
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The use of organolithium reagents for the synthesis of 4-aryl-2-phenylpyridines and their corresponding iridium(III) complexes†
Ross
Davidson
,
Yu-Ting
Hsu
,
Thomas
Batchelor
,
Dmitry
Yufit
and
Andrew
Beeby
*
Department of Chemistry, Durham University, South Rd, Durham, DH1 3LE, UK. E-mail: andrew.beeby@durham.ac.uk
First published on 27th June 2016
Abstract
A versatile palladium-free route for the synthesis of 4-aryl-substituted phenylpyridines (ppy), starting from tert-butyl 4-oxopiperidine-1-carboxylate, is reported. Reaction with an aryllithium, followed by trifluoroacetic acid dehydration/deprotection and oxidation with 2-iodoylbenzoic acid and finally phenylation, gave 4 ligands (L1–4H): 2,4-diphenylpyridine, 4-(4-methoxyphenyl)-2-phenylpyridine, 2-phenyl-4-(o-tolyl)pyridine and 4-mesityl-2-phenylpyridine. These ligands were coordinated to iridium to give the corresponding Ir(L)2(A) complexes (Ir1–7), where A = ancillary ligand acetylacetate or 2-picolinate. This was used to demonstrate that, through a combination of ancillary ligand choice and torsional twisting between the 4-aryl substituents of the ppy ligands, it is possible to tune the phosphorescent emission of the complexes in the range 502–560 nm.
Introduction
Complexes containing the iridium(III)bis(2-phenylpyridine) motif form some of the most popular and versatile, small-molecule emitters used in OLEDs, owing to their high phosphorescence quantum yields (PLQYs, Φ),1–3 short emission lifetimes4 and ease of synthesis.5–10 The phosphorescent emission is enhanced by strong spin–orbit coupling of iridium and the mixed MLCT/LC nature of the emission process in these complexes. Significantly, this allows for the harvesting of both singlet and triplet excitons within an OLED, theoretically allowing 100% internal quantum, and since their first deployment in these devices by Forrest et al., they have been the subject of numerous patents and papers.11 In addition, this class of complexes has been reported for use in biological systems as phosphorescent labels and in a myriad of other applications where their relatively long-lived (μs) emission can be readily differentiated from sample auto-fluorescence by simple time-gating methods.12–21 Their emission spectra can be readily tuned by altering either the ancillary or 2-phenylpyridine (ppy) ligand. The two most common ancillary ligands used are acetoacetate (acac, Ir(ppy)2(acac), λem = 516 nm)22 and picolinate (pic, Ir(ppy)2(pic), λem = 506 nm),23 with the emission of pic-based complexes typically being slightly blue shifted relative to acac analogues. However, there are now numerous other bidentate and monodentate ligands known that can induce more significant red or blue shifts than these two examples.24–33 There are also many examples where the ppy core has been modified to induce blue or red shifts in the emission spectrum.23,34–40 Adding electron-withdrawing groups to the phenyl ring induces a stabilisation of the HOMO and a concomitant blue shifting; conversely, adding electron-withdrawing groups to the pyridine ring lowers the energy of the LUMO and brings about a red shift.41,42 However, it is also known that extending the conjugation of any part of the ligand typically results in a red shift of emission (e.g. 2,4-diphenylpyridine). Recently, Kozhevnikov et al. have demonstrated that the addition of a twist between the 4-phenyl substituent and the ppy core by replacing the 4-phenyl group with a mesitylene (mes) maintained the high PLQY associated with a ppy complex and prevented red shifting.3 The addition of the two ortho-methyl groups offers enough steric hindrance to twist the mesityl out of plane of the pyridine, effectively breaking the conjugation. An added benefit of the addition of the mes ring is that it reduced intermolecular π–π interactions and greatly increased the solubility of the complexes, making them more readily solution processable.Despite the advances made in the behaviour of the final complexes, the synthesis of the ligands remains complex, and little has been done to expand the existing methods of preparing ppy ligands substituted at the 4-position of the pyridine ring. To date, synthesis of the ppy ligand can be broken down into two main strategies: (i) modifying an existing pyridine ring or (ii) forming the central pyridine ring with the desired substituents attached. The first approach can be achieved by an assortment of reactions often involving palladium-catalysed reactions, for example, Suzuki–Miyaura,3 Stille,43 Kumada44 or Negishi45 reactions with substituted 2- or 4-halo-pyridines or 2-chloropyridines. This approach is very popular because these types of reactions offer reliable modular chemistry with high yields, with a minimal number of steps. Although tolerant of a wide range of functional groups, these reactions are typically air sensitive and require palladium catalysts which can be prohibitively expensive, preventing large-scale production. There also currently exists a small range of infrequently used techniques to add a phenyl group to a pyridine, such as reacting with an aryl lithium or Grignard reagent or an aromatic diazonium salt. In the case of the latter reaction, arylation occurs at all free positions of the pyridine ring, whilst the former are both selective for the 2 position. However, when unsubstituted pyridine is used the phenylation occurs at the 2- and 4-positions of the pyridine. Once the 4-position of the pyridine is blocked with a substituent, phenylation exclusively occurs at the 2-position e.g. the reaction of 4-(phenylethynyl)pyridine with phenyllithium produces 2-phenyl-4-(phenylethynyl)pyridine with a yield of only 26%.46 The second approach, forming the pyridine ring with the substituents attached, tends to involve a specific chemistry unique to each molecule. The most commonly used technique for this is Kröhnke synthesis, which involves reacting a cinnamaldehyde derivative with a N-phenylacylpyridinium iodide in the presence of an ammonia source.47,48 Whilst this route can be performed on a large scale in an open reaction vessel and requires comparatively simple purifications it can give a wide range of yields dependent on the molecules being produced and requires the synthesis of the substituted cinnamaldehydes, which can be non-trivial. Recently, Guzel demonstrated the functionalisation of N-acyl-2,3-dihydropyridones using a nucleophilic attack of organocerium reagents followed by oxidative aromatisation to give the corresponding substituted ppy.49 However, in our lab the nucleophilic addition using organocerium reagents resulted in a Michael addition, substituting at the 6-position of the tetrahydropyridone.
In this work we describe a facile route to synthesize a range of 4-aryl-2-phenylpyridines without the use of palladium catalysis. We then go on to demonstrate how these modifications to the pyridyl ring of the ppy ligand can result in enhanced photophysical properties of the iridium(III) complexes.
Experimental section
General details
NMR spectra were recorded in deuterated solvent solutions on a Varian VNMRS-600 spectrometer and referenced against solvent resonances (1H, 13C). ESMS data were recorded on a TQD mass spectrometer (Waters Ltd, UK) in either acetonitrile or methanol, GCMS data were recorded on a Trace GCMS (ThermoFinnigan) GCMS recorded in DCM, ASAP data were recorded on a Xevo QTOF (Waters) high resolution, accurate mass tandem mass spectrometer equipped with an Atmospheric Pressure Gas Chromatography (APGC) system and an Atmospheric Solids Analysis Probe (ASAP). MALDI data were recorded on a Bruker Autoflex II ToF/FoF spectrometer. Microanalyses were performed by the Elemental Analysis Service, London Metropolitan University, UK or Elemental Microanalysis Service, Durham University, UK. All chemicals were sourced from standard suppliers.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DCM: acetone and the solvent was removed to give an orange solid. Crystals were grown by the slow evaporation of a DCM/methanol solution. Yield: 0.23 g (74%). 1H-NMR (CD2Cl2): δ 8.75 (dd (J = 6, 1 Hz), 1H), 8.28 (ddd (J = 8, 2, 1 Hz)), 8.16 (dd (J = 2,1 Hz), 1H), 8.11 (dd (J = 2, 1 Hz), 1H), 7.93 (td (J = 8, 2 Hz), 1H), 7.84 (ddd (J = 5, 2, 1 Hz), 1H), 7.78–7.74 (m, 6H), 7.58–7.55 (m, 5H), 7.52–7.49 (m, 2H), 7.41 (dd (J = 6, 2 Hz)), 7.37 (ddd (J = 8, 5, 1 Hz), 1H), 7.22 (dd (J = 6, 2 Hz), 1H), 7.00–6.96 (m, 2H), 6.85–6.83 (m 2H), 6.53 (d (J = 8 Hz), 1H), 6.34 (d (J = 8 Hz), 1H) ppm. 13C-NMR (CD2Cl2): δ 172.4, 168.7, 167.6, 152.6, 149.7, 149.5, 148.7, 148.5, 148.2, 147.5, 144.3, 144.2, 137.6, 137.2, 137.1, 132.5, 129.8, 129.7, 129.6, 129.4, 129.1, 128.0, 124.4, 124.0, 121.5, 121.0, 120.5, 120.4, 116.8, 116.2 ppm. MALDI: m/z 775.2 [M]+. Anal. Calc. for C40H28IrN3O2: C, 62.00; H, 3.64; N 5.42%. Found: C, 61.87; H, 3.56; N, 5.39%.
:1 DCM: acetone and the solvent was removed to give an orange solid. Yield: 0.25 g (77%). 1H-NMR (CD2Cl2): δ 8.49 (d (J = 6 Hz), 2H), 8.07 (d (J = 2 Hz), 2H), 7.81 (d (J = 8 Hz), 4H), 7.68 (dd (J = 8, 1 Hz), 2H), 7.41 (dd (J = 6, 1 Hz), 2H), 7.10 (d (J = 8 Hz), 4H), 6.87 (td (J = 7, 1 Hz), 2H), 6.71 (td (J = 7, 1 Hz), 2H), 6.35 (dd (J = 8, 1 Hz), 2H), 5.33 (s, 1H), 3.91 (s, 6H), 1.83 (s, 6H) ppm. 13C-NMR (CD2Cl2): δ 206.3, 184.6, 168.0, 161.0, 148.8, 148.0, 147.5, 145.2, 133.2, 129.5, 128.6, 128.3, 123.6, 120.6, 119.2, 115.4, 114.5, 100.2, 55.4, 30.5, 28.3 ppm. MALDI: m/z 812.2 [M]+. Anal. Calc. for C41H35IrN2O2·1¼CH2Cl2: C, 57.27; H, 4.27; N 3.16%. Found: C, 57.20; H, 4.30; N, 3.16%.
:1 DCM: acetone and the solvent was removed to give an orange solid. Crystals were grown by the slow evaporation of a DCM/methanol solution. Yield: 0.23 g (74%). 1H-NMR (CD2Cl2): δ 8.75 (dd (J = 6, 1 Hz), 1H), 8.28 (ddd (J = 8, 2, 1 Hz)), 8.16 (dd (J = 2,1 Hz), 1H), 8.11 (dd (J = 2, 1 Hz), 1H), 7.93 (td (J = 8, 2 Hz), 1H), 7.84 (ddd (J = 5, 2, 1 Hz), 1H), 7.78–7.74 (m, 6H), 7.58–7.55 (m, 5H), 7.52–7.49 (m, 2H), 7.41 (dd (J = 6, 2 Hz)), 7.37 (ddd (J = 8, 5, 1 Hz), 1H), 7.22 (dd (J = 6, 2 Hz), 1H), 7.00–6.96 (m, 2H), 6.85–6.83 (m 2H), 6.53 (d (J = 8 Hz), 1H), 6.34 (d (J = 8 Hz), 1H) ppm. 13C-NMR (CD2Cl2): δ 172.4, 168.7, 167.6, 152.6, 149.7, 149.5, 148.7, 148.5, 148.2, 147.5, 144.3, 144.2, 137.6, 137.2, 137.1, 132.5, 129.8, 129.7, 129.6, 129.4, 129.1, 128.0, 124.4, 124.0, 121.5, 121.0, 120.5, 120.4, 116.8, 116.2 ppm. MALDI: m/z 775.2 [M]+. Anal. Calc. for C40H28IrN3O2: C, 62.00; H, 3.64; N 5.42%. Found: C, 61.87; H, 3.56; N, 5.39%.
:1 DCM: acetone and the solvent was removed to give an orange solid. Yield: 0.25 g (77%). 1H-NMR (CD2Cl2): δ 8.49 (d (J = 6 Hz), 2H), 8.07 (d (J = 2 Hz), 2H), 7.81 (d (J = 8 Hz), 4H), 7.68 (dd (J = 8, 1 Hz), 2H), 7.41 (dd (J = 6, 1 Hz), 2H), 7.10 (d (J = 8 Hz), 4H), 6.87 (td (J = 7, 1 Hz), 2H), 6.71 (td (J = 7, 1 Hz), 2H), 6.35 (dd (J = 8, 1 Hz), 2H), 5.33 (s, 1H), 3.91 (s, 6H), 1.83 (s, 6H) ppm. 13C-NMR (CD2Cl2): δ 206.3, 184.6, 168.0, 161.0, 148.8, 148.0, 147.5, 145.2, 133.2, 129.5, 128.6, 128.3, 123.6, 120.6, 119.2, 115.4, 114.5, 100.2, 55.4, 30.5, 28.3 ppm. MALDI: m/z 812.2 [M]+. Anal. Calc. for C41H35IrN2O2·1¼CH2Cl2: C, 57.27; H, 4.27; N 3.16%. Found: C, 57.20; H, 4.30; N, 3.16%.
Instrumentation
All the photophysical measurements of iridium complexes were performed using DCM as the solvent.The UV-visible spectra were recorded on a Unicam UV2-100 spectrometer operated with the Unicam Vison software in quartz cuvettes with path length l = 1 cm.
Excitation and emission photoluminescence spectra were recorded on a Horiba Jobin Yvon SPEX Fluorolog 3-22 spectrofluorometer. Solutions of the complexes (Ir1–7) were made in dichloromethane and degassed by repeated freeze–pump–thaw cycles using a turbomolecular pump until the pressure was stable in quartz cuvettes, l = 1 cm. The solutions had absorbance below 0.10 to minimise inner filter effects. PLQYs were measured in degassed dichloromethane referenced to quinine sulfate in 0.1 M H2SO4 (ϕF: 0.546, ref. 50); full details are given in the ESI.†
Emission lifetimes were recorded by exciting the sample with the output of a pulsed laser diode which produced a 1 kHz train of pulses of 20 ns duration at 405 nm. The luminescence was collected at 90° and focused onto the entrance slit of a monochromator (Bentham TM-300 V). The emission was detected by a photon-counting PMT and the arrival times of photons at the detector were determined using a digital oscilloscope (NI-5133) configured as a virtual multichannel by LabVIEW. The data were transferred to a PC and analysed using non-linear regression to a single exponential decay, and the quality of fit established by reduced χ2 and random residuals. The samples were degassed by repeated freeze–pump–thaw cycles in duplicates. The decay data were fitted to a single exponential function.
Electrochemical analyses of the iridium complexes were carried out using a PalmSens EmStat2 potentiometer, with platinum working, platinum counter and platinum pseudo reference electrodes, from solutions in DCM containing 0.1 M supporting electrolyte (tetrabutylammonium hexafluorophosphate, TBAPF6), scan rate = 100 mV s−1. The ferrocene/ferrocenium couple was used as the internal reference.
Computational
Density functional theory (DFT) calculations were carried out using the Gaussian 09 package (Gaussian, Inc.),51 and all results were displayed using GaussView52 and GaussSum.53 All calculations used the B3LYP level set employing three different basis sets, SDD and 6-31G(d)/LANL2DZ, geometrically optimised in a DCM solvent field using the SCRF–PCM method. A comparison was made between the calculated bond lengths and X-ray data to determine which was the most accurate basis set, finding SDD to be the best suited for these complexes (see the ESI†).X-ray crystallography
The single crystal X-ray data for compounds Ir3, Ir5 and Ir7 were collected on a Bruker SMART CCD 6000 3-circle diffractometer (fine-focus sealed tube, graphite monochromator) and for compounds [7][TFA] and Ir1 – on an Agilent XCalibur 4-circle κ-diffractometer (Sapphire-3 CCD detector, fine-focus sealed tube, graphite monochromator). The data for compound 4 were collected on a Bruker D8Venture 3-circle diffractometer (Photon100 CMOS detector, IμS microsource, focusing mirrors). All data were collected using λMoKα radiation (λ = 0.71073 Å) at 120.0(2) K, and the temperature on the crystals was maintained by using Cryostream (Oxford Cryosystems) open-flow nitrogen cryostats. The structures were solved by direct methods and refined against F2 with full-matrix least-squares using OLEX254 software. Anisotropic displacement parameters were employed for the non-hydrogen atoms. All H-atoms were added at calculated positions and refined by the use of riding models. The crystallographic and refinement parameters are listed in the ESI.† Crystallographic data for the structures have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication CCDC-1474217–1474222.
Results and discussion
Synthesis
Each of the ligands (L1H–L4H) was prepared by a four-step synthesis as shown in Scheme 1, with an overall yield of 42% (L1H)–27% (L4H). In the first step the commercially available tert-butyl 4-oxopiperidine-1-carboxylate (pip-Boc) was reacted with an aryl to form the respective tertiary alcohol (compounds 1–4). | ||
| Scheme 1 Schematic synthesis of ligands L1H–L4H and complexes Ir1–Ir7; (i) ArLi and THF, where Ar = phenyl (-Ph), 4-anisole (-ani), 2-toluene (-tol) or 2-mesitylene (-mes); (ii) TFA; (iii) 2-iodylbenzoic acid and DMSO; (iv) PhLi and THF; (v) IrCl3·3H2O, ethoxyethanol and water; (vi) K2CO3, ethoxyethanol and 2-picolinic acid [Ir1 (Ar = -Ph), Ir2 (Ar = -ani), Ir4 (Ar = -tol), Ir6 (Ar = -mes)] where L∩L = pic or [Ir3 (Ar = -ani), Ir5 (Ar = -tol), Ir7 (Ar = -mes)] where L∩L = acac. | ||
Deprotection and dehydration of this product to form the respective alkenes (compounds 5–8) were achieved readily by refluxing the tertiary alcohol in TFA, adapted from Chen's method.55 This step occurred at the same rate and at a similar yield (83–90%) independent of the aryl group's ability to planarise with the alkene, suggesting that the aryl group is not offering significant stabilisation of the carbocation that may be formed during the reaction. The tetrahydropyridines were not sufficiently stable, when neutral, to obtain consistent elemental analysis results; however, their identity was confirmed by 1H and 13C NMR, mass-spectrometry, and the crystal structure of the trifluoroacetic acid salt of 7 ([7][TFA]). Aromatisation of the tetrahydropyridine to give the respective substituted pyridines (9–12) was achieved by oxidisation using 2-iodoylbenzoic acid as demonstrated by Nicolaou.56 We have found that using the free-base of the tetrahydropyridine, aromatisation occurred in under 4 hours, while Nicolaou reported that at least 48 hours were required for the hydrogen chloride salts under similar conditions. Attempts were made to combine the dehydration/deprotection and aromatisation steps by adding IBX to the TFA reaction mixture; however these yielded only the alkene. Both of these points reinforce that IBX oxidation is significantly hindered by the addition of acid.
Finally, the 2-phenyl group was introduced by the reaction of the substituted pyridine with four equivalents of phenyllithium (LiPh) in dry THF to form the respective ppy (L1H–L4H). The reaction mixtures were initially cooled to −78 °C for 1 hour followed by allowing it to warm to room temperature for an additional 5 hours by an adaptation of Abramovitch's method.57 Commercially available LiPh was employed in this work although we note that we have also investigated the in situ generation of LiPh by a halo-metal exchange with n-butyllithium and bromobenzene. This resulted in very low yields <10%, which we attribute to side reactions between the product and the 1-bromobutane formed in the metal exchange. This can be resolved by the use of tert-butyllithium where the tert-butylbromide produced does not produce by-products and results in yields equivalent to those using the commercially available LiPh.46,57,58
Dry solvents and an inert atmosphere are only required in steps 1 and 4 whilst steps 2 and 3 were performed in open air. Finally, in each of the steps’ reactions, purification was achieved by crystallisation or filtration through a silica plug, making the synthesis simple and readily scalable to multi-gram quantities.
The iridium complexes (Ir1–Ir7) were prepared using conventional conditions reacting the chosen ligand (LnH) with IrCl3·3H2O in an ethoxyethanol/water solution to give the corresponding iridium μ-chloride dimer that was isolated and subsequently treated with the ancillary ligands in the presence of K2CO3 to yield the final complexes.
Molecular structures
The unit cell of 4 contains three independent molecules with virtually identical conformation (see Fig. 1a and b). The structure shows a hydroxy-group and a mes-ring attached to C3 of the Boc-protected piperidine and is consistent with the NMR, mass-spectrometry and elemental analyses. The piperidine ring is in the chair conformation and the mes ring is almost perpendicular to the mean plane of the heterocycle (the torsion angle C2–C3–C11–C16 is in the range 84.9(2)–99.53(18)°). The OH⋯O(carbonyl) hydrogen bonds link molecules 4 into double columns along the c-axis. | ||
| Fig. 1 Crystal structure of 4; hydrogen atoms have been removed for clarity (a) and overlay of 3 independent molecules (b). Thermal ellipsoids are displayed at 50% probability. | ||
The structure [7][TFA] (see Fig. 2) shows that the tertiary hydroxy-group in 3 has been dehydrated to form an alkene bond between C3 and C4, and the corresponding bond length is 1.392(2) Å. The Boc protecting group has also been removed to give the secondary amine, protonated in this structure, giving the tetrahydropyridine consistent with NMR and mass-spectrometry. The rings in the cation [7] are also almost perpendicular to the torsion angle (C4–C3–C6–C7) of 72.98(17)°. This value is higher than that in the 4-phenyl-1,2,3,6-tetrahydropyridium chloride reported by Chakrabarti where the corresponding torsion angle is 32.9(8)°,59 reflecting the higher steric demand of the tolyl group. The cations and anions in the structure [7][TFA] are linked by N–H⋯O hydrogen bonds into chains along the a-axis.
 | ||
| Fig. 2 Crystal structure of [7][TFA]. Thermal ellipsoids are displayed at 50% probability. | ||
Each unit cell in the structure Ir1 contains a single complex and two methanol molecules (selected bond lengths and angles are given in the ESI†). The structure shows that the iridium centre has ‘N3OC2’ coordination in trans orientation with respect to the two L1 pyridines 1 and is consistent with the NMR and mass-spectrometry data collected. Ir–Nppy (2.032(6) and 2.031(6) Å), Ir–NPic (2.153(6) Å), Ir–Opic (2.172(3) Å) and Ir–Cppy (2.020(7) and 2.006(5) Å) are similar to Ir(ppy)2(pic).23 The phenyl ring is not coplanar with the central pyridine ring of the ppy ligand; the corresponding torsion angles (C2–C3–C12–C13) and (C22–C23–C32–C33) are equal to 21.0(9) and 27.8(6)° respectively. Given that Ir1 shows a similar coordination behaviour to that of other Ir(ppy)2(pic) we can assume that Ir2, Ir4 and Ir6 coordinate in a similar fashion (Fig. 3).23,40
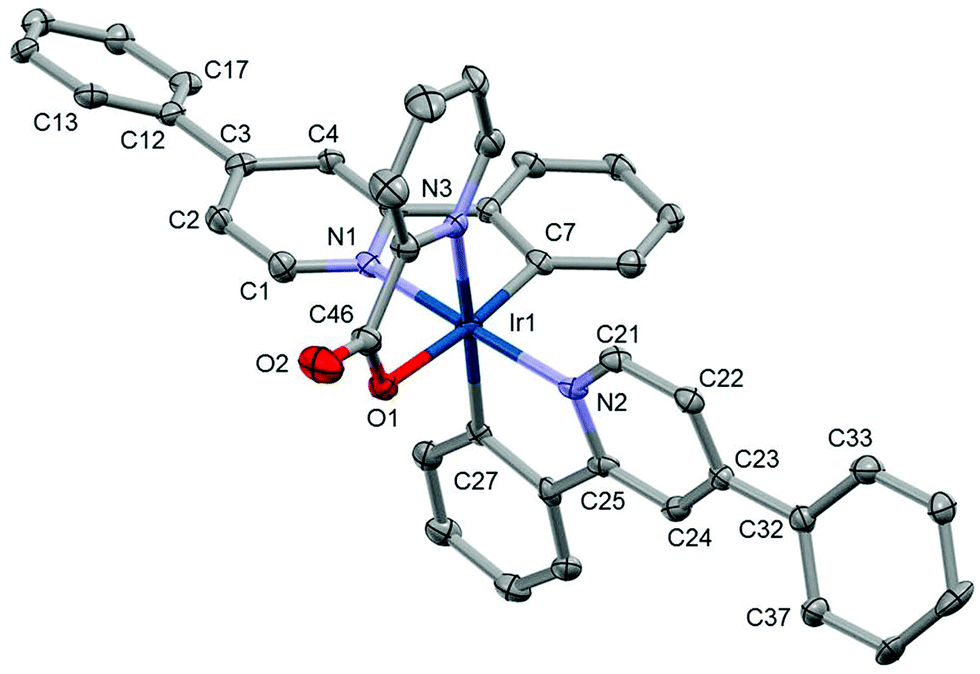 | ||
| Fig. 3 Crystal structure of Ir1; solvents and hydrogen atoms have been removed for clarity. Thermal ellipsoids are displayed at 50% probability. | ||
The unit cell of Ir5 contains two crystallographically independent molecules of Ir5 with slightly different orientation of one of the terminal tolyl groups (selected bond lengths and angles are given in the ESI†). The structure shows that the iridium atom has a ‘N2C2O2’ coordination sphere with the ppy pyridines trans to each other, typical of Ir(ppy)2(acac) based complexes. The bond lengths of Ir–N (2.034(4) and 2.044(4) Å), Ir–C (1.993(4) and 1.989(5) Å), and Ir–O (2.142(4) and 2.149(3) Å) are similar to those of Ir(ppy)2(acac). The dihedral angles between tolyl and ppy rings are in the range 39.0(6)–55.2(5)°. Therefore, it is logical to assume that complex Ir4 also has a similar range of tolyl-ppy dihedral angles (≥38°).
Ir3 (see the ESI†) crystallises with one molecule of Ir3 and dichloromethane per unit cell. As with Ir4, the iridium centre of Ir3 has a coordination environment analogous to that of Ir(ppy)2(acac); however, the methoxy group of the anisole prevents many of the π–π and π–H interactions. As a result the anisole–pyridine dihedral angles are only 23.7(5) and 24.0(6)° (Fig. 4).
 | ||
| Fig. 4 Crystal structure of Ir5; hydrogen atoms have been removed for clarity. Thermal ellipsoids are displayed at 50% probability. | ||
Each unit cell in a crystal of Ir7 contains a single molecule of Ir7. L4 and the acac coordinate to the iridium centre in a similar fashion to complexes Ir3 and Ir5 (see Fig. 5), consistent with the NMR and mass-spectrometry. However, the dihedral angles between the ppy and mes rings (61.5(8) and 62.8(8)°) are significantly higher than those in Ir5 presumably due to the increased steric hindrance from an additional methyl group.
 | ||
| Fig. 5 Crystal structure of Ir7; hydrogen atoms have been removed for clarity. Thermal ellipsoids are displayed at 50% probability. | ||
Computational
A brief investigation was performed using ab initio calculations to study the electronic structures of Ir1–Ir7 and iridium(III)bis(2,4-diphenylpyridine)(acetylacetonate) (Ir(Phppy)2(acac)). Initial geometries for complexes were based on the crystallographic structures of Ir1, Ir3, Ir5 and Ir7. The highest-occupied-molecular-orbital (HOMO) energy for each of the pic complexes is similar (−2.076 to −2.053 eV). The acac complexes also show similar behaviour, which is due to the HOMO consisting of 47% Ir(5d) and 48% ppy(π*) for the acac complexes and 44% Ir(5d) and 52% ppy(π*) with negligible contributions from the aromatic group for either the acac or pic complexes. The lowest-unoccupied-molecular-orbitals (LUMOs) showed a significant difference in behaviour – the pic complexes Ir2, Ir4 and Ir6 have a LUMO consisting of 90–96% pic(π*) while Ir1 consists of 75% pic(π*) with a 20% Ph(π*) contribution. This high level of localisation to the pic results in the aromatic substitution having little effect on the behaviour of the ligand's photophysical behaviour. The LUMO for the acac complexes Ir(Phppy)2(acac), Ir3 and Ir5, each are predominantly ppy(π*) (73–82%) with a contribution from the substituted aromatic group (14–23%) but the Ir7 has negligible contributions from the mes group due to the twisting, significantly reducing the conjugation of the molecule (Fig. 6). | ||
| Fig. 6 LUMO orbital diagrams: (a) Ir(Phppy)2(acac), (b) Ir6. | ||
Electrochemistry
Cyclic voltammograms were recorded for all of the complexes in 0.1 M TBAPF6 in dichloromethane, and referenced against ferrocene (i.e. E½ FeCp2/[FeCp2]+ = 0.00 V). Each of the iridium complexes (Ir1–Ir7) displays a single oxidation wave primarily attributed to the Ir(III)/Ir(IV) couple. The oxidation potential pic complexes (Ir1, Ir2, Ir4 and Ir6) show almost no change as a result of the variation in the aromatic groups attached (i.e.Ir2E½ = 0.47 V and Ir6E½ = 0.50 V; see Table 1). This suggests that oxidation is localised to the Ir(ppy)2(pic) centre with no involvement of the aromatic groups attached to the ppy pyridine. The acac complexes (Ir5 and Ir7) have oxidation potentials identical to those of Ir(ppy)2(acac) (E½ = 0.33 V) and Ir(Phppy)2(acac) (E½ = 0.34 V) approximately 150 mV lower than the pic analogue, but Ir3 has an oxidation potential 50 mV lower than that of the other acac complexes as a result of the addition of the electron-donating group (OMe), suggesting a greater involvement of the ppy ligand in the oxidation of this complex.| Compound | E OX, VFc/Fc+ | Absorption (ε), nm (×103 L mol−1 cm−1) | λ emission, nm | PLQY | Lifetime, μS | k r, 105 s−1 | k nr, 105 s−1 | Pure radiative lifetime (τ0), μs |
|---|---|---|---|---|---|---|---|---|
| The radiative kr and non-radiative knr values in neat film were calculated according to the equations: kr = Φ/τ and knr = (1 − Φ)/τ, from the quantum yields Φ and the lifetime τ values. | ||||||||
| Ir(ppy)2(pic)23 | 0.51 | 506 | 0.03 | <0.1 | ||||
| Ir(ppy)2(acac)60 | 0.33 (ref. 22) | 516 | 0.90 | 1.43 | ||||
| Ir(Phppy)2(acac) | 0.34 | 264 (74.9), 353 (21.6), 463 (br, 5.4) | 560 | 0.75 | 1.25 | 6.00 | 2.00 | 1.66 |
| Ir1 | 0.48 | 271 (65.5), 336 (24.6), 410 (8.1) | 531 | 0.73 | 0.98 | 7.45 | 2.76 | 1.34 |
| Ir2 | 0.47 | 278 (56.6), 297 (62.8), 342 (37.6), 414 (9.7) | 522 | 0.65 | 0.69 | 9.42 | 5.07 | 1.06 |
| Ir3 | 0.29 | 267 (57.0), 299 (68.0), 354 (29.0), 467 (4.2) | 550 | 0.47 | 0.70 | 6.71 | 7.57 | 1.48 |
| Ir4 | 0.49 | 269 (54.1), 333 (17.9), 402 (6.6), 454 (4.0) | 521 | 0.51 | 0.72 | 7.08 | 6.81 | 1.41 |
| Ir5 | 0.34 | 261 (67.8), 348 (18.0), 411 (7.8), 463 (4.9) | 542 | 0.52 | 0.93 | 5.59 | 5.16 | 1.79 |
| Ir6 | 0.50 | 269 (57.1), 331 (17.0), 401 (7.4), 445 (4.6) | 504 | 0.30 | 0.61 | 5.00 | 11.10 | 2.00 |
| Ir7 | 0.34 | 261 (57.9), 344 (11.6), 410 (6.7), 460 (4.5) | 520 | 0.62 | 1.13 | 5.44 | 3.33 | 1.84 |
Photophysical properties
Absorption spectra were recorded for all complexes in DCM, spectra are shown in Fig. 7, and the data are summarized in Table 1. Each of the complexes shows absorption bands below 300 nm, based on the literature attributed to π → π* transitions; the higher intensity bands 350–400 nm are associated with 1MLCT transitions and the remaining lower intensity bands 400–500 nm are assigned to 3MLCT transitions. These are similar for both the pic (Ir-1, 4 and 6) and acac (Ir(Phppy)2(acac), 5 and 7); however, both Ir2 and Ir3 show notable enhancement of the π → π* band ca. 300 nm and the 1MLCT band ca. 350 nm with no significant difference to the 3MLCT. | ||
| Fig. 7 Absorption spectra of the iridium complexes Ir1–7 recorded in DCM, (a) Ir-1, 2, 4 and 6; (b) Ir(PhPPy)2(acac), 3, 5 and 7. | ||
The pic complexes (Ir1, Ir2, Ir4 and Ir6) each show emission peaks ranging from 504 nm (Ir6) to 531 nm (Ir1) while the acac complexes (Ir(Phppy)2(acac), Ir3, Ir5 and Ir7) show emission peaks ranging from 520 nm (Ir7) to 560 nm (Ir(Phppy)2(acac)); see Table 1. Based on the electrochemical data the acac-based complexes appear to be more significantly influenced by the substitution of the ppy ligand than the pic complexes. DFT calculations suggest that this is the result of the LUMO having more significant contributions from the ancillary ligand (pic) than from the ppy substituents. In both classes of complexes the addition of an electron-donating group (OMe) blue shifted the emission by ca. 10 nm, consistent with the previous reports by De Angelis.41 A more notable difference occurs when a slight twist is added to the complex by the addition of an ortho-tolyl group. Based on crystallographic data, the angle between the ppy and tolyl groups is >38°, significantly reducing the conjugation between these two groups. For the acac complex (Ir5) the emission was blue shifted by 18 nm (593 cm−1) relative to that of Ir(Phppy)2(acac), exceeding that of the addition of a simple electron-donating group. However, for the pic complex the blue shift was only 10 nm (361 cm−1) analogous to that of (Ir2), likely due to the degree of localisation of the LUMO on this pic group. Upon replacement of the tolyl by mesityl the emission spectra of both classes of complexes were notably blue shifted relative to the unsubstituted analogues, consistent with the results reported by Kozhevnikov.3 In addition, as the emission of the complexes was blue shifted both the lifetimes and PLQYs for the pic complexes decreased but for the acac complexes no trend was observed. Ir6 (emitting at 520 nm) has a PLQY 0.62 greater than that of Ir2 and Ir4, but much lower than those of Ir(ppy)2(acac) (0.90) and Ir(Phppy)2(acac) (0.75), with the emission lifetimes behaving in the same manner. This can be explained by examination of the kr and knr decay constants; while the kr values only range from 5.00 to 9.42 × 105 s−1 for all of the complexes, the knr values increase from 2.76 × 105 s−1 (Ir1) to 11.10 × 105 s−1 (Ir6) while the acac complexes only range from 3.33 × 105 s−1 (Ir7) to 7.57 × 105 s−1 (Ir3). The combination of the structureless emission and the pure-radiative lifetimes (τ0) being 1.06 and 2.00 μS for complexes Ir1–5 indicates that the transition is purely MLCT3 in nature; however, complexes Ir6 and Ir7 show a structured emission with Ir6λemiss max = 504 nm with a shoulder at ca. 530 nm and Ir7λemiss max = 520 nm with a shoulder at ca. 540 nm; this is attributed to the combination of both MLCT3 and LC3. The intersystem crossing is more significant for Ir6 than complexes Ir1–5, as is supported by the high knr explains its PLQY of 0.30 (Fig. 8).
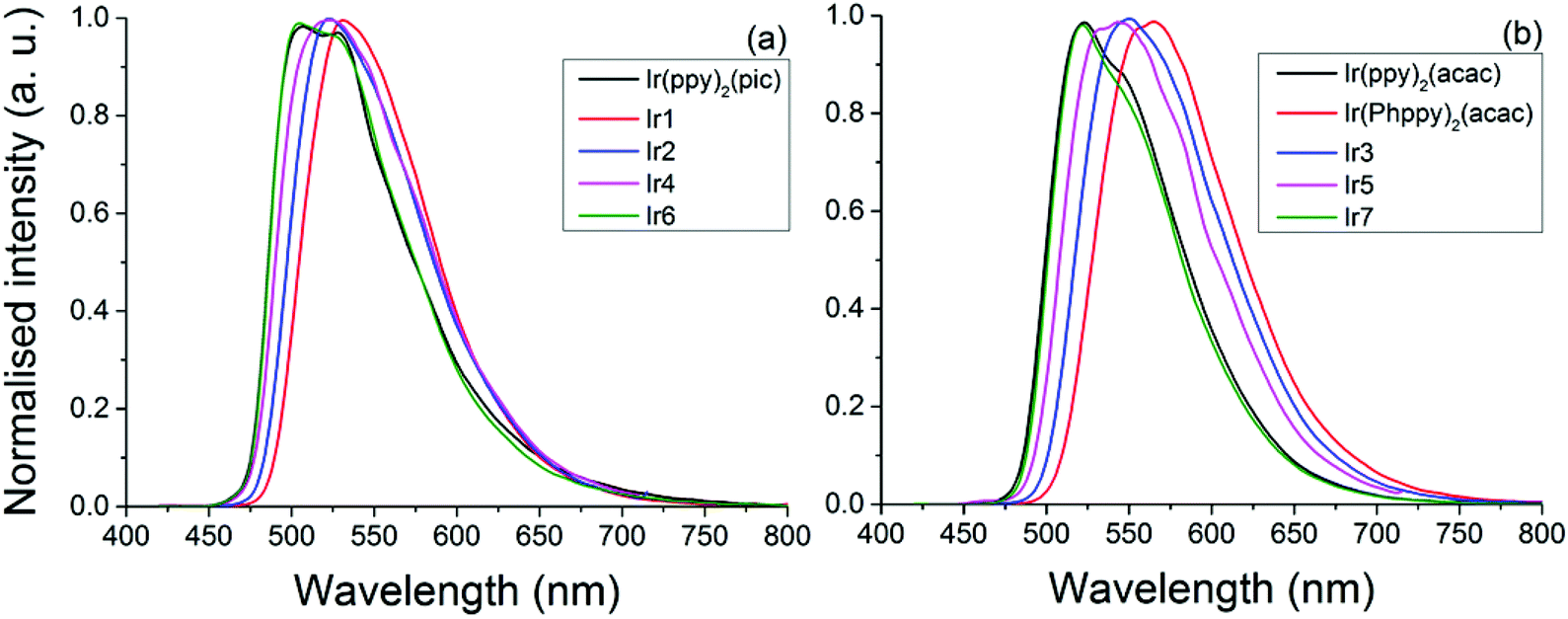 | ||
| Fig. 8 Emission spectra of complexes recorded in DCM, excited at 360 (Ir2, Ir4 and Ir5) and 410 nm (Ir1, Ir3, Ir6, Ir7 and Ir(Phppy)2(acac)): (a) pic complexes Ir1, Ir2, Ir4, Ir6 and (b) acac complexes Ir(Phppy)2(acac), Ir3, Ir5 and Ir7. | ||
Conclusion
Using facile lithiation, dehydration/deprotection, oxidation and phenylation steps we have synthesised 2,4-diphenylpyridine (L1H) and three new ligands (L2–4H), substituted at the 4-position of the ppy pyridine with phenyl, p-anisole, o-tolyl and mesityl groups. This demonstrates a new general route to synthesize 4-aryl-2-phenylpyridine ligands using low-cost reagents without the use of air-sensitive palladium catalysts or costly dihalopyridines or pyridine-4-boronic acid derivatives. The process could be readily adapted to employ alternative aryl lithium reagents or even a reaction of the pyridine with aryl diazonium salts or Grinard reagents in the final arylation step to furnish 2-aryl-4-aryl′-pyridines. Ongoing work seeks to investigate the synthesis and deployment of 2-aryl piperidones as starting materials using these established routes.Using the ligands L1–4H, seven new iridium(III) complexes were made with both pic and acac as ancillary ligands (Ir1–Ir7). X-ray crystal structures of Ir1, Ir3, Ir5 and Ir7 demonstrate that the dihedral angle between the aromatic ring and the ppy moiety can be controlled by the inclusion of sterically hindering methyl groups, but the addition of a single o-methyl group only induces an angle of 39.0°, compared to the 2,6-dimethyl groups in the mesityl derivative resulting in an angle of 61.5° in the crystal structure. This difference in dihedral angle and reduced conjugation with the pyridine group are reflected in the emission spectra of the complexes being blue shifted, particularly for the acac-based complexes. The LUMO of the pic complexes was primarily located on the pic – as a result, the reduction resulted in only modest blue shifts of the emission.
Acknowledgements
RJD gratefully acknowledge the EPSRC (EP/K007548/1) for funding this work.References
- C.-H. Yang, Y.-M. Cheng, Y. Chi, C.-J. Hsu, F.-C. Fang, K.-T. Wong, P.-T. Chou, C.-H. Chang, M.-H. Tsai and C.-C. Wu, Angew. Chem., Int. Ed., 2007, 46, 2418–2421 CrossRef CAS PubMed.
- A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971–12979 CrossRef CAS PubMed.
- V. N. Kozhevnikov, Y. Zheng, M. Clough, H. A. Al-Attar, G. C. Griffiths, K. Abdullah, S. Raisys, V. Jankus, M. R. Bryce and A. P. Monkman, Chem. Mater., 2013, 25, 2352–2358 CrossRef CAS.
- T. Hofbeck and H. Yersin, Inorg. Chem., 2010, 49, 9290–9299 CrossRef CAS PubMed.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H.-E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef CAS PubMed.
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, in Photochemistry and Photophysics of Coordination Compounds II, ed. V. Balzani and S. Campagna, Springer, Berlin, Heidelberg, 2007, vol. 281, ch. 131, pp. 143–203 Search PubMed.
- Y. You and S. Y. Park, J. Am. Chem. Soc., 2005, 127, 12438–12439 CrossRef CAS PubMed.
- R. M. Edkins, A. Wriglesworth, K. Fucke, S. L. Bettington and A. Beeby, Dalton Trans., 2011, 40, 9672–9678 RSC.
- K. A. McGee and K. R. Mann, Inorg. Chem., 2007, 46, 7800–7809 CrossRef CAS PubMed.
- E. Baranoff, B. F. E. Curchod, J. Frey, R. Scopelliti, F. Kessler, I. Tavernelli, U. Rothlisberger, M. Grätzel and M. K. Nazeeruddin, Inorg. Chem., 2012, 51, 215–224 CrossRef CAS PubMed.
- M. A. Baldo, M. E. Thompson and S. R. Forrest, Nature, 2000, 403, 750–753 CrossRef CAS PubMed.
- L. Mosca, R. S. Khnayzer, M. S. Lazorski, E. O. Danilov, F. N. Castellano and P. Anzenbacher, Chem. – Eur. J., 2015, 21, 4056–4064 CrossRef CAS PubMed.
- K. Y. Zhang, H.-W. Liu, M.-C. Tang, A. W.-T. Choi, N. Zhu, X.-G. Wei, K.-C. Lau and K. K.-W. Lo, Inorg. Chem., 2015, 54, 6582–6593 CrossRef CAS PubMed.
- K. J. Castor, K. L. Metera, U. M. Tefashe, C. J. Serpell, J. Mauzeroll and H. F. Sleiman, Inorg. Chem., 2015, 54, 6958–6967 CrossRef CAS PubMed.
- A. Wragg, M. R. Gill, L. McKenzie, C. Glover, R. Mowll, J. A. Weinstein, X. Su, C. Smythe and J. A. Thomas, Chem. – Eur. J., 2015, 21, 11865–11871 CrossRef CAS PubMed.
- K. Qiu, H. Huang, B. Liu, Y. Liu, P. Zhang, Y. Chen, L. Ji and H. Chao, J. Mater. Chem. B, 2015, 3, 6690–6697 RSC.
- C. Jin, J. Liu, Y. Chen, L. Zeng, R. Guan, C. Ouyang, L. Ji and H. Chao, Chem. – Eur. J., 2015, 21, 12000–12010 CrossRef CAS PubMed.
- Z. Wu, J. Mu, Q. Wang, X. Chen, L. Jensen, C. Yi and M.-J. Li, J. Organomet. Chem., 2015, 791, 175–182 CrossRef CAS.
- D.-L. Ma, M. Wang, B. He, C. Yang, W. Wang and C.-H. Leung, ACS Appl. Mater. Interfaces, 2015, 7, 19060–19067 Search PubMed.
- P. Alam, G. Kaur, A. Sarmah, R. K. Roy, A. R. Choudhury and I. R. Laskar, Organometallics, 2015, 34, 4480–4490 CrossRef CAS.
- S. Lin, B. He, C. Yang, C.-H. Leung, J.-L. Mergny and D.-L. Ma, Chem. Commun., 2015, 51, 16033–16036 RSC.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, R. Kwong, I. Tsyba, M. Bortz, B. Mui, R. Bau and M. E. Thompson, Inorg. Chem., 2001, 40, 1704–1711 CrossRef CAS PubMed.
- M. Xu, R. Zhou, G. Wang, Q. Xiao, W. Du and G. Che, Inorg. Chim. Acta, 2008, 361, 2407–2412 CrossRef CAS.
- F. Zhang, D. Ma, L. Duan, J. Qiao, G. Dong, L. Wang and Y. Qiu, Inorg. Chem., 2014, 53, 6596–6606 CrossRef CAS PubMed.
- Y. Si, S. Zhang, N. Qu, G. Luan and Z. Wu, New J. Chem., 2015, 39, 4147–4153 RSC.
- X. Huixia, S. Peng, Z. Dan, Y. Tingting, H. Yuying, W. Hua, S. Heping and X. Bingshe, New J. Chem., 2015, 39, 5293–5299 RSC.
- T. Kim, J. Lee, S. U. Lee and M. H. Lee, Organometallics, 2015, 34, 3455–3458 CrossRef CAS.
- K. Hasan, A. K. Pal, T. Auvray, E. Zysman-Colman and G. S. Hanan, Chem. Commun., 2015, 51, 14060–14063 RSC.
- W. Cho, J. Yoon, G. Sarada, T. Giridhar, Y. Liu, Z.-Q. Jiang, Y. H. Son, B. Y. Kang, J. H. Kwon and S.-H. Jin, Dyes Pigm., 2015, 123, 132–138 CrossRef CAS.
- H. Gao, S. Xia, R. Zhang, Y. Zhao, W. Wang, Z. An and H. Qi, J. Electroanal. Chem., 2015, 755, 71–76 CrossRef CAS.
- T. F. Mastropietro, M. La Deda, N. Godbert, L. Ricciardi, E. I. Szerb, M. Ghedini and I. Aiello, J. Organomet. Chem., 2015, 786, 55–62 CrossRef CAS.
- R. Tao, J. Qiao, G. Zhang, L. Duan, L. Wang and Y. Qiu, J. Phys. Chem. C, 2012, 116, 11658–11664 CrossRef CAS.
- G. Li, J. Lumin., 2011, 131, 184–189 CrossRef CAS.
- J. Zhao, Y. Yu, X. Yang, X. Yan, H. Zhang, X. Xu, G. Zhou, Z. Wu, Y. Ren and W.-Y. Wong, ACS Appl. Mater. Interfaces, 2015, 7, 24703–24714 Search PubMed.
- M. Zaarour, A. Singh, C. Latouche, J. A. G. Williams, I. Ledoux-Rak, J. Zyss, A. Boucekkine, H. Le Bozec, V. Guerchais, C. Dragonetti, A. Colombo, D. Roberto and A. Valore, Inorg. Chem., 2013, 52, 7987–7994 CrossRef CAS PubMed.
- T. Kim, H. Kim, K. M. Lee, Y. S. Lee and M. H. Lee, Inorg. Chem., 2013, 52, 160–168 CrossRef CAS PubMed.
- M. Xu, R. Zhou, G. Wang and J. Yu, Inorg. Chim. Acta, 2009, 362, 2183–2188 CrossRef CAS.
- G. Zhou, C.-L. Ho, W.-Y. Wong, Q. Wang, D. Ma, L. Wang, Z. Lin, T. B. Marder and A. Beeby, Adv. Funct. Mater., 2008, 18, 499–511 CrossRef CAS.
- V. V. Grushin, N. Herron, D. D. LeCloux, W. J. Marshall, V. A. Petrov and Y. Wang, Chem. Commun., 2001, 1494–1495 RSC.
- E. Baranoff, B. F. E. Curchod, F. Monti, F. Steimer, G. Accorsi, I. Tavernelli, U. Rothlisberger, R. Scopelliti, M. Grätzel and M. K. Nazeeruddin, Inorg. Chem., 2012, 51, 799–811 CrossRef CAS PubMed.
- F. De Angelis, S. Fantacci, N. Evans, C. Klein, S. M. Zakeeruddin, J.-E. Moser, K. Kalyanasundaram, H. J. Bolink, M. Grätzel and M. K. Nazeeruddin, Inorg. Chem., 2007, 46, 5989–6001 CrossRef CAS PubMed.
- D. Di Censo, S. Fantacci, F. De Angelis, C. Klein, N. Evans, K. Kalyanasundaram, H. J. Bolink, M. Grätzel and M. K. Nazeeruddin, Inorg. Chem., 2008, 47, 980–989 CrossRef CAS PubMed.
- S. Gronowitz, P. Björk, J. Malm and A.-B. Hörnfeldt, J. Organomet. Chem., 1993, 460, 127–129 CrossRef CAS.
- P. Walla and C. O. Kappe, Chem. Commun., 2004, 564–565, 10.1039/B313887A.
- S. Sase, M. Jaric, A. Metzger, V. Malakhov and P. Knochel, J. Org. Chem., 2008, 73, 7380–7382 CrossRef CAS PubMed.
- R. M. Edkins, S. L. Bettington, A. E. Goeta and A. Beeby, Dalton Trans., 2011, 40, 12765–12770 RSC.
- J.-P. Djukic, C. Michon, A. Maisse-François, R. Allagapen, M. Pfeffer, K. H. Dötz, A. De Cian and J. Fischer, Chem. – Eur. J., 2000, 6, 1064–1077 CrossRef CAS.
- I. Sasaki, L. Vendier, A. Sournia-Saquet and P. G. Lacroix, Eur. J. Inorg. Chem., 2006, 2006, 3294–3302 CrossRef.
- M. Guzel, J. Watts, M. McGilvary, M. Wright and S. Kiren, Tetrahedron Lett., 2015, 56, 5275–5277 CrossRef CAS.
- W. H. Melhuish, J. Phys. Chem., 1961, 65, 229–235 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, O. K. Y. Honda, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, A.1 edn, 2009.
- R. Dennington, T. Keith and J. Millam, GaussView, Version 5, Semichem Inc., Shawnee Mission KS, 2009 Search PubMed.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. Chen, H. Xia, Y. Cai, D. Ma, J. Yuan and C. Yuan, Bioorg. Med. Chem. Lett., 2011, 21, 234–239 CrossRef CAS PubMed.
- K. C. Nicolaou, C. J. N. Mathison and T. Montagnon, J. Am. Chem. Soc., 2004, 126, 5192–5201 CrossRef CAS PubMed.
- R. A. Abramovitch and C.-S. Giam, Can. J. Chem., 1962, 40, 213–219 CrossRef CAS.
- M. Lepeltier, T. K.-M. Lee, K. K.-W. Lo, L. Toupet, H. Le Bozec and V. Guerchais, Eur. J. Inorg. Chem., 2007, 2007, 2734–2747 CrossRef.
- C. Chakrabarti, R. Sengupta and J. K. Dattagupta, Z. Kristallogr., 1996, 211, 971 CAS.
- P. I. Djurovich, D. Murphy, M. E. Thompson, B. Hernandez, R. Gao, P. L. Hunt and M. Selke, Dalton Trans., 2007, 3763–3770 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data for compounds 4, [7 + H]+[TFA]−, Ir1·2CH3OH, Ir3·CH2Cl2, Ir5, and Ir7. NMR spectra and selected bond parameters. CCDC 1474217–1474222. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01461e |
| This journal is © The Royal Society of Chemistry 2016 |