
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A ruthenium racemisation catalyst for the synthesis of primary amines from secondary amines†
Dennis
Pingen
a,
Çiğdem
Altıntaş‡
c,
Max
Rudolf Schaller‡
d and
Dieter
Vogt
*b
aChemical Materials Science, Department of Chemistry, University of Konstanz, Universitätsstrasse 10, 78457 Konstanz, Germany
bEaStCHEM, School of Chemistry, University of Edinburgh, King's Buildings, Joseph Black Building, West Mains Road, Edinburgh EH9 3JJ, Scotland, UK. E-mail: D.Vogt@ed.ac.uk
cDepartment of Chemical & Biological Engineering, Koç University Rumelifeneri Yolu, Sariyer 34450 Istanbul, Turkey. E-mail: caltintas@ku.edu.tr
dFraunhofer-Institut für Keramische Technologien und Systeme IKTS, Winterbergstraße 28, 01277 Dresden, Germany. E-mail: max.rudolf.schaller@ikts.fraunhofer.de
First published on 10th June 2016
Abstract
A Ru-based half sandwich complex used in amine and alcohol racemization reactions was found to be active in the splitting of secondary amines to primary amines using NH3. Conversions up to 80% along with very high selectivities were achieved. However, after about 80% conversion the catalyst lost activity. Similar to Shvo's catalyst, the complex might deactivate under the influence of ammonia. It was revealed that not NH3 but mainly the primary amine is responsible for the deactivation.
Introduction
Primary amines are valuable building blocks in industrial chemistry; they are the main building blocks for a large variety of polymers, surfactants, corrosion inhibitors and fine-chemicals.1,2 For the production of primary amines, waste-free, selective protocols are highly desired with special emphasis on renewable resources such as bio-alcohols.3 However, current industrial (heterogeneously catalysed) syntheses of amines from alcohols inevitably give mixtures of primary, secondary, and tertiary amines.4 Current homogeneously catalysed alcohol amination reactions were shown to be very selective towards primary amines, barely producing any secondary amines. Although the current homogeneous catalysts, developed by Milstein,5 Beller6 and Vogt7 are very selective towards primary amines, anticipation on the industrial synthesis of primary amines also requires the development of catalysts for the splitting of secondary (and tertiary) amines. Reusing secondary amines in the synthesis of primary amines will reduce the waste stream in the total production. At the moment, only one example of a homogeneous catalyst has been reported that efficiently catalysed the splitting of secondary and tertiary amines.8The splitting of secondary amines is expected to proceed via the ‘Hydrogen Shuttling’ concept; dehydrogenation of the secondary amine forming the dialkylimine. This subsequently undergoes nucleophilic attack from NH3 resulting in primary amine and primary imine. The primary imine will then be hydrogenated to produce another equivalent of primary amine (Scheme 1).
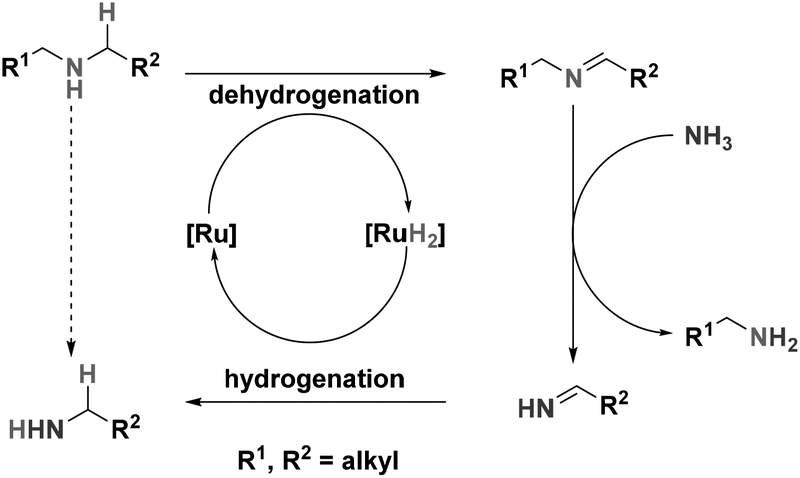 | ||
| Scheme 1 ‘Hydrogen Shuttling’ in the splitting of secondary amines to primary amines. | ||
The system reported by Beller and coworkers uses Shvo's catalyst in the splitting of secondary and tertiary amines. This catalyst has previously been used in various reactions, one of which is amine racemization.9 As the racemization of amines by Shvo's catalyst and related systems proceeds via initial dehydrogenation of the amine,10 it is anticipated that similar systems will also be active in the splitting of amines.
Results and discussion
Ruthenium half sandwich complexes bearing a pentaphenylcyclopentadienyl (CpPh5) moiety (Fig. 1) were investigated in the splitting of secondary amines with ammonia. Complexes 1–3 have been employed before in the racemization of alcohols.9–12 In addition, complex 1 was also used in the racemization of amines.9,10 Complex 2 did show activity as well, albeit a long induction period was noticed before activity was observed.11 Complex 3 was developed as a more electron rich variant of complex 1 but was only active in alcohol racemization.12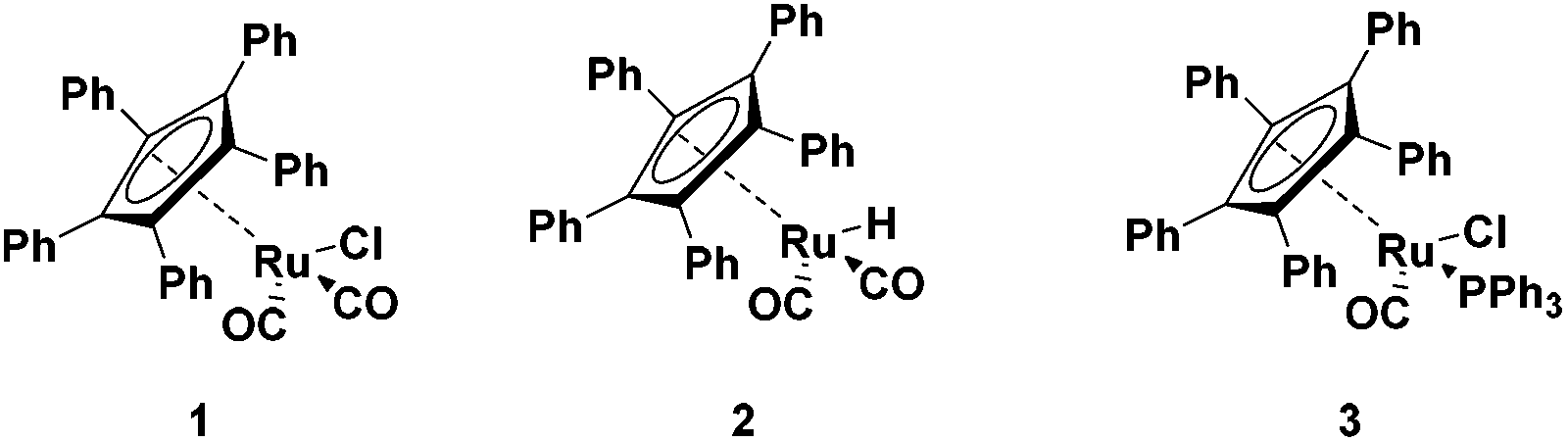 | ||
| Fig. 1 Half sandwich Ru(CpPh5) complexes investigated in this study. | ||
Initial reactions with complexes 1 and dicyclohexylamine as model substrate were performed employing a catalyst loading of 2 mol% at 150 °C (ESI Table S5†). Under these conditions only 40% conversion could be achieved (99% selectivity, see ESI†). Increasing the temperature to 170 °C significantly improved the conversion without affecting the selectivity (Table 1). As it can be anticipated that the excess of NH3 applied will have significant influence on the amine splitting, the amount of NH3 was varied.
| Entry | Complex | Time (h) | NH3(l) equiv. | Conv.a (%) | Yield prim. aminea (%) | Prim. amine selectivity (%) |
|---|---|---|---|---|---|---|
| Complex (2 mol%), dicyclohexylamine (1.5 mmol), tert-amyl alcohol (3 mL), NH3(l), 170 °C.a Standard deviation over 3 experiments.b 0.75 mL MTBE as co-solvent.c 2 mol% KOtBu added. | ||||||
| 1 | 1 | 24 | 120 | 81.5 ± 5 | 76.5 ± 4 | 94 |
| 2 | 2 | 24 | 120 | 22.5 ± 2 | 22.5 ± 2 | 100 |
| 3 | 3 | 24 | 120 | 26.5 ± 2 | 25 ± 2 | 94 |
| 4 | 1 + KOtBuc | 23.75 | 120 | 0 | 0 | 0 |
| 5 | 1 | 21 | 12 | 54 ± 3 | 53 ± 3 | 98 |
| 6 | 2 | 21 | 12 | 13 ± 1 | 11 ± 1 | 85 |
| 7 | 3 | 21.5 | 12 | 68.5 ± 4 | 62 ± 3 | 90 |
| 8 | 1 + KOtBuc | 21.5 | 12 | 23.5 ± 2 | 23.5 ± 2 | 100 |
Complex 1 is the most active with 120 eq. of NH3. Up to 80% conversion and a selectivity of 94% to the primary amine was achieved (entry 1). Complexes 2 and 3 both showed low activity under these conditions (entries 2 and 3). The situation was different at a 10-fold lower excess of NH3 (12 eq., entries 5–7). Now complex 3 showed the highest conversion of 68% and complex 1 still gave reasonable conversion (54%). Complex 2 was the least active under both conditions (entries 2 and 6). The addition of base has been shown to be beneficial in activating complex 1 in the alcohol racemization.13 However, addition of KOtBu with 120 eq. of NH3 completely deactivated the catalyst (entry 4), while with 12 eq. of NH3 still some conversion was achieved; though lower than without the addition of base (entry 8).
The results reported in Table 1 confirmed the expected strong effect of the excess amount of ammonia on the performance in catalysis. Complex 1 appeared to be the most active and therefore, the effect of the NH3 excess was investigated in more detail for this complex (Table 2).
| Entry | Time (h) | NH3 (l) equiv. | Conversiona (%) | Yield prim. aminea (%) | Prim. amine selectivity |
|---|---|---|---|---|---|
| Complex 1 (2 mol%), dicyclohexylamine (1.5 mmol), tert-amyl alcohol (3 ml), NH3(l), 170 °C.a Standard deviation over 3 experiments. | |||||
| 1 | 23.5 | 120 | 81.5 ± 5 | 76.5 ± 4 | 94 |
| 2 | 21 | 60 | 82.5 ± 3 | 78 ± 2 | 94 |
| 3 | 23.5 | 40 | 61 ± 3 | 58.5 ± 3 | 96 |
| 4 | 24 | 20 | 51 ± 3 | 47.5 ± 2 | 94 |
| 5 | 21 | 12 | 54 ± 3 | 53 ± 2 | 98 |
| 6 | 23.5 | 4 | 16.5 ± 1 | 15.5 ± 1 | 95 |
The data in Table 2 confirm that a large excess of NH3 is required (60–120 eq.) in order to achieve good conversion. However, it is worth noting that the selectivity was very high (>94%) in all cases, even at very low excess of NH3. Under the optimised conditions complex 1 was now used for a range of substrates (Table 3).
| Entry | Substrate | NH3 (l) (eq.) | Conv.a (%) | Prim. amine (%)/selec.a (%) | Other (imine/secondary imine/tertiary amine/nitrile) |
|---|---|---|---|---|---|
| Complex 1 (2 mol%, 0.03 mmol), substrate (1.5 mmol), t-amylalcohol (3 mL), NH3 (2.5 mL for 60 eq., 5 mL for 120 eq.), 170 °C, 23.5 h.a Standard deviation over 3 experiments.b Based on cyclohexylamine.c Based on aniline.d Single experiments, not in triplo. | |||||
| 1 | Dicyclohexylamine | 60 | 82.5 ± 3 | 78 ± 2/94 | 5% dicyclohexylimine |
| 2 | N-Methyl cyclohexyl-amineb | 60 | 73 ± 2 | 58 ± 2/79 | 15.5 dicyclohexylamine |
| 3 | N-Isopropyl cyclohexylamineb | 60 | 81 ± 5 | 74 ± 4/91 | 7.5% dicyclohexylamine |
| 4 | Dihexylamine | 60 | 43.5 ± 1 | 25 ± 1/56 | 11% trihexylamine, 8.5% hexanenitrile |
| 5 | Dioctylamine | 60 | 80 ± 4 | 0/0 | 43.5% dioctylimine, 20% dioctylenamine, 16% octanenitrile, |
| 6 | Dioctylamine | 120 | 50 ± 4 | 30 ± 3/60 | 4.5% octylimine, 12.5% dioctylimine, 3% octanenitrile |
| 7 | Dibenzylamine | 120 | 25 ± 3 | 19 ± 4/76 | 3% dibenzylimine, 2.5% benzonitrile, |
| 8 | Dibenzylamine | 60 | 23 ± 2 | 0/0 | 2.5% benzylimine, 17.5% dibenzylimine, 3% benzonitrile, |
| 9 | N-Methylanilinec,d | 120 | 2.5 | 1/39 | 1.5% methyleneaniline |
| 10 | N-Methylanilinec,d | 60 | 2 | 1.5/75 | 0.5% methyleneaniline |
| 11 | Trioctylamined | 120 | 60 | 13.5/22 | 43% dioctylamine, 3.5% octanenitrile |
| 12 | Trihexylamined | 120 | 67.5 | 12/17 | 2.5% hexylimine, 41.5% dihexylimine, 8% dihexylamine, 3.5% hexanenitrile |
For amines bearing bulkier secondary alkyl substituents the conversion and selectivity was generally high (entries 1–3). Linear secondary amines resulted in lower conversion but also lower selectivity (entries 4–6). The lower selectivity can be in part attributed to the formation of nitriles as a side reaction. Surprisingly, dioctylamine initially gave no primary amine, mainly secondary imine (entry 5). Increasing the excess of NH3 resulted in lower conversion but gave primary amine in reasonable selectivity (entry 6). Dibenzylamine also resulted in low conversion, whereas a higher NH3 loading also appeared to be beneficial for the selectivity (entries 7 and 8). In case of N-methylaniline, barely any conversion was observed. A larger excess of NH3 brought no improvement (entries 9 and 10). Reacting tertiary amines also leads to fairly low selectivity; the substrate has to undergo 2 steps before the final product can be formed. Moreover, it is likely that dehydrogenation of a tertiary amine is more difficult as it has to proceed either via the intermediate enamine or iminium species, which makes the formation of the secondary amine or imine more difficult.
As the results show, the maximum conversion reached was limited in all cases, which might be due to catalyst deactivation. In fact Beller and co-workers have shown earlier that the related Shvo's catalyst was inhibited by the coordination of NH3 as well as by primary amines.14 They showed that this inhibition became partially reversible at higher temperature. However, even at 150 °C or higher, conversions were generally lower than 85%.8 In our case for complex 1 increasing the reaction temperature to 170 °C did increase the conversion, though never reaching more than 80%. Hence we conclude that we are dealing with a different cause of deactivation and decided to study this in more detail. In order to see if we are dealing with product inhibition by primary amines, 20 mol% of cyclohexylamine were added at the start and the reaction was monitored in time (Fig. 2). The reaction was significantly slower and gave approximately 15–20% lower conversion.
 | ||
| Fig. 2 Catalyst inhibition by primary amine (A) compared to the reaction without additional primary amine (B). Conditions: Complex 1 (2 mol%, 0.09 mmol), dicyclohexylamine (4.5 mmol), tamylalcohol (9 mL), NH3(l) (2.5 mmol, 90 mmol, 60 eq.), 170 °C. Cyclohexylamine (20 mol%, 0.9 mmol) added (A). ■ = dicyclohexylamine, ● = cyclohexylamine, ▲ = dicyclohexylimine. Data points for A have been normalized to the actually produced cyclohexylamine. | ||
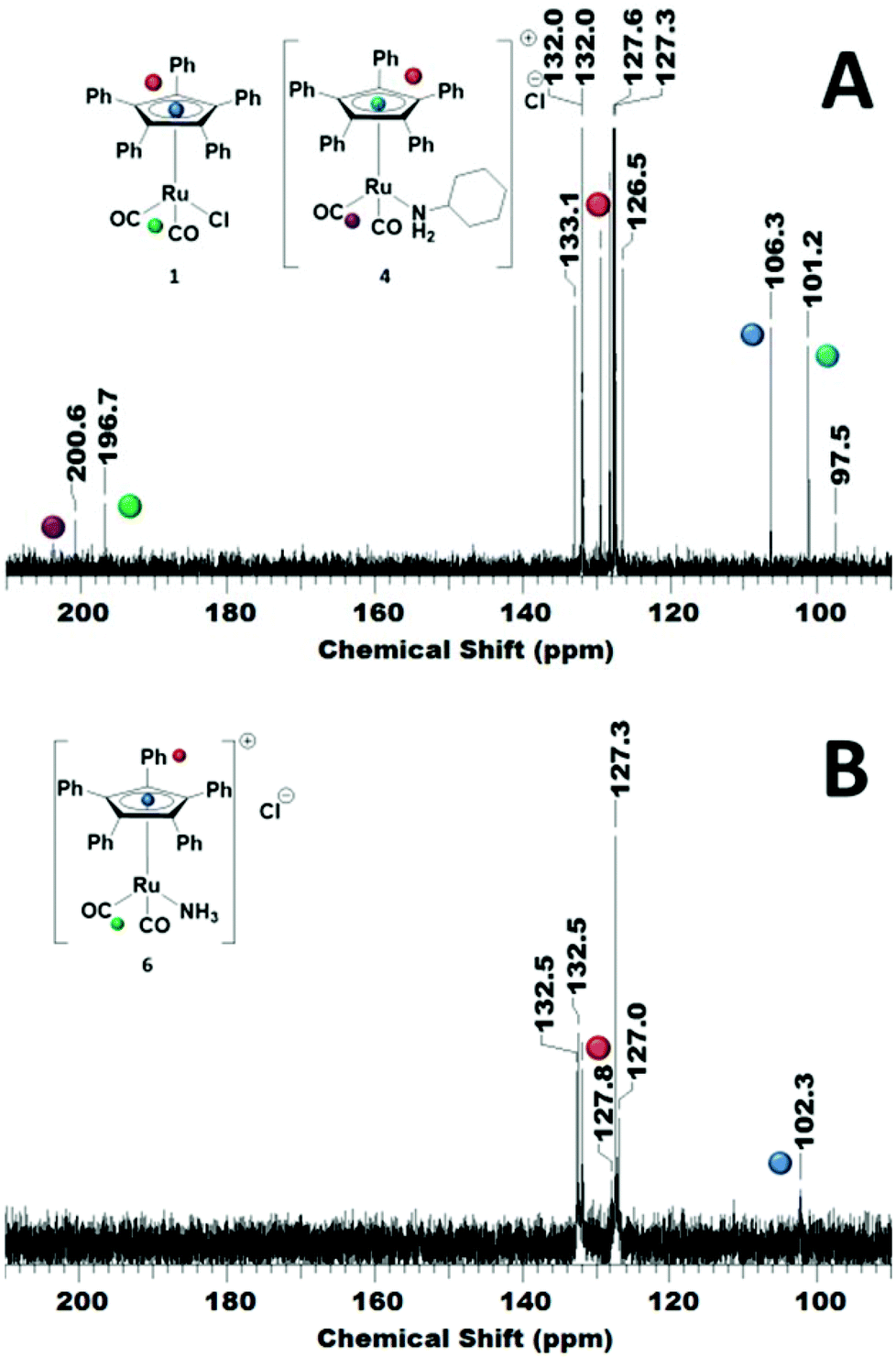
In order to reveal the inhibiting effect of the primary amine, the reaction of cyclohexylamine with complex 1 was monitored by 13C NMR. The Cp ring carbon atoms give a characteristic carbon shift and can probe a change at the Ru centre (106 ppm, Fig. 3A). The resonance at about 197 ppm corresponds to the CO ligands, which can also be used as a probe for changes in the coordination sphere of Ru. The signals between 127–132 ppm belong to the phenyl groups on the Cp ring.
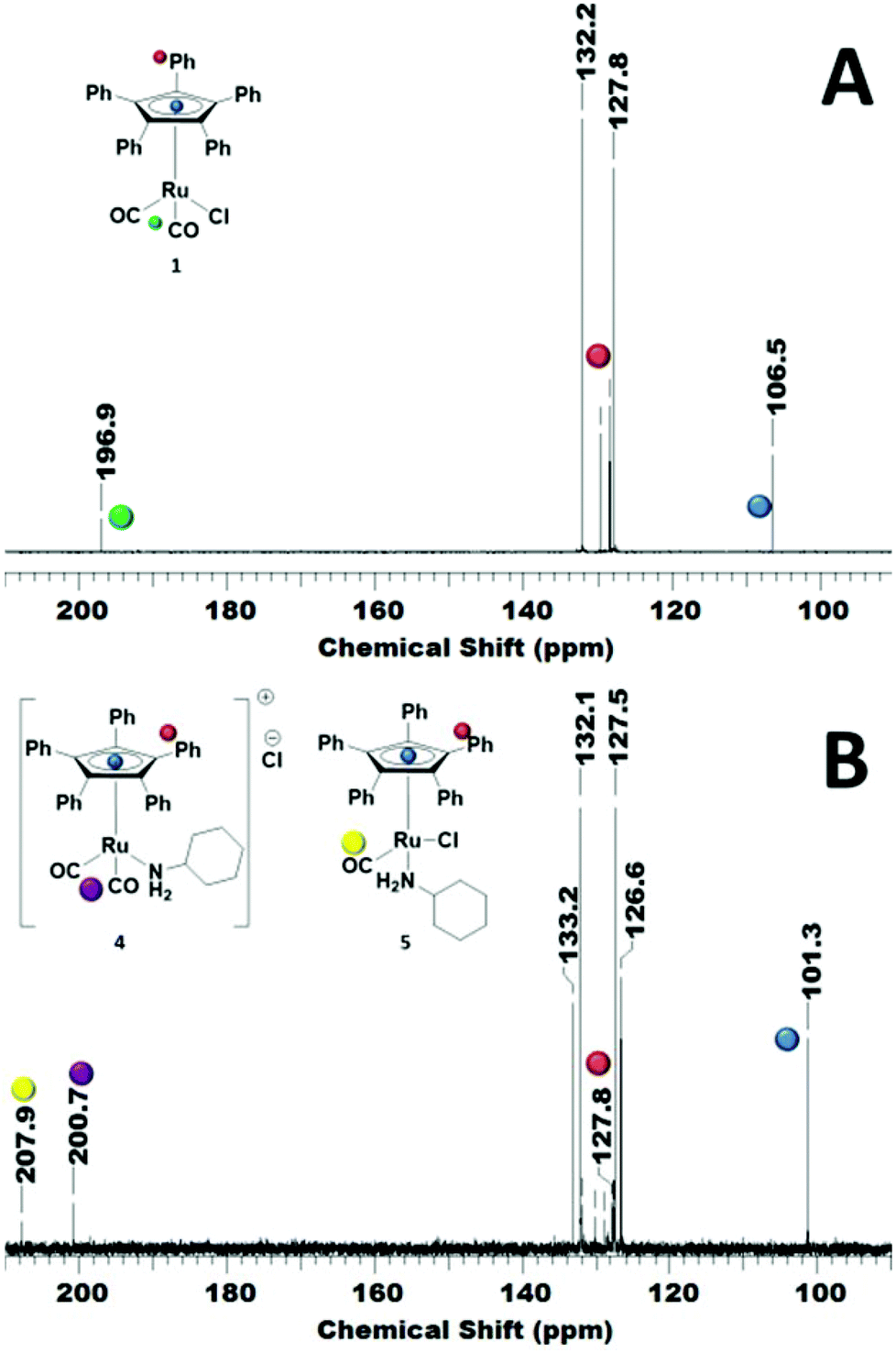 | ||
| Fig. 3 13C NMR (125.7 MHz, 298 K, CDCl3) spectrum of complex 1 showing the distinctive Cp carbon shift at 106 ppm (upper spectrum, A), and the resulting complex mixture after treatment with 10 equivalents of cyclohexylamine after 18 h at r.t. (lower spectrum, B). | ||
The addition of primary amine slowly led to an upfield shift of the Cp ring carbon atoms in the 13C NMR. After 18 h at r.t., the peak at 106 ppm had completely disappeared and a new peak at 101 ppm was observed (Fig. 3B). There are now 2 carbonyl signals observed, one for the monocarbonyl complex 5 in which one CO of the original complex is replaced by the amine15 and the other one for the cationic dicarbonyl complex 4, in which the chloride has been replaced by the amine.16
Investigating the reversibility of the amine coordination and substitution, the mixture consisting of complex 1 and cyclohexylamine was warmed to 60 °C for 1 h. After this time, the peak at 106 ppm is observed again, indicating that cyclohexylamine coordination is indeed reversible (Scheme 2). The process of amine coordination is apparently reversible at already fairly low temperature, and is therefore unlikely to be the cause of catalyst deactivation. However, a small new peak emerges at 97 ppm (Fig. 4A).
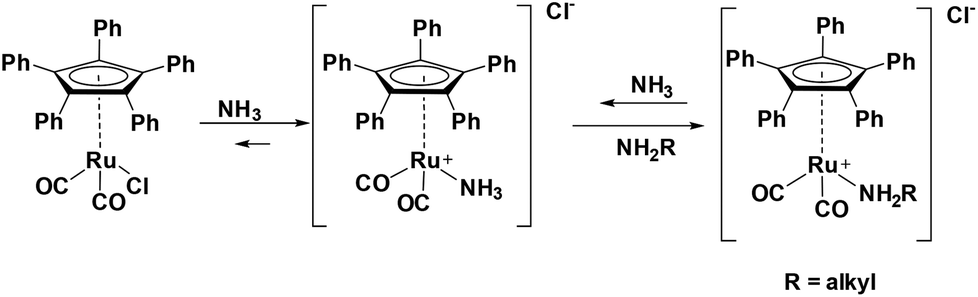 | ||
| Scheme 2 Reversible deactivation of Ru(CpPh5)(CO)2Cl by NH3 and primary amine, similar to the deactivation of Shvo's catalyst.14 | ||
 | ||
| Fig. 4 13C NMR (125.73 MHz, 298 K, CDCl3) spectrum of Ru(CpPh5)(CO)2Cl in the presence of 10 eq. dicyclohexylamine and 10 eq. cyclohexylamine heated to 60 °C for 4 h. | ||
A similar treatment of complex 1 was repeated with ammonia instead of cyclohexylamine. A solution of complex 1 was placed in a 10 mL stainless steel autoclave, which was subsequently charged with NH3. After stirring the solution for 1 h at 170 °C the autoclave was opened and the excess of NH3 was released. The solution was transferred to a Wilmad-Young NMR tube and a spectrum was recorded (Fig. 4B).
Fig. 4B reveals that a similar reaction occurred; again, a shift of the Cp carbons was observed, now from 106 to 102 ppm. When then 10 eq. of cyclohexylamine were added to the NH3 adduct complex, the NH3 was immediately replaced for the cyclohexylamine. If this type of coordination was indeed the cause of deactivation, one would expect this process to be irreversible.
To exclude deactivation by the starting material in catalysis, dicyclohexylamine, a reaction with 10 eq. of dicyclohexylamine was performed. Because conversion up to 80% is observed, it is not expected that this deactivates the catalyst. In addition, the bulkiness of the substrate might even hamper coordination to the complex if not under the exact reaction conditions. Fig. S8 (ESI†) shows the complex after 18 h at room temperature in the presence of dicyclohexylamine. However, after addition of 10 eq. cyclohexylamine to this mixture and heating for 4 h at 60 °C, the peaks at 101 and 97 ppm were observed again. In addition, a peak at 170 ppm was observed (Fig. 5). The large downfield shift indicates that this originates from a carbonyl compound. However, the only carbonyl source in the mixture are the carbonyl ligands on the complex itself. It might be possible that one of the carbonyls undergoes nucleophilic attack by cyclohexylamine.17,18 This is in agreement with previous other carbonyl complexes, which were used in the carbonylation of amines.19 Moreover, complexes similar to those used in this study have been shown to be susceptible for nucleophilic attack by even less nucleophilic compounds.20
 | ||
| Fig. 5 13C NMR (125.73 MHz, 298 K, CDCl3) spectrum of complex 1 after addition of cyclohexylamine and heating to 60 °C for 1 h (upper spectrum, A) and spectrum of complex 1 after reaction with NH3 at 170 °C for 1 h (lower spectrum), due to a low concentration, the carbonyl signals could not be observed. | ||
Upon nucleophilic attack of the primary amine on one of the CO ligands, a carbamoyl complex is formed.21 The carbamoyl ligand neutralizes the cationic complex. The remaining proton in the amide bond can be removed by the excess of amines present in solution (Scheme 3). The carbamoyl complex appears to be very stable and does not react back (catalyst deactivation). The remaining Ru complex remains in solution though most likely in some dimeric form, which is typical for these types of ruthenium complexes.22
 | ||
| Scheme 3 Attack of primary amine on CO in the complex, followed by deprotonation of (secondary) amine. | ||
In the 13C NMR spectra, distinct signals of the carbon in the CN bond of cyclohexyl groups were observed. Compared to free cyclohexylamine, this showed a downfield shift (see ESI, Fig. S11†). In addition, the reaction was also monitored by in situ IR. The carbonyl vibrations at 2010 and 2050 cm−1 seem to disappear equally. This indicates that there is no difference between the carbonyls in terms of reactivity for the nucleophilic attack of the amine, and that the original starting complex disappears. Upon heating the mixture to 40 °C, upcoming peaks at 1710 and 1620 cm−1 are observed over longer reaction times. These regions are typical for amide/formamide vibrations. The increase of these peaks over time indicates the formation of carbamoyl and formamide derivatives from CO (Fig. 6). The band at 1700 cm−1 increases fast in the beginning and remains strong, also indicating the formation of an amide that most likely remains coordinated to Ru. The band at 1620 cm−1 confirms this as well.
 | ||
| Fig. 6 In situ FT-IR spectra of the reaction of Ru(CpPh5)(CO)2Cl with 10 eq. cyclohexylamine at 40 °C in the region of 2150 to 1550 cm−1 (upper spectrum), and in the region of 2600 to 2200 cm−1 (lower spectrum). | ||
Another indication for the nucleophilic attack of the amine to the carbonyl is seen in the 2400 cm−1 region. Here a strong band is seen almost from the start, which later becomes less intense. This region indicates the presence of ammonium ions. The decrease in intensity indicates that the ammonium is deprotonated, forming the inactive complex as stable species. The conversion of secondary n-alkylamines was found to be lower. It is likely that n-alkylamines react even more readily with the carbonyl moieties due to less steric hindrance. Performing the same reactions with n-hexylamine, revealed it was indeed fast, as the 13C-signal at 106 ppm was not observed at all (Fig. S11 and S12 in ESI†). In addition, the resulting carbamoyl peak after heating has a slightly different shift (164.7 ppm), indicating that this is a product of a reaction of the amine with a carbonyl ligand. So far, the spectroscopic techniques used suggest a carbamoyl complex, though not certain in what form exactly. Therefore we performed mass spectrometry on the complex used in the in situ IR experiments. In this higher mass complexes (mass higher than the monometallic complex) were found, showing that indeed multimetallic, mostly bimetallic, species are formed upon degradation. Also, when complex 1 was refluxed in toluene in the presence of 40 eq. cyclohexylamine, the resulting complex displayed no carbonyl signals anymore, and only a single peak at 170 ppm was found (Fig. 7).
 | ||
| Fig. 7 13C NMR (125.73 MHz, 298 K, CDCl3) spectrum of Ru(CpPh5)(CO)2Cl (1) after refluxing in toluene for 24 h in the presence of 40 equivalents of cyclohexylamine. | ||
In addition, complex 3 was also subjected to the deactivation experiments. Complex 3 showed initially less activity, though showed fairly good conversion at low NH3 loading. As the reason of deactivation is clearer now, it might be that this complex just deactivates much faster. On the other hand, it might still be possible that the complex is just less active. In this case, also the PPh3 can be monitored by means of 31P NMR. Again it was found that a reaction occurs upon addition of primary amine. However, it is also observed now that PPh3 remains coordinated (free PPh3 would show up at −5 ppm). The deactivation still occurs at a fairly low temperature relative to the reaction temperature, though it only starts at 90 °C after longer reaction times compared to 60 °C for complex 1. This suggests that the complex is more stable, though just less active (see ESI† for further details).
Conclusions
We have shown that a catalyst previously applied in amine and alcohol racemization is also an active catalyst in the splitting of secondary and tertiary amines with ammonia. However, full conversion could not be achieved, which is most likely due to deactivation of the catalyst. In this, it seems that deactivation is mainly caused by the primary amine product. One possibility is that the deactivation proceeds via nucleophilic attack of the primary amine to a carbonyl moiety on the metal complex. The resulting carbamoyl species has been identified by NMR, in situ FTIR and mass spectrometry.Experimental
All work was carried out under standard Schlenk conditions under argon; all solvents were dried, degassed and purged with Ar prior to use. All used glassware was pre-dried at 120 °C and heated with a heat gun while purged with Ar prior to use. Chemicals were purchased from Sigma-Aldrich and Strem and were used as received. All reactions were performed in a magnetically stirred home-made stainless steel autoclave equipped with manometer, temperature controller and sampling unit (50 μL samples). 1H, 13C, 31P NMR spectra were recorded on a Bruker Avance 400 MHz with broad band inverse detection probe and a Bruker Avance 500 MHz with dual channel cryo probe optimized for 13C/1H (DCH). Chemical shifts are reported in ppm using TetraMethylSilane (TMS) as reference. All NMR experiments were performed under inert atmosphere in airtight Wilmad Young NMR quartz tubes. GC-analyses were performed on a Shimadzu GC-2010 equipped with an Ultra-2 column. Ammonia of purity grade N4.7 was used and was introduced to the autoclaves using a Bronkhorst liquiFlow mass-flow controller (MFC). FT-IR spectra were recorded in situ on a Shimadzu 8300 equipped with a home-made autoclave with a built-in ZnS path length window. Complexes 1, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10c and complex 312 were synthesized according to literature procedures.
10c and complex 312 were synthesized according to literature procedures.
General procedure for the splitting of secondary amines
A 75 ml stainless-steel autoclave is charged with 2 mol% of the appropriate complex and purged with Ar. Secondary amine and t-amylalcohol were added via syringe. The autoclave is closed and liquid NH3 was added via a mass flow controller (MFC). The mixture was heated to 170 °C for the 23.5 h time.Procedure for the NMR reactions
In situ FT-IR monitoring
In a home-made stainless steel autoclave equipped with a ZnS path length cell, 6 mL CHCl3 (dry degassed) was placed and a background was recorded for further use. At the same time, complex 1 (0.1 mmol, 63.8 mg) was weighed into a Schlenk tube and dissolved in dry degassed CHCl3. The autoclave was emptied and dried before purging it with Ar again. The solution of complex 1 was transferred to the autoclave and a spectrum was recorded again. After this, cyclohexylamine (1 mmol, 115 μL) was added and the autoclave was sealed and heated to 40 °C. Spectra were recorded with 15 minutes time intervals.Acknowledgements
We would like to thank Ton Staring for his technical assistance. This research has been in part funded by the Netherlands Ministry of Economic Affairs and the Netherlands Ministry of Education, Culture, and Sciences within the framework of the CatchBio program.Notes and references
- R. J. Palmer, in Encyclopedia of Polymer Science and Technology, John Wiley & Sons, Inc., 2002 Search PubMed.
- K. Eller, E. Henkes, R. Rossbacher and H. Höke, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- (a) K. S. Hayes, Appl. Catal., A, 2001, 221, 187–195 CrossRef CAS; (b) S. A. Lawrence, Amines: Synthesis, Properties and Applications, 2004 Search PubMed; (c) A. Simplício, J. Clancy and J. Gilmer, Molecules, 2008, 13, 519–547 CrossRef.
- C. Gunanathan and D. Milstein, Angew. Chem., Int. Ed., 2008, 47, 8661–8664 CrossRef CAS PubMed.
- (a) S. Imm, S. Bähn, L. Neubert, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8126–8129 CrossRef CAS PubMed; (b) S. Imm, S. Bähn, M. Zhang, L. Neubert, H. Neumann, F. Klasovsky, J. Pfeffer, T. Haas and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 7599–7603 CrossRef CAS PubMed.
- (a) D. Pingen, C. Müller and D. Vogt, Angew. Chem., Int. Ed., 2010, 49, 8130–8133 CrossRef CAS PubMed; (b) D. Pingen, O. Diebolt and D. Vogt, ChemCatChem, 2013, 5, 2905–2912 CrossRef CAS.
- S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann and M. Beller, Chem. – Eur. J., 2011, 17, 4705–4708 CrossRef PubMed.
- J. Paetzold and J. E. Backvall, J. Am. Chem. Soc., 2005, 127, 17620–17621 CrossRef CAS PubMed.
- (a) G. Csjernyik, K. Bogár and J.-E. Bäckvall, Tetrahedron Lett., 2004, 45, 6799–6802 CrossRef CAS; (b) B. Martín-Matute, M. Edin, K. Bogár and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2004, 43, 6535–6539 CrossRef PubMed; (c) B. Martín-Matute, M. Edin, K. Bogár, B. Kaynak and J.-E. Bäckvall, J. Am. Chem. Soc., 2005, 127, 8817–8825 CrossRef PubMed.
- M. C. Warner and J.-E. Bäckvall, Acc. Chem. Res., 2013, 46, 2545–2555 CrossRef CAS PubMed.
- S.-B. Ko, B. Baburaj, M.-J. Kim and J. Park, J. Org. Chem., 2007, 72, 6860–6864 CrossRef CAS PubMed.
- B. Martín-Matute, J. B. Åberg, M. Edin and J. E. Bäckvall, Chem. – Eur. J., 2007, 13, 6063–6072 CrossRef PubMed.
- D. Hollmann, H. Jiao, A. Spannenberg, S. Bähn, A. Tillack, R. Parton, R. Altink and M. Beller, Organometallics, 2009, 28, 473–479 CrossRef CAS.
- (a) W. Baratta, E. Herdtweck, W. A. Herrmann, P. Rigo and J. Schwarz, Organometallics, 2002, 21, 2101–2106 CrossRef CAS; (b) M. Kanthak, A. Aniol, M. Nestola, K. Merz, I. M. Oppel and G. Dyker, Organometallics, 2011, 30, 215–229 CrossRef CAS; (c) E. A. Nyawade, H. B. Friedrich and B. Omondi, Inorg. Chim. Acta, 2014, 415, 44–51 CrossRef CAS.
- A. Scherer, T. Mukherjee, F. Hampel and J. A. Gladysz, Organometallics, 2014, 33, 6709–6722 CrossRef CAS.
- A. Aballay, G. E. Buono-Core, F. Godoy, A. H. Klahn, A. Ibañez and M. T. Garland, J. Organomet. Chem., 2009, 694, 3749–3752 CrossRef CAS.
- (a) M. V. Ovchinnikov and R. J. Angelici, J. Am. Chem. Soc., 2000, 122, 6130–6131 CrossRef CAS; (b) M. Baghbanzadeh and C. O. Kappe, Aust. J. Chem., 2009, 62, 244–249 CrossRef CAS.
- (a) Y. Tsuji, T. Ohsumi, T. Kondo and Y. Watanabe, J. Organomet. Chem., 1986, 309, 333–344 CrossRef CAS; (b) G. Bitsi and G. Jenner, J. Organomet. Chem., 1987, 330, 429–435 CrossRef CAS; (c) G. Liu, M. Hakimifard and M. Garland, J. Mol. Catal. A: Chem., 2001, 168, 33–37 CrossRef CAS.
- (a) T. O. Dairo, A. Ellern, R. J. Angelici and L. K. Woo, Organometallics, 2014, 33, 2266–2276 CrossRef CAS; (b) M. C. Warner, O. Verho and J.-E. Backvall, J. Am. Chem. Soc., 2011, 133, 2820–2823 CrossRef CAS PubMed; (c) N. Kawasake, K. Masuzoe, F. Ozawa and A. Yamamoto, J. Organomet. Chem., 1989, 361, C37–C40 CrossRef.
- R. J. Angelici and R. W. Brink, Inorg. Chem., 1973, 12, 1067–1071 CrossRef CAS.
- (a) S. Fabre, P. Kalck and G. Lavigne, Angew. Chem., Int. Ed., 1997, 36, 1092–1095 CrossRef CAS; (b) J. G. Bullitt, F. A. Cotton and T. J. Marks, Inorg. Chem., 1972, 11, 671–676 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6dt01525e |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2016 |