
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The contrasting catalytic efficiency and cancer cell antiproliferative activity of stereoselective organoruthenium transfer hydrogenation catalysts†
Ying
Fu‡§
a,
Carlos
Sanchez-Cano‡
a,
Rina
Soni‡
a,
Isolda
Romero-Canelon
a,
Jessica M.
Hearn
b,
Zhe
Liu¶
a,
Martin
Wills
*a and
Peter J.
Sadler
*a
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: p.j.sadler@warwick.ac.uk
bWarwick Systems Biology Centre, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK
First published on 25th April 2016
Abstract
The rapidly growing area of catalytic ruthenium chemistry has provided new complexes with potential as organometallic anticancer agents with novel mechanisms of action. Here we report the anticancer activity of four neutral organometallic RuII arene N-tosyl-1,2-diphenylethane-1,2-diamine (TsDPEN) tethered transfer hydrogenation catalysts. The enantiomers (R,R)-[Ru(η6-C6H5(CH2)3-TsDPEN-N-Me)Cl] (8) and (S,S)-[Ru(η6-C6H5(CH2)3-TsDPEN-N-Me)Cl] (8a) exhibited higher potency than cisplatin against A2780 human ovarian cancer cells. When the N-methyl was replaced by N–H, i.e. to give (R,R)-[Ru(η6-Ph(CH2)3-TsDPEN-NH)Cl] (7) and (S,S)-[Ru(η6-Ph(CH2)3-TsDPEN-NH)Cl] (7a), respectively, anticancer activity decreased >5-fold. Their antiproliferative activity appears to be linked to their ability to accumulate in cells, and their mechanism of action might involve inhibition of tubulin polymerisation. This appears to be the first report of the potent anticancer activity of tethered RuII arene complexes, and the structure–activity relationship suggests that the N-methyl substituents are important for potency. In the National Cancer Institute 60-cancer-cell-line screen, complexes 8 and 8a exhibited higher activity than cisplatin towards a broad range of cancer cell lines. Intriguingly, in contrast to their potent anticancer properties, complexes 8/8a are poor catalysts for asymmetric transfer hydrogenation, whereas complexes 7/7a are effective asymmetric hydrogenation catalysts.
Introduction
Platinum-group metal complexes, including those of Ru,1,2 Rh,3,4 Pd,5,6 Ir,7–10 Os,11–13 and Pt,14–18 have shown potent anticancer activity in vitro and in vivo. Among them, the anticancer activity of organometallic half-sandwich RuII complexes has been well studied recently.19–27 Although these RuII complexes share a similar general half-sandwich structure,28–30 their mechanisms of action vary,27,31–33 as illustrated by the following examples. (1) Complex DW1 (Scheme 1, compound 1), developed by Meggers et al., is a potent inhibitor of glycogen synthase kinase (GSK-3β); and induces apoptosis of cancer cells through a p53 dependent pathway.34 (2) [Ru(η6-biphenyl)(ethylenediamine)]PF6 (Scheme 1, compound 2) is believed to target DNA; the labile Ru–Cl bond can undergo hydrolysis to form Ru–OH2 intracellularly, whereas this hydrolysis is suppressed extracellularly.35 The hydrolysis step generates a reactive site on the Ru centre which can bind to DNA bases (e.g. guanine) and lead to DNA double-strand breaks and apoptosis. (3) [Ru(η6-arene)Cl2(pta)] {PTA = 1,3,5-triaza-7-phosphatricyclo-[3.3.1.1]decane} (Scheme 1, compound 3) exhibits promising anti-metastatic effects in vitro and in vivo.36,37 (4) Ruthenium arene paullone complexes, e.g. (η6-p-cymene){9-bromo-6-[(α-picolyl-κN)imino-κN]-7,12-dihydroindolo[3,2-d][1]benzazepine}chlororuthenium(II) chloride (Scheme 1, compound 4) are putative cyclin-dependent kinase (CDK) inhibitors, showing potent anticancer activity in vitro.38 (5) [Ru(η6-arene)(TsEn)Cl] (Scheme 1, compound 5) is able to target cancer cells by altering the NADH/NAD+ ratio in cells.27 There is much current interest in exploring alternative profiles of reactivity for ruthenium arene complexes. Progress in optimising activity has centred on modifying the arene,39,40 the monodentate ligand,41–43 or chelating ligand;44–51 and multinuclear ruthenium arene complexes can also exhibit potent anticancer activity.52–54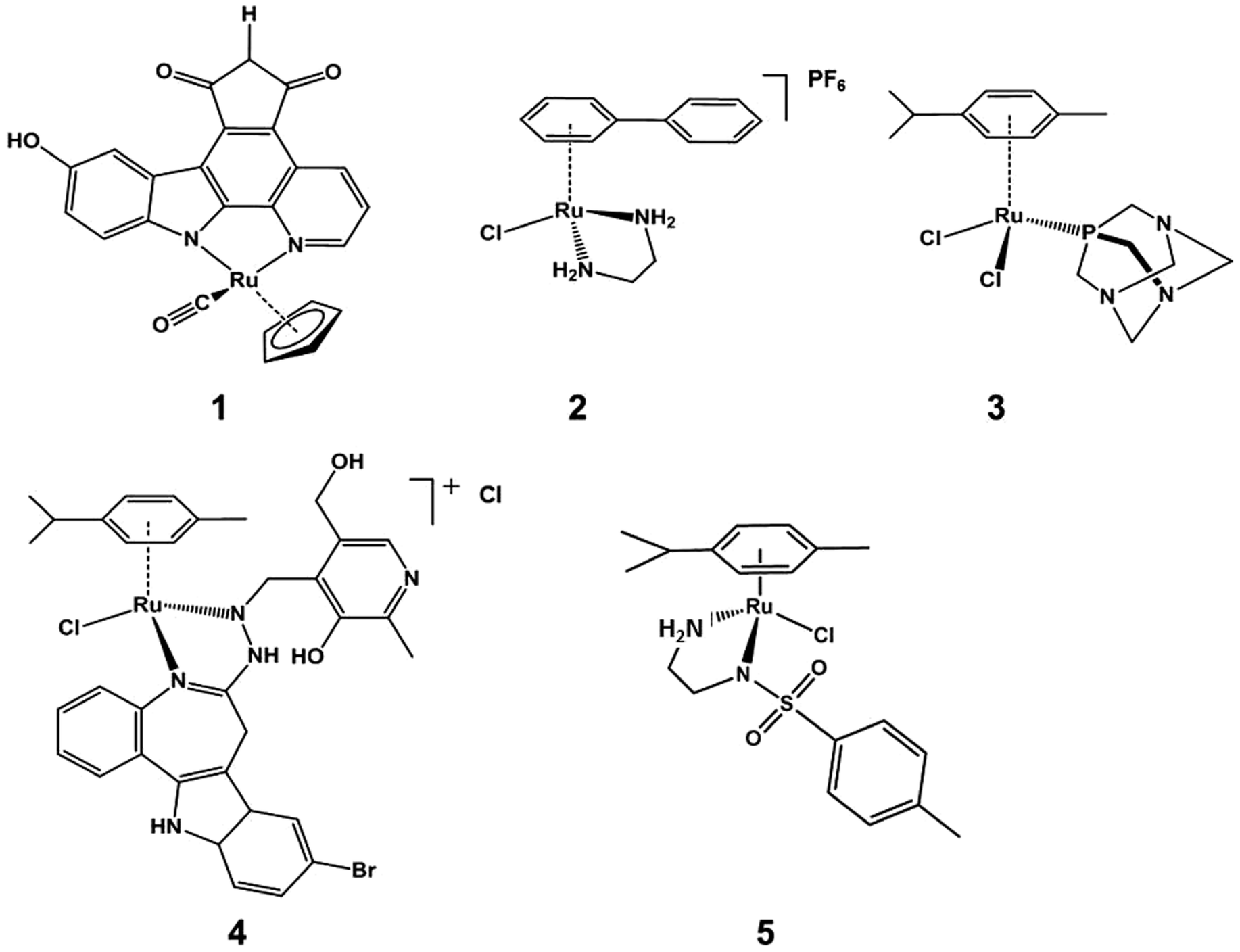 | ||
| Scheme 1 Examples of organometallic half-sandwich RuII complexes which exhibit anticancer activity. 1. DW1 (the R enantiomer of the DW 1/2 racemic mixture); 2. [Ru(η6-biphenyl)(ethylenediamine)]PF6; 3. [Ru(η6-p-cymene)Cl2(pta)] (PTA = 1,3,5-triaza-7-phosphatricyclo-[3.3.1.1]decane); 4. (η6-p-cymene){9-bromo-6-[(α-picolyl-κN)imino-κN]-7,12-dihydroindolo[3,2-d][1]benzazepine}chlororuthenium(II) chloride; 5. [Ru(η6-p-cymene)(TsEn)Cl] (TsEn = p-methybenzene). | ||
Organometallic half-sandwich RuII complexes have also been shown to be outstanding catalysts for asymmetric transfer hydrogenation of ketones and imines. Noyori and co-workers55,56 first reported the synthesis and use of compound N-[(1R,2R)-1,2-diphenylethyl]-4-methylbenzenesulfonamide (p-cymene)ruthenium chloride 6 (Scheme 2) as a catalyst for the asymmetric hydrogenation of ketones and imines giving products in high enantiomeric excesses, using a number of reducing agents including hydrogen gas, isopropanol/base, and formic acid/triethylamine (FA/TEA).57,58 Some RuII arene complexes are active both as hydrogenation catalysts and as anticancer agents.27,59,60
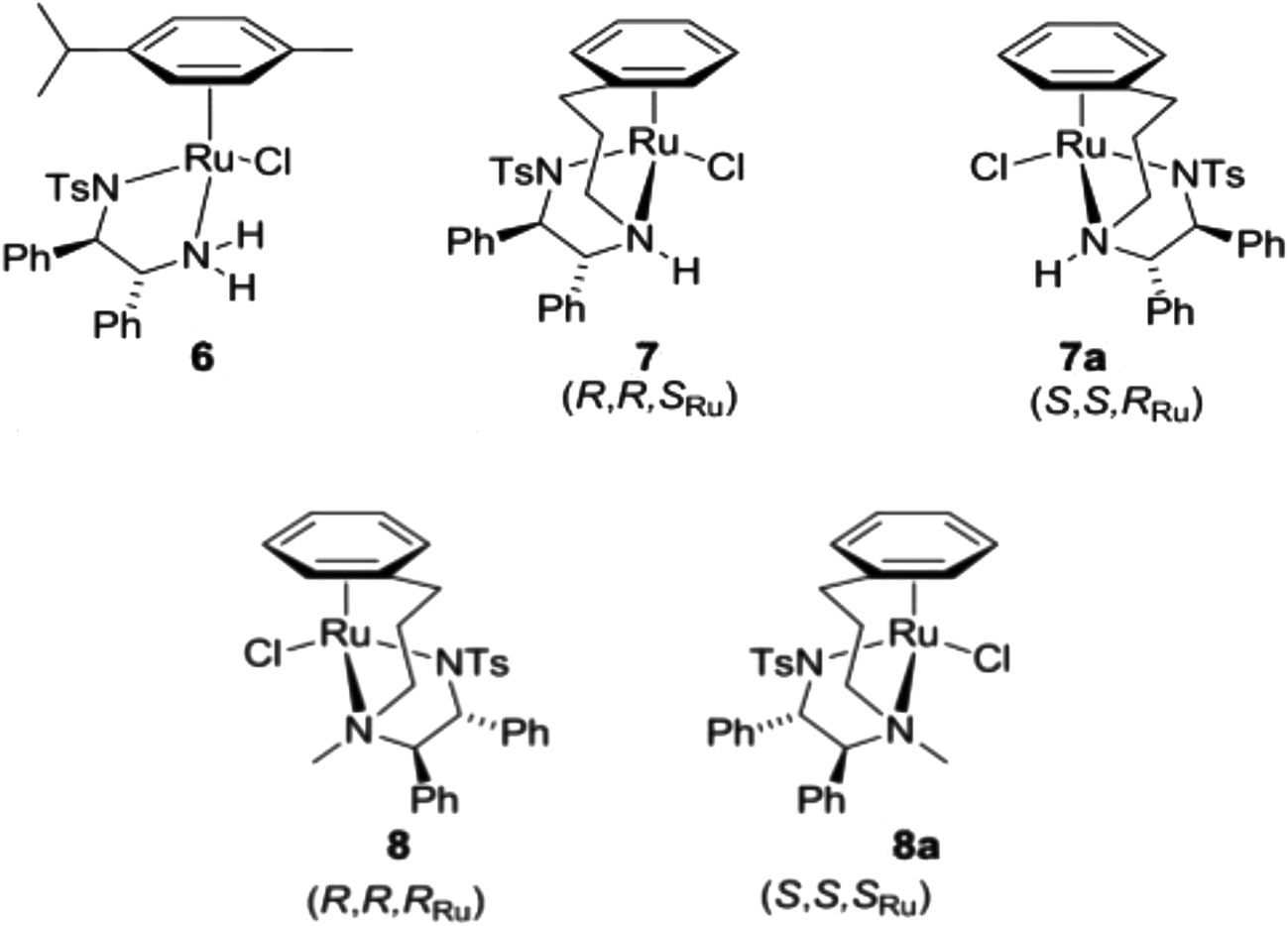 | ||
| Scheme 2 Half-sandwich RuII complexes containing a TsDPEN ligand; complexes 7/7a and 8/8a were studied in this work. | ||
There are four major connections between reported organometallic half-sandwich RuII complexes in the apparently distinct fields of anticancer activity and catalysis. (i) Structural similarity: they have the general structure [Ru(η6-arene)(YZ)(X)], where YZ is typically a chelating diamine ligand (e.g. ethylenediamine, N-tosylethylenediamine)61 and X is a halide (e.g. Cl);62 (ii) the monodentate ligand, which has a similar function in both cases, is normally a leaving group for hydride formation or coordination of biomolecules to the RuII centre, for the catalytic and anticancer effects, respectively;63–65 (iii) the chelating ligand, which often plays an important role in the selectivity of anticancer targets1 and regioselectivity in catalysis;66–68 (iv) chirality, which is important for both of the two families of complexes, plays a vital role in substrate and target recognition.69–71
‘Tethered’ complexes such {N-[(1R,2R)-1,2-diphenylethyl-2-(3-phenylpropylamino)]-4-methylbenzenesulfonamide}ruthenium chloride (Scheme 2, compound 7) are known to be particularly effective catalysts, capable of performing the enantioselective reduction of ketones and imines through an asymmetric transfer hydrogenation (ATH) mechanism, illustrated in Scheme 3.77 Further experiments using compounds carrying TsDPEN derivatives with one alkyl substituent on the basic nitrogens (Scheme 2, compound 8) have revealed the importance of the N–H bond for ketone reduction.72 This is presumably due to lack of the –NH⋯OC interaction required for the postulated six membered cyclic transition state of the ketone reduction process (Scheme 3a).73 The hydrides illustrated in Scheme 3 are known to be formed under the reaction conditions.63,74,75 In contrast, compounds 8/8a are still effective for imine reduction under similar conditions as predicted, but with lower reactivity when compared with catalysts 7/7a. As a result of these observations, coupled with studies reported by others,76,77 imine reduction is speculated to take place via the transition state illustrated in Scheme 3b. In this ‘open’ transition state, no N–H⋯N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond is present, hence the N-alkylated catalysts 8 remain active.78 However, increased steric hindrance imposed by the larger methyl group (replacing the H atom) serve to lower their activity. Interestingly, the use of complexes 6 and 7 showing the same chiral configuration at the ligand for the reduction of ketones and imines results in formation of products of opposite configuration.
C bond is present, hence the N-alkylated catalysts 8 remain active.78 However, increased steric hindrance imposed by the larger methyl group (replacing the H atom) serve to lower their activity. Interestingly, the use of complexes 6 and 7 showing the same chiral configuration at the ligand for the reduction of ketones and imines results in formation of products of opposite configuration.
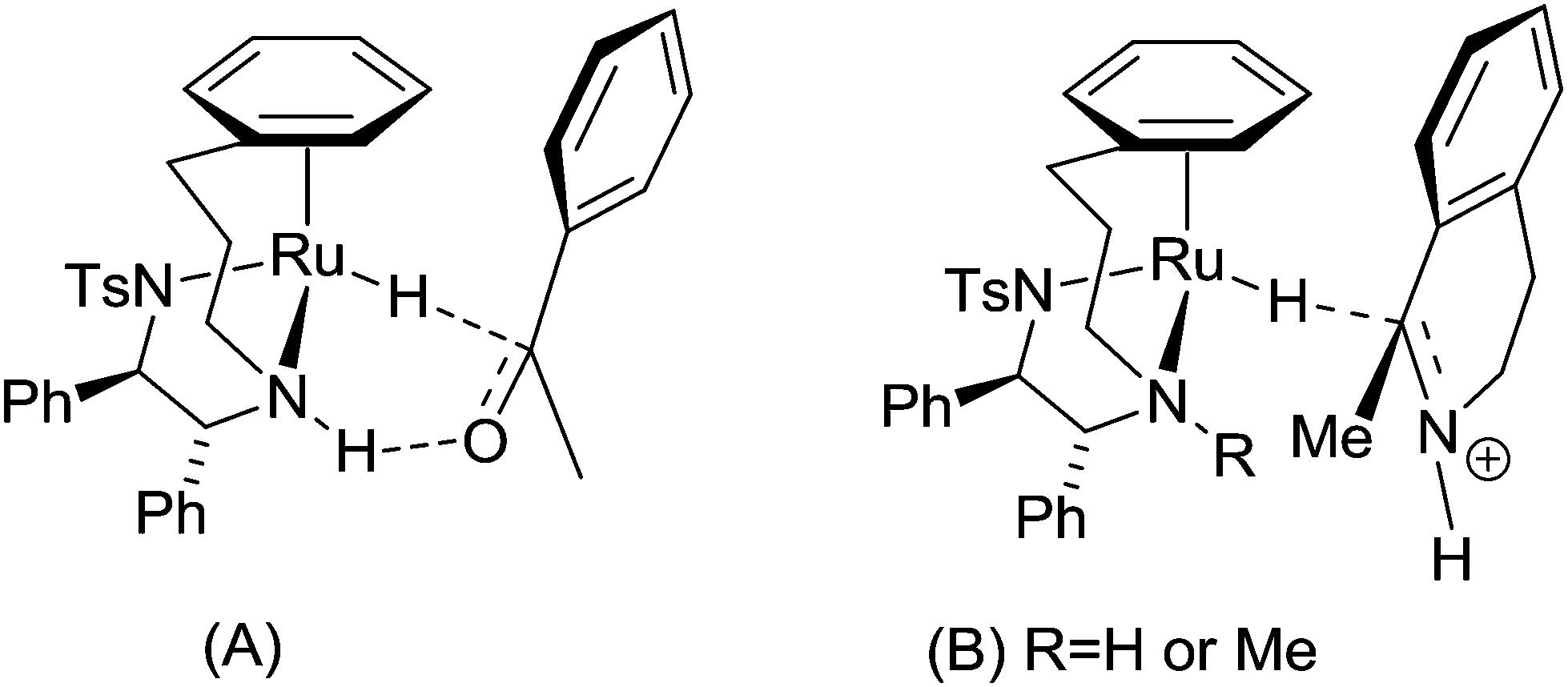 | ||
| Scheme 3 (A) Mechanism of hydrogen transfer from the hydride derived from 7 to a ketone, in which the hydrogen bond from the N–H is required for ketone reduction. Complex 8, lacking this N–H bond, are inactive in ketone reduction. (B) Mechanism of hydrogen transfer from the hydride derived from 7 or 8 to an imine. The ‘open’ nature of the transition state, without an intramolecular hydrogen bond, allows both complexes to be effective in the reduction of imines. Complex 8 is, however, less active than 7 due to the additional steric hindrance imposed by the methyl group compared to a proton. | ||
In recent years, the fast growing field of catalysis using ruthenium complexes has generated exciting new leads for anticancer research, e.g. the Hoveyda–Grubbs catalyst was demonstrated by Ott et al. to exhibit anticancer activity in MCF-7 breast and HT29 colon cancer cell lines.79 We also have recently reported a series of ruthenium complexes structurally similar to Noyori catalyst 6 that show anticancer properties in A2780 ovarian carcinoma cells. They reduce NAD+ to NADH catalytically using formate as an hydride source,60 and their antiproliferative activity is markedly increased when cells are co-incubated with the complex and formate (Scheme 1, compound 5).27
The only previous investigation of the anticancer activity of tethered complexes used the amine-tethered RuIIarene [η6:η1-C6H5(CH2)nNH2)RuCl2] (n = 2 or 3). These complexes could form monofunctional adducts on DNA, but did not possess high anticancer activity in vitro. Here we explore the anticancer properties of catalytically active complexes 7/7a and 8/8a. The tethered arene structure found in complexes 7 and 8 restricts the movement of the N,N-chelated ligand TsDPEN within the coordination sphere of the stereogenic ruthenium centre. This greatly increases the conformational stability of the complexes and hence comparisons of the anticancer activity of the enantiomers may aid understanding of the role of chirality, if any, in the mode of action of this class of complexes.
Results
Complexes 7 and 8 (Scheme 2) are enantiomerically-pure and show different catalytic efficiency. Each of the four complexes illustrated in Scheme 2 exists predominantly in the diastereoisomeric form shown. To assign the chirality at each ruthenium centre, Cahn–Ingold–Prelog priority rules (CIP system) were used to define the priority sequence of ligands attached to the RuII centre: η6-C6 > Cl > CHN-Ts > CHNH. According to the sequence rule of the R/S system, the configurations of the RuII centre in these four chiral ruthenium arene complexes are: 7 = SRu, 7a = RRu, 8 = RRu, 8a = SRu. The chirality details for the two pairs of ruthenium diastereoisomers are: (SRu, RC, RC)-7, (RRu, SC, SC)-7a, (RRu, RC, RC)-8 and (SRu, SC, SC)-8a (Table 1A).| (B) | |||
|---|---|---|---|
| Complexb | GI50 (μM) | TGI (μM) | LC50 (μM) |
| a Colchicine and vinblastine IC50 values reported previously.93 b Complexes 7 and 7a did not show significant antiproliferative activity (i.e. GI50 > 10 μM) in the 60 cell lines tested by the NCI. Cisplatin data from NCI/DTP screening: March 2012, 48 h incubation. c The maximum concentration tested by the NCI. | |||
| 7 | >10c | NA | NA |
| 7a | >10c | NA | NA |
| 8 | 1.62 | 4.16 | 14.45 |
| 8a | 1.62 | 4.07 | 18.62 |
| Cisplatin | 1.49 | 9.33 | 44.0 |
Antiproliferative activity
The potential to inhibit the growth of cultured cancer cells provides a useful initial screening of anticancer activity. Table 1A shows the ability of complexes 7 and 8 to inhibit the proliferation of A2780 human ovarian carcinoma cells (GI50 values in μM). It is notable that the most efficient catalysts show the least anticancer activity. Complexes 7 and 7a were 5–10 fold less active than 8 and 8a, which were more active than cisplatin. Complexes containing the RR isomers of the TsDPEN-based chelated ligand showed better activity than complexes with the SS isomer.The four RuII complexes were further tested in the NCI-60 panel.96,97 Their potency was comparable to that measured in A2780 cells (Tables 1B and S1†). Enantiomers 8 and 8a showed similar potency with the same mean GI50 value of 1.62 μM. This activity was similar to that of cisplatin (mean GI50 value = 1.49 μM). However, 7 and 7a were much less active than 8, 8a and cisplatin with mean GI50 values >10 μM.
GI50 mean graphs
Selectivity in the NCI-60 screen can be displayed in mean graphs, which plot positive and negative bars for each cell line relative to a central line which represents the mean GI50 (Fig. 1). Negative values projecting to the right represent higher cellular sensitivities for 8 or 8a compared with the mean value, whereas positive values projecting to the left represent lower cell line sensitivities for 8 or 8a compared to the average. It can be seen in Fig. 1 that the selectivities of 8 and 8a for the different cell lines are similar. | ||
| Fig. 1 Overlay of mean graphs for antiproliferative activity (GI50 values) of ruthenium complexes 8 and 8a in the NCI-60 cell line screen. The positive and negative values are plotted along a vertical line that represents the mean response of all the cell lines in the panel to 8 and 8a. Bars to the right (log scale) indicate activity higher than the mean. | ||
Complex 8 showed a broad spectrum of activity, with GI50 values ranging from 0.13 to 11.7 μM, with the highest activity in CCRF-CEM (leukemia), K-562 (leukemia), NCI-H522 (non-small cell lung), SW-620 (colon), DU-145 (prostate) and MDA-MB-168 (breast) cell lines, with GI50 values in the nM–μM range. The total growth inhibition (TGI) values ranged from 1.3 to 33.9 μM; and 50% lethal concentration (LC50) values ranging from 3.7 to >100 μM. Complex 8a also showed a broad spectrum of activity, with GI50 values ranging from 0.26 to 8.3 μM, with the highest anticancer activity in K-562 (leukemia), NCI-H522 (non-small cell lung), HCT-116 (colon), U251 (central nervous system) and MDA-MB-168 (breast) cell lines, again with GI50 values in the nM–μM range. The TGI and LC50 values ranged from 1.6 to 21.6 μM and 4.8 μM to >100 μM, respectively. In comparison, the renal cell lines in general showed low sensitivity to both 8 and 8a. This is a common observation in screening as these cell lines are chemoresistant, likely owing to their high levels of multi-drug resistant proteins.80
COMPARE analysis
The patterns of sensitivity in the GI50, TGI and LC50 mean graphs for 8 and 8a, were compared to the patterns of sensitivity for compounds populating the NCI/NIH databases using the COMPARE algorithm.97 The top 10 positive correlations in both the Standard Agents and Synthetic Agents database were considered, with Pearson correlation coefficients (r) > 0.6. Tables S2 and S3† show the results of this analysis. The correlations are to an agent (mercaptoacetate) that can interact with (and in some cases cleave) DNA, to two agents that can interact with RNA machinery (rifamycin SV and caracemide), to one anti-angiogenic compound (withaferin A), and an agent which inhibits flavoproteinthioredoxin reductase (pleurotin). All other positive correlations were <0.6 and therefore not deemed significant. This analysis indicates that there are very few significant correlations with either database and therefore provides little insight into the mechanism of action (MoA). However, it does suggest that catalysts 8 and 8a have novel MoAs compared to the other compounds populating the NCI/NIH database.We assessed quantitatively the similarity in mean graphs of 8 and 8a (Table S4†), giving correlation values at 0.868 (GI50 mean graph), 0.945 (TGI mean graph) and 0.942 (LC50 mean graph). These highly significant correlations confirm the observations in Fig. 1 and suggest that the mirror-image complexes 8 and 8a have the same MoA. The lack of correlation of activity for either of these compounds to the square-planar PtII drug cisplatin confirms that even though they have similar potencies, their MoA is different, and is reflected in the A2780 and NCI-60 screening.
Interactions with nucleobases and aqueous stability
Since DNA is a potential target for transition metal anticancer drugs,81,82 reactions of complex 8a with nucleobases 9-ethylguanine (9-EtG) and 9-methyladenine (9-MeA) were investigated by 1H NMR spectroscopy. No reaction between these nucleobases and the Ru complex was observed after 24 h at 310 K (Fig. S1 and S2†). The NMR data showed that these DNA bases bind very weakly, and that the arene and N,N′-chelated ligands are strongly bound in aqueous solution. Substitution of the monodentate ligand would be expected to be facile since the 16-electron catalyst is known to be readily formed.78Interactions with coenzyme NADH
In recent years, a number of half-sandwich RuII, OsII arene and IrIII cyclopentadienyl anticancer complexes have been shown to oxidise reduced coenzyme NADH to NAD+ with formation of corresponding metal hydride complexes.27,83,84 Consequently, reactions of complex 8a with NADH were investigated. However, no reaction between NADH and the complex was observed after 3 h at 298 K (Fig. S3†). In contrast, formation of a ruthenium hydride derived from 8 can be observed when the complex is treated with formic acid.78However, the antiproliferative activity of complexes 7/8 was increased by 5–25% upon co-incubation with sodium formate (Fig. S4†). This improvement is quite dramatic for complexes 7 and 7a (25% reduced cell proliferation when co-incubated with 2 mM formate), but less significant for the less-efficient catalysts; 8 and 8a (5%).
Cell cycle arrest
Since complexes 7/8 did not interact with the cellular targets previously observed for related ruthenium complexes, we analysed their effect over the cell cycle of A2780 cancer cells by propidium iodide staining and flow cytometry. Treatment with 2 μM 7/8 led to different effects; 7 and 7a induced S and G2/M arrest, while 8 and 8a arrested the cell cycle of A2780 in G2/M (Fig. 2).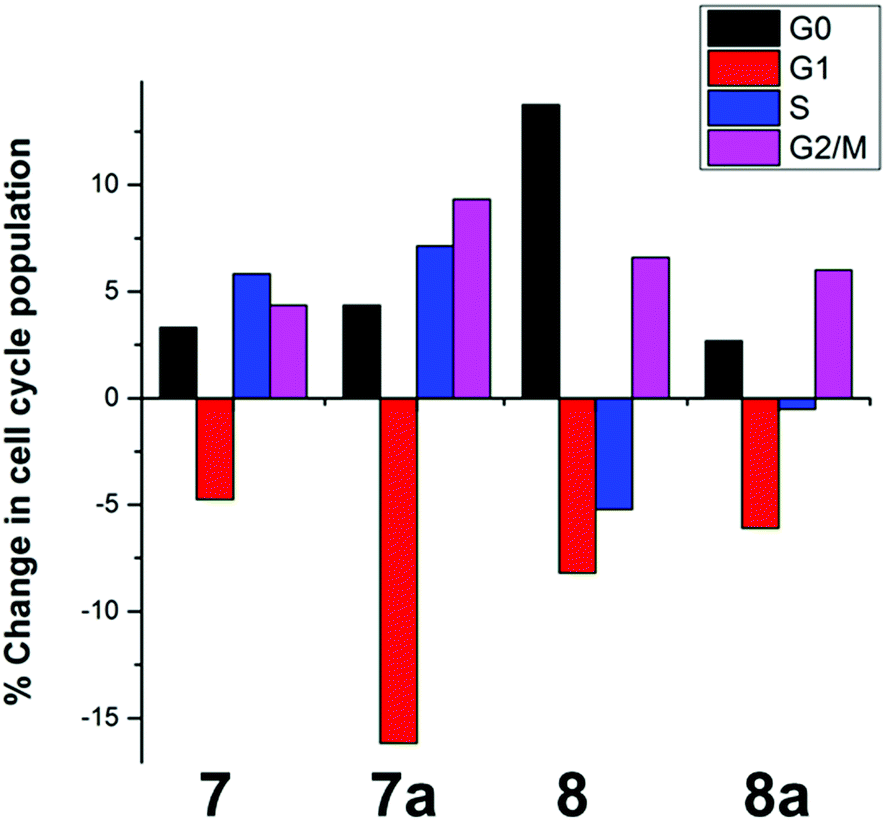 | ||
| Fig. 2 Changes in the cell cycle of A2780 human ovarian carcinoma cells after 24 h treatment with 2 μM of complexes 7, 7a, 8 and 8a. | ||
Cellular accumulation
Cellular uptake plays an important role in antiproliferative activity.85 A2780 cells were treated for 24 h with different concentrations (2 μM or GI50 concentration) of complexes 7/8, and the cell accumulation of Ru was measured using ICP-MS. Fig. 3 shows that 8 and 8a were accumulated to a much greater extent compared to 7 and 7a when cells were incubated with the same concentration of the complexes (2 μM, Fig. 3A). Furthermore, A2780 cells accumulated similar amounts of Ru when treated with concentrations of the complexes that induced the same cell growth inhibition (GI50 concentrations, Fig. 3B).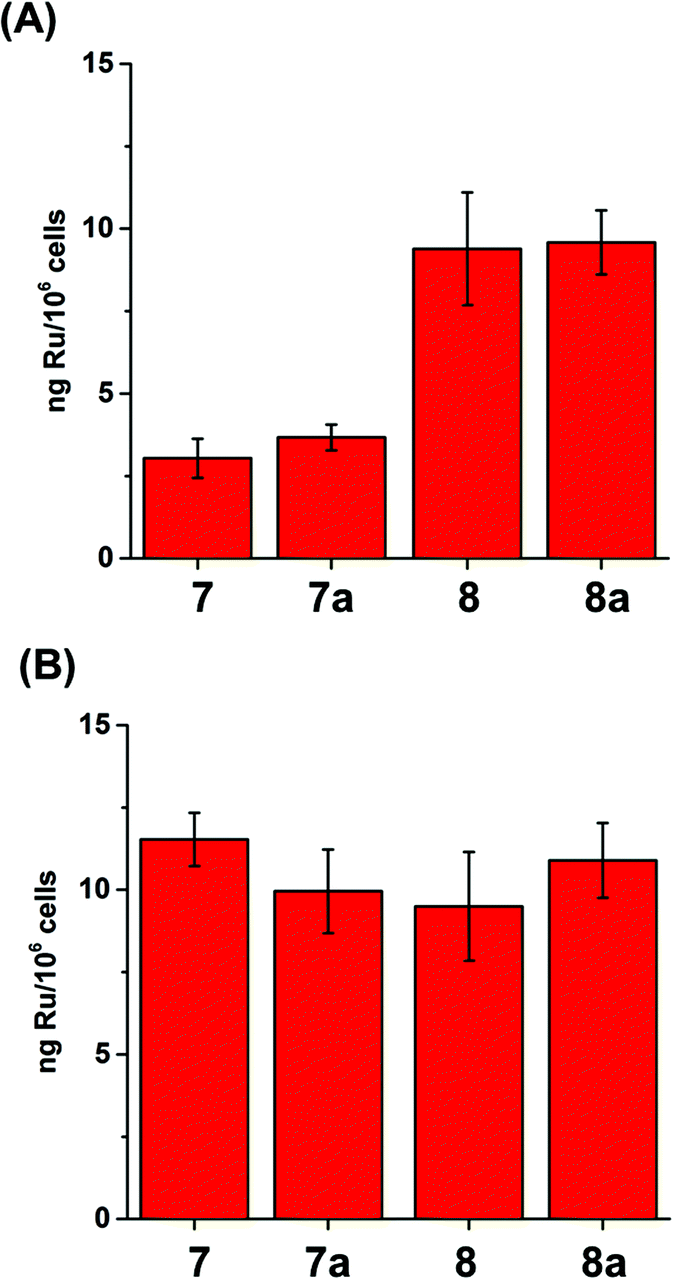 | ||
| Fig. 3 Accumulation of Ru in A2780 human ovarian carcinoma cells (expressed as ng Ru/106 cells) after 24 h treatment with (A) 2 μM and (B) GI50 concentrations of 7/7a or 8/8a. | ||
When the compartmentalisation of Ru within the cells was determined, we found that most of the Ru was in the cytosol, with less Ru located in the membranes (although significantly high amounts for 8 and 8a) and in the cytoskeleton, and little in the nucleus (Fig. 4 and S5†). This finding also supports the conclusion that the complexes in this family do not target nuclear DNA, in contrast to e.g. ethylenediamine ruthenium arene anticancer complexes.41,42 Similar amounts of Ru from 8 and 8a were found in the various fractions when A2780 cells were treated with GI50 (1.2 and 1.8 μM, respectively) or 2 μM concentrations (Fig. 4B), but significantly increased amounts of Ru from 7 and 7a when cells were treated with GI50 concentrations of 7 and 7a, which are 2.8× and 6.9×, respectively, higher than 2 μM (Fig. 4A).
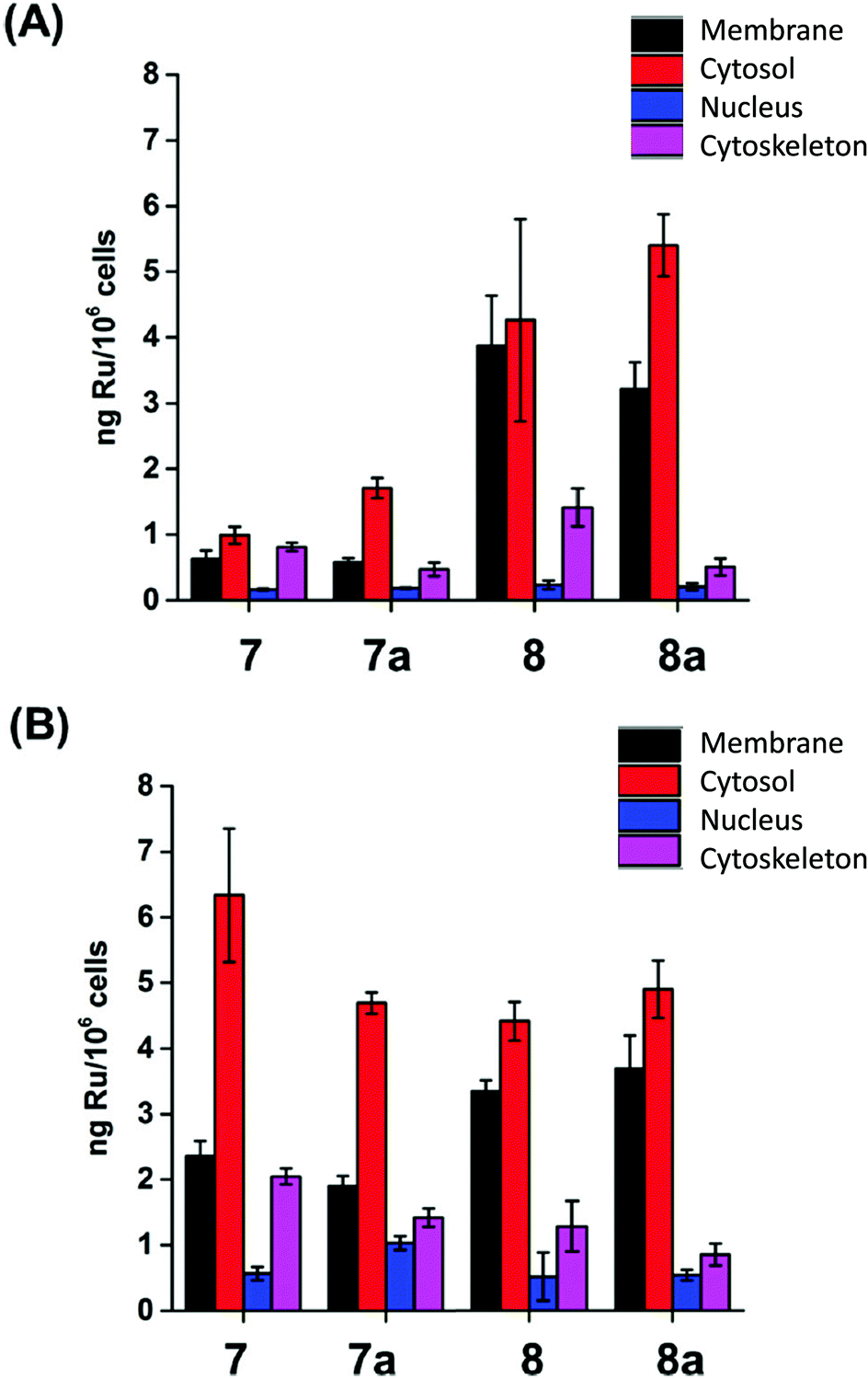 | ||
| Fig. 4 Ru content of the cellular fractions of A2780 ovarian carcinoma cells (expressed in ng Ru/106 cells) after 24 h treatment with (A) 2 μM and (B) GI50 concentrations of 7/8. | ||
Microtubule polymerisation inhibition
The data for cellular distribution show that cells treated with complexes 7/8 accumulate up to 25% of the drug in the cytoskeletal fraction (Fig. 4, S5 and S6†). This unusual pattern strongly indicates that complexes 7/8 may target cytoskeletal proteins including tubulin (protein component of microtubules), actin (component of microfilaments) and lamin (component of intermediate filaments). It is known that G2/M arrest can be induced by tubulin-targeting molecules; our flow cytometry analysis confirmed this suggestion (Fig. S7†).86,87 To test this hypothesis, we investigated the ability of complexes 7/8 to affect tubulin polymerisation using a fluorescence-based assay. Fig. 5 and Table 1 show that 7/8 inhibit tubulin polymerisation with potency following the trend 7 > 7a > 8 > 8a. The IC50 values (concentrations which cause 50% polymerisation inhibition) vary from 1.1 to 15.3 μM which is at the same order of magnitude as the GI50 values against cancer cell lines. More importantly, a correlation between the ability of the complexes to inhibit microtubule polymerisation, and the amount of complex found in the cytoskeleton of treated cells (at GI50 concentration) is observed (Fig. 6).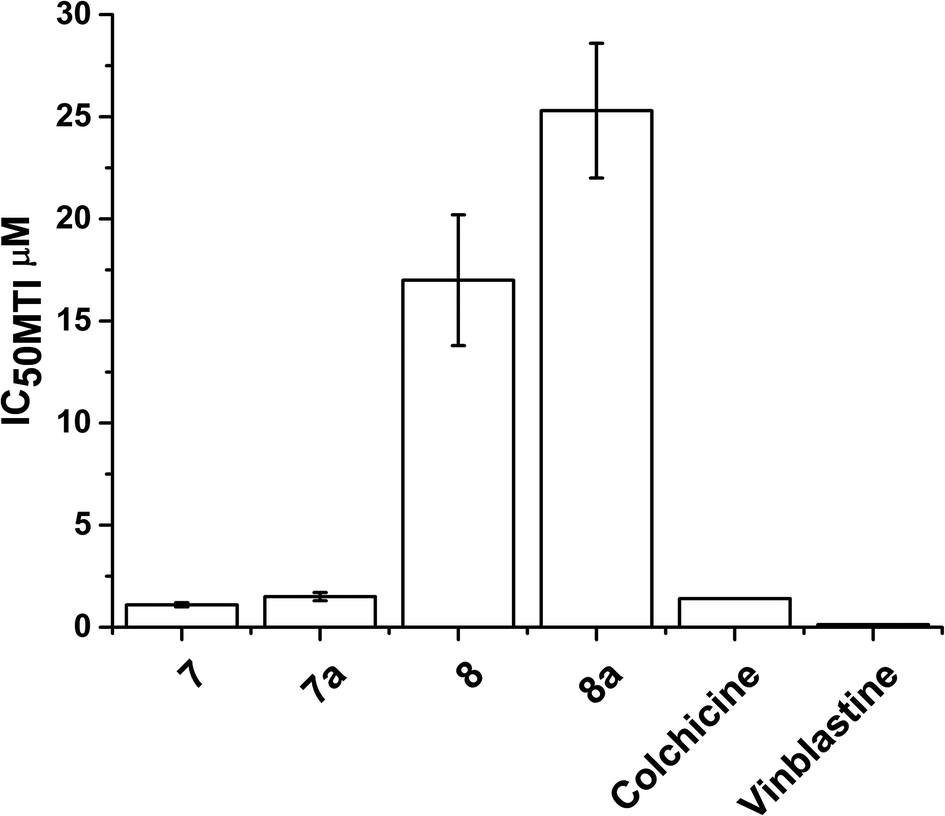 | ||
| Fig. 5 IC50 values (μM) for inhibition of microtubule polymerization inhibition (MTI) in vitro by complexes 7/8 compared to colchicine and vinblastine (values taken from ref. 93). | ||
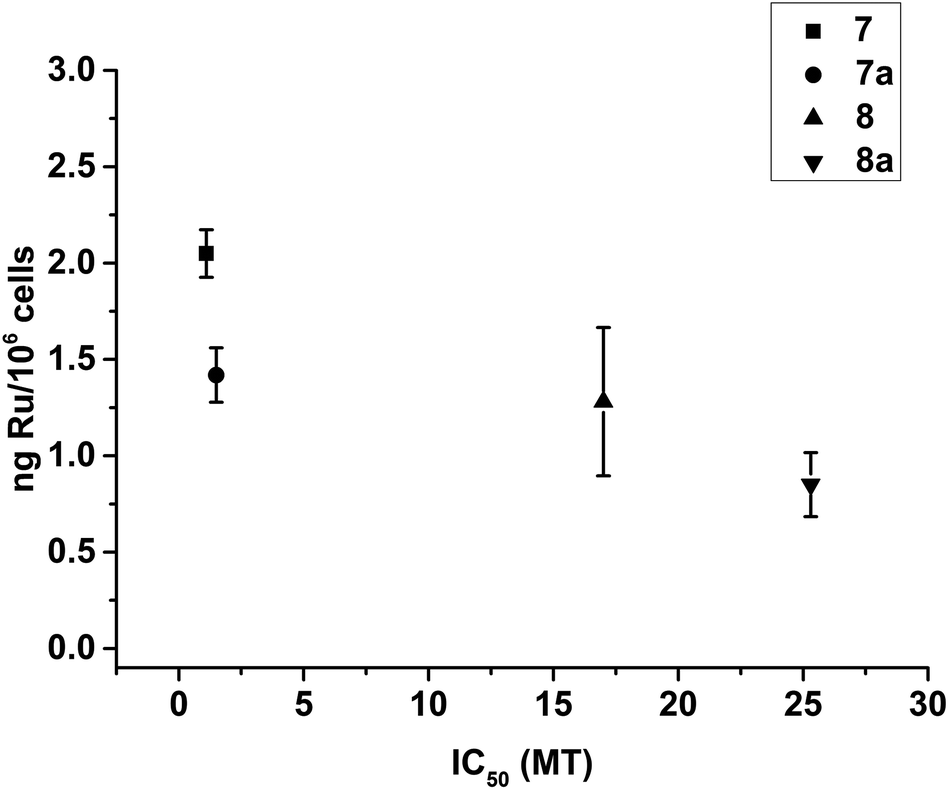 | ||
| Fig. 6 Plot showing the correlation between the ability of complexes 7–8 to inhibit the microtubule polymerisation in vitro (expressed as IC50/μM), and the amount of Ru found in the cytoskeleton of A2780 cells treated with GI50 concentrations of the complexes (expressed as ng Ru/106 cells). | ||
Discussion
Our experiments show that 7–8 inhibit the proliferation of A2780 ovarian carcinoma cells with different efficiency. The N-methyl complexes 8 (RRu, RC, RC) and 8a (SRu, SC, SC) are more active than cisplatin, whereas the N–H complexes 7 (SRu, RC, RC) and 7a (RRu, SC, SC) are less potent. The trend in the antiproliferative properties (8, 8a > 7 > 7a) is the inverse of that for catalytic activity. Remarkably, subtle structural differences between the four complexes result in dramatic differences in their anticancer activity.(i) The GI50 values in A2780 ovarian carcinoma cells of [Ru(η6-benzene-(CH2)3-TsDPEN-N-X)Cl] complexes (7/8) increase up to an order of magnitude (5–13 μM for 7/7a compared to 1–2 μM for 8/8a) when the N–H group located at the basic nitrogen atom in the (η6-benzene-(CH2)3-TsDPEN-N-X) chelating ligand (7/7a) is replaced by an N-methyl group (8/8a). A similar trend was also observed for the NCI-60 cell line panel; the mean GI50 values for 7 and 7a were >10 μM. This observation suggests that an NH group on the chelated ligand is not essential for antiproliferative activity, unlike the ethylenediamine series [Ru(η6-arene)(ethylenediamine)Cl]PF6 where it appears to play a role in H-bonding to the C6O of guanine when bound to DNA via N7.88
(ii) Although COMPARE analysis suggests that enantiomers probably have a similar mode of action, complexes containing RR-ligands (7 and 8) show higher activity than SS-isomers (7a and 8a), suggesting that ligand chirality influences their biological activity. Such effects might arise for example if (chiral) proteins are the targets. Additionally, the NCI-60 panel screening for 8 and 8a show that both enantiomers possess a differential selectivity towards individual cell lines (although they showed the same mean GI50 value).
This antiproliferative activity for 7–8 is not the result of the interaction of the complexes with NADH or nucleotides (as observed for structurally-related complexes).27,41,42 Perhaps it is not surprising that Ru centres which can form relatively stable 16-electron complexes (pseudo-5-coordinate) bind relatively weakly to 6th ligands such as DNA bases. Nevertheless, a decrease in the proliferation of A2780 cells was observed when 7 and 8 were co-incubated with a hydride source (sodium formate);60 suggesting that these compounds might be able to perform catalytic reactions inside cells. This increase in activity follows the trend previously observed for the catalytic properties of the complexes (7 ≫ 8), and is significant for 7 and 7a (25% decrease in cell growth), making these complexes promising hydrogen-activated prodrugs. However, 8 and 8a are still more efficient in inhibiting the proliferation of A2780 cells, even in presence of sodium formate.
The difference in activity of 7 and 8 can be partially explained by differences in the cellular accumulation of the complexes. More Ru is found in A2780 ovarian carcinoma cells treated with 2 μM of the more active 8 or 8a than in cells treated with the same concentration of the less active 7 or 7a. Additionally, similar quantities of Ru were found in A2780 cells treated with GI50 concentrations of 7/7a or 8/8a. The less efficient accumulation (perhaps uptake) of 7 and 7a by cells might be a key factor in lowering their antiproliferative activity.
The cellular targets for 7/8 are most likely to be found in the cytosol of treated cells, where the drugs mainly accumulate. However, up to 25% of the Ru taken up by cells is found in the cytoskeleton (Fig. 4, S5 and S6†). Based on the cellular distribution data and G2/M phase cell cycle arrest observed, we hypothesised that targets could also be located in the cytoskeleton of the treated cells.
Microtubules are cycloskeletal polymers that show dynamic behaviour, polymerising and depolymerising multiple times upon hydrolysis of GTP.89 They form a scaffold that is in constant reorganisation, and yet serves as a stable source of polarity information across the cell, separating the replicated chromosomes between the two daughter cells during cell division through the formation of the mitotic spindle.90 This makes them important targets for the treatment of cancer, as disruption of the dynamic nature of microtubules stops cell proliferation and leads to apoptosis. Drugs such as taxanes (e.g. taxol) which stabilise microtubules and vinca alkaloids (e.g. vinblastine) which inhibit the formation of microtubules, are already used clinically as anticancer chemotherapeutic agents.91 The TsDPEN ligand in complexes 7/8 shows structural similarities with the active conformation of a new series of light activated compounds capable of inhibiting the formation of microtubules by mimicking colchicine.92 Complexes 7 and 7a inhibit the polymerisation of microtubules in vitro (IC50 of 1.1 μM and 1.5 μM) as effectively as colchicine or vinblastine (clinically-used microtubule targeting antimitotic drugs; GI50 1.4 μM, and 0.13 μM respectively);93 whereas 8 and 8a are less potent (GI50 = 40.6 μM, 17 μM and 25.3 μM, respectively).
Furthermore, there is a correlation between the ability of the complexes to inhibit the polymerisation of microtubules in vitro and the quantity of Ru found in the cytoskeleton (Fig. 6). Our data indicate that 7/8 might be able to interact with microtubules in treated cells. This could lead to the observed G2/M phase cell cycle arrest, ultimately inhibiting proliferation of cells.
There is increasing interest in the activity of specific enantiomers of chiral tubulin-targeting agents. For example, natural colchicine has an aS,7S-absolute configuration, and is much more potent than its enantiomer.94 These data indicate the importance of the configuration of these agents.95 As found for the tethered Ru complexes studied in this work. Our results show that the chirality of the backbone or ruthenium centre individually is not critical for inhibiting tubulin polymerization. However, the combination of chiralities is important for maintaining the potency of these tethered ruthenium diastereoisomers. In addition, although 7 and 7a show stronger inhibition of tubulin polymerisation than 8 and 8a, the trend is different from that of the antiproliferation. This suggests that other cellular targets, perhaps located in the cytosol or membranes of cells, could have an important role in the biological activity of such compounds.
Conclusions
Complexes 7/7a and 8/8a are Noyori-like ruthenium arene transfer hydrogenation catalysts capable of the enantioselective reduction of ketones and imines through an ATH mechanism following the trend 7 ≫ 8 in catalytic efficiency. Our experiments show that their antiproliferative potency towards cancer cells follows the trend 8, 8a >7 >7a. Complex 7 carrying the RR isomer of the TsDPEN derivative exhibits higher activity than the SS derivative 7a. Unlike related ‘piano-stool’ ruthenium anticancer complexes,33,41,42 the antiproliferative potency of these Noyori-like ruthenium catalysts appears not to be related to their NADH catalytic activity or to DNA binding. The antiproliferative activity (GI50 values) of 7/7a and 8/8a appears to be related to their ability to accumulate in cancer cells, and is at least partially linked to their accumulation in the cytoskeleton, and inhibition of the polymerisation of microtubules.Methods
Materials
Complexes 7/8 were synthesised as previously described.75,92 The A2780 human ovarian carcinoma cell line was purchased from European Collection of Animal Cell Cultures (ECACC), Salisbury, UK), RPMI-1640, as well as, foetal bovine serum, L-glutamine, penicillin/streptomycin mixture, trypsin, trypsin/EDTA, phosphate buffered saline (PBS) were purchased from GE Healthcare. Propidium iodide (>94%) and RNAse A were obtained from Sigma Aldrich.Cell culture
A2780 human ovarian carcinoma cells were used between passages 5 and 18 and grown in Roswell Park Memorial Institute medium (RPMI-1640) supplemented with 10% of foetal calf serum, 1% of 2 mM glutamine and 1% penicillin/streptomycin. They were grown as adherent monolayers at 310 K in a 5% CO2 humidified atmosphere and passaged at approximately 70–80% confluence.In vitro growth inhibition assays
The antiproliferative activity of complexes 7–8 was determined in A2780 ovarian cancer cells. Briefly, 96-well plates were used to seed 5000 cells per well. The plates were left to pre-incubate with drug-free medium at 310 K for 48 h before adding different concentrations of the compounds to be tested. A drug exposure period of 24 h was allowed. After this, supernatants were removed by suction and each well was washed with PBS. A further 48 h was allowed for the cells to recover in drug-free medium at 310 K. The SRB assay was used to determine cell viability. GI50 values, as the concentration which causes 50% cell death, were determined as duplicates of triplicates in two independent sets of experiments and their standard deviations were calculated.Cell viability modulation by co-administration of sodium formate
Cell viability assays were carried out with complexes 7/8 in A2780 ovarian cancer cells, as described above with the following modifications: a fixed concentration of each Ru complex equal to 1/5 GI50 was used in co-administration with three different concentrations of sodium formate or sodium acetate (0.5, 1.0 and 2.0 mM). To prepare the stock solution of the drug, the complex was dissolved in 5% DMSO and, diluted in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 0.9% saline:cell culture medium. This stock was further diluted using RPMI-1640 until working concentrations were achieved. Separately, stock solutions of sodium formate were prepared in saline. The complex and formate were added to each well independently, but within 5 min of each other. The SRB assay was used to determine cell viability. Cell proliferation was studied as duplicates of triplicates in two independent sets of experiments and their standard deviations were calculated.
:1 mixture of 0.9% saline:cell culture medium. This stock was further diluted using RPMI-1640 until working concentrations were achieved. Separately, stock solutions of sodium formate were prepared in saline. The complex and formate were added to each well independently, but within 5 min of each other. The SRB assay was used to determine cell viability. Cell proliferation was studied as duplicates of triplicates in two independent sets of experiments and their standard deviations were calculated.
NCI-60 cell screening
The protocols used in the National Cancer Institute (NCI) screens have been described previously.96 Briefly, cells were treated with complex for 48 h at five concentrations ranging from 0.01 to 100 μM. Three endpoints were calculated: GI50 (the concentration to inhibit cell growth by 50%): TGI (the concentration to inhibit cell growth by 100%), LC50 (the concentration which kills 50% of the original cell count), MG-MID (full-panel mean-graph midpoint). The data reported for cisplatin were from the most up-to-date NCI/DTP screening conducted in March 2012.Mean graphs and COMPARE analysis
Mean graphs were constructed by plotting positive and negative values along a vertical line, representing the mean response over all cell lines in the panel (mean GI50). This is calculated in 3 steps: (1) the GI50 value for each cell line tested against each compound is converted to its corresponding log10 GI50 value; (2) log10GI50 values are averaged; (3) each log10 GI50 value is subtracted from the average to create a delta value. These positive and negative deltas are plotted along a vertical line which represents the mean response of all the 60 cell lines in the NCI panel. Projections to the right indicate cell lines with susceptibility that exceeds the mean, projections to the left indicate cell lines with lower susceptibility. The COMPARE analysis quantitatively compares the mean graph patterns of compounds against the NCI/DTP Standard Agents Database (a collection of 171 known anticancer compounds to provide preliminary indications on a possible mechanism of action) and the Synthetic Agents Database (>40000 pure natural and synthetic compounds).97 For this analysis, we assessed the top 10 returned hits in each database, for positive and negative correlations. High positive Pearson correlations (r ≥ 0.6) to agents in each database may indicate similar anti-cancer mechanism.
Interactions with nucleobases
The reaction of complex 8a (1.5 mM) with the nucleobases 9-ethylguanine (9-EtG) and 9-methyladenine (9-MeA) typically involved addition of a solution containing 1 mol equiv. of nucleobase in D2O to an equilibrium solution of complex 8a in 25% MeOD-d4/75% D2O (v/v). 1H NMR spectra of these solutions were recorded at 310 K after 10 min and 24 h.Interactions with NADH
The reaction of complex 8a (1.5 mM) with NADH typically involved addition of a solution containing 3 mol equiv. of NADH in H2O to an equilibrium solution of complex 8a in 25% MeOD-d4/75% H2O (v/v). 1H NMR spectra of these solutions were recorded at 298 K after 10 min and 3 h.Ruthenium accumulation in whole cancer cells
Cell accumulation studies for metal complexes 7 and 8 were conducted on A2780 ovarian cells. Briefly, 10 × 106 cells were seeded on 145 mm petri dishes. After 24 h of pre-incubation time in drug-free medium at 310 K, the complexes were added to give final concentrations equal to GI50 or 2 μM and a further 24 h of drug exposure was allowed. After this time, cells were treated with trypsin, counted, and cell pellets were collected. Each pellet was digested overnight in concentrated nitric acid (73%) at 353 K; the resulting solutions were diluted with double-distilled water to a final concentration of 5% HNO3 and the amount of Ru taken up by the cells was determined by ICP-MS (Agilent technologies 7500 series). Data acquisition was carried out in ICP-MS top B.03.05 and analysis on offline Data analysis B.03.05). These experiments did not include any cell recovery time in drug-free media; they were carried out in triplicate and the standard deviations were calculated.Ruthenium distribution in cancer cells
Cell pellets were obtained as described above, and were fractionated using the Fraction PREP kit from BioVision according to the supplier's instructions. Each sample was digested overnight in concentrated nitric acid (73%) and the amount of Ru taken up by the cells was determined by ICP-MS. Data acquisition was carried out in ICP-MS top B.03.05 and analysis on offline Data analysis B.03.05). These experiments were all carried out in triplicate and the standard deviations were calculated.Cell cycle analysis
A2780 ovarian cancer cells were seeded in a 6-well plate using 1.0 × 106 cells per well. They were pre-incubated in drug-free media at 310 K for 24 h, after which complexes were added at concentrations equal to 2 μM (or 100 nM for Colchicine and Taxol controls). After 24 h of drug exposure, supernatants were removed by suction and cells were washed with PBS. Finally, cells were harvested using trypsin. DNA staining was achieved after cold ethanol fixation by re-suspending the cell pellets in PBS containing propidium iodide (PI) and RNAse A. Cell pellets were re-suspended in PBS before being analysed by flow cytometry using the maximum excitation of PI-bound DNA at 536 nm, and its emission at 617 nm. Data were processed using Flowjo software. These experiments were carried out in triplicate, although only selected histograms are shown, full numerical data and statistical analysis can be found in the ESI.†Microtubule polymerisation assays
The effect of complexes 7 and 8 on tubulin polymerisation was determined using a fluorescence-based tubulin polymerisation assay (BK011P; Cytoskeleton, Inc.) according to the supplier's instructions. Measurements were performed in a plate reader GloMax®-Multi+ Detection System with Temperature Control (E9032; Promega) equipped with a fluorescence module (E8051; Promega) using the UV fluorescence optical kit (λexc 365 nm; λem 410–460 nm). IC50 values, as the concentration which causes 50% polymerisation inhibition, were determined as duplicates.Acknowledgements
We thank the ERC (grant no. 247450 BIOINCMED), EPSRC (grant no. EP/F034210/1) and Science City/EU ERDF/AWM (MaXis mass spectrometer and the X-ray diffractometer) for funding. Dr Michael Khan (Life Sciences) for provision of facilities for cell culture and the National Cancer Institute (NCI) for 60-cancer-cell-line screening. We also thank Warwick University for support of Rina Soni and Ying Fu through Warwick Postgraduate Research Studentships, and the BBSRC for support of Jessica Hearn (PhD studentship).References
- B. K. Keppler, M. Henn, U. M. Juhl, M. R. Berger, R. Niebl and F. E. Wagner, in Ruthenium and Other Non-Platinum Metal Complexes in Cancer Chemotherapy, Springer, Berlin, Heidelberg, 1989, vol. 10, pp. 41–69 Search PubMed.
- A. Bergamo, S. Zorzet, B. Gava, A. Sorc, E. Alessio, E. Iengo and G. Sava, Anticancer Drugs, 2000, 11, 665–672 CrossRef CAS PubMed.
- M. Gras, B. Therrien, G. Süss-Fink, A. Casini, F. Edafe and P. J. Dyson, J. Organomet. Chem., 2010, 695, 1119–1125 CrossRef CAS.
- Y. Geldmacher, M. Oleszak and W. S. Sheldrick, Inorg. Chim. Acta, 2012, 393, 84–102 CrossRef CAS.
- A. M. Krause-Heuer, P. Leverett, A. Bolhuis and J. R. Aldrich-Wright, Aust. J. Chem., 2012, 65, 860–873 CrossRef CAS.
- M. Carreira, R. Calvo-Sanjuán, M. Sanaú, I. Marzo and M. Contel, Organometallics, 2012, 31, 5772–5781 CrossRef CAS PubMed.
- A. Kastl, A. Wilbuer, A. L. Merkel, L. Feng, P. Di Fazio, M. Ocker and E. Meggers, Chem. Commun., 2012, 48, 1863–1865 RSC.
- J. M. Hearn, I. Romero-Canelón, B. Qamar, Z. Liu, I. Hands-Portman and P. J. Sadler, ACS Chem. Biol., 2013, 8, 1335–1343 CrossRef CAS PubMed.
- R. Cao, J. Jia, X. Ma, M. Zhou and H. Fei, J. Med. Chem., 2013, 56, 3636–3644 CrossRef CAS PubMed.
- X. Song, Y. Qian, R. Ben, X. Lu, H.-L. Zhu, H. Chao and J. Zhao, J. Med. Chem., 2013, 56, 6531–6535 CrossRef CAS PubMed.
- K. Suntharalingam, T. C. Johnstone, P. M. Bruno, W. Lin, M. T. Hemann and S. J. Lippard, J. Am. Chem. Soc., 2013, 135, 14060–14063 CrossRef CAS PubMed.
- J. Maksimoska, D. S. Williams, G. E. Atilla-Gokcumen, K. S. M. Smalley, P. J. Carroll, R. D. Webster, P. Filippakopoulos, S. Knapp, M. Herlyn and E. Meggers, Chem. – Eur. J., 2008, 14, 4816–4822 CrossRef PubMed.
- Y. Fu, A. Habtemariam, A. M. Pizarro, S. H. van Rijt, D. J. Healey, P. A. Cooper, S. D. Shnyder, G. J. Clarkson and P. J. Sadler, J. Med. Chem., 2010, 53, 8192–8196 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- J. Z. Zhang, N. S. Bryce, A. Lanzirotti, C. K. J. Chen, D. Paterson, M. D. de Jonge, D. L. Howard and T. W. Hambley, Metallomics, 2012, 4, 1209–1217 RSC.
- G. Y. Park, J. J. Wilson, Y. Song and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11987–11992 CrossRef CAS PubMed.
- P. J. Bednarski, K. Korpis, A. F. Westendorf, S. Perfahl and R. Grünert, Philos. Trans. R. Soc., A, 2013, 371, 20120118 CrossRef PubMed.
- L. Messori, A. Casini, C. Gabbiani, E. Michelucci, L. Cubo, C. Ríos-Luci, J. M. Padrón, C. Navarro-Ranninger and A. G. Quiroga, ACS Med. Chem. Lett., 2010, 1, 381–385 CrossRef CAS PubMed.
- G. Gasser and N. Metzler-Nolte, Curr. Opin. Chem. Biol., 2012, 16, 84–91 CrossRef CAS PubMed.
- C. G. Hartinger and P. J. Dyson, Chem. Soc. Rev., 2009, 38, 391–401 RSC.
- I. Bratsos, E. Mitri, F. Ravalico, E. Zangrando, T. Gianferrara, A. Bergamo and E. Alessio, Dalton Trans., 2012, 41, 7358–7371 RSC.
- G. S. Smith and B. Therrien, Dalton Trans., 2011, 40, 10793–10800 RSC.
- V. Brabec and O. Nováková, Drug Resist. Updates, 2006, 9, 111–122 CrossRef CAS PubMed.
- A. Bergamo and G. Sava, Dalton Trans., 2011, 40, 7817–7823 RSC.
- G. Suss-Fink, Dalton Trans., 2010, 39, 1673–1688 RSC.
- C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677–5685 CrossRef CAS.
- J. J. Soldevila-Barreda, I. Romero-Canelon, A. Habtemariam and P. J. Sadler, Nat. Commun., 2015, 6, 6582 CrossRef CAS PubMed.
- W. H. Ang, Z. Grote, R. Scopelliti, L. Juillerat-Jeanneret, K. Severin and P. J. Dyson, J. Organomet. Chem., 2009, 694, 968–972 CrossRef CAS.
- V. B. Arion, A. Dobrov, S. Goschl, M. A. Jakupec, B. K. Keppler and P. Rapta, Chem. Commun., 2012, 48, 8559–8561 RSC.
- W. Kandioller, C. G. Hartinger, A. A. Nazarov, C. Bartel, M. Skocic, M. A. Jakupec, V. B. Arion and B. K. Keppler, Chem. – Eur. J., 2009, 15, 12283–12291 CrossRef CAS PubMed.
- S. Das, S. Sinha, R. Britto, K. Somasundaram and A. G. Samuelson, J. Inorg. Biochem., 2010, 104, 93–104 CrossRef CAS PubMed.
- S. M. Meier, M. Novak, W. Kandioller, M. A. Jakupec, V. B. Arion, N. Metzler-Nolte, B. K. Keppler and C. G. Hartinger, Chem. – Eur. J., 2013, 19, 9297–9307 CrossRef CAS PubMed.
- F. Barragán, P. López-Senín, L. Salassa, S. Betanzos-Lara, A. Habtemariam, V. Moreno, P. J. Sadler and V. Marchán, J. Am. Chem. Soc., 2011, 133, 14098–14108 CrossRef PubMed.
- K. S. M. Smalley, R. Contractor, N. K. Haass, A. N. Kulp, G. E. Atilla-Gokcumen, D. S. Williams, H. Bregman, K. T. Flaherty, M. S. Soengas, E. Meggers and M. Herlyn, Cancer Res., 2007, 67, 209–217 CrossRef CAS PubMed.
- H. Chen, J. A. Parkinson, S. Parsons, R. A. Coxall, R. O. Gould and P. J. Sadler, J. Am. Chem. Soc., 2002, 124, 3064–3082 CrossRef CAS PubMed.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS PubMed.
- W. Han Ang and P. J. Dyson, Eur. J. Inorg. Chem., 2006, 2006, 4003–4018 CrossRef.
- W. F. Schmid, R. O. John, V. B. Arion, M. A. Jakupec and B. K. Keppler, Organometallics, 2007, 26, 6643–6652 CrossRef CAS.
- A. K. Renfrew, A. D. Phillips, E. Tapavicza, R. Scopelliti, U. Rothlisberger and P. J. Dyson, Organometallics, 2009, 28, 5061–5071 CrossRef CAS.
- Y. Lin, Y. Huang, W. Zheng, F. Wang, A. Habtemariam, Q. Luo, X. Li, K. Wu, P. J. Sadler and S. Xiong, J. Inorg. Biochem., 2013, 128, 77–84 CrossRef CAS PubMed.
- L. Oehninger, M. Stefanopoulou, H. Alborzinia, J. Schur, S. Ludewig, K. Namikawa, A. Munoz-Castro, R. W. Koster, K. Baumann, S. Wolfl, W. S. Sheldrick and I. Ott, Dalton Trans., 2013, 42, 1657–1666 RSC.
- A. A. Nazarov, D. Gardini, M. Baquie, L. Juillerat-Jeanneret, T. P. Serkova, E. P. Shevtsova, R. Scopelliti and P. J. Dyson, Dalton Trans., 2013, 42, 2347–2350 RSC.
- R. Mitra, S. Das, S. Shinde, S. Sinha, K. Somasundaram and A. G. Samuelson, Chem. – Eur. J., 2012, 18, 12278–12291 CrossRef CAS PubMed.
- W. H. Ang, E. Daldini, C. Scolaro, R. Scopelliti, L. Juillerat-Jeannerat and P. J. Dyson, Inorg. Chem., 2006, 45, 9006–9013 CrossRef CAS PubMed.
- W. Ginzinger, G. Mühlgassner, V. B. Arion, M. A. Jakupec, A. Roller, M. Galanski, M. Reithofer, W. Berger and B. K. Keppler, J. Med. Chem., 2012, 55, 3398–3413 CrossRef CAS PubMed.
- S. J. Lucas, R. M. Lord, R. L. Wilson, R. M. Phillips, V. Sridharan and P. C. McGowan, Dalton Trans., 2012, 41, 13800–13802 RSC.
- F. Schmitt, N. P. E. Barry, L. Juillerat-Jeanneret and B. Therrien, Bioorg. Med. Chem. Lett., 2012, 22, 178–180 CrossRef CAS PubMed.
- Q. Wu, C. Fan, T. Chen, C. Liu, W. Mei, S. Chen, B. Wang, Y. Chen and W. Zheng, Eur. J. Med. Chem., 2013, 63, 57–63 CrossRef CAS PubMed.
- A. Valente, M. H. Garcia, F. Marques, Y. Miao, C. Rousseau and P. Zinck, J. Inorg. Biochem., 2013, 127, 79–81 CrossRef CAS PubMed.
- I. Romero-Canelón, L. Salassa and P. J. Sadler, J. Med. Chem., 2013, 56, 1291–1300 CrossRef PubMed.
- L. He, S.-Y. Liao, C.-P. Tan, R.-R. Ye, Y.-W. Xu, M. Zhao, L.-N. Ji and Z.-W. Mao, Chem. – Eur. J., 2013, 19, 12152–12160 CrossRef CAS PubMed.
- N. P. E. Barry, O. Zava, J. Furrer, P. J. Dyson and B. Therrien, Dalton Trans., 2010, 39, 5272–5277 RSC.
- A. Dubey, J. W. Min, H. J. Koo, H. Kim, T. R. Cook, S. C. Kang, P. J. Stang and K.-W. Chi, Chem. – Eur. J., 2013, 19, 11622–11628 CrossRef CAS PubMed.
- A. Mishra, H. Jung, J. W. Park, H. K. Kim, H. Kim, P. J. Stang and K.-W. Chi, Organometallics, 2012, 31, 3519–3526 CrossRef CAS PubMed.
- N. Uematsu, A. Fujii, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 4916–4917 CrossRef CAS.
- T. Ohkuma, N. Utsumi, K. Tsutsumi, K. Murata, C. Sandoval and R. Noyori, J. Am. Chem. Soc., 2006, 128, 8724–8725 CrossRef CAS PubMed.
- S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 7562–7563 CrossRef CAS.
- R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS.
- Y. Yan, M. Melchart, A. Habtemariam, A. A. Peacock and P. Sadler, J. Biol. Inorg. Chem., 2006, 11, 483–488 CrossRef CAS PubMed.
- J. J. Soldevila-Barreda, P. C. A. Bruijnincx, A. Habtemariam, G. J. Clarkson, R. J. Deeth and P. J. Sadler, Organometallics, 2012, 31, 5958–5967 CrossRef CAS.
- M. Ito, T. Ootsuka, R. Watari, A. Shiibashi, A. Himizu and T. Ikariya, J. Am. Chem. Soc., 2011, 133, 4240–4242 CrossRef CAS PubMed.
- J. Hannedouche, G. J. Clarkson and M. Wills, J. Am. Chem. Soc., 2004, 126, 986–987 CrossRef CAS PubMed.
- K.-J. Haack, S. Hashiguchi, A. Fujii, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed. Engl., 1997, 36, 285–288 CrossRef CAS.
- H.-K. Liu, S. J. Berners-Price, F. Wang, J. A. Parkinson, J. Xu, J. Bella and P. J. Sadler, Angew. Chem., Int. Ed., 2006, 118, 8333–8336 CrossRef.
- F. Wang, A. Habtemariam, E. P. L. van der Geer, R. Fernández, M. Melchart, R. J. Deeth, R. Aird, S. Guichard, F. P. A. Fabbiani, P. Lozano-Casal, I. D. H. Oswald, D. I. Jodrell, S. Parsons and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18269–18274 CrossRef CAS PubMed.
- T. Ikariya, K. Murata and R. Noyori, Org. Biomol. Chem., 2006, 4, 393–406 CAS.
- S. Gladiali and E. Alberico, Chem. Soc. Rev., 2006, 35, 226–236 RSC.
- T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300–1308 CrossRef CAS PubMed.
- J. K. Barton and E. Lolis, J. Am. Chem. Soc., 1985, 107, 708–709 CrossRef CAS.
- C. Qian, J.-Q. Wang, C.-L. Song, L.-L. Wang, L.-N. Ji and H. Chao, Metallomics, 2013, 5, 844–854 RSC.
- D. Sun, Y. Liu, D. Liu, R. Zhang, X. Yang and J. Liu, Chem. – Eur. J., 2012, 18, 4285–4295 CrossRef CAS PubMed.
- B. Zhao, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2013, 52, 4744–4788 CrossRef CAS PubMed.
- J. E. Martins, G. J. Clarkson and M. Wills, Org. Lett., 2009, 11, 847–850 CrossRef CAS PubMed.
- D. A. Alonso, P. Brandt, S. J. M. Nordin and P. G. Andersson, J. Am. Chem. Soc., 1999, 121, 9580–9588 CrossRef CAS.
- M. Yamakawa, H. Ito and R. Noyori, J. Am. Chem. Soc., 2000, 122, 1466–1478 CrossRef CAS.
- L. Evanno, J. Ormala and P. M. Pihko, Chem. – Eur. J., 2009, 15, 12963–12967 CrossRef CAS PubMed.
- M. R. Ringenberg and T. R. Ward, Chem. Commun., 2011, 47, 8470–8476 RSC.
- R. Soni, F. K. Cheung, G. C. Clarkson, J. E. D. Martins, M. A. Graham and M. Wills, Org. Biomol. Chem., 2011, 9, 3290–3294 CAS.
- L. Oehninger, H. Alborzinia, S. Ludewig, K. Baumann, S. Wölfl and I. Ott, ChemMedChem, 2011, 6, 2142–2145 CrossRef CAS PubMed.
- D. Türk, M. D. Hall, B. F. Chu, J. A. Ludwig, H. M. Fales, M. M. Gottesman and G. Szakács, Cancer Res., 2009, 69, 8293–8301 CrossRef PubMed.
- J.-H. Wen, C.-Y. Li, Z.-R. Geng, X.-Y. Ma and Z.-L. Wang, Chem. Commun., 2011, 47, 11330–11332 RSC.
- J. Reedijk, Metallomics, 2012, 4, 628–632 RSC.
- S. Betanzos-Lara, Z. Liu, A. Habtemariam, A. M. Pizarro, B. Qamar and P. J. Sadler, Angew. Chem., Int. Ed., 2012, 51, 3897–3900 CrossRef CAS PubMed.
- Y. Fu, M. J. Romero, A. Habtemariam, M. E. Snowden, L. Song, G. J. Clarkson, B. Qamar, A. M. Pizarro, P. R. Unwin and P. J. Sadler, Chem. Sci., 2012, 3, 2485–2494 RSC.
- Y. Fu, A. Habtemariam, A. M. Basri, D. Braddick, G. J. Clarkson and P. J. Sadler, Dalton Trans., 2011, 40, 10553–10562 RSC.
- K. C. Morrison and P. J. Hergenrother, Anal. Biochem., 2012, 420, 26–32 CrossRef CAS PubMed.
- S. Tcherniuk, S. Deshayes, V. Sarli, G. Divita and A. Abrieu, Chem. Biol., 2011, 18, 631–641 CrossRef CAS PubMed.
- H. Chen, J. A. Parkinson, S. Parsons, R. A. Coxall, R. O. Gould and P. J. Sadler, J. Am. Chem. Soc., 2002, 124, 3064–3082 CrossRef CAS PubMed.
- T. Hawkins, M. Mirigian, M. S. Yasar and J. L. Ross, J. Biomech., 2010, 43, 23–30 CrossRef PubMed.
- E. Karsenti and I. Vernos, Science, 2001, 294, 543–547 CrossRef CAS PubMed.
- M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253–265 CrossRef CAS PubMed.
- M. Borowiak, W. Nahaboo, M. Reynders, K. Nekolla, P. Jalinot, J. Hasserodt, M. Rehberg, M. Delattre, S. Zahler, A. Vollmar, D. Trauner and O. Thorn-Seshold, Cell, 2015, 162, 403–411 CrossRef CAS PubMed.
- H. Prinz, P. Schmidt, K. J. Bohm, S. Baasner, K. Muller, E. Unger, M. Gerlach and E. G. Gunther, J. Med. Chem., 2009, 52, 1284–1294 CrossRef CAS PubMed.
- Q. Shi, P. VerdierPinard, A. Brossi, E. Hamel, A. T. McPhail and K. H. Lee, J. Med. Chem., 1997, 40, 961–966 CrossRef CAS PubMed.
- R. Bai, M. C. Roach, S. K. Jayaram, J. Barkoczy, G. R. Pettit, R. F. Luduena and E. Hamel, Biochem. Pharmacol., 1993, 45, 1503–1515 CrossRef CAS PubMed.
- S. L. Holbeck, J. M. Collins and J. H. Doroshow, Mol. Cancer Ther., 2010, 9, 1451–1460 CrossRef CAS PubMed.
- K. D. Paull, R. H. Shoemaker, L. Hodes, A. Monks, D. A. Scudiero, L. Rubinstein, J. Plowman and M. R. Boyd, J. Natl. Cancer Inst., 1989, 81, 1088–1092 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6dt01242f |
| ‡ These authors contributed equally to this work. |
| § Current address: Laboratory of Molecular Biology, Center for Cancer Research, NCI, Building 37, Room 5011, Bethesda, MD 20892-4264. |
| ¶ Current address: Qufu Normal University, China. |
| This journal is © The Royal Society of Chemistry 2016 |