 3). | ||
The cell proliferation study reveals that compounds with ligand LB, which bear sulfur (1B and 2B) as well as ligand LB by itself have cytotoxic effects on the model cell line of hormone dependent breast cancer, MCF-7, with more than 50-fold higher cytotoxic effects observed for LB and 1B compared to 2B. In contrast, compounds with ligand LA, which are bearing oxygen, have the opposite effect. The increase in activity by several orders of magnitude upon O→S substitution was previously observed in a number of cases and was mainly attributed to the increased stability of the ruthenium complexes with O,S-chelating ligands in comparison with their O,O-analogs.36,38,49,50 Additionally, the chloride ligands in complex 1B undergo hydrolysis which enhances direct interactions with proteins as well as nucleic acids through coordination at the metal centre which may interfere with cell proliferation. AKR1C enzymes are also expressed in the MCF-7 cell line56 where AKR1C3 catalyzes the formation of the most potent estrogen mitogen estradiol57 while AKR1C1 catalyzes metabolism of progesterone to pro-proliferative 5α-pregnanes.56 Complex 1B potently inhibits both AKR1C1 and AKR1C3 as well as displays high toxicity on MCF-7 hormone dependent breast cancer cell line, which suggests a potential relation of the compound–enzyme interaction and cytotoxicity.
Conclusions
In summary, we have successfully synthesized and characterized two organometallic and two trithiacyclononane-bearing ruthenium coordination compounds of pyrithione (LB) and its oxygen analog 2-hydroxypyridine N-oxide (LA). We have determined their structure in the solid state by means of single crystal X-ray diffraction and evaluated the potential of both ligands and the four ruthenium complexes as prospective anticancer agents. The compounds were tested for the inhibition of three enzymes of the aldo–keto reductase subfamily 1C (AKR1C1–C3), which are involved in the development and/or progression of a plethora of cancers, including breast cancer. A kinetic study assisted with docking simulations revealed that these compounds show different mechanisms of action: (i) combined reversible and irreversible inhibition (1A, 1B, 2A) by binding to the active and peripheral sites, respectively and (ii) irreversible inhibition by binding to the peripheral site (LB, 2B). Our study identified the complexes 1A and 1B as the most potent reversible/irreversible inhibitors of AKR1C enzymes among the six compounds evaluated. Both organometallic complexes 1A and 1B displayed a relatively high degree of selectivity towards AKR1C1 (AKR1C1 > AKR1C3 ≫ AKR1C2), which is a remarkable feature considering only seven amino acid difference between AKR1C1 and AKR1C2 enzymes. To the best of our knowledge Ru complexes reported in this and our previous study41 represent the first quasi-irreversible inhibitors of AKR1C enzymes.Additionally, B series compounds also displayed cytotoxic effects on the model cell line of hormone-dependent breast cancer, MCF-7; where LB and 1B showed EC values in the low μM range, while the ruthenium complex bearing the sulfur macrocycle 9aneS32B had lower cytotoxic effects. On the other hand, A series compounds had no cytotoxic or cytostatic effects. Our results thus support the idea that there might be an association between anticancer activity and AKR1C enzyme inhibition for compound 1B, whereas for other compounds such correlation was not observed. As the organoruthenium pyrithione complex 1B shows potent AKR1C inhibition and high cytotoxic effects on the model breast cancer cells it represents a prime candidate for future research, as a lead compound in molecular design as well as in mechanistic studies, which would help elucidating the role of AKR1C enzymes in carcinogenesis.
Acknowledgements
This study was supported by junior researcher grants to K. T., M. S., the Erasmus grant for E. E. C., the programme grant P1-0175 (I. T.), the postdoctoral research grant Z1-6735 to J. K. and the J3-4135 grant to T. L. R., all from the Slovenian Research Agency. The EN→FIST Centre of Excellence, Dunajska 156, SI-1000 Ljubljana, Slovenia, is acknowledged for the use of the SuperNova diffractometer. The authors thank Dr T. M. Penning (University of Pennsylvania, School of Medicine, Philadelphia, PA, USA) for the pcDNA3-AKR1C1 and pcDNA3-AKR1C2 constructs, and Dr Jerzy Adamski (Helmholtz Zentrum Munchen, German Research Centre for Environmental Health, GmbH, Neuherberg, Germany) for the pGeX-AKR1C3 construct.References
- B. Rosenberg, L. Vancamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS PubMed.
- Bioinorganic Medicinal Chemistry, ed. E. Alessio, Wiley-VCH Verlag & Co. KGaA, Weinheim, Germany, 2011 Search PubMed.
- J. S. Ferlay, I. Soerjomataram, R. Dikshit, S. Eser, C. Mathers, M. Rebelo, D. M. Parkin, D. Forman and F. Bray, Int. J. Cancer, 2015, 136, E359–E386 CrossRef CAS PubMed.
- T. Lanišnik Rižner and T. M. Penning, Steroids, 2014, 79, 49–63 CrossRef PubMed.
- T. M. Penning and J. E. Drury, Arch. Biochem. Biophys., 2007, 464, 241–250 CrossRef CAS PubMed.
- T. M. Penning and M. C. Byrns, Ann. N. Y. Acad. Sci., 2009, 1155, 33–42 CrossRef CAS PubMed.
- H. B. Deng, M. Adikari, H. K. Parekh and H. Simpkins, Cancer Chemother. Pharmacol., 2004, 54, 301–307 CrossRef CAS PubMed.
- J. Chen, M. Adikari, R. Pallai, H. K. Parekh and H. Simpkins, Cancer Chemother. Pharmacol., 2008, 61, 979–987 CrossRef CAS PubMed.
- C.-C. Chen, C.-B. Chu, K.-J. Liu, C.-Y. F. Huang, J.-Y. Chang, W.-Y. Pan, H.-H. Chen, Y.-H. Cheng, M.-F. Lee, C.-C. Kuo and L.-T. Chen, Biochem. Pharmacol., 2013, 86, 872–887 CrossRef CAS PubMed.
- W. Q. Ding and S. E. Ling, IUBMB Life, 2009, 61, 1013–1018 CrossRef CAS PubMed.
- C. J. Chandler and I. H. Segel, Antimicrob. Agents Chemother., 1978, 14, 60–68 CrossRef CAS PubMed.
- P. Krejčová, P. Kučerová, G. I. Stafford, A. K. Jäger and R. Kubec, J. Ethnopharmacol., 2014, 154, 176–182 CrossRef PubMed.
- D. P. Martin, P. G. Blachly, J. A. McCammon and S. M. Cohen, J. Med. Chem., 2014, 57, 7126–7135 CrossRef CAS PubMed.
- M. Qiu, Y. Chen, Y. Chu, S. Song, N. Yang, J. Gao and Z. Wu, Antiviral Res., 2013, 100, 44–53 CrossRef CAS PubMed.
- M. Tailler, L. Senovilla, E. Lainey, S. Thépot, D. Métivier, M. Sébert, V. Baud, K. Billot, P. Fenaux, L. Galluzzi, S. Boehrer, G. Kroemer and O. Kepp, Oncogene, 2012, 31, 3536–3546 CrossRef CAS PubMed.
- E. Rudolf and M. Červinka, BioMetals, 2010, 23, 339–354 CrossRef CAS PubMed.
- A. A. Nazarov, S. M. Meier, O. Zava, Y. N. Nosova, E. R. Milaeva, C. G. Hartinger and P. J. Dyson, Dalton Trans., 2015, 44, 3614–3623 RSC.
- M. Schmidlehner, L. S. Flocke, A. Roller, M. Hejl, M. A. Jakupec, W. Kandioller and B. K. Keppler, Dalton Trans., 2016, 45, 724–733 RSC.
- A. Bergamo and G. Sava, Dalton Trans., 2011, 40, 7817–7823 RSC.
- L. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC.
- S. Leijen, S. A. Burgers, P. Baas, D. Pluim, M. Tibben, E. Van Werkhoven, E. Alessio, G. Sava, J. H. Beijnen and J. H. M. Schellens, Invest. New Drugs, 2015, 33, 201–214 CrossRef CAS PubMed.
- J. Du, E. L. Zhang, Y. Zhao, W. Zheng, Y. Zhang, Y. Lin, Z. Y. Wang, Q. Luo, K. Wu and F. Y. Wang, Metallomics, 2015, 7, 1573–1583 RSC.
- H. Q. Lai, Z. N. Zhao, L. L. Li, W. J. Zheng and T. F. Chen, Metallomics, 2015, 7, 439–447 RSC.
- J. Yuan, Z. N. Lei, X. Wang, F. Zhu and D. B. Chen, Metallomics, 2015, 7, 896–907 RSC.
- Q. Wu, J. T. He, W. J. Mei, Z. Zhang, X. H. Wu and F. Y. Sun, Metallomics, 2014, 6, 2204–2212 RSC.
- R. Hudej, I. Turel, M. Kandušer, J. Ščančar, S. Kranjc, G. Serša, D. Miklavčič, M. A. Jakupec, B. K. Keppler and M. Čemažar, Anticancer Res., 2010, 30, 2055–2063 CAS.
- R. Hudej, D. Miklavčič, M. Čemažar, V. Todorovič, G. Serša, A. Bergamo, G. Sava, A. Martinčič, J. Ščančar, B. K. Keppler and I. Turel, J. Membr. Biol., 2014, 247, 1239–1251 CrossRef CAS PubMed.
- C. M. Clavel, P. Nowak-Sliwinska, E. Paunescu, A. W. Griffioen and P. J. Dyson, Chem. Sci., 2015, 6, 2795–2801 RSC.
- S. Betanzos-Lara, L. Salassa, A. Habtemariam, O. Novakova, A. M. Pizarro, G. J. Clarkson, B. Liskova, V. Brabec and P. J. Sadler, Organometallics, 2012, 31, 3466–3479 CrossRef CAS.
- A. Habtemariam, M. Melchart, R. Fernandez, S. Parsons, I. D. H. Oswald, A. Parkin, F. P. A. Fabbiani, J. E. Davidson, A. Dawson, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2006, 49, 6858–6868 CrossRef CAS PubMed.
- S. B. Murray, V. M. Babak, G. C. Hartinger and J. P. Dyson, Coord. Chem. Rev., 2016, 306, 86–114 CrossRef.
- S. Seršen, J. Kljun, K. Kryeziu, R. Panchuk, B. Alte, W. Körner, P. Heffeter, W. Berger and I. Turel, J. Med. Chem., 2015, 58, 3984–3996 CrossRef PubMed.
- H. Bregman, P. J. Carroll and E. Meggers, J. Am. Chem. Soc., 2006, 128, 877–884 CrossRef CAS PubMed.
- J. Kljun, A. J. Scott, T. Lanišnik Rižner, J. Keiser and I. Turel, Organometallics, 2014, 33, 1594–1601 CrossRef CAS.
- J. Kljun, I. Bratsos, E. Alessio, G. Psomas, U. Repnik, M. Butinar, B. Turk and I. Turel, Inorg. Chem., 2013, 52, 9039–9052 CrossRef CAS PubMed.
- R. Hudej, J. Kljun, W. Kandioller, U. Repnik, B. Turk, C. G. Hartinger, B. K. Keppler, D. Miklavčič and I. Turel, Organometallics, 2012, 31, 5867–5874 CrossRef CAS.
- A. Rilak, I. Bratsos, E. Zangrando, J. Kljun, I. Turel, Z. D. Bugarčić and E. Alessio, Dalton Trans., 2012, 41, 11608–11618 RSC.
- J. Kljun, A. K. Bytzek, W. Kandioller, C. Bartel, M. A. Jakupec, C. G. Hartinger, B. K. Keppler and I. Turel, Organometallics, 2011, 30, 2506–2512 CrossRef CAS PubMed.
- I. Turel, J. Kljun, F. Perdih, E. Morozova, V. Bakulev, N. Kasyanenko, J. A. W. Byl and N. Osheroff, Inorg. Chem., 2010, 49, 10750–10752 CrossRef CAS PubMed.
- A. Martinčič, M. R. J. Vidmar, I. Turel, B. K. Keppler and J. Ščančar, J. Chromatogr. A, 2014, 1371, 168–176 CrossRef PubMed.
- K. Traven, M. Sinreih, J. Stojan, S. Seršen, J. Kljun, J. Bezenšek, B. Stanovnik, I. Turel and T. Lanišnik Rižner, Chem.-Biol. Interact., 2015, 234, 349–359 CrossRef CAS PubMed.
- I. Bratsos, E. Mitri, F. Ravalico, E. Zangrando, T. Gianferrara, A. Bergamo and E. Alessio, Dalton Trans., 2012, 41, 7358–7371 RSC.
- P. Brožič, T. Šmuc, S. Gobec and T. Lanišnik Rižner, Mol. Cell. Endocrinol., 2006, 259, 30–42 CrossRef PubMed.
- J. Lee, X. Cheng, J. M. Swails, M. S. Yeom, P. K. Eastman, J. A. Lemkul, S. Wei, J. Buckner, J. C. Jeong, Y. Qi, S. Jo, V. S. Pande, D. A. Case, C. L. Brooks III, A. D. MacKerell Jr., J. B. Klauda and W. Im, J. Chem. Theory Comput., 2016, 12, 405–413 CrossRef CAS PubMed.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- T. Lanišnik Rižner, Front. Pharmacol., 2012, 3, 34 Search PubMed.
- A. F. A. Peacock, M. Melchart, R. J. Deeth, A. Habtemariam, S. Parsons and P. J. Sadler, Chem. – Eur. J., 2007, 13, 2601–2613 CrossRef CAS PubMed.
- F. Y. Wang, A. Habtemariam, E. P. L. Van der Geer, R. Fernandez, M. Melchart, R. J. Deeth, R. Aird, S. Guichard, F. P. A. Fabbiani, P. Lozano-Casal, I. D. H. Oswald, D. I. Jodrell, S. Parsons and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18269–18274 CrossRef CAS PubMed.
- W. Kandioller, C. G. Hartinger, A. A. Nazarov, M. L. Kuznetsov, R. O. John, C. Bartel, M. A. Jakupec, V. B. Arion and B. K. Keppler, Organometallics, 2009, 28, 4249–4251 CrossRef CAS.
- W. Kandioller, C. G. Hartinger, A. A. Nazarov, C. Bartel, M. Skocic, M. A. Jakupec, V. B. Arion and B. K. Keppler, Chem. – Eur. J., 2009, 15, 12283–12291 CrossRef CAS PubMed.
- I. Bratsos, S. Jedner, A. Bergamo, G. Sava, T. Gianferrara, E. Zangrando and E. Alessio, J. Inorg. Biochem., 2008, 102, 1120–1133 CrossRef CAS PubMed.
- H. Tabe, K. Fujita, S. Abe, M. Tsujimoto, T. Kuchimaru, S. Kizaka-Kondoh, M. Takano, S. Kitagawa and T. Ueno, Inorg. Chem., 2015, 54, 215–220 CrossRef CAS PubMed.
- J. Sun, Y. Huang, C. Zheng, Y. Zhou, Y. Liu and J. Liu, Biol. Trace Elem. Res., 2015, 163, 266–274 CrossRef CAS PubMed.
- G. Ferraro, L. Massai, L. Messori and A. Merlino, Chem. Commun., 2015, 51, 9436–9439 RSC.
- O. El-Kabbani, U. Dhagat and A. Hara, J. Steroid Biochem. Mol. Biol., 2011, 125, 105–111 CrossRef CAS PubMed.
- J. P. Wiebe and M. J. Lewis, BMC Cancer, 2003, 3, 9 CrossRef PubMed.
- M. C. Byrns, L. Duan, S. H. Lee, I. A. Blair and T. M. Penning, J. Steroid Biochem. Mol. Biol., 2010, 118, 177–187 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1444456–1444459. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00668j |
| This journal is © The Royal Society of Chemistry 2016 |